


Epigenetic Regulation of Stromal and Immune Cells and Therapeutic Targets in the Tumor Microenvironment

 ,
,
Abstract
1. Background
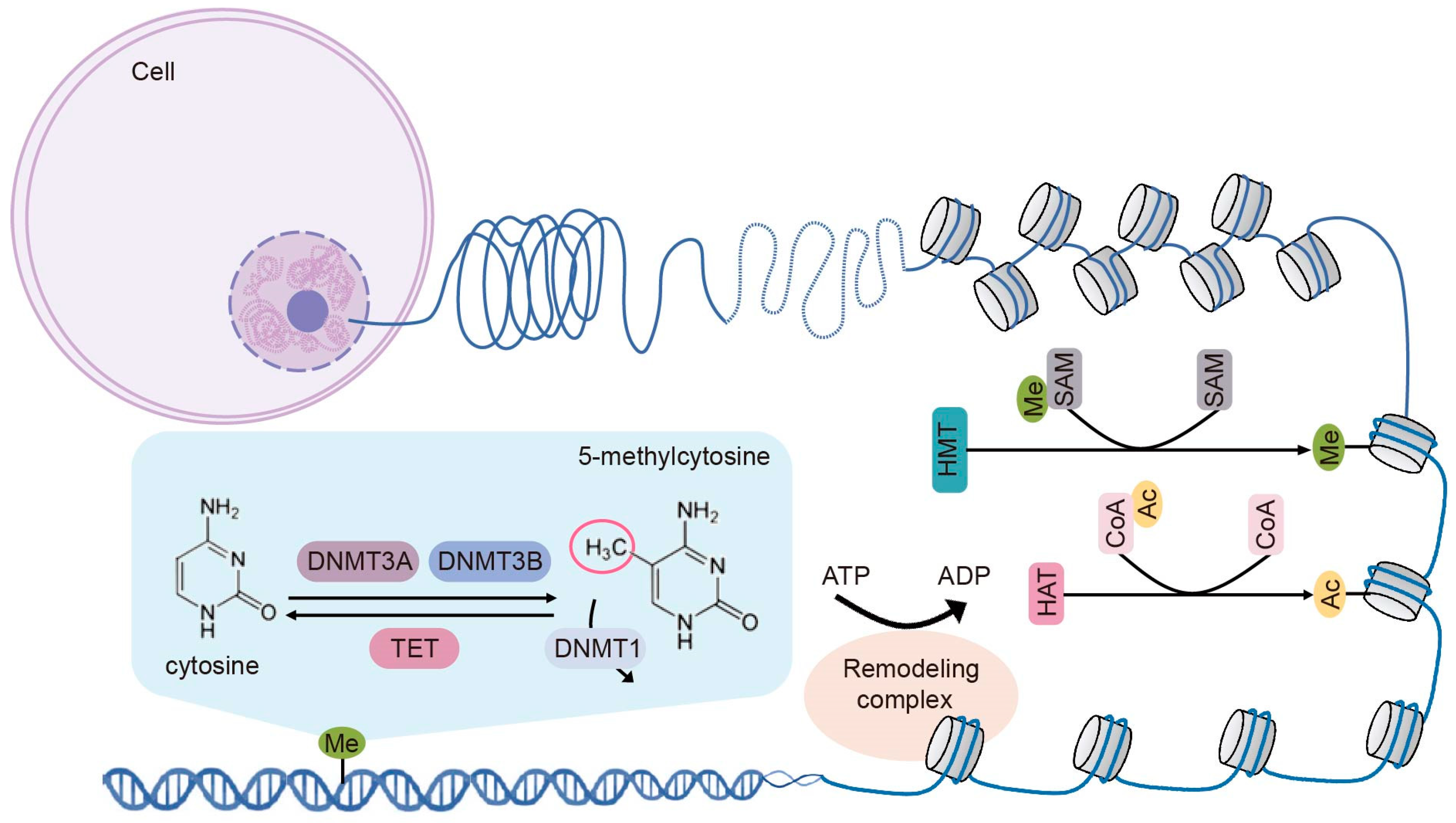
2. Fundamental Epigenetic Modifications
2.1. DNA Methylation
2.2. Histone Modifications
2.2.1. Histone Methylation
2.2.2. Histone Acetylation
2.3. Chromatin Remodeling
3. Basic Cellular Components of the TME
3.1. Cancer-Associated Fibroblasts
3.2. Tumor-Associated Macrophages
3.3. Myeloid-Derived Suppressor Cells
3.4. Tumor-Infiltrating Lymphocytes
4. Epigenetic Modifications in TME
4.1. Epigenetic Modifications of CAFs
4.1.1. DNA Methylation in CAFs
4.1.2. Histone Modifications and Chromatin Remodeling in CAFs
4.2. Epigenetic Modulations of TAMs
4.2.1. DNA Methylation in TAMs
4.2.2. Histone Modifications in TAMs
4.3. Epigenetic Modulations of MDSCs
4.3.1. DNA Methylation in MDSCs
4.3.2. Histone Modifications and Chromatin Remodeling in MDSCs
4.4. Epigenetic Modulations of TIL T Cells
4.4.1. DNA Methylation in TIL T Cells
4.4.2. Histone Modifications and Chromatin Remodeling in TIL T Cells
5. Epigenetic Therapies Target TME
5.1. DNA Methyltransferase Inhibitor
5.2. Histone Modification Inhibitors
5.3. Chromatin Remodeler Modulators
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Seferbekova, Z.; Lomakin, A.; Yates, L.R.; Gerstung, M. Spatial biology of cancer evolution. Nat. Rev. Genet. 2023, 24, 295–313. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Yu, D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol. Ther. 2021, 221, 107753. [Google Scholar] [CrossRef] [PubMed]
- Arneth, B. Tumor Microenvironment. Medicina 2019, 56, 15. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Harvey, Z.H.; Chen, Y.; Jarosz, D.F. Protein-Based Inheritance: Epigenetics beyond the Chromosome. Mol. Cell 2018, 69, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Recillas-Targa, F. Cancer Epigenetics: An Overview. Arch. Med. Res. 2022, 53, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Mendes, B.B.; Sousa, D.P.; Conniot, J.; Conde, J. Nanomedicine-based strategies to target and modulate the tumor microenvironment. Trends Cancer 2021, 7, 847–862. [Google Scholar] [CrossRef]
- Bates, S.E. Epigenetic Therapies for Cancer. N. Engl. J. Med. 2020, 383, 650–663. [Google Scholar] [CrossRef]
- Yang, J.; Xu, J.; Wang, W.; Zhang, B.; Yu, X.; Shi, S. Epigenetic regulation in the tumor microenvironment: Molecular mechanisms and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 210. [Google Scholar] [CrossRef] [PubMed]
- Hogg, S.J.; Beavis, P.A.; Dawson, M.A.; Johnstone, R.W. Targeting the epigenetic regulation of antitumour immunity. Nat. Rev. Drug Discov. 2020, 19, 776–800. [Google Scholar] [CrossRef] [PubMed]
- Dai, E.; Zhu, Z.; Wahed, S.; Qu, Z.; Storkus, W.J.; Guo, Z.S. Epigenetic modulation of antitumor immunity for improved cancer immunotherapy. Mol. Cancer 2021, 20, 171. [Google Scholar] [CrossRef]
