Abstract
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal cancers, characterized by a dense and immunosuppressive tumor microenvironment. With the limited actions of drugs and conventional monovalent antibodies, the success of existing cancer therapies is restricted so far. Recently, bispecific antibodies (BsAbs) have emerged as a promising therapeutic platform, capable of overcoming the limitations of current PDAC treatments by engaging T cells and delivering drugs to multiple targets in a selective manner. Furthermore, the recruitment of additional payloads expands their therapeutic potential, offering more selective drug delivery and presenting new possibilities for treating PDAC. However, a limited number of relevant studies and a lack of comprehensive research have hindered trials for the development of BsAbs and bispecific antibody-drug conjugates (BsADCs) in PDAC therapeutics. This review aims to provide the characteristics of BsAbs and BsADCs and their recent applications in PDAC treatment. Additionally, frequent targets of PDAC treatments will be discussed to suggest how to design BsAbs and BsADCs for PDAC treatments.
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal malignancies, with a dismal prognosis despite decades of clinical efforts. Particularly, PDAC is the most common type of pancreatic cancer, accounting for more than 90% of cases [1,2]. According to recent Surveillance, Epidemiology, and End Results (SEER) data, the 5-year relative survival rate of PDAC remains approximately 12–13% in the United States, far below the average of other types of cancers [3]. Primarily, it might be attributed to the difficulty of early detection of PDAC, since early symptoms in PDAC are either absent or nonspecific. Therefore, over 80% of patients are diagnosed at an advanced or metastatic stage when surgical resection is no longer feasible. Additionally, the majority of patients are considered to be surgically unresectable due to complicated vascular involvement and the high dependency of PDAC in cases of locally advanced growth. Therefore, even among the 20% or fewer patients who undergo resection, recurrence rates remain high, and 5-year survival rarely exceeds 30% [4]. Thus, anti-cancer treatments are more important for PDAC than other types of cancer.
Despite substantial efforts in PDAC treatments, the clinical outcomes of conventional therapies for PDAC have been disappointing thus far due to its unique tumor microenvironment (TME). Molecularly, pancreatic cancer is characterized by nearly universal Kirsten Rat Sarcoma Viral Oncogene Homolog (KRAS) mutations (>90%), and high expressions of multidrug resistance genes. These features collectively limit the efficacy of conventional chemotherapies. For example, standard chemotherapy regimens such as FOLFIRINOX or gemcitabine with nab-paclitaxel extend the median survival to only 10–12 months in PDAC [5]. Furthermore, desmoplastic stroma and poorly organized tumor vasculature have also remained significant challenges in the treatment of PDAC. These abnormal anatomical features obstruct the diffusion of oxygen and drugs and impede the infiltration of immune cells, resulting in poor drug delivery efficiency and immune responses. Therefore, immune checkpoint inhibitors (anti-PD-1/PD-L1, anti-CTLA-4) have also shown minimal benefit in pancreatic cancer due to the highly immunosuppressive TME [6]. As a result, pancreatic cancer remains a “silent killer” with significant therapeutic resistance and unmet medical needs.
Bispecific antibodies (BsAbs) have recently been developed as next-generation agents for treating refractory cancers. BsAbs are engineered immunoglobulins that can bind to two different targets or epitopes simultaneously. By targeting two mechanisms concurrently, BsAbs can enhance tumor selectivity and reduce the likelihood of tumor escape or therapeutic resistance. For example, T-cell-engaging BsAbs not only can redirect cytotoxic lymphocytes to attack tumor cells but also reprogram the immunosuppressive TME by using the opposite side [7]. It is noteworthy that BsAbs offer diverse therapeutic approaches unattainable with conventional chemotherapeutic drugs or antibodies.
Similarly, Bispecific antibody–drug conjugates (BsADCs) represent an innovative extension of BsAbs, combining multiple binding affinities with potent cytotoxic payloads. The primary premise of BsADCs is that co-recognition of two antigens can enable more selective drug delivery and uptake in tumor cells, mitigating the off-tumor toxicities seen with traditional single-target ADCs [8]. In other words, a BsADC is designed to exert cytotoxic effects predominantly on cells co-expressing both target antigens, thereby sparing normal tissues that express only one [9]. Collectively, these advances underscore the potential of BsAbs and BsADCs to surmount clinical and molecular challenges of PDAC, and they provide a strong rationale for pursuing these approaches as next-generation strategies to improve outcomes in treating PDAC [10,11]. However, the applicability of BsADCs for the treatment of PDAC has not been the focus of much discussion to date, due to the limited literature so far. In this research, we thus comprehensively review the definition, structure, and mechanisms of action of BsAbs and BsADCs for treating PDAC. Furthermore, we evaluate their preclinical and clinical applications in PDAC and discuss their key achievements and remaining limitations. Finally, we discuss how these platforms can address unmet clinical needs in pancreatic cancer treatment and propose future directions [10,11].
2. BsAbs and Their Therapeutic Potential in PDAC
2.1. Classification of BsAbs
BsAbs are engineered proteins that recognize two distinct antigens or epitopes within a single molecule. This dual recognition enhances therapeutic specificity and efficacy in oncology and immune-mediated diseases. Unlike monoclonal antibodies (mAbs), which act on a single target, BsAbs provide dual targeting capabilities, enabling either complex mechanisms of action or direct immune cell redirection, thus positioning them as a promising next-generation therapeutic strategy [10]. The structure of BsAbs dictates their mechanisms of action. IgG-like BsAbs are suited for effector-mediated functions and dual-receptor blockade, whereas non-IgG-like BsAbs are optimized for T-cell-mediated immune activation. Based on their structural framework, BsAbs are broadly categorized into IgG-like and non-IgG-like types as shown in Figure 1. Due to their structural properties, there are significant differences between BsAbs in terms of immune activation, serum half-life and tissue penetration [12].
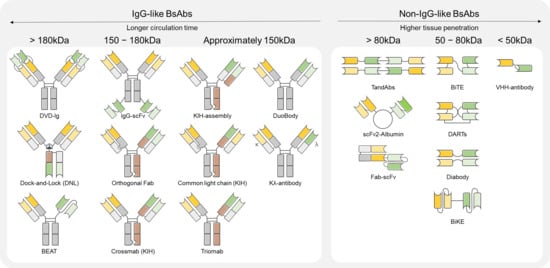
Figure 1.
Types of BsAbs by molecular weights.
2.1.1. IgG-like BsAbs
IgG-like BsAbs retain the conventional IgG backbone and incorporate the Fc domain, facilitating effector functions such as antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) through interaction with Fc receptors [12]. This architecture is advantageous for enhancing serum half-life and ensuring molecular stability as a therapeutic antibody. A representative platform within this category is the Knobs-into-Holes (KIH) technology, developed to prevent nonspecific heavy chain pairing by promoting selective heterodimerization of the Fc region. Amivantamab (EGFR×MET), a KIH-based BsAb, has shown efficacy in non-small-cell lung cancer (NSCLC) with EGFR mutations and MET amplification by inhibiting both pathways. It acts via multiple mechanisms, including Fc-mediated ADCC and receptor internalization [13]. Additionally, it has been reported to function via Fc-independent mechanisms, effectively inducing therapeutic responses even under ligand-dependent TME [14]. On the other hand, CrossMab technology resolves heavy/light chain mispairing by domain crossover to ensure stable bi-specificity [12]. Faricimab (VEGF-A×Ang-2) outperformed VEGF monotherapy by extending treatment intervals while maintaining vision [15,16]. Other IgG-like platforms include Duobody and Dual-Variable Domain Immunoglobulin (DVD-Ig), both of which are engineered to enhance specificity while maintaining the favorable pharmacological properties of classical IgG structures. Duobody antibodies are generated via controlled Fab-arm exchange by recombining half-molecules of two monospecific antibodies. This approach prevents chain mispairing while preserving Fc effector functions and pharmacokinetics. Several Duobody-based candidates are under clinical development for solid tumors and hematological malignancies. [10] Meanwhile, DVD-Ig antibodies consist of tandemly arranged variable domains (VH-VL) within each Fab arm, enabling simultaneous binding to two distinct epitopes. This bivalent configuration either enhances overall binding affinity or blocks two independent pathways synergistically. DVD-Ig molecules have shown promise in inflammatory and autoimmune diseases, and their potential is being explored in dual-targeted cancer immunotherapy [17]. Overall, IgG-like BsAbs have longer half-life that offers leverage effector functions. However, their large size over 150 kDa can hinder penetration into the dense PDAC tissues [18].
2.1.2. Non-IgG-like BsAbs
Non-IgG-like BsAbs lack the Fc domain and are generally constructed by linking two single-chain variable fragments (scFvs). This format offers advantages such as smaller molecular size, improved tissue penetration, and flexible spatial arrangement between target-binding domains [19]. A representative non-IgG-like platform is the bispecific T-cell engager (BiTE), composed of two scFvs targeting CD3 and a tumor antigen. It redirects T cells to tumor cells to form immune synapses and induce MHC-independent cytotoxicity. Blinatumomab (CD19×CD3), the first BiTE-based therapeutic approved for clinical use, has demonstrated efficacy in treating acute lymphoblastic leukemia (ALL) by redirecting T cells for tumor cell lysis [18]. Despite its potent T-cell activation and rapid onset of action, the lack of an Fc domain results in a short half-life, necessitating continuous infusion [20]. The Dual Affinity Re-Targeting (DART) platform represents an evolution of BiTEs with enhanced structural stability by cross-linking scFvs and stabilizing the configuration through disulfide bonds. This architecture improves binding affinity and selectivity while optimizing interchain alignment for efficient T-cell redirection. Flotetuzumab (CD123×CD3), a DART-based BsAb, targets CD123-positive tumor cells in relapsed or high-risk acute myeloid leukemia (AML) and has shown promising responses in early-phase clinical trials [18,21]. In addition, TandAb is a symmetric BsAbs in which two diabody structures are linked in tandem, resulting in four antigen-binding sites. This architecture enhances binding avidity and provides more stable immune cell redirection, while exhibiting a longer serum half-life compared to conventional BiTE molecules. TandAbs are utilized in tumor-cell-killing mechanisms by engaging either CD3 or NK cell receptors, and several candidates have already advanced into clinical trials [22,23]. Bi-nanobody is an ultra-small BsAbs format constructed by linking two or more camelid-derived single-domain antibodies (nanobodies). Due to its small molecular size, non-IgG-like BsAbs typically offers excellent tumor tissue penetration, low immunogenicity, and rapid tissue diffusion, making it suitable for diverse applications such as imaging diagnostics, drug delivery, and immune modulation. More recently, bispecific nanobody strategies that recognize specific antigen pairs within the TME have been explored to overcome immune suppression [24,25].
2.2. Functions of BsAbs
2.2.1. T-Cell Engagement
T cell engagement is the most representative function of BsAbs. By binding a T-cell (usually via CD3 on T cells) with one arm and a tumor-associated antigen with the other. This in-trans cell bridging recruits T cells to tumor cells, triggering T-cell-mediated killing of the tumor [18]. For example, blinatumomab (CD19×CD3), the first approved BiTE, is a tandem scFv fragment that can efficiently connect T cells to B-leukemia cells. It lacks an Fc (no effector function), but its small size enables agile T-cell redirection albeit with the need for continuous infusion [26]. Later, Newer T-cell engagers use IgG-like formats for better pharmacokinetics—e.g., epcoritamab (CD20×CD3) and glofitamab (CD20×CD3) are full-length IgG-like bi-specifics approved in 2022–2023. These IgG-like engagers often utilize an IgG4 or Fc-mutated backbone to minimize FcγR interactions, preventing unwanted killing of T cells (since an active IgG1 Fc could opsonize the T cell when the BsAb brings T cells and tumor together) [27]. For example, epcoritamab was engineered using Genmab’s DuoBody platform (IgG4-based), while glofitamab was engineered using Roche’s CrossMab technology. Both were created to form stable asymmetric IgG BsAbs that bridge CD3 on T cells to CD20 on B cells.
2.2.2. Dual Immune Checkpoint Inhibition
Dual immune checkpoint inhibitory BsAbs are engineered to block two inhibitory receptors or ligands in tandem, with the goal of overcoming checkpoint redundancy and adaptive resistance that limit single-agent PD-1/PD-L1 or CTLA-4 therapies. Conceptually, dual blockade can operate in-cis on the same effector cell or in parallel across complementary inhibitory axes in TME, thereby amplifying T-cell activation and anti-tumor effector functions beyond monotherapy or simple antibody combinations. Recently, Bielski et al. report that anti-VISTA/anti-PD-L1 BsAbs for mitigating acquired resistance [28]. They constructed anti-VISTA/anti-PD-L1 BsAbs were constructed in three formats, symmetric IgG-HC-scFv, asymmetric Fab-scFv-Fc, and a 2×scFv design, and benchmarked against each parental monoclonal and their combination across endometrial (RL95-2), pancreatic (PANC-1), and breast (BT-20) models. In the preclinical study, Fc-based BsAbs produced significantly higher tumor-cell lysis and greater secretion of pro-inflammatory mediators (e.g., Interferon-gamma (IFN-γ), Tumor necrosis factor-alpha (TNFα), Granzyme B) than either monotherapy or the antibody combination, with the symmetric IgG-HC-scFv format emerging as most promising [28]. Collectively, these results indicate that dual immune-checkpoint blockade integrated within a single bispecific scaffold can surpass the activity of separate agents while mechanistically aligning with the need to counter checkpoint redundancy in solid tumors such as pancreatic cancer.