- Song, H.; Liu, D.; Dong, S.; Zeng, L.; Wu, Z.; Zhao, P.; Zhang, L.; Chen, Z.-S.; Zou, C. Epitranscriptomics and epiproteomics in cancer drug resistance: Therapeutic implications. Signal Transduct. Target. Ther. 2020, 5, 193. [Google Scholar] [CrossRef] [PubMed]
- Turk, A.; Čeh, E.; Calin, G.A.; Kunej, T. Multiple omics levels of chronic lymphocytic leukemia. Cell Death Discov. 2024, 10, 293. [Google Scholar] [CrossRef] [PubMed]
- Mattei, A.L.; Bailly, N.; Meissner, A. DNA methylation: A historical perspective. Trends Genet. 2022, 38, 676–707. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Li, E.; Bestor, T.H.; Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992, 69, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Shimbo, T.; Song, X.; Wade, P.A.; Min, J. Proteins That Read DNA Methylation. Adv. Exp. Med. Biol. 2022, 1389, 269–293. [Google Scholar]
- Bray, J.K.; Dawlaty, M.M.; Verma, A.; Maitra, A. Roles and Regulations of TET Enzymes in Solid Tumors. Trends Cancer 2021, 7, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA methylation landscape of cancer. Trends Genet. 2021, 37, 1012–1027. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, Z.; Jia, J.; Du, T.; Zhang, N.; Tang, Y.; Fang, Y.; Fang, D. Overview of Histone Modification. Adv. Exp. Med. Biol. 2021, 1283, 1–16. [Google Scholar]
- Neganova, M.E.; Klochkov, S.G.; Aleksandrova, Y.R.; Aliev, G. Histone modifications in epigenetic regulation of cancer: Perspectives and achieved progress. Semin. Cancer Biol. 2022, 83, 452–471. [Google Scholar] [CrossRef]
- Mushtaq, A.; Mir, U.S.; Hunt, C.R.; Pandita, S.; Tantray, W.W.; Bhat, A.; Pandita, R.K.; Altaf, M.; Pandita, T.K. Role of Histone Methylation in Maintenance of Genome Integrity. Genes 2021, 12, 1000. [Google Scholar] [CrossRef]
- Li, Y.; Chen, X.; Lu, C. The interplay between DNA and histone methylation: Molecular mechanisms and disease implications. EMBO Rep. 2021, 22, e51803. [Google Scholar] [CrossRef]
- Husmann, D.; Gozani, O. Histone lysine methyltransferases in biology and disease. Nat. Struct. Mol. Biol. 2019, 26, 880–889. [Google Scholar] [CrossRef] [PubMed]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hu, Y.; Zhang, B.; Liang, X.; Li, X. The JMJD Family Histone Demethylases in Crosstalk Between Inflammation and Cancer. Front. Immunol. 2022, 13, 881396. [Google Scholar] [CrossRef] [PubMed]
- Shvedunova, M.; Akhtar, A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2022, 23, 329–349. [Google Scholar] [CrossRef]
- Minisini, M.; Mascaro, M.; Brancolini, C. HDAC-driven mechanisms in anticancer resistance: Epigenetics and beyond. Cancer Drug Resist. 2024, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Schizas, D.; Mastoraki, A.; Naar, L.; Tsilimigras, D.I.; Katsaros, I.; Fragkiadaki, V.; Karachaliou, G.-S.; Arkadopoulos, N.; Liakakos, T.; Moris, D. Histone Deacetylases (HDACs) in Gastric Cancer: An Update of their Emerging Prognostic and Therapeutic Role. Curr. Med. Chem. 2020, 27, 6099–6111. [Google Scholar] [CrossRef]
- Gold, S.; Shilatifard, A. Therapeutic targeting of BET bromodomain and other epigenetic acetylrecognition domain-containing factors. Curr. Opin. Genet. Dev. 2024, 86, 102181. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D. To slide or not to slide: Key role of the hexasome in chromatin remodeling revealed. Nat. Struct. Mol. Biol. 2024, 31, 742–746. [Google Scholar] [CrossRef]
- Mashtalir, N.; D’avino, A.R.; Michel, B.C.; Luo, J.; Pan, J.; Otto, J.E.; Zullow, H.J.; McKenzie, Z.M.; Kubiak, R.L.; Pierre, R.S.; et al. Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 2018, 175, 1272–1288.e20. [Google Scholar] [CrossRef] [PubMed]
- Malone, H.A.; Roberts, C.W.M. Chromatin remodellers as therapeutic targets. Nat. Rev. Drug Discov. 2024, 23, 661–681. [Google Scholar] [CrossRef] [PubMed]
- Kadoch, C. Diverse compositions and functions of chromatin remodeling machines in cancer. Sci. Transl. Med. 2019, 11, eaay1018. [Google Scholar] [CrossRef]
- de Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef] [PubMed]
- Toninelli, M.; Rossetti, G.; Pagani, M. Charting the tumor microenvironment with spatial profiling technologies. Trends Cancer 2023, 9, 1085–1096. [Google Scholar] [CrossRef]
- Elhanani, O.; Ben-Uri, R.; Keren, L. Spatial profiling technologies illuminate the tumor microenvironment. Cancer Cell 2023, 41, 404–420. [Google Scholar] [CrossRef]
- Polak, R.; Zhang, E.T.; Kuo, C.J. Cancer organoids 2.0: Modelling the complexity of the tumour immune microenvironment. Nat. Rev. Cancer 2024, 24, 523–539. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Desbois, M.; Wang, Y. Cancer-associated fibroblasts: Key players in shaping the tumor immune microenvironment. Immunol. Rev. 2021, 302, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Tsoumakidou, M. The advent of immune stimulating CAFs in cancer. Nat. Rev. Cancer 2023, 23, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Tan, Z.W.; Zhu, P.; Tan, N.S. Cancer-associated fibroblasts in tumor microenvironment—Accomplices in tumor malignancy. Cell Immunol. 2019, 343, 103729. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; McAndrews, K.M.; Kalluri, R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat. Rev. Clin. Oncol. 2021, 18, 792–804. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Tumor-associated macrophages. Curr. Biol. 2020, 30, R246–R248. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 323–338.e6. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; Sureka, N.; Ahuja, S.; Aden, D.; Zaheer, S.; Zaheer, S. Tumor-associated macrophages: A sentinel of innate immune system in tumor microenvironment gone haywire. Cell Biol. Int. 2024, 48, 1406–1449. [Google Scholar] [CrossRef] [PubMed]
- McWhorter, R.; Bonavida, B. The Role of TAMs in the Regulation of Tumor Cell Resistance to Chemotherapy. Crit. Rev. Oncog. 2024, 29, 97–125. [Google Scholar] [CrossRef]
- Li, S.; Sheng, J.; Zhang, D.; Qin, H. Targeting tumor-associated macrophages to reverse antitumor drug resistance. Aging 2024, 16, 10165–10196. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Hashemi Goradel, N.; Farhood, B.; Salehi, E.; Nashtaei, M.S.; Khanlarkhani, N.; Khezri, Z.; Majidpoor, J.; Abouzaripour, M.; Habibi, M.; et al. Macrophage polarity in cancer: A review. J. Cell Biochem. 2019, 120, 2756–2765. [Google Scholar] [CrossRef]