2.2.3. Dual Tumor Antigen Targeting
BsAbs designed for dual tumor antigen targeting represent an innovative approach to improve therapeutic specificity while minimizing off-target toxicities. Unlike monospecific antibodies, these molecules are engineered to simultaneously bind two distinct antigens that are co-expressed on the same tumor cell. This strategy enables in-cis recognition, where binding requires the concurrent presence of both targets, thereby reducing nonspecific interactions with normal tissues [29,30]. In addition, dual antigen binding can induce avidity effects, whereby the combined affinity enhances tumor selectivity compared to single-target engagement [31]. Mechanistically, dual targeting BsAbs can exert in-cis antagonism or neutralization of parallel oncogenic pathways, thus reducing the likelihood of resistance arising from compensatory signaling [30,32]. This principle has been exemplified in clinical settings: amivantamab, which targets EGFR and MET, demonstrates the potential of dual receptor blockade in overcoming resistance mechanisms in lung cancer, while faricimab, targeting VEGF-A and Ang-2, illustrates the utility of dual ligand neutralization in modulating angiogenic pathways [33,34]. Collectively, these cases underscore the therapeutic value of dual tumor antigen targeting BsAbs as next-generation modalities in oncology and beyond.
2.2.4. Receptor Clustering
Receptor clustering represents a distinct mechanism of BsAbs, whereby a single molecule simultaneously binds two receptors and enforces their spatial proximity. This process functions as a synthetic agonist, promoting receptor cross-linking and subsequent signaling cascades [31]. Typically, clustering occurs in-cis, with both receptors located on the same cell surface, and can be exploited to selectively activate immune co-stimulatory receptors or to trigger apoptotic pathways in tumor cells. A major challenge of this approach is the risk of systemic activation, which may result in severe immune-related toxicities; thus, conditional activation is essential for clinical translation [31,35]. To address this limitation, BsAbs have been engineered as conditional agonists, inducing receptor clustering only under tumor-associated conditions. A prototypical example is acasunlimab (GEN1046/BNT311), an IgG-like DuoBody BsAbs that targets PD-L1 on tumor cells and 4-1BB on T cells. This design ensures that 4-1BB clustering and activation occur only in the presence of PD-L1, thereby coupling checkpoint blockade with localized T-cell co-stimulation in the TME [35]. Its Fc domain is further silenced to prevent non-specific cross-linking and Fc-mediated effector functions, enhancing safety. Similarly, tetravalent PD-L1×4-1BB constructs such as ATG-101 have demonstrated broad therapeutic windows in preclinical studies, eliciting strong anti-tumor immunity while avoiding systemic cytokine release and hepatotoxicity [36,37]. Additional strategies include CD28×tumor antigen BsAbs, which deliver co-stimulatory signals exclusively upon binding to tumor cells, thereby improving both selectivity and therapeutic safety [38]. Collectively, receptor clustering BsAbs illustrate how rational antibody engineering can integrate checkpoint inhibition and conditional co-stimulation into a single molecule. Among the various designs, Fc-silenced IgG-like formats remain the preferred architecture, balancing conditional agonist activity with extended serum half-life and favorable pharmacokinetic properties.
2.2.5. Cytokine or Cofactor Mimicking
Cytokine- or cofactor-mimicking BsAbs are designed to replicate the functional activity of natural cytokines or cofactors. These molecules achieve their effect by simultaneously binding two distinct targets—either proteins or receptor subunits—and thereby enforcing an interaction that would normally require a soluble mediator. Through this mechanism, BsAbs can activate downstream biochemical pathways that are otherwise impaired in disease states [10]. One clinically validated example is emicizumab (Hemlibra®), an IgG4 BsAb that simultaneously binds activated factor IX (FIXa) and factor X (FX). By mimicking the cofactor activity of factor VIII, emicizumab restores tenase complex formation in patients with hemophilia A, thereby normalizing thrombin generation and blood clotting [39,40]. Its Fc-silenced IgG4 backbone reduces unwanted immune effector activity, and its extended half-life supports convenient subcutaneous dosing. Importantly, emicizumab represents the first FDA-approved cofactor-mimetic BsAb, highlighting the therapeutic potential of this strategy [39]. Beyond coagulation, cytokine-mimetic BsAbs are being investigated in oncology. For example, IL-15 superagonist constructs have been developed to bridge IL-15 with IL-15Rβγ selectively within the TME, enhancing local T and NK cell activity while limiting systemic toxicity [41]. Other approaches include BsAbs engineered to induce receptor dimerization in the absence of a natural ligand, effectively substituting for cytokine-mediated signaling [10]. Taken together, cytokine- and cofactor-mimicking BsAbs demonstrate how rational antibody engineering can recreate essential biological functions without reliance on native cofactors. Owing to the need for long-term pharmacological activity, most of these molecules adopt IgG-like architectures, which provide stability, extended serum half-life, and suitability for chronic therapeutic use [10].
2.3. Examples of BsAbs in PDAC Treatments
Currently, a few BsAbs are in clinical trials as PDAC treatments. Table 1 summarizes these agents, their targets and mechanisms, and their current development status, along with key findings from studies to date.

Table 1.
Examples of BsAbs in PDAC treatment.
Based on recent clinical findings, BsAbs in PDAC have shown several notable achievements. As shown in Figure 2, four bispecific antibodies (BsAbs) have been developed to target multiple antigens, and two BsAbs have been developed to engage T cells. For example, zenocutuzumab achieved clinical success in a subset of patients harboring NRG1 fusions, representing the first FDA approval of a BsAb for pancreatic cancer [51]. This milestone demonstrates that dual-targeting antibodies can provide meaningful benefits in carefully defined patient populations. Furthermore, preclinical studies have validated T-cell redirection strategies, showing that BsAbs can recruit cytotoxic lymphocytes into TME and directly induce tumor cell killing [52]. These findings suggest that BsAbs are not only able to overcome immune evasion but may also expand into broader applications, including their potential role in companion diagnostics and advanced drug delivery systems. In addition, BsAbs are being actively explored in combination with conventional chemotherapies and immune checkpoint inhibitors, aiming to enhance therapeutic efficacy and overcome resistance to monotherapies [53].
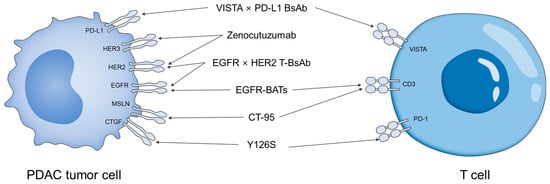
Figure 2.
Schematic illustration of BsAbs in PDAC and their target ligands.
Nevertheless, important limitations must also be acknowledged. The highly immunosuppressive TME, characterized by dense stroma, hypoperfusion, and suppressive immune cell populations, continues to hinder immune activation and drug penetration [52]. Antigen heterogeneity further complicates therapeutic applicability, as expression patterns vary across tissues and patients, limiting the breadth of treatment. Safety concerns remain critical, including cytokine release syndrome, immune-related adverse events, and the burden of intravenous administration [52]. Clinical data are still insufficient, with most findings derived from preclinical or early-phase studies, and overall survival outcomes remain uncertain [52]. From a developmental perspective, the structural complexity of BsAbs leads to high manufacturing costs, challenging scalability, and accessibility [53]. Finally, resistance mechanisms, such as downregulation of target antigen expression or secretion of immunosuppressive cytokines, may compromise the long-term durability of therapeutic responses [53].
Meanwhile, neither Abs nor ADCs have yet received clinical approval for PDAC treatment (Table 2) [54]. It is noteworthy that Abs and ADCs are being developed to target PDAC-specific TAAs, such as CLDN18.2, MUC1, uPAR, EphA2, CEACAM5, Trop2. Currently, the majority of BsAbs are used to treat PDAC target common antigens found in cancer, such as HERs, EGFR, and PD-L1, rather than being specific to PDAC [30,55]. This is because common cancer antigens are targets for which the clinical safety and efficacy of Abs or ADCs has already been extensively validated [56]. These antigens have the advantage of a relatively predictable toxicity profile, enabling a rapid transition from preclinical to clinical trials. Furthermore, targets that are widely expressed across multiple solid tumours (e.g., breast, lung, colorectal, and pancreatic cancers) are preferred due to their broad applicability and significant commercial and clinical potential [30,56]. This is because the inherently complex structure of BsAbs, compared to standard antibodies, results in higher development costs. Furthermore, the target for BsAbs must be selected with due consideration of their unique mechanism of action [57].
Table 2.
Abs and ADCs in clinical trials for PDAC.
Table 2.
Abs and ADCs in clinical trials for PDAC.
| Types | Names | Targets | Disease | Company | Development Stage | NCT. | Ref. |
|---|---|---|---|---|---|---|---|
| Abs | HuMab-5B1 (MVT-5873) | CA19-9 | Pancreatic ductal adenocarcinoma, biliary cancers | Sorrento/MabVax Therapeutics | Phase 1 | NCT02672917 | [58,59] |
| ensituximab (NPC-1C/NEO-102) | MUC5AC-related tumor antigen | Refractory colorectal and pancreatic cancer | Precision Biologics/Neogenix | Phase 1/2 | NCT01834235 | [60,60] | |
| ADCs | IBI343 | CLDN18.2 | Pancreatic ductal adenocarcinoma (PDAC), biliary tract cancer (BTC) | Innovent Biologics | Phase 1 | NCT05458219 | [61,62] |
| EBC-129 | CEACAM5/CEACAM6 | Pancreatic ductal adenocarcinoma (PDAC) | Experimental Drug Development Centre | Phase 1 | NCT05701527 | [63,64] | |
| OMTX705 | FAP | Advanced pancreatic adenocarcinoma | Phase 1b | NCT05547321 | [65,66] | ||
| uPAR-ADC | uPAR | Pancreatic ductal adenocarcinoma (PDAC) | Oncomatryx/TFS HealthScience | Preclinical | no NCT | [67] | |
| GPC-1 ADC | GPC-1 | GPC1-positive pancreatic cancer | University of Copenhagen/Genmab | Preclinical | no NCT | [68] | |
| TR1801-ADC | c-MET | KRAS-mutated PDAC/resistant tumors | Kyowa Kirin/Osaka University | Preclinical | no NCT | [69] |
3. Bispecific ADCs and Their Therapeutic Potential in PDAC
3.1. Definition of BsADCs
BsADCs are structures comprising an antibody, a linker and a payload, which integrates the capacity of antibody–drug conjugates (ADCs) to deliver cytotoxic drugs with the dual-targeting capability of BsAbs. With the ability of BsAbs, BsADCs can recognize two antigens or epitopes simultaneously, enhancing tumor specificity and increasing intracellular uptake efficiency. This dual-targeting mechanism enables precise recognition of malignant cells while reducing nonspecific binding to normal tissues, thereby minimizing off-target toxicity [11,70]. Structurally, BsADCs have the potential to simultaneously inhibit multiple cancer-related signaling pathways by simultaneously targeting cell surface ligands and intracellular metabolic pathways. Therefore, BsADCs have emerged as a next-generation therapeutic platform by mitigating tumor heterogeneity and drug resistance, which have challenged conventional chemotherapies and mAb therapies [70,71].
3.1.1. Linkers of BsADCs
In BsADCs, the linkers serve as a structural cornerstone that connects the antibody to the cytotoxic payload, critically influencing pharmacological behavior, in vivo stability, and overall therapeutic efficacy [11,71]. Linkers are broadly classified into cleavable and non-cleavable types, and this classification fundamentally influences drug release mechanisms and tissue specificity [11].
Cleavable linkers are designed to selectively dissociate in response to the unique biochemical environment within tumor cells. Prominent triggers include acidic pH, high concentrations of glutathione (GSH), and lysosomal enzymes such as cathepsin B, which are typically more pronounced in tumor cells than in normal tissues. Leveraging these conditions, cleavable linkers enable payload release predominantly within tumor cells, thereby minimizing off-target toxicity while maximizing therapeutic specificity and efficacy [70,72].
Non-cleavable linkers possess high chemical stability, resisting degradation under both extracellular and intracellular physicochemical conditions. In BsADCs containing non-cleavable linkers, the conjugate is internalized into the cell, where lysosomal degradation of the antibody releases the payload. At this stage, the payload remains covalently bound to amino acid residues of the antibody, typically via thioether bonds [11]. This structural feature significantly enhances systemic stability and effectively prevents premature drug leakage. However, since the released payload exists in a chemically modified form, its intracellular cytotoxic activity may sometimes be reduced [11.