- Rajkumari, S.; Singh, J.; Agrawal, U.; Agrawal, S. Myeloid-derived suppressor cells in cancer: Current knowledge and future perspectives. Int. Immunopharmacol. 2024, 142, 112949. [Google Scholar] [CrossRef]
- Lasser, S.A.; Ozbay Kurt, F.G.; Arkhypov, I.; Utikal, J.; Umansky, V. Myeloid-derived suppressor cells in cancer and cancer therapy. Nat. Rev. Clin. Oncol. 2024, 21, 147–164. [Google Scholar] [CrossRef]
- Adeshakin, A.O.; Liu, W.; Adeshakin, F.O.; Afolabi, L.O.; Zhang, M.; Zhang, G.; Wang, L.; Li, Z.; Lin, L.; Cao, Q.; et al. Regulation of ROS in myeloid-derived suppressor cells through targeting fatty acid transport protein 2 enhanced anti-PD-L1 tumor immunotherapy. Cell Immunol. 2021, 362, 104286. [Google Scholar] [CrossRef]
- Jachetti, E.; Sangaletti, S.; Chiodoni, C.; Ferrara, R.; Colombo, M.P. Modulation of PD-1/PD-L1 axis in myeloid-derived suppressor cells by anti-cancer treatments. Cell Immunol. 2021, 362, 104301. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Teng, D.; Yang, L.; Xu, X.; Chen, J.; Jiang, T.; Feng, A.Y.; Zhang, Y.; Frederick, D.T.; Gu, L.; et al. Myeloid-derived itaconate suppresses cytotoxic CD8+ T cells and promotes tumour growth. Nat. Metab. 2022, 4, 1660–1673. [Google Scholar] [CrossRef]
- Azzaoui, I.; Uhel, F.; Rossille, D.; Pangault, C.; Dulong, J.; Le Priol, J.; Lamy, T.; Houot, R.; Le Gouill, S.; Cartron, G.; et al. T-cell defect in diffuse large B-cell lymphomas involves expansion of myeloid-derived suppressor cells. Blood 2016, 128, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Kapor, S.; Radojković, M.; Santibanez, J.F. Myeloid-derived suppressor cells: Implication in myeloid malignancies and immunotherapy. Acta Histochem. 2024, 126, 152183. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Lin, A.; Jiang, A.; Zhang, C.; Zhang, J.; Cheng, Q.; Luo, P.; Bai, Y. CTLs heterogeneity and plasticity: Implications for cancer immunotherapy. Mol. Cancer 2024, 23, 58. [Google Scholar] [CrossRef] [PubMed]
- Maimela, N.R.; Liu, S.; Zhang, Y. Fates of CD8+ T cells in Tumor Microenvironment. Comput. Struct. Biotechnol. J. 2019, 17, 1–13. [Google Scholar] [CrossRef]
- Speiser, D.E.; Chijioke, O.; Schaeuble, K.; Münz, C. CD4+ T cells in cancer. Nat. Cancer 2023, 4, 317–329. [Google Scholar] [CrossRef]
- Ruterbusch, M.; Pruner, K.B.; Shehata, L.; Pepper, M. In Vivo CD4+ T Cell Differentiation and Function: Revisiting the Th1/Th2 Paradigm. Annu. Rev. Immunol. 2020, 38, 705–725. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T cells in tumor microenvironment: New mechanisms, potential therapeutic strategies and future prospects. Mol. Cancer 2020, 19, 116. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, T.; Gu, J.; Lu, L. Targeting the metabolism of tumor-infiltrating regulatory T cells. Trends Immunol. 2023, 44, 598–612. [Google Scholar] [CrossRef]
- Kyrysyuk, O.; Wucherpfennig, K.W. Designing Cancer Immunotherapies That Engage T Cells and NK Cells. Annu. Rev. Immunol. 2023, 41, 17–38. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Rebuffet, L.; Narni-Mancinelli, E.; Cornen, S.; Igarashi, R.Y.; Fantin, V.R. Natural killer cell therapies. Nature 2024, 626, 727–736. [Google Scholar] [CrossRef]
- Ruffin, A.T.; Cillo, A.R.; Tabib, T.; Liu, A.; Onkar, S.; Kunning, S.R.; Lampenfeld, C.; Atiya, H.I.; Abecassis, I.; Kürten, C.H.L.; et al. B cell signatures and tertiary lymphoid structures contribute to outcome in head and neck squamous cell carcinoma. Nat. Commun. 2021, 12, 3349. [Google Scholar] [CrossRef]
- Downs-Canner, S.M.; Meier, J.; Vincent, B.G.; Serody, J.S. B Cell Function in the Tumor Microenvironment. Annu. Rev. Immunol. 2022, 40, 169–193. [Google Scholar] [CrossRef]
- Qayoom, H.; Sofi, S.; Mir, M.A. Targeting tumor microenvironment using tumor-infiltrating lymphocytes as therapeutics against tumorigenesis. Immunol. Res. 2023, 71, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef] [PubMed]
- Saw, P.E.; Chen, J.; Song, E. Targeting CAFs to overcome anticancer therapeutic resistance. Trends Cancer 2022, 8, 527–555. [Google Scholar] [CrossRef]
- Madar, S.; Goldstein, I.; Rotter, V. ‘Cancer associated fibroblasts’—More than meets the eye. Trends Mol. Med. 2013, 19, 447–453. [Google Scholar] [CrossRef]
- Su, S.-F.; Ho, H.; Li, J.-H.; Wu, M.-F.; Wang, H.-C.; Yeh, H.-Y.; Kuo, S.-W.; Chen, H.-W.; Ho, C.-C.; Li, K.-C. DNA methylome and transcriptome landscapes of cancer-associated fibroblasts reveal a smoking-associated malignancy index. J. Clin. Investig. 2021, 131, e139552. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Wang, F.; Gao, X.; Zhao, H.; Zhang, J.; Wang, N.; Liu, Z.; Yan, X.; Jin, J.; Ba, Y.; et al. Integrated analysis of genome-wide DNA methylation and cancer-associated fibroblasts identified prognostic biomarkers and immune checkpoint blockade in lower grade gliomas. Front. Oncol. 2022, 12, 977251. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Maié, T.; Cramer, T.; Costa, I.G.; Wagner, W. Cancer-associated fibroblasts reveal aberrant DNA methylation across different types of cancer. Clin. Epigenet. 2024, 16, 164. [Google Scholar] [CrossRef]
- Pidsley, R.; Lawrence, M.G.; Zotenko, E.; Niranjan, B.; Statham, A.; Song, J.; Chabanon, R.M.; Qu, W.; Wang, H.; Richards, M.; et al. Enduring epigenetic landmarks define the cancer microenvironment. Genome Res. 2018, 28, 625–638. [Google Scholar] [CrossRef]
- Halperin, C.; Hey, J.; Weichenhan, D.; Stein, Y.; Mayer, S.; Lutsik, P.; Plass, C.; Scherz-Shouval, R. Global DNA Methylation Analysis of Cancer-Associated Fibroblasts Reveals Extensive Epigenetic Rewiring Linked with RUNX1 Upregulation in Breast Cancer Stroma. Cancer Res. 2022, 82, 4139–4152. [Google Scholar] [CrossRef] [PubMed]