Therefore, linkers selection in BsADCs design requires comprehensive consideration of TME, the internalization capacity of the target antigen, the structural sensitivity of the payload, and its pharmacodynamic properties. These factors collectively act as pivotal variables that determine the balance between efficacy and stability in the therapeutic system [11,72].
3.1.2. Payload of BsADCs
The payload of BsADCs must exhibit sufficient potency to induce apoptosis within target cells while also maintaining chemical stability in conjugation with the antibody and preserving both activity and structural integrity in the linker-bound state [73,74]. The most widely employed payload classes are microtubule inhibitors (monomethyl auristatin E, MMAE; monomethyl auristatin F, MMAF), DNA-damaging agents (calicheamicin; pyrrolobenzodiazepine, PBD), and topoisomerase I inhibitors (SN-38; deruxtecan, DXd). The features of each payload class are shown in Table 3.
Table 3.
Payload candidates of BsADCs for PDAC treatments.
These agents exert strong cytotoxic effects even at extremely low concentrations, and notably, topoisomerase I inhibitors have recently emerged as pivotal payloads in the development of diverse BsADCs [11,92]. In clinical applications, trastuzumab deruxtecan (DXd) and sacituzumab govitecan (SN-38) have demonstrated improvements in progression-free and overall survival in HER2-low breast cancer and metastatic triple-negative breast cancer, respectively, thereby validating the clinical utility of the topoisomerase I inhibitor class [93,94]. Payload selection should not be based solely on cytotoxic potency but must also incorporate considerations of antibody internalization capacity, linker release mechanisms, and physicochemical properties, alongside the optimization of pharmacokinetics and pharmacodynamics (PK/PD) [73,74].
3.2. Rationales of BsADCs in PDAC Treatments
BsADCs are composed of an antibody scaffold bearing two antigen-binding sites conjugated via a linker to a cytotoxic payload, thereby combining the targeting precision of antibodies (Abs, BsAbs) with the drug delivery capability of ADCs to enhance therapeutic efficacy [74,95]. Additionally, BsADCs offer significant advantages, as a therapeutic strategy to block complex oncogenic signaling pathways. While a single antibody can inhibit only one pathway, BsADCs are capable of simultaneously suppressing parallel or compensatory pathways, thereby enhancing both the intensity and durability of antitumor effects. This dual-pathway inhibition provides a critical strategic benefit for tumor therapies that rely on multi-pathway targeting [96].
Compared to Abs, BsADCs are designed to recognize combinations of antigens that are co-expressed on the surface of tumor cells. It can remarkably reduce the likelihood of nonspecific binding to normal cells while markedly improving tumor specificity [96,97]. In particular, by simultaneously attacking cell populations expressing different antigens, it can effectively reduce treatment evasion caused by antigen heterogeneity and antigen loss, which are commonly observed in PDAC. As a result, BsADCs can minimize off-target toxicity and enable more precise tumor targeting [11]. Furthermore, in cases where antigen expression is downregulated or lost due to mutations, conventional antibody-based therapeutics often fail to sustain drug activity. By contrast, BsAbs can effectively control compensatory signaling activation that is difficult to inhibit with single antibodies by simultaneously blocking different signaling pathways (e.g., EGFR–HER2, MET–HER3) [54]. In PDAC, downstream signaling pathways like KRAS mutations remain active after antibody blockade, readily leading to resistance. However, the dual-target approach simultaneously suppresses parallel upstream pathways, slowing the development of resistance and enhancing the durability of therapeutic response [98]. This dual-target format also confers advantages in overcoming therapeutic resistance [65].
Meanwhile, BsADCs possess a distinct cytotoxic mechanism by releasing their payloads intracellularly through receptor-mediated internalization and lysosomal linker cleavage. For cytotoxic payloads to exert their therapeutic effects, efficient and stable intracellular uptake is essential, and the structural design of BsADCs effectively enable this process. Dual-targeting further enhances this function by pairing antigens with slow or negligible internalization capacity to those with rapid internalization, thereby promoting uptake and trafficking of the entire complex. Consequently, BsADCs can expand their therapeutic reach to target populations that are largely inaccessible to conventional ADCs or Abs [74,95,99].
Additionally, BsADCs can target the heterogenous TME that is intractable for conventional ADCs. Intertumoral heterogeneity in antigen expression is one of the key factors limiting the therapeutic efficacy of single-antibody-based treatments. By simultaneously recognizing distinct antigens, BsADCs enable comprehensive targeting of diverse tumor cell subtypes, thereby contributing to consistent drug efficacy even within complex TME [30]. When antigen heterogeneity restricts the effects of Abs or BsAbs, BsADCs can broaden coverage across multiple subtypes. When paired with payload–linker combinations capable of exerting bystander effects, BsADCs can indirectly induce cytotoxicity in antigen-negative cells [75,100]. Moreover, for targets that are poorly internalized, BsADCs can incorporate an internalizing antigen as a co-target, thereby securing alternative pathways for drug uptake. This strategy is regarded as a valid approach to expanding therapeutic options for low-activity targets, which are often unaddressed by conventional Abs or BsAbs [101].
Furthermore, from a pharmacokinetic perspective, bispecific antibodies (BsAbs) can enhance tissue penetration and speed of action by employing small formats, such as BiTE and DART. Certain structures can also optimise drug distribution and accumulation within the body by modulating half-life [31,73]. In PDAC, which has a hypovascular structure, such modifications to half-life and binding affinity can improve drug accessibility within the tumour [102]. Compared to monoclonal antibodies or ADCs, which generally exhibit consistent distribution patterns, BsAbs/BsADCs have the potential advantage of leveraging structural diversity to finely tune tissue penetration, binding strength and range of action [73].
In summary, BsAbs and BsADCs are being evaluated as flexible design platforms that can simultaneously overcome complex antigen expression heterogeneity and resistance mechanisms in PDAC, thus surpassing single-antibody-based therapies. The dual-target strategy, recognizing two targets in parallel or complementarily, is gaining attention as a key approach to overcoming the limitations of existing therapies, such as antigen loss, resistance and toxicity, and maximising therapeutic efficacy in PDAC.
3.3. Recent Progress of BsADCs in Anticancer Therapies
To date, no BsADCs for the treatment of PDAC are currently in clinical trials. Meanwhile, several BsADCs have been used in anti-cancer therapies, as shown in Table 4. Notably, a few BsADCs have reached a stage of clinical advancement, such as AZD9592 (EGFR×c-MET), M1231 (MUC1×EGFR), and BL-B01D1 (EGFR×HER3) [103,104,105]. AZD9592 (tilatamig samrotecan) is an IgG1 BsAbs, targeting EGFR and c-MET, conjugated through a cleavable peptide linker to a CPT (camptothecin)-class topoisomerase I inhibitor (AZ14170132, samrotecan). Preclinical studies showed that AZD9592 correlated with target expression and induced DNA double-strand break markers such as γH2AX in EGFR–MET co-expressing solid tumors [106,107,108]. Currently, a multinational first-in-human (FIH) phase I study (EGRET, NCT05647122) is evaluating both monotherapy and combinations with dose-escalation and expansion cohorts. According to recent studies, the clinical practice remains in the stage of assessing safety, pharmacokinetics/pharmacodynamics (PK/PD), and preliminary antitumor activity, with efficacy outcomes such as objective response rate (ORR) not having been reported so far [103,107].
M1231 consists of an MUC1×EGFR BsAb conjugated via a Val-Cit-PABA cathepsin-cleavable linker (drug–antibody ratio [109] ≈ 4) to SC209, a hemiasterlin-derived microtubule inhibitor. In preclinical patient-derived xenograft (PDX) models of non-small-cell lung cancer (NSCLC) and esophageal squamous cell carcinoma (ESCC), M1231 exhibited potent antitumor activity including complete remission (CR). Mechanistically, M1231 showed enhanced co-internalization and lysosomal trafficking compared to monospecific bivalent antibodies [110]. A phase I clinical trial (NCT04695847) is currently in the dose-escalation and expansion phase, focusing mainly on NSCLC and ESCC. At this stage, the available data emphasize tolerability, recommended dose exploration (RDE/MTD), and preliminary efficacy signals, however, definitive clinical outcomes have not yet been established [104].
Among them, BL-B01D1 represents the most advanced clinical program. This EGFR×HER3 BsAb is conjugated at a high drug–antibody ratio (~8) via a cathepsin B–cleavable linker to Ed-04, a topoisomerase I inhibitor. In a phase I (a/b) analysis, an ORR of 34% (60/174; 95% CI 27–42) was reported, thereby establishing a recommended phase II dose (RP2D) of 2.5 mg/kg administered on days 1 and 8 of a 3-week cycle. In terms of safety, treatment-related grade ≥3 adverse events were observed in 71% of patients including neutropenia (47%), anemia (39%), leukopenia (39%), and thrombocytopenia (32%). In addition, treatment-related mortality occurred in 2% of patients, and a single case of interstitial lung disease (ILD) was reported. [105]. Subsequently, the platform has been extended to additional indications, with efficacy signals observed in an ESCC phase Ib cohort. Furthermore, multiple global expansion studies are being conducted, including combination regimens in renal cell carcinoma with axitinib×pembrolizumab (NCT06962787) [9,111,112].
Taken together, these three agents represent the expanding clinical scope of BsADCs utilizing topoisomerase I inhibitor- and microtubule inhibitor-based payloads. Notably, BL-B01D1 is currently the most advanced program, with validated dosing, scheduling, and objective response data [105].
Table 4.
BsADCs in clinical trials for anticancer therapies.
Table 4.
BsADCs in clinical trials for anticancer therapies.