- Mathot, P.; Grandin, M.; Devailly, G.; Souaze, F.; Cahais, V.; Moran, S.; Campone, M.; Herceg, Z.; Esteller, M.; Juin, P.; et al. DNA methylation signal has a major role in the response of human breast cancer cells to the microenvironment. Oncogenesis 2017, 6, e390. [Google Scholar] [CrossRef] [PubMed]
- Albrengues, J.; Bourget, I.; Pons, C.; Butet, V.; Hofman, P.; Tartare-Deckert, S.; Feral, C.C.; Meneguzzi, G.; Gaggioli, C. LIF mediates proinvasive activation of stromal fibroblasts in cancer. Cell Rep. 2014, 7, 1664–1678. [Google Scholar] [CrossRef] [PubMed]
- Albrengues, J.; Bertero, T.; Grasset, E.; Bonan, S.; Maiel, M.; Bourget, I.; Philippe, C.; Herraiz Serrano, C.; Benamar, S.; Croce, O.; et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat. Commun. 2015, 6, 10204. [Google Scholar] [CrossRef]
- Xiao, Q.; Zhou, D.; Rucki, A.A.; Williams, J.; Zhou, J.; Mo, G.; Murphy, A.; Fujiwara, K.; Kleponis, J.; Salman, B.; et al. Cancer-Associated Fibroblasts in Pancreatic Cancer Are Reprogrammed by Tumor-Induced Alterations in Genomic DNA Methylation. Cancer Res. 2016, 76, 5395–5404. [Google Scholar] [CrossRef]
- Becker, L.M.; O’Connell, J.T.; Vo, A.P.; Cain, M.P.; Tampe, D.; Bizarro, L.; Sugimoto, H.; McGow, A.K.; Asara, J.M.; Lovisa, S.; et al. Epigenetic Reprogramming of Cancer-Associated Fibroblasts Deregulates Glucose Metabolism and Facilitates Progression of Breast Cancer. Cell Rep. 2020, 31, 107701. [Google Scholar] [CrossRef] [PubMed]
- Tyan, S.-W.; Hsu, C.-H.; Peng, K.-L.; Chen, C.-C.; Kuo, W.-H.; Lee, E.Y.-H.P.; Shew, J.-Y.; Chang, K.-J.; Juan, L.-J.; Lee, W.-H. Breast cancer cells induce stromal fibroblasts to secrete ADAMTS1 for cancer invasion through an epigenetic change. PLoS ONE 2012, 7, e35128. [Google Scholar] [CrossRef] [PubMed]
- Eckert, M.A.; Coscia, F.; Chryplewicz, A.; Chang, J.W.; Hernandez, K.M.; Pan, S.; Tienda, S.M.; Nahotko, D.A.; Li, G.; Blaženović, I.; et al. Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts. Nature 2019, 569, 723–728. [Google Scholar] [CrossRef]
- Yamamoto, J.; Han, Q.; Inubushi, S.; Sugisawa, N.; Hamada, K.; Nishino, H.; Miyake, K.; Kumamoto, T.; Matsuyama, R.; Bouvet, M.; et al. Histone methylation status of H3K4me3 and H3K9me3 under methionine restriction is unstable in methionine-addicted cancer cells, but stable in normal cells. Biochem. Biophys. Res. Commun. 2020, 533, 1034–1038. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, Y.; Hattori, N.; Iida, N.; Takeshima, H.; Maeda, M.; Kiyono, T.; Sekine, S.; Seto, Y.; Ushijima, T. SAA1 is upregulated in gastric cancer-associated fibroblasts possibly by its enhancer activation. Carcinogenesis 2021, 42, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tu, K.; Liu, D.; Guo, L.; Chen, Y.; Li, Q.; Maiers, J.L.; Liu, Z.; Shah, V.H.; Dou, C.; et al. p300 Acetyltransferase Is a Cytoplasm-to-Nucleus Shuttle for SMAD2/3 and TAZ Nuclear Transport in Transforming Growth Factor β-Stimulated Hepatic Stellate Cells. Hepatology 2019, 70, 1409–1423. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, X.-Y.; Zhang, P.; He, T.-C.; Han, J.-H.; Zhang, R.; Lin, J.; Fan, J.; Lu, L.; Zhu, W.-W.; et al. Cancer-derived exosomal HSPC111 promotes colorectal cancer liver metastasis by reprogramming lipid metabolism in cancer-associated fibroblasts. Cell Death Dis. 2022, 13, 57. [Google Scholar] [CrossRef]