| Development Stage | BsADCs (Targets) | Payload | Disease | Company | NCT. | Ref. |
|---|---|---|---|---|---|---|
| Preclinical stage | BVX001 (CD7×CD33) | Monomethyl auristatin F (MMAF) | Acute Myeloid Leukemia (CD7+/CD33+) | BiVictriX Therapeutics | × | [113,114] |
| VBC103 (Trop2×Nectin4) | Topoisomerase I inhibitor | Urothelial Carcinoma (metastatic), Triple-Negative Breast Cancer (+ Othesrs) | VelaVigo, Avenzo Therapeutics | × | [115] | |
| Phase1 | AZD9592 (EGFR×c-MET) | Topoisomerase I inhibitor (“samrotocan”) | Advanced solid tumors (EGFR×c-MET co-expressing; NSCLC focus) | AstraZeneca | NCT05647122 | [103,116] |
| ZW-49 (HER2 (ECD2×ECD4)) | Auristatin (microtubule inhibitor) | HER2-expressing solid tumors (breast, gastric, etc.) | Zymeworks | NCT03821233 | [117,118] | |
| JSKN0016 (HER3×TROP2) | Topoisomerase I inhibitor | Advanced solid tumors (basket, incl. lung, breast) | Alphamab Oncology | NCT06868732 | [119,120] | |
| M1231 (MUC1×EGFR) | SC209 (microtubule disruptor) | Advanced solid tumors (dose escalation); NSCLC & ESCC (expansion cohorts) | EMD Serono, Sutro Biopharma | NCT04695847 | [104,110] | |
| DM001 (EGFR×TROP2) | Monomethyl auristatin E (MMAE) | Advanced solid tumors (breast, EGFR-mut/wt NSCLC, gastric, esophageal, colorectal) | XADCera | NCT06475937 | [121,122] | |
| DM005 (EGFR×c-MET) | Topoisomerase I inhibitor | Advanced solid tumors (NSCLC, H&N, GI, etc.) | Doma Biopharm | NCT06515990 | [123,124,125] | |
| BL-B01D1 (EGFR×HER3) | Ed-04 (topoisomerase I inhibitor) | Multiple solid tumors (NSCLC, breast, etc.) | Sichuan Baili, SystImmune | NCT05983432 | [9,105,126] | |
| BL-B01D1 (EGFR×HER3) | Ed-04 (topoisomerase I inhibitor) | Advanced solid tumors (Phase 1a dose escalation); Expansion in ESCC and other GI cancers | Sichuan Baili, SystImmune | NCT05262491 | [9,105,127] | |
| BL-B16D1 (EGFR×HER3) | Monomethyl auristatin E (MMAE) | Advanced solid tumors (all-comers) | Sichuan Baili, SystImmune | NCT06475131 | [128] | |
| BL-B16D1 (EGFR×HER3) | Monomethyl auristatin E (MMAE) | Head & Neck Squamous Cell Carcinoma (+ others) | Sichuan Baili, SystImmune | NCT06469008 | [129] | |
| BL-B16D1 (EGFR×HER3) | Monomethyl auristatin E (MMAE) | HER2-negative Breast Cancer (+ others) | Sichuan Baili, SystImmune | NCT06493864 | [130] | |
| IBI-3001 (B7-H3×EGFR) | Undisclosed | Advanced solid tumors (B7-H3 & EGFR co-expressing) | Innovent Biologics | NCT06349408 | [131,132,133] | |
| Phase1/2 | REGN5093-M114 (MET biparatopic) | Maytansine derivative (M24) | Non-Small-cell Lung Cancer (NSCLC; MET-overexpressing; advanced) | Regeneron Pharmaceuticals | NCT04982224 | [134,135] |
| MEDI4276 (HER2 (ECD2×ECD4) | AZ13599185 (microtubule inhibitor) | Breast & Gastric Cancer (HER2-overexpressing; advanced) | MedImmune, AstraZeneca | NCT02576548 | [136,137] | |
| GEN1286 (EGFR×c-MET) | Topoisomerase I inhibitor | Advanced solid tumors (ovarian, NSCLC, gastric, etc.) | Genmab | NCT06685068 | [138,139] | |
| VBC101- (EGFR×c-Met) | Monomethyl auristatin E (MMAE) | Solid tumors co-expressing EGFR & MET | VelaVigo Bio | NCT07136779 | [140,141,142] | |
| Phase2 | BL-B01D1 (EGFR×HER3) | Ed-04 (topoisomerase I inhibitor) | Renal Cell Carcinoma (locally advanced or metastatic) | Sichuan Baili, Bristol Myers Squib | NCT06962787 | [9,105,143] |
| Phase3 | TQB2102 (HER2 (ECD2×ECD4)) | Topoisomerase I inhibitor | Breast Cancer (HER2+, early stage, neoadjuvant) | Chia Tai Tianqing | NCT07043725 | [144,145] |
| JSKN003 (HER2 biparatopic) | Topoisomerase I inhibitor | Ovarian Cancer (HER2-expressing, platinum-resistant) | lphamab Oncology | NCT06751485 | [146] | |
| JSKN003 (HER2 biparatopic) | Topoisomerase I inhibitor | HER2-positive Breast Cancer (advanced, post-T-DM1) | lphamab Oncology | NCT06846437 | [147,148] | |
| BL-B01D1 (EGFR×HER3) | Ed-04 (topoisomerase I inhibitor) | Triple-Negative Breast Cancer (metastatic) | Sichuan Baili, Bristol Myers Squib | NCT06382142 | [9,105,149] | |
| BL-B01D1 (EGFR×HER3) | Ed-04 (topoisomerase I inhibitor) | HR+/HER2– Metastatic Breast Cancer | Sichuan Baili, Bristol Myers Squib | NCT06343948 | [9,105,150] | |
| BL-B01D1 (EGFR×HER3) | Ed-04 (topoisomerase I inhibitor) | Esophageal squamous cell carcinoma (2L, post-IO) | Sichuan Baili, Bristol Myers Squib | NCT06304974 | [9,105,151] |
3.4. Recent Progress of BsADCs for PDAC Treatments
To date, the development of BsADCs for PDAC treatment has remained in the early stages, but several drug candidates have demonstrated potential in preclinical studies (Figure 3). For example, EpCAM×CLDN3 BsADC, engineered using CrossMab and Knobs-into-Holes technologies and conjugated with DXd via a GGFG-based cleavable linker, exhibited potent cytotoxicity with an IC50 of 0.72 μg/mL in EpCAM+/CLDN3+ cell lines and induced significant tumor regression in xenograft models while maintaining favorable safety and pharmacokinetic stability [109]. Notably, it shows low activity in EpCAM-high/CLDN3-low models, suggesting the rationale for AND-gating-based specificity providing reduced off-target toxicity. Another example is HER3×MUC1 BsADC (DM002 series). It was developed in two formats, DM002-vcMMAE and DM002-BLD1102, both of which enhanced co-internalization and intracellular drug delivery, resulting in excellent antitumor efficacy in cell-line-derived xenograft and patient-derived xenograft tumor models. DM002-vcMMAE consistently outperformed its parental ADC, while DM002-BLD1102 maintained or exceeded efficacy in vcMMAE-resistant models, suggesting that payload class switching may overcome acquired resistance [152,153]. In the meantime, mesothelin-targeting ADC (DMOT4039A) demonstrated partial responses in a subset of pancreatic cancer patients in a phase I trial, with a manageable safety profile, thereby validating mesothelin as a promising target in pancreatic cancer [154]. Therefore, these findings highlight that BsADCs have achieved clear preclinical and early clinical successes, providing a rationale for their further exploration in PDAC.
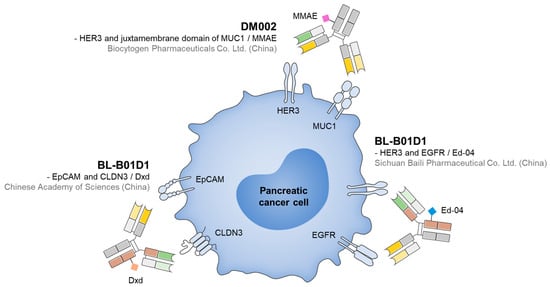
Figure 3.
Representative BsADCs for PDAC treatments, along with their target ligands and payloads.
Among the ongoing programs, EGFR×HER3 BsADC (BL-B01D1) represents the most clinically advanced candidate. In a phase I trial including 174 evaluable patients with advanced solid tumors, BL-B01D1 achieved an ORR of 34% and established an RP2D of 2.5 mg/kg on Days 1 and 8 of a 3-week cycle. Safety outcomes were generally manageable, and some patients experienced durable responses [105]. These data support the translational feasibility of BsADCs in overcoming resistance and broadening the therapeutic utility of ADC platforms in PDAC [36].
4. Discussion and Future Perspectives
Through the efforts of many researchers, the potential for treating PDAC using BsAbs and BsADCs has been validated, and reports demonstrating its clinical utility are gradually increasing [54,56]. Nevertheless, the unique tumor microenvironment and biological characteristics of PDAC have hindered the practical application of BsADCs thus far. For example, the dense ECM and elevated interstitial pressure in PDAC markedly reduce the diffusion and tissue penetration of large antibody molecules and their payloads, resulting in poor intratumoral drug distribution [102]. Moreover, the poor vascular perfusion and hypovascularity of PDAC further restrict antibody delivery compared with highly vascularized cancers such as breast or lung tumors [155]. In addition, tumour antigens are heterogeneous, and inconsistent co-expression of dual targets limits the reliability of dual-targeting of BsADCs (AND strategy) and conditional or sequential targeting (OR strategies) [54]. Moreover, frequent targets, such as TROP2, MUC1, and mesothelin, are also expressed at low levels in normal tissues, therefore raising concerns of off-target toxicities [156,157]. Furthermore, PDAC exhibits a “cold tumor” immune phenotype characterized by low T-cell infiltration and a predominance of immunosuppressive cell populations such as regulatory T cells, tumor-associated macrophages (TAMs), and myeloid-derived suppressor cells (MDSCs), which collectively limit the efficacy of T-cell-engaging BsAbs [158]. Finally, reduced tumor perfusion decreases overall antibody delivery efficiency, while nonspecific interstitial binding within the fibrotic stroma prolongs tissue residence time, leading to payload leakage and increased off-tumor toxicity [159]. Collectively, these factors complicate pharmacokinetic (PK) predictability and narrow the effective dose window. Meanwhile, payload-related toxicities, including ILD observed with DXd-based agents, further constrain the therapeutic window [156]. Resistance mechanisms such as antigen downregulation, drug efflux, and enhanced DNA repair also limit durable responses [160,161,162]. In this regard, despite their potent payloads and encouraging preclinical activity, ADCs have so far produced disappointing results in clinical trials for PDAC. Taken together, while BsADCs have shown significant promise in both preclinical and early clinical contexts, overcoming biological and technical hurdles remains essential to realize their therapeutic potential in PDAC [54].
First, a deep understanding of the in vivo behavior of engineered nanomaterials is required [163]. Unlike biogenic mAbs, BsAbs and BsADCs are heterogeneous nanomaterials. Therefore, their intrinsic behaviors as antibodies, such as binding to Fc receptors or Fc recycling, may differ significantly from those of mAbs. Therefore, in-depth studies on the distribution behavior of BsAbs and BsADCs under various in vivo conditions, as well as their in vivo distribution in PDAC models, appear necessary [102,164].
Second, strategies to overcome the demoplastic and immunosuppressive environment of pancreatic cancer must be considered. Beyond T-cell-engaging strategies, which have shown clinically successful outcomes, introducing additional strategies to enhance T cell function and control the function of cancer-associated fibroblasts that generate tolerogenic signals could provide a novel approach to maximize the synergistic effects of the cytotoxic payload and immunosurveillance effects of BsADCs [159].
Third, consideration is needed for strategies to overcome the low perfusion and high interstitial pressure in pancreatic cancer. For PDAC, where particle extravasation into tissue is inefficient, binding to claudins or specific binding to albumin could increase accumulation rates in pancreatic tissue. This, coupled with extended half-lives, could provide BsAbs and BsADCs with more opportunities to bind their targets [165,166]. Another example is increased tissue penetration through fragmentation. The nanosatellite strategy, one of the penetration strategies for nanomaterials, involves components fragmenting into smaller sizes specifically within the target tissue, enabling effective penetration into the tissue interior [167]. Similarly, if the linkers connecting the fragments composing BsAbs are designed to be selectively cleavable within the TME, large structures like IgG-like BsAbs or TandAbs could be expected to dissociate into smaller bispecific fragments within the microenvironment, thereby enhancing penetration efficiency into solid tumors.
Author Contributions
Conceptualization, W.U. and M.K.; writing—original draft, H.S. and D.G.; investigation, S.Y.J. and S.H.; writing—review and editing, W.U., H.S. and V.Q.N.; funding acquisition, W.U. and M.K. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by National Research Foundation of Korea (NRF) grants funded by the Korean government (MSIT, No. RS-2023-00242186, RS-2025-25400013, and RS-2025-16069638). This work was supported by the Pukyong National University Research Fund in 2024 (202411970001).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—Clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Masugi, Y. The desmoplastic stroma of pancreatic cancer: Multilayered levels of heterogeneity, clinical significance, and therapeutic opportunities. Cancers 2022, 14, 3293. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.; Xu, D.; Liao, M.M.; Sun, Y.; Bao, W.d.; Yao, F.; Ma, L. Barriers and opportunities in pancreatic cancer immunotherapy. npj Precis. Oncol. 2024, 8, 199. [Google Scholar] [CrossRef]
- Hosein, A.N.; Dougan, S.K.; Aguirre, A.J.; Maitra, A. Translational advances in pancreatic ductal adenocarcinoma therapy. Nat. Cancer 2022, 3, 272–286. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Arias-Pinilla, G.A.; Modjtahedi, H. Therapeutic Application of Monoclonal Antibodies in Pancreatic Cancer: Advances, Challenges and Future Opportunities. Cancers 2021, 13, 1781. [Google Scholar] [CrossRef]
- Kontermann, R.E.; Brinkmann, U. Bispecific antibodies. Drug Discov. Today 2015, 20, 838–847. [Google Scholar] [CrossRef]
- Luo, M.; Wang, X.; Yu, G.; Ji, J.; Li, L.; Song, F. Development of a bispecific antibody–drug conjugate targeting EpCAM and CLDN3 for the treatment of multiple solid tumors. Exp. Hematol. Oncol. 2025, 14, 33. [Google Scholar] [CrossRef]
- Liu, C.; Liu, D.; Ji, Y.; Sun, M.; Gao, S.; Ma, X.; Zhong, D.; Zhu, J.; Cao, Y.; Qi, C.; et al. A bispecific antibody–drug conjugate targeting EGFR and HER3 in metastatic esophageal squamous cell carcinoma: A phase 1b trial. Nat. Med. 2025. In press. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Wang, Z.; Wang, Y. Bispecific antibody drug conjugates: Making 1+1>2. Acta Pharm. Sin. B 2024, 14, 1965–1986. [Google Scholar] [CrossRef]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. MAbs 2017, 9, 182–212. [Google Scholar] [CrossRef]
- Cho, B.C.; Simi, A.; Sabari, J.; Vijayaraghavan, S.; Moores, S.; Spira, A. Amivantamab, an Epidermal Growth Factor Receptor (EGFR) and Mesenchymal-epithelial Transition Factor (MET) Bispecific Antibody, Designed to Enable Multiple Mechanisms of Action and Broad Clinical Applications. Clin. Lung Cancer 2023, 24, 89–97. [Google Scholar] [CrossRef]
- Rivera-Soto, R.; Henley, B.; Pulgar, M.A.; Lehman, S.L.; Gupta, H.; Perez-Vale, K.Z.; Weindorfer, M.; Vijayaraghavan, S.; Yao, T.W.S.; Laquerre, S.; et al. Amivantamab efficacy in wild-type EGFR NSCLC tumors correlates with levels of ligand expression. npj Precis. Oncol. 2024, 8, 192. [Google Scholar] [CrossRef] [PubMed]
- Agostini, H.; Abreu, F.; Baumal, C.R.; Chang, D.S.; Csaky, K.G.; Demetriades, A.M.; Kodjikian, L.; Lim, J.I.; Margaron, P.; Monés, J.M.; et al. Faricimab for neovascular age-related macular degeneration and diabetic macular edema: From preclinical studies to phase 3 outcomes. Graefe’s Arch. Clin. Exp. Ophthalmol. 2024, 262, 3437–3451. [Google Scholar] [CrossRef]
- Khanani, A.M.; Patel, S.S.; Ferrone, P.J.; Osborne, A.; Sahni, J.; Grzeschik, S.; Basu, K.; Ehrlich, J.S.; Haskova, Z.; Dugel, P.U. Efficacy of Every Four Monthly and Quarterly Dosing of Faricimab vs Ranibizumab in Neovascular Age-Related Macular Degeneration: The STAIRWAY Phase 2 Randomized Clinical Trial. JAMA Ophthalmol. 2020, 138, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Ying, H.; Grinnell, C.; Bryant, S.; Miller, R.; Clabbers, A.; Bose, S.; McCarthy, D.; Zhu, R.R.; Santora, L. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat. Biotechnol. 2007, 25, 1290–1297. [Google Scholar] [CrossRef]
- Tian, Z.; Liu, M.; Zhang, Y.; Wang, X. Bispecific T cell engagers: An emerging therapy for management of hematologic malignancies. J. Hematol. Oncol. 2021, 14, 75. [Google Scholar] [CrossRef]
- Han, L.; Wang, K.; Jiang, Z.; Guo, X.; Yu, J. Recent development in bispecific antibody immunotherapy for hematological malignancies. Crit. Rev. Oncol./Hematol. 2025, 212, 104752. [Google Scholar] [CrossRef]
- Mocquot, P.; Mossazadeh, Y.; Lapierre, L.; Pineau, F.; Despas, F. The pharmacology of blinatumomab: State of the art on pharmacodynamics, pharmacokinetics, adverse drug reactions and evaluation in clinical trials. J. Clin. Pharm. Ther. 2022, 47, 1337–1351. [Google Scholar] [CrossRef] [PubMed]
- Duell, J.; Lammers, P.E.; Djuretic, I.; Chunyk, A.G.; Alekar, S.; Jacobs, I.; Gill, S. Bispecific Antibodies in the Treatment of Hematologic Malignancies. Clin. Pharmacol. Ther. 2019, 106, 781–791. [Google Scholar] [CrossRef]
- Reusch, U.; Burkhardt, C.; Fucek, I.; Le Gall, F.; Le Gall, M.; Hoffmann, K.; Knackmuss, S.H.; Kiprijanov, S.; Little, M.; Zhukovsky, E.A. A novel tetravalent bispecific TandAb (CD30/CD16A) efficiently recruits NK cells for the lysis of CD30+ tumor cells. MAbs 2014, 6, 728–739. [Google Scholar] [CrossRef]
- Shastri, T.; Trabolsi, A.; Arumov, A.; Schatz, J.H. Bispecific Antibodies in Hematologic Malignancies: Attacking the Frontline. BioDrugs 2025, 39, 793–814. [Google Scholar] [CrossRef]
- Muyldermans, S. Nanobodies: Natural single-domain antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef]
- Li, H.; Zhou, Q.; Cao, N.; Hu, C.; Wang, J.; He, Y.; Jiang, S.; Li, Q.; Chen, M.; Gong, L.; et al. Nanobodies and their derivatives: Pioneering the future of cancer immunotherapy. Cell Commun. Signal. 2025, 23, 271. [Google Scholar] [CrossRef]
- Köhnke, T.; Krupka, C.; Tischer, J.; Knösel, T.; Subklewe, M. Increase of PD-L1 expressing B-precursor ALL cells in a patient resistant to the CD19/CD3-bispecific T cell engager antibody blinatumomab. J. Hematol. Oncol. 2015, 8, 111. [Google Scholar] [CrossRef]
- Pang, X.; Huang, Z.; Zhong, T.; Zhang, P.; Wang, Z.M.; Xia, M.; Li, B. Cadonilimab, a tetravalent PD-1/CTLA-4 bispecific antibody with trans-binding and enhanced target binding avidity. MAbs 2023, 15, 2180794. [Google Scholar] [CrossRef]
- Bielski, P.; Barczyński, J.; Mikitiuk, M.; Myrcha, M.; Rykała, K.; Boon, L.; Gąsior, W.; Hec-Gałązka, A.; Holak, T.A.; Sitar, T. The bispecific antibody targeting VISTA and PD-L1 shows enhanced tumor inhibitory activity in pancreatic, endometrial and breast cancers compared to mono- and combination immune checkpoint blockade. Front. Immunol. 2025, 16, 1486799. [Google Scholar] [CrossRef]
- Oslund, R.C.; Holland, P.M.; Lesley, S.A.; Fadeyi, O.O. Therapeutic potential of cis-targeting bispecific antibodies. Cell Chem. Biol. 2024, 31, 1473–1489. [Google Scholar] [CrossRef]
- Huang, S.; Van Duijnhoven, S.M.J.; Sijts, A.J.A.M.; Van Elsas, A. Bispecific antibodies targeting dual tumor-associated antigens in cancer therapy. J. Cancer Res. Clin. Oncol. 2020, 146, 3111–3122. [Google Scholar] [CrossRef]
- Kontermann, R. Dual targeting strategies with bispecific antibodies. MAbs 2012, 4, 182–197. [Google Scholar] [CrossRef]
- Fan, G.; Wang, Z.; Hao, M.; Li, J. Bispecific antibodies and their applications. J. Hematol. Oncol. 2015, 8, 130. [Google Scholar] [CrossRef]
- Luo, J. Amivantamab in EGFR Exon 20 Insertion Mutated NSCLC Progression on Platinum Chemotherapy: Initial Results from the CHRYSALIS Phase 1 Study. J. Clin. Oncol. 2021, 39, 3391–3402. [Google Scholar]
- Khanani, A.M.; Kotecha, A.; Chang, A.; Chen, S.J.; Chen, Y.; Guymer, R.; Heier, J.S.; Holz, F.G.; Iida, T.; Ives, J.A. TENAYA and LUCERNE: Two-year results from the phase 3 neovascular age-related macular degeneration trials of faricimab with treat-and-extend dosing in year 2. Ophthalmology 2024, 131, 914–926. [Google Scholar] [CrossRef]
- Muik, A.; Garralda, E.; Altintas, I.; Gieseke, F.; Geva, R.; Ben-Ami, E.; Maurice-Dror, C.; Calvo, E.; LoRusso, P.M.; Alonso, G.; et al. Preclinical characterization and phase I trial results of a bispecific antibody targeting PD-L1 and 4-1BB (GEN1046) in patients with advanced refractory solid tumors. Cancer Discov. 2022, 12, 1248–1265. [Google Scholar] [CrossRef]
- Cheng, L.S.; Zhu, M.; Gao, Y.; Liu, W.T.; Yin, W.; Zhou, P.; Zhu, Z.; Niu, L.; Zeng, X.; Zhang, D.; et al. An Fc-muted bispecific antibody targeting PD-L1 and 4-1BB induces antitumor immune activity in colorectal cancer without systemic toxicity. Cell. Mol. Biol. Lett. 2023, 28, 47. [Google Scholar] [CrossRef]
- Li, T.; Niu, M.; Zhou, J.; Wu, K.; Yi, M. The enhanced antitumor activity of bispecific antibody targeting PD-1/PD-L1 signaling. Cell Commun. Signal. 2024, 22, 179. [Google Scholar] [CrossRef]
- Warwas, K.M.; Meyer, M.; Gonçalves, M.; Moldenhauer, G.; Bulbuc, N.; Knabe, S.; Luckner-Minden, C.; Ziegelmeier, C.; Heussel, C.P.; Zörnig, I.; et al. Co-Stimulatory Bispecific Antibodies Induce Enhanced T Cell Activation and Tumor Cell Killing in Breast Cancer Models. Front. Immunol. 2021, 12, 719116. [Google Scholar] [CrossRef]
- Kitazawa, T.; Shima, M. Emicizumab, a humanized bispecific antibody to coagulation factors IXa and X with a factor VIIIa-cofactor activity. Int. J. Hematol. 2020, 111, 20–30. [Google Scholar] [CrossRef]
- Oldenburg, J.; Mahlangu, J.N.; Kim, B.; Schmitt, C.; Callaghan, M.U.; Young, G.; Santagostino, E.; Kruse-Jarres, R.; Negrier, C.; Kessler, C. Emicizumab prophylaxis in hemophilia A with inhibitors. N. Engl. J. Med. 2017, 377, 809–818. [Google Scholar] [CrossRef]
- Sindaco, P.; Pandey, H.; Isabelle, C.; Chakravarti, N.; Brammer, J.E.; Porcu, P.; Mishra, A. The role of interleukin-15 in the development and treatment of hematological malignancies. Front. Immunol. 2023, 14, 1141208. [Google Scholar] [CrossRef]
- Blair, H.A. Zenocutuzumab: First Approval. Drugs 2025, 85, 591–597. [Google Scholar] [CrossRef]
- Mbbs, S.R.; Mbbs, U.F.; Waheed, A.; Mbbs, M.K.B.; Md, A.B.; Mbbs, H.N.; Gul, M.H.; Mbbs, A.A.; Mbbs, Z.I.; Wardak, A.B. Unveiling the Potential of Zenocutuzumab: A Breakthrough in NSCLC and Pancreatic Adenocarcinoma Treatment. Oncology 2025, 7, 298–300. [Google Scholar]
- Schram, A.M.; Goto, K.; Kim, D.W.; Macarulla, T.; Hollebecque, A.; O’Reilly, E.M.; Ou, S.H.I.; Rodon, J.; Rha, S.Y.; Nishino, K.; et al. Efficacy of zenocutuzumab in NRG1 fusion–positive cancer. N. Engl. J. Med. 2025, 392, 566–576. [Google Scholar] [CrossRef]
- Long, A.W.; Xu, H.; Santich, B.H.; Guo, H.; Hoseini, S.S.; de Stanchina, E.; Cheung, N.K.V. Heterodimerization of T cell engaging bispecific antibodies to enhance specificity against pancreatic ductal adenocarcinoma. J. Hematol. Oncol. 2024, 17, 20. [Google Scholar] [CrossRef]
- Chen, S.; Zhao, Y.; Cao, M.; Peng, W.; Huang, H.; Yang, Y.; Liang, J.; Chen, W.; Bai, S.; Zhou, Q. Anti-CTGF/PD-1 bispecific antibody Y126S restrains desmoplastic and immunosuppressive microenvironment in pancreatic cancer. J. Immunother. Cancer 2025, 13, e012144. [Google Scholar] [CrossRef]
- Lum, L.G.; Thakur, A.; Choi, M.; Deol, A.; Kondadasula, V.; Schalk, D.; Fields, K.; Dufrense, M.; Philip, P.; Dyson, G. Clinical and immune responses to anti-CD3 x anti-EGFR bispecific antibody armed activated T cells (EGFR BATs) in pancreatic cancer patients. Oncoimmunology 2020, 9, 1773201. [Google Scholar] [CrossRef] [PubMed]
- Phase Ib/II Treatment of Advanced Pancreatic Cancer with Anti-CD3 x Anti-EGFR-Bispecific Antibody Armed Activated T-Cells (BATs). 2017. Available online: https://clinicaltrials.gov/study/NCT03269526 (accessed on 30 September 2025).
- Suurs, F.V.; Lorenczewski, G.; Bailis, J.M.; Stienen, S.; Friedrich, M.; Lee, F.; van der Vegt, B.; de Vries, E.G.E.; de Groot, D.J.A.; Lub-de Hooge, M.N. Mesothelin/CD3 Half-Life–Extended Bispecific T-Cell Engager Molecule Shows Specific Tumor Uptake and Distributes to Mesothelin and CD3-Expressing Tissues. J. Nucl. Med. 2021, 62, 1797–1804. [Google Scholar] [CrossRef]
- Phase 1a/1b Study of CT-95 in Advanced Cancers Associated with Mesothelin Expression. 2024. Available online: https://clinicaltrials.gov/study/NCT06756035 (accessed on 14 October 2025).
- United States Food and Drug Administration. FDA Grants Accelerated Approval to Zenocutuzumab-Zbco for Non-Small Cell Lung Cancer and Pancreatic Adenocarcinoma Harboring NRG1 Gene Fusions. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-zenocutuzumab-zbco-non-small-cell-lung-cancer-and-pancreatic (accessed on 4 December 2024).