- Aprelikova, O.; Yu, X.; Palla, J.; Wei, B.R.; John, S.; Yi, M.; Stephens, R.; Simpson, R.M.; Risinger, J.I.; Jazaeri, A.; et al. The role of miR-31 and its target gene SATB2 in cancer-associated fibroblasts. Cell Cycle 2010, 9, 4387–4398. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.E.; Procopio, M.-G.; Ghosh, S.; Jo, S.-H.; Goruppi, S.; Magliozzi, F.; Bordignon, P.; Neel, V.; Angelino, P.; Dotto, G.P. Convergent roles of ATF3 and CSL in chromatin control of cancer-associated fibroblast activation. J. Exp. Med. 2017, 214, 2349–2368. [Google Scholar] [CrossRef]
- Campos Gudiño, R.; McManus, K.J.; Hombach-Klonisch, S. Aberrant HMGA2 Expression Sustains Genome Instability That Promotes Metastasis and Therapeutic Resistance in Colorectal Cancer. Cancers 2023, 15, 1735. [Google Scholar] [CrossRef] [PubMed]
- Strell, C.; Norberg, K.J.; Mezheyeuski, A.; Schnittert, J.; Kuninty, P.R.; Moro, C.F.; Paulsson, J.; Schultz, N.A.; Calatayud, D.; Löhr, J.M.; et al. Stroma-regulated HMGA2 is an independent prognostic marker in PDAC and AAC. Br. J. Cancer 2017, 117, 65–77. [Google Scholar] [CrossRef]
- Zong, Y.; Huang, J.; Sankarasharma, D.; Morikawa, T.; Fukayama, M.; Epstein, J.I.; Chada, K.K.; Witte, O.N. Stromal epigenetic dysregulation is sufficient to initiate mouse prostate cancer via paracrine Wnt signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E3395–E3404. [Google Scholar] [CrossRef]
- Yang, X.; Wang, X.; Liu, D.; Yu, L.; Xue, B.; Shi, H. Epigenetic regulation of macrophage polarization by DNA methyltransferase 3b. Mol. Endocrinol. 2014, 28, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Huang, C.; Ma, T.-T.; Bian, E.-B.; He, Y.; Zhang, L.; Li, J. SOCS1 hypermethylation mediated by DNMT1 is associated with lipopolysaccharide-induced inflammatory cytokines in macrophages. Toxicol. Lett. 2014, 225, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Pan, X.; Fujiwara, K.; Jurcak, N.; Muth, S.; Zhou, J.; Xiao, Q.; Li, A.; Che, X.; Li, Z.; et al. Pancreatic cancer cells render tumor-associated macrophages metabolically reprogrammed by a GARP and DNA methylation-mediated mechanism. Signal Transduct. Target. Ther. 2021, 6, 366. [Google Scholar] [CrossRef]
- Huang, Y.; Tian, C.; Li, Q.; Xu, Q. TET1 Knockdown Inhibits Porphyromonas gingivalis LPS/IFN-γ-Induced M1 Macrophage Polarization through the NF-κB Pathway in THP-1 Cells. Int. J. Mol. Sci. 2019, 20, 2023. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Abreu-Rodriguez, I.; Ye, S.; Gay, S.; Distler, O.; Neidhart, M.; Karouzakis, E. TET1 is an important transcriptional activator of TNFα expression in macrophages. PLoS ONE 2019, 14, e0218551. [Google Scholar] [CrossRef]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.-L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Cull, A.H.; Snetsinger, B.; Buckstein, R.; Wells, R.A.; Rauh, M.J. Tet2 restrains inflammatory gene expression in macrophages. Exp. Hematol. 2017, 55, 56–70.e13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef]