- Shen, L.; Schaefer, A.; Huckaby, J.; Wolf, W.; Lai, S.K. Bispecific Siglec-15/T cell antibody (STAB) activates T cells and suppresses pancreatic ductal adenocarcinoma and non-small cell lung tumors in vivo. Theranostics 2025, 15, 5529–5542. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Wang, X.; Niu, M.; Wang, M.; Zhou, J.; Wu, K.; Yi, M. Bispecific antibody targeting TGF-β and PD-L1 for synergistic cancer immunotherapy. Front. Immunol. 2023, 14, 1196970. [Google Scholar] [CrossRef] [PubMed]
- Wittwer, N.L.; Brown, M.P.; Liapis, V.; Staudacher, A.H. Antibody drug conjugates: Hitting the mark in pancreatic cancer? J. Exp. Clin. Cancer Res. 2023, 42, 280. [Google Scholar] [CrossRef]
- Yu, X.; Zhang, J.; Liu, J.; Yang, J.; Yue, J.; Sun, Y.; Pan, Y.; Sun, M.; Qin, Y.; Shen, L. 132MO Anti-claudin18. 2 (CLDN18. 2) antibody-drug conjugate (ADC) IBI343 in patients (pts) with advanced pancreatic ductal adenocarcinoma (PDAC): Updated results from a phase I study. Ann. Oncol. 2024, 35, S1456. [Google Scholar] [CrossRef]
- Farhangnia, P.; Khorramdelazad, H.; Nickho, H.; Delbandi, A.A. Current and future immunotherapeutic approaches in pancreatic cancer treatment. J. Hematol. Oncol. 2024, 17, 40. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Han, Y.; Fang, Y.; Ma, P.; Zhou, J.; Jiang, Y.; Xing, S.; Tang, Q.; Hou, Y.; Wang, S.; et al. Antibody-drug conjugates: Current challenges and innovative solutions for precision cancer therapy. Med 2025, 6, 100849. [Google Scholar] [CrossRef]
- Phase 1 Safety and Tolerability Study of Human Monoclonal Antibody 5B1 (MVT-5873) with Expansion in Subjects with Pancreatic Cancer or Other CA19-9 Positive Malignancies. 2016. Available online: https://clinicaltrials.gov/study/NCT02672917 (accessed on 14 October 2025).
- O’Reilly, E.M.; Wang, J.S.Z.; Yu, K.H.; Lowery, M.A.; Varghese, A.M.; Bendell, J.C.; Borazanci, E.H.; Estrella, H.; Fowler, K.; Hoskins, M. Abstract LB-B25: Preliminary phase I data comparing HuMab-5B1 (MVT-5873), a monoclonal antibody targeting sLea, as a single agent and in combination with first line nab-paclitaxel and gemcitabine in patients with CA19-9 positive pancreatic cancer. Mol. Cancer Ther. 2018, 17, LB-B25. [Google Scholar] [CrossRef]
- Huffman, B.M.; Mallick, A.B.; Horick, N.K.; Wang-Gillam, A.; Hosein, P.J.; Morse, M.; Beg, M.S.; Murphy, J.E.; Schlechter, B.L.; Sanoff, H.; et al. Abstract A019: A multicenter randomized phase II study of gemcitabine and nab-paclitaxel versus gemcitabine and nab-paclitaxel with a MUC5AC antibody (NPC-1C) in advanced pancreatic cancer previously treated with FOLFIRINOX (NCT01834235). Cancer Res. 2022, 82, A019. [Google Scholar] [CrossRef]
- A First-in-human Study of IBI343 in Subjects with Locally Advanced Unresectable or Metastatic Solid Tumors. 2025. Available online: https://clinicaltrials.gov/study/NCT05458219 (accessed on 27 April 2025).
- Yu, X.; Ying, J.; Enxiao, L.; Zhou, A.; Sun, Y.; Yue, J.; Ruan, J.; Zhang, J.; Shen, L.; Du, J.; et al. Claudin18.2 (CLDN18.2) expression and efficacy in pancreatic ductal adenocarcinoma (PDAC): Results from a phase I dose expansion cohort evaluating IBI343. J. Clin. Oncol. 2025, 43, 4017. [Google Scholar] [CrossRef]
- A Phase 1A/B Study to Evaluate the Safety and Tolerability of EBC-129 as a Single Agent and in Combination with Pembrolizumab in Advanced Solid Tumours. 2023. Available online: https://clinicaltrials.gov/study/NCT05701527 (accessed on 4 September 2025).
- Lentz, R.W.; Ng, M.C.H.; Yong, W.-P.; Meric-Bernstam, F.; Singh, I.; Srirangam, V.; Cometa, J.; Blanchard, S.; Nellore, R.; Shah, K.J.; et al. Clinical activity of EBC-129, a first-in class, anti N256-glycosylated CEACAM5 and CEACAM6 antibody-drug conjugate (ADC), in patients with pancreatic ductal adenocarcinoma (PDAC) in a phase 1 study. J. Clin. Oncol. 2025, 43, 4018. [Google Scholar] [CrossRef]
- Phase 1 Dose-Escalation Trial of OMTX705, an Anti-Fibroblast Activation Protein Antibody-Drug Conjugate, as Single Agent and in Combination with Pembrolizumab in Patients with Advanced Solid Tumors. 2022. Available online: https://clinicaltrials.gov/study/NCT05547321 (accessed on 27 April 2025).
- Torres-Jiménez, J.; Paisan, A.; Ponz-Sarvise, M.; Bockorny, B.; Gil-Martin, M.; Cubedo, R.; Paz-Ares, L.G.; Landa-Magdalena, A.; Wulf, G.M.; Sàbat, P.; et al. First-in-human phase 1 dose escalation trial of OMTX705, a novel anti-fibroblast activation protein (FAP) antibody drug conjugate (ADC), in monotherapy and in combination with pembrolizumab in patients with solid tumors. J. Clin. Oncol. 2025, 43, 3028. [Google Scholar] [CrossRef]
- Metrangolo, V.; Blomquist, M.H.; Dutta, A.; Gårdsvoll, H.; Krigslund, O.; Nørregaard, K.S.; Jürgensen, H.J.; Ploug, M.; Flick, M.J.; Behrendt, N. Targeting uPAR with an antibody-drug conjugate suppresses tumor growth and reshapes the immune landscape in pancreatic cancer models. Sci. Adv. 2025, 11, eadq0513. [Google Scholar] [CrossRef]
- Ghosh, S.; Huda, P.; Fletcher, N.; Campbell, D.; Thurecht, K.J.; Walsh, B. Clinical development of an anti-GPC-1 antibody for the treatment of cancer. Expert Opin. Biol. Ther. 2022, 22, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Gymnopoulos, M.; Betancourt, O.; Blot, V.; Fujita, R.; Galvan, D.; Lieuw, V.; Nguyen, S.; Snedden, J.; Stewart, C.; Villicana, J. TR1801-ADC: A highly potent cMet antibody–drug conjugate with high activity in patient-derived xenograft models of solid tumors. Mol. Oncol. 2020, 14, 54–68. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yu, X.; Wang, X.; Yuan, K.; Wang, G.; Hu, L.; Zhang, G.; Pei, W.; Wang, L.; Sun, C. Bispecific antibodies in cancer therapy: Target selection and regulatory requirements. Acta Pharm. Sin. B 2023, 13, 3583–3597. [Google Scholar] [CrossRef]
- Wang, R.; Hu, B.; Pan, Z.; Mo, C.; Zhao, X.; Liu, G.; Hou, P.; Cui, Q.; Xu, Z.; Wang, W.; et al. Antibody–Drug Conjugates (ADCs): Current and future biopharmaceuticals. J. Hematol. Oncol. 2025, 18, 51. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Nam, S.M.; Moon, A. Antibody-drug conjugates and bispecific antibodies targeting cancers: Applications of click chemistry. Arch. Pharm. Res. 2023, 46, 131–148. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Riccardi, F.; Dal Bo, M.; Macor, P.; Toffoli, G. A comprehensive overview on antibody-drug conjugates: From the conceptualization to cancer therapy. Front. Pharmacol. 2023, 14, 1274088. [Google Scholar] [CrossRef]
- Li, F.; Emmerton, K.K.; Jonas, M.; Zhang, X.; Miyamoto, J.B.; Setter, J.R.; Nicholas, N.D.; Okeley, N.M.; Lyon, R.P.; Benjamin, D.R.; et al. Intracellular Released Payload Influences Potency and Bystander-Killing Effects of Antibody-Drug Conjugates in Preclinical Models. Cancer Res. 2016, 76, 2710–2719. [Google Scholar] [CrossRef]
- Study of XB002 in Subjects with Solid Tumors (JEWEL-101). 2023. Available online: https://clinicaltrials.gov/study/NCT04925284 (accessed on 4 April 2025).
- de Vries, E.G.E.; Rüschoff, J.; Lolkema, M.; Tabernero, J.; Gianni, L.; Voest, E.; de Groot, D.J.A.; Castellano, D.; Erb, G.; Naab, J.; et al. Phase II study (KAMELEON) of single-agent T-DM1 in patients with HER2-positive advanced urothelial bladder cancer or pancreatic cancer/cholangiocarcinoma. Cancer Med. 2023, 12, 12071–12083. [Google Scholar] [CrossRef]
- Qian, Y.; Gong, Y.; Fan, Z.; Luo, G.; Huang, Q.; Deng, S.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. Molecular alterations and targeted therapy in pancreatic ductal adenocarcinoma. J. Hematol. Oncol. 2020, 13, 130. [Google Scholar] [CrossRef]
- Spiliopoulou, P.; Kasi, A.; Abushahin, L.; Cardin, D.; Lenz, H.-J.; Dayyani, F.; Messersmith, W.; Ezenwajiaku, N.; Oberstein, P.; Paluri, R.; et al. Phase Ib study of anetumab ravtansive in combination with immunotherapy or immunotherapy plus chemotherapy in mesothelin-enriched advanced pancreatic adenocarcinoma: NCI10208. J. Clin. Oncol. 2022, 40, 4136. [Google Scholar] [CrossRef]
- Golfier, S.; Kopitz, C.; Kahnert, A.; Heisler, I.; Schatz, C.A.; Stelte-Ludwig, B.; Mayer-Bartschmid, A.; Unterschemmann, K.; Bruder, S.; Linden, L.; et al. Anetumab Ravtansine: A Novel Mesothelin-Targeting Antibody–Drug Conjugate Cures Tumors with Heterogeneous Target Expression Favored by Bystander Effect. Mol. Cancer Ther. 2014, 13, 1537–1548. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Leal, A.D.; Parikh, A.; Ryan, D.P.; Wang, S.; Bahamon, B.; Gupta, N.; Moss, A.; Pye, J.; Miao, H.; et al. A phase I, first-in-human study of TAK-164, an antibody-drug conjugate, in patients with advanced gastrointestinal cancers expressing guanylyl cyclase C. Cancer Chemother. Pharmacol. 2023, 91, 291–300. [Google Scholar] [CrossRef]
- Yao, H.-P.; Zhao, H.; Hudson, R.; Tong, X.M.; Wang, M.H. Duocarmycin-based antibody–drug conjugates as an emerging biotherapeutic entity for targeted cancer therapy: Pharmaceutical strategy and clinical progress. Drug Discov. Today 2021, 26, 1857–1874. [Google Scholar] [CrossRef]
- MGC018 with or Without MGA012 in Advanced Solid Tumors. 2023. Available online: https://clinicaltrials.gov/study/NCT03729596 (accessed on 31 July 2025).
- Zammarchi, F.; Havenith, K.; Chivers, S.; Hogg, P.; Bertelli, F.; Tyrer, P.; Janghra, N.; Reinert, H.; Hartley, J.; Berkel, P. Preclinical Development of ADCT-601, a Novel Pyrrolobenzodiazepine Dimer-based Antibody–drug Conjugate Targeting AXL-expressing Cancers. Mol. Cancer Ther. 2022, 21, 582–593. [Google Scholar] [CrossRef] [PubMed]
- Zammarchi, F.; Havenith, K.; Chivers, S.; Hogg, P.W.; Britten, C.; Dissanayake, S.; Tyrer, P.; Bertelli, F.; Hutchinson, I.; Masterson, L.; et al. Abstract 2792A: Preclinical activity of ADCT-601, a novel pyrrolobenzodiazepine (PBD) dimer-based antibody-drug conjugate (ADC) targeting AXL-expressing tumors. Cancer Res. 2018, 78, 2792A. [Google Scholar] [CrossRef]
- Valsasina, B.; Orsini, P.; Terenghi, C.; Ocana, A. Present Scenario and Future Landscape of Payloads for ADCs: Focus on DNA-Interacting Agents. Pharmaceuticals 2024, 17, 1338. [Google Scholar] [CrossRef]
- Dotan, E.; Cohen, S.; Starodub, A.; Lieu, C.; Messersmith, W.; Simpson, P.; Guarino, M.; Marshall, J.; Goldberg, R.; Hecht, J.; et al. Phase I/II Trial of Labetuzumab Govitecan (Anti-CEACAM5/SN-38 Antibody-Drug Conjugate) in Patients with Refractory or Relapsing Metastatic Colorectal Cancer. J. Clin. Oncol. 2017, 35, 3338–3346. [Google Scholar] [CrossRef]
- Sharkey, R.; Govindan, S.; Cardillo, T.; Donnell, J.; Xia, J.; Rossi, E.; Chang, C.H.; Goldenberg, D. Selective and Concentrated Accretion of SN-38 with a CEACAM5-Targeting Antibody–Drug Conjugate (ADC), Labetuzumab Govitecan (IMMU-130). Mol. Cancer Ther. 2018, 17, 196–203. [Google Scholar] [CrossRef]
- Ocean, A.J.; Starodub, A.N.; Bardia, A.; Vahdat, L.T.; Isakoff, S.J.; Guarino, M.; Messersmith, W.A.; Picozzi, V.J.; Mayer, I.A.; Wegener, W.A.; et al. Sacituzumab govitecan (IMMU-132), an anti-Trop-2-SN-38 antibody-drug conjugate for the treatment of diverse epithelial cancers: Safety and pharmacokinetics. Cancer 2017, 123, 3843–3854. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Makker, V.; Oaknin, A.; Oh, D.Y.; Banerjee, S.; González-Martín, A.; Jung, K.H.; Ługowska, I.; Manso, L.; Manzano, A.; et al. Efficacy and Safety of Trastuzumab Deruxtecan in Patients with HER2-Expressing Solid Tumors: Primary Results from the DESTINY-PanTumor02 Phase II Trial. J. Clin. Oncol. 2024, 42, 47–58. [Google Scholar] [CrossRef]
- Okajima, D.; Yasuda, S.; Maejima, T.; Karibe, T.; Sakurai, K.; Aida, T.; Toki, T.; Yamaguchi, J.; Kitamura, M.; Kamei, R.; et al. Datopotamab Deruxtecan, a Novel TROP2-directed Antibody-drug Conjugate, Demonstrates Potent Antitumor Activity by Efficient Drug Delivery to Tumor Cells. Mol. Cancer Ther. 2021, 20, 2329–2340. [Google Scholar] [CrossRef] [PubMed]
- Conilh, L.; Sadilkova, L.; Viricel, W.; Dumontet, C. Payload diversification: A key step in the development of antibody–drug conjugates. J. Hematol. Oncol. 2023, 16, 3. [Google Scholar] [CrossRef]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S. Trastuzumab deruxtecan in previously treated HER2-low advanced breast cancer. N. Engl. J. Med. 2022, 387, 9–20. [Google Scholar] [CrossRef]
- Bardia, A.; Hurvitz, S.A.; Tolaney, S.M.; Loirat, D.; Punie, K.; Oliveira, M.; Brufsky, A.; Sardesai, S.D.; Kalinsky, K.; Zelnak, A.B. Sacituzumab govitecan in metastatic triple-negative breast cancer. N. Engl. J. Med. 2021, 384, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: The “biological missile” for targeted cancer therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef]
- Herrera, M.; Pretelli, G.; Desai, J.; Garralda, E.; Siu, L.L.; Steiner, T.M.; Au, L. Bispecific antibodies: Advancing precision oncology. Trends Cancer 2024, 10, 893–919. [Google Scholar] [CrossRef]
- Oostindie, S.C.; Rinaldi, D.A.; Zom, G.G.; Wester, M.J.; Paulet, D.; Al-Tamimi, K.; van der Meijden, E.; Scheick, J.R.; Wilpshaar, T.; de Jong, B.; et al. Logic-gated antibody pairs that selectively act on cells co-expressing two antigens. Nat. Biotechnol. 2022, 40, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yuting, L.; Tang, S.C.; To, K.F.; Li, B.; Chan, S.; Wong, C.H. The molecular mechanism underlying KRAS regulation on STK31 expression in Pancreatic ductal adenocarcinoma. Cancer Sci. 2023, 115, 3288–3304. [Google Scholar] [CrossRef]
- Balamkundu, S.; Liu, C.-F. Lysosomal-cleavable peptide linkers in antibody–drug conjugates. Biomedicines 2023, 11, 3080. [Google Scholar] [CrossRef]
- Staudacher, A.H.; Brown, M.P. Antibody drug conjugates and bystander killing: Is antigen-dependent internalisation required? Br. J. Cancer 2017, 117, 1736–1742. [Google Scholar] [CrossRef] [PubMed]
- de Goeij, B.E.; Vink, T.; Ten Napel, H.; Breij, E.C.; Satijn, D.; Wubbolts, R.; Miao, D.; Parren, P.W. Efficient payload delivery by a bispecific antibody–drug conjugate targeting HER2 and CD63. Mol. Cancer Ther. 2016, 15, 2688–2697. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed]
- First in Human Study of AZD9592 in Solid Tumors (EGRET). 2022. Available online: https://clinicaltrials.gov/study/NCT05647122 (accessed on 3 October 2025).
- A Phase I Open Label First in Human Dose Escalation and Expansion Study of the Bispecific Anti-Mucin 1—Epidermal Growth Factor Receptor Antibody Drug Conjugate M1231 as a Single Agent in Participants with Advanced Solid Tumors. 2021. Available online: https://clinicaltrials.gov/study/NCT04695847 (accessed on 7 July 2023).
- Ma, Y.; Huang, Y.; Zhao, Y.; Zhao, S.; Xue, J.; Yang, Y.; Fang, W.; Guo, Y.; Han, Y.; Yang, K.; et al. BL-B01D1, a first-in-class EGFR-HER3 bispecific antibody-drug conjugate, in patients with locally advanced or metastatic solid tumours: A first-in-human, open-label, multicentre, phase 1 study. Lancet Oncol. 2024, 25, 901–911. [Google Scholar] [CrossRef]
- McGrath, L.; Zheng, Y.; Christ, S.; Sachs, C.C.; Khelifa, S.; Windmüller, C.; Sweet, S.; Kim, Y.J.; Sutton, D.; Sulikowski, M.; et al. Abstract 5737: Evaluation of the relationship between target expression and in vivo anti-tumor efficacy of AZD9592, an EGFR/c-MET targeted bispecific antibody drug conjugate. Cancer Res. 2023, 83, 5737. [Google Scholar] [CrossRef]
- Aggarwal, C.; Azzoli, C.G.; Spira, A.I.; Solomon, B.J.; Le, X.; Rolfo, C.; Planchard, D.; Felip, E.; Wu, Y.-L.; Ahn, M.-J.; et al. EGRET: A first-in-human study of the novel antibody-drug conjugate (ADC) AZD9592 as monotherapy or combined with other anticancer agents in patients (pts) with advanced solid tumors. J. Clin. Oncol. 2023, 41, TPS3156. [Google Scholar] [CrossRef]
- Hsu, R.; Benjamin, D.J. A narrative review of antibody-drug conjugates in EGFR-mutated non-small cell lung cancer. Front. Oncol. 2023, 13, 1252652. [Google Scholar] [CrossRef]
- Hana, C.; Thaw Dar, N.N.; Galo Venegas, M.; Vulfovich, M. Claudins in cancer: A current and future therapeutic target. Int. J. Mol. Sci. 2024, 25, 4634. [Google Scholar] [CrossRef]
- Knuehl, C.; Toleikis, L.; Dotterweich, J.; Ma, J.; Kumar, S.; Ross, E.; Wilm, C.; Schmitt, M.; Grote, H.J.; Amendt, C. Abstract 5284: M1231 is a bispecific anti-MUC1xEGFR antibody-drug conjugate designed to treat solid tumors with MUC1 and EGFR co-expression. Cancer Res. 2022, 82, 5284. [Google Scholar] [CrossRef]
- Study of Izalontamab Brengitecan (BMS-986507) Versus Platinum-Pemetrexed for EGFR-mutated Non-small Cell Lung Cancer After Failure of EGFR TKI Therapy (IZABRIGHT-Lung01). 2025. Available online: https://clinicaltrials.gov/study/NCT07100080 (accessed on 16 October 2025).
- PH-139: A Phase I Safety and Pharmacokinetic Study of Gamitrinib Administered Intravenously to Patients with Advanced Cancer. 2021. Available online: https://clinicaltrials.gov/study/NCT04827810 (accessed on 29 June 2025).
- Vitale, M.; Griffiths, H.B.S.; Jones, N.; Mistry, M.; Sangster, J.; Patterson, C.; O’Halloran, S.E.; Blanco-Gomez, A.; Schon, O.; Thorn, T. BVX001, a Bispecific ADC Targeting CD7-Positive AML with Favorable Toxicity Profile, Exhibits Significant Efficacy in Primary Patient Samples and PDX-Models. Blood 2024, 144, 2773. [Google Scholar] [CrossRef]
- Wahner, A. FDA Grants Orphan Drug Designation to BVX001 in AML. Available online: https://www.onclive.com/view/fda-grants-orphan-drug-designation-to-bvx001-in-aml (accessed on 17 April 2024).
- Wang, W.; Li, J.; Guan, L.; Xu, M.; Yin, Q. VBC103. Available online: https://www.adcreview.com/drugmap/vbc103/ (accessed on 30 August 2025).
- AstraZeneca. WCLC Advance Ambition: AstraZeneca Medicine for More than Half of All Patients Treated for Lung Cancer by 2030. Available online: https://www.astrazeneca.com/media-centre/press-releases/2023/wclc-advance-ambition-astrazeneca-medicine-for-more-than-half-of-all-patients-treated-for-lung-cancer-2030.html (accessed on 30 August 2025).
- A Dose Finding Study of ZW49 in Patients with HER2-Positive Cancers. 2019. Available online: https://www.clinicaltrials.gov/study/NCT03821233 (accessed on 29 January 2025).
- Oh, D.Y.; Bedard, P.L.; Lee, K.W.; Han, H.S.; Kang, Y.K.; Miller, W.H.; Rha, S.Y.; Kim, J.H.; Dotan, E.; Liao, C.Y.; et al. Abstract B130: Phase 1 study of Zanidatamab Zovodotin (ZW49): Safety Profile and Recommended Dose (RD) in patients with Human Epidermal Growth Factor 2 (HER2)-positive solid cancers. Mol. Cancer Ther. 2023, 22, B130. [Google Scholar] [CrossRef]
- Evaluation of JSKN016 Combination Therapy in Subjects with Advanced Non-Small Cell Lung Cancer: A Phase Ib Study. 2025. Available online: https://clinicaltrials.gov/study/NCT06868732 (accessed on 11 March 2025).
- Alphamab Oncology. Pipeline Programs: JSKN016. Available online: https://www.alphamabonc.com/en/pipeline/jskn016.html (accessed on 30 August 2025).
- Li, Z.; Shang, C.; Guan, X.; An, G.; Guo, Y.; Zhang, E.; Lin, Q.; Yang, Y. Abstract LB215: A first-in-class anti-TROP2/EGFR bispecific antibody-drug conjugate, DM001, exhibits potent anti-tumor efficacy. Cancer Res. 2023, 83, LB215. [Google Scholar] [CrossRef]
- A Phase I, Multicenter, Open-label, First-in-Human, Dose Escalation and Expansion Study of DM001 in Patients with Advanced Solid Tumors. 2024. Available online: https://clinicaltrials.gov/study/NCT06475937 (accessed on 17 April 2025).
- A Phase 1, Multicenter, Open-Label, First-in-Human, Dose Escalation and Expansion Study of DM005 in Patients with Advanced Solid Tumors. 2024. Available online: https://clinicaltrials.gov/study/NCT06515990 (accessed on 15 January 2025).
- Wei, H.; Dai, W.; Yu, W.; Ma, S.; Zhu, L.; Cheng, X.; Chen, R.; Shen, Y. DM005: An anti-EGFR/c-MET bispecific antibody-drug conjugate for advanced solid tumors. J. Clin. Oncol. 2025, 43, e15010. [Google Scholar] [CrossRef]
- Han, Y.; Dai, W.; Shang, C.; Li, Z.; Han, Z.; Li, J.; Cui, Z.; An, G.; Hao, W.; Liu, Y.; et al. 1150 DM005, an EGFR × MET bispecific antibody-drug conjugate, showed robust anti-tumor activity in PDX models. J. Immunother. Cancer 2023, 11, A1–A1731. [Google Scholar] [CrossRef]
- Evaluate BL-B01D1 in Patients with Metastatic or Unresectable Non-Small Cell Lung Cancer (NSCLC) and Other Solid Tumors. 2023. Available online: https://clinicaltrials.gov/study/NCT05983432 (accessed on 6 August 2025).
- A Phase I Clinical Study to Evaluate the Safety, Tolerability, Pharmacokinetic Characteristics and Preliminary Efficacy of BL-B01D1 in Patients with Locally Advanced or Metastatic Gastrointestinal Tumor and Other Solid Tumor. 2022. Available online: https://clinicaltrials.gov/study/NCT05194982 (accessed on 26 September 2025).
- A Study of BL-B16D1 in Patients with Locally Advanced or Metastatic Solid Tumors. 2024. Available online: https://clinicaltrials.gov/study/NCT06475131 (accessed on 18 September 2025).
- A Phase I Clinical Study to Evaluate the Safety, Tolerability, Pharmacokinetics and Preliminary Efficacy of BL-B16D1 in Patients with Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma and Other Solid Tumors. 2024. Available online: https://clinicaltrials.gov/study/NCT06469008 (accessed on 14 October 2025).
- A Study of BL-B16D1 in Patients with Unresectable Locally Advanced or Metastatic Breast Cancer and Other Solid Tumors. 2025. Available online: https://clinicaltrials.gov/study/NCT06493864 (accessed on 18 September 2025).
- A Phase 1 Study of IBI3001 in Participants with Unresectable, Locally Advanced or Metastatic Solid Tumors. 2024. Available online: https://clinicaltrials.gov/study/NCT06349408 (accessed on 27 January 2025).
- Guan, J.; Zhang, X.; Wu, W.; Cao, L.; Liao, Z.; Zhu, T.; Liu, C.; Zhou, S.; Lu, J.; Li, N.; et al. Abstract LB055: IBI3001: A potentially first-in-class site-specifically conjugated B7-H3/EGFR bispecific ADC for multiple solid tumors. Cancer Res. 2024, 84, LB055. [Google Scholar] [CrossRef]
- Institute, N.C. anti-B7-H3/anti-EGFR Bispecific Antibody-Drug Conjugate IBI3001. Available online: https://www.cancer.gov/publications/dictionaries/cancer-drug/def/anti-b7-h3-anti-egfr-bispecific-antibody-drug-conjugate-ibi3001 (accessed on 30 August 2025).
- Drilon, A.E.; Awad, M.M.; Gadgeel, S.M.; Villaruz, L.C.; Sabari, J.K.; Perez, J.; Daly, C.; Patel, S.; Li, S.; Seebach, F.A.; et al. A phase 1/2 study of REGN5093-M114, a METxMET antibody-drug conjugate, in patients with mesenchymal epithelial transition factor (MET)-overexpressing NSCLC. J. Clin. Oncol. 2022, 40, TPS8593. [Google Scholar] [CrossRef]
- A Phase 1/2 Study of REGN5093-M114 (METxMET Antibody-Drug Conjugate) in Patients with MET Overexpressing Advanced Cancer. 2021. Available online: https://clinicaltrials.gov/study/NCT04982224 (accessed on 28 February 2025).
- A Phase 1/2 Study of MEDI4276 in Adults Subjects with Select HER2-expressing Advanced Solid Tumors (MEDI4276). 2018. Available online: https://clinicaltrials.gov/study/NCT02576548 (accessed on 18 July 2019).
- Pegram, M.D.; Hamilton, E.P.; Tan, A.R.; Storniolo, A.M.; Balic, K.; Rosenbaum, A.I.; Liang, M.; He, P.; Marshall, S.; Scheuber, A.; et al. First-in-Human, Phase 1 Dose-Escalation Study of Biparatopic Anti-HER2 Antibody–Drug Conjugate MEDI4276 in Patients with HER2-positive Advanced Breast or Gastric Cancer. Mol. Cancer Ther. 2021, 20, 1442–1453. [Google Scholar] [CrossRef]
- A Phase 1/2 Study of GEN1286 in Patients with Advanced Solid Tumors. 2024. Available online: https://clinicaltrials.gov/study/NCT06685068 (accessed on 8 October 2025).
- Xiao, Y.; Huang, H.; Yin, J.; Qiu, X.; Wang, L.; Liu, H.; Zhao, B.; Chen, Z. Abstract 6580: A Novel EGFR x cMET bispecific ADC PRO1286 demonstrated broad antitumor activity and promising tolerability in preclinical models. Cancer Res. 2024, 84, 6580. [Google Scholar] [CrossRef]
- Phase 1/2A Open-Label Clinical Trial Evaluating Vbc101, an Egfr and Cmet Targeted BI-Specific Antibody Drug Conjugate. 2025. Available online: https://clinicaltrials.gov/study/NCT07136779 (accessed on 22 August 2025).
- A Phase I, Multicentre, Open-Label, First-in-Human, Dose Escalation and Expansion Study of DM002 in Patients with Advanced Solid Tumors. 2024. Available online: https://adisinsight.springer.com/trials/700379138 (accessed on 22 April 2025).
- Wang, W.; Li, J.; Ma, L.; Xu, M.; Yin, Q. Abstract LB043: VBC101-F11: An innovative EGFR/cMet bispecific antibody drug conjugate (ADC) targeting key oncogenic drivers in solid tumors. Cancer Res. 2024, 84, LB043. [Google Scholar] [CrossRef]
- A Phase II Clinical Study to Evaluate the Efficacy and Safety of BL-B01D1+ Axitinib Without or with Pembrolizumab (BL-B01D1+ Axitinib ± Pembrolizumab) in Patients with Locally Advanced or Metastatic Renal Cancer. 2025. Available online: https://clinicaltrials.gov/study/NCT06962787 (accessed on 28 August 2025).
- A Randomized, Open-label, Multicenter, Parallel-controlled Phase III Clinical Trial to Evaluate the Efficacy and Safety of TQB2102 for Injection Versus TCbHP in Neoadjuvant Treatment of Breast Cancer with Positive HER2 Expression. 2025. Available online: https://clinicaltrials.gov/study/NCT07043725 (accessed on 23 September 2025).
- Li, J.; Zhang, Q.; Zeng, X.; Zhang, W.; Chen, L.; Wu, J.; Liu, G.; Wang, Z.; Hu, X.; Shao, Z. Efficacy and safety of neoadjuvant TQB2102 in women with locally advanced or early HER2-positive breast cancer: A randomized, open-label, multi-centre phase 2 trial. J. Clin. Oncol. 2025, 43, 591. [Google Scholar] [CrossRef]
- A Randomized, Open-Label, Parallel-Controlled, Multi-center Phase III Study of JSKN003 Versus Investigator-Choice Chemotherapy for Platinum-Resistant, Relapsed Epithelial Ovarian, Primary Peritoneal, or Fallopian Tube Cancer. 2024. Available online: https://www.clinicaltrials.gov/study/NCT06751485 (accessed on 31 December 2024).
- A Randomized, Controlled, Open-Label, Multicenter, Phase 3 Study to Compare the Efficacy and Safety of JSKN003 Versus Trastuzumab Emtansine (T-DM1) for HER2-Positive, Advanced Breast Cancer Subjects. 2025. Available online: https://clinicaltrials.gov/study/NCT06846437 (accessed on 24 March 2025).
- Oncology, A. Alphamab Oncology Presented Multiple Clinical Data of Anti-HER2 Biparatopic ADC JSKN003 at the 2025 ASCO Annual Meeting. Available online: https://www.prnewswire.com/apac/news-releases/alphamab-oncology-presented-multiple-clinical-data-of-anti-her2-biparatopic-adc-jskn003-at-the-2025-asco-annual-meeting-302471380.html (accessed on 30 August 2025).
- A Phase III Randomized Controlled Clinical Study Comparing BL-B01D1 with Chemotherapy of Physician’s Choice in Patients with Unresectable Locally Advanced or Metastatic Triple-Negative Breast Cancer After Taxane Failure. 2024. Available online: https://adisinsight.springer.com/trials/700373045 (accessed on 4 May 2025).
- A Phase III Randomized Controlled Clinical Study Comparing BL-B01D1 with Chemotherapy of Physician’s Choice in Patients with Unresectable Locally Advanced, Recurrent, or Metastatic HR+HER2- Breast Cancer. 2025. Available online: https://clinicaltrials.gov/study/NCT06343948 (accessed on 25 June 2025).
- A Study Comparing BL-B01D1 with Chemotherapy of Physician’s Choice in Patients with Recurrent or Metastatic Esophageal Squamous Cell Carcinoma. 2025. Available online: https://clinicaltrials.gov/study/NCT06304974 (accessed on 2 May 2025).
- Zhang, Y.; Shang, C.; Wang, N.; An, G.; Zhang, E.; Lin, Q.; Yang, Y. Abstract LB214: A first-in-class bispecific antibody-drug conjugate (DM002) targeting HER3 and the juxtamembrane domain of MUC1. Cancer Res. 2023, 83, LB214. [Google Scholar] [CrossRef]
- Zhang, Y.; Shang, C.; Wang, N.; An, G.; Guo, C.; An, W.F.; Yang, Y. Preclinical Efficacy of DM002, a bispecific HER3× MUC1 antibody-drug conjugate with a novel DNA topoisomerase I inhibitor, in solid tumor models. Cancer Res. 2024, 84, 2621. [Google Scholar] [CrossRef]
- Weekes, C.D.; Lamberts, L.E.; Borad, M.J.; Voortman, J.; McWilliams, R.R.; Diamond, J.R.; De Vries, E.G.; Verheul, H.M.; Lieu, C.H.; Kim, G.P. Phase I study of DMOT4039A, an antibody–drug conjugate targeting mesothelin, in patients with unresectable pancreatic or platinum-resistant ovarian cancer. Mol. Cancer Ther. 2016, 15, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Zhang, J.; Wang, Z.; Lan, H.; Hou, J.; Zhang, N.; Wang, X.; Lu, H. Targeting Trop-2 in cancer: Recent research progress and clinical application. Biochim. Et Biophys. Acta (BBA)-Rev. Cancer 2023, 1878, 188902. [Google Scholar] [CrossRef]
- Cazes, A.; Betancourt, O.; Esparza, E.; Mose, E.S.; Jaquish, D.; Wong, E.; Wascher, A.A.; Tiriac, H.; Gymnopoulos, M.; Lowy, A.M. A MET Targeting antibody–drug conjugate overcomes gemcitabine resistance in pancreatic cancer. Clin. Cancer Res. 2021, 27, 2100–2110. [Google Scholar] [CrossRef]
- Schmiechen, Z.C.; Stromnes, I.M. Mechanisms governing immunotherapy resistance in pancreatic ductal adenocarcinoma. Front. Immunol. 2021, 11, 613815. [Google Scholar] [CrossRef]
- Erkan, M.; Hausmann, S.; Michalski, C.W.; Fingerle, A.A.; Dobritz, M.; Kleeff, J.; Friess, H. The role of stroma in pancreatic cancer: Diagnostic and therapeutic implications. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 454–467. [Google Scholar] [CrossRef]
- Li, S.; Zhao, X.; Fu, K.; Zhu, S.; Pan, C.; Yang, C.; Wang, F.; To, K.K.W.; Fu, L. Resistance to antibody-drug conjugates: A review. Acta Pharm. Sin. B 2025, 15, 737–756. [Google Scholar] [CrossRef]
- He, J.; Zeng, X.; Wang, C.; Wang, E.; Li, Y. Antibody–drug conjugates in cancer therapy: Mechanisms and clinical studies. MedComm. 2024, 5, e671. [Google Scholar] [CrossRef]
- Kalim, M.; Chen, J.; Wang, S.; Lin, C.; Ullah, S.; Liang, K.; Ding, Q.; Chen, S.; Zhan, J. Intracellular trafficking of new anticancer therapeutics: Antibody–drug conjugates. Drug Des. Dev. Ther. 2017, 11, 2265–2276. [Google Scholar] [CrossRef]
- Ward, E.S.; Devanaboyina, S.C.; Ober, R.J. Targeting FcRn for the modulation of antibody dynamics. Mol. Immunol. 2015, 67, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef]
- Um, W.; Park, J.; Youn, A.; Cho, H.; Lim, S.; Lee, J.W.; Yoon, H.Y.; Lim, D.-K.; Park, J.H.; Kim, K. A Comparative Study on Albumin-Binding Molecules for Targeted Tumor Delivery through Covalent and Noncovalent Approach. Bioconjugate Chem. 2019, 30, 3107–3118. [Google Scholar] [CrossRef] [PubMed]
- Park, A.-R.; Jung, E.-Y.; Shin, H.H.; Park, J.-H.; Lim, S.I.; Lee, D.-H. Single-chain variable fragments targeting domain II of human serum albumin for enhanced circulatory half-life via albumin association. Int. J. Biol. Macromol. 2025, 322, 146933. [Google Scholar] [CrossRef] [PubMed]
- Son, S.; Deepagan, V.G.; Shin, S.; Ko, H.; Min, J.; Um, W.; Jeon, J.; Kwon, S.; Lee, E.S.; Suh, M. Ultrasmall gold nanosatellite-bearing transformable hybrid nanoparticles for deep tumor penetration. Acta Biomater. 2018, 79, 294–305. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).