- Pan, W.; Zhu, S.; Qu, K.; Meeth, K.; Cheng, J.; He, K.; Ma, H.; Liao, Y.; Wen, X.; Roden, C.; et al. The DNA Methylcytosine Dioxygenase Tet2 Sustains Immunosuppressive Function of Tumor-Infiltrating Myeloid Cells to Promote Melanoma Progression. Immunity 2017, 47, 284–297.e5. [Google Scholar] [CrossRef] [PubMed]
- Kittan, N.A.; Allen, R.M.; Dhaliwal, A.; Cavassani, K.A.; Schaller, M.; Gallagher, K.A.; Carson, W.F.; Mukherjee, S.; Grembecka, J.; Cierpicki, T.; et al. Cytokine induced phenotypic and epigenetic signatures are key to establishing specific macrophage phenotypes. PLoS ONE 2013, 8, e78045. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Luo, M.; Shao, B.; Yang, J.; Tong, A.; Wang, R.; Liu, Y.; Jun, R.; Liu, T.; Yi, T.; et al. Phosphatidylserine released from apoptotic cells in tumor induces M2-like macrophage polarization through the PSR-STAT3-JMJD3 axis. Cancer Commun. 2022, 42, 205–222. [Google Scholar] [CrossRef] [PubMed]
- Xun, J.; Du, L.; Gao, R.; Shen, L.; Wang, D.; Kang, L.; Zhang, Z.; Zhang, Y.; Yue, S.; Feng, S.; et al. Cancer-derived exosomal miR-138-5p modulates polarization of tumor-associated macrophages through inhibition of KDM6B. Theranostics 2021, 11, 6847–6859. [Google Scholar] [CrossRef]
- Wang, X.; Chen, S.; He, J.; Chen, W.; Ding, Y.; Huang, J.; Huang, J. Histone methyltransferases G9a mediated lipid-induced M1 macrophage polarization through negatively regulating CD36. Metabolism 2021, 114, 154404. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Han, Z.; Wu, Z.; Xia, Y.; Yang, G.; Yin, Y.; Ren, W. GABA regulates IL-1β production in macrophages. Cell Rep. 2022, 41, 111770. [Google Scholar] [CrossRef]
- Tokarz, P.; Płoszaj, T.; Regdon, Z.; Virág, L.; Robaszkiewicz, A. PARP1-LSD1 functional interplay controls transcription of SOD2 that protects human pro-inflammatory macrophages from death under an oxidative condition. Free Radic. Biol. Med. 2019, 131, 218–224. [Google Scholar] [CrossRef]
- Sobczak, M.; Strachowska, M.; Gronkowska, K.; Karwaciak, I.; Pułaski, Ł.; Robaszkiewicz, A. LSD1 Facilitates Pro-Inflammatory Polarization of Macrophages by Repressing Catalase. Cells 2021, 10, 2465. [Google Scholar] [CrossRef]
- Mazzarella, L.; Santoro, F.; Ravasio, R.; Fumagalli, V.; Massa, P.E.; Rodighiero, S.; Gavilán, E.; Romanenghi, M.; Duso, B.A.; Bonetti, E.; et al. Inhibition of the lysine demethylase LSD1 modulates the balance between inflammatory and antiviral responses against coronaviruses. Sci. Signal 2023, 16, eade0326. [Google Scholar] [CrossRef]
- Sun, P.; Zhang, S.-J.; Maksim, S.; Yao, Y.-F.; Liu, H.-M.; Du, J. Epigenetic Modification in Macrophages: A Promising Target for Tumor and Inflammation-associated Disease Therapy. Curr. Top. Med. Chem. 2019, 19, 1350–1362. [Google Scholar] [CrossRef]
- Tan, A.H.Y.; Tu, W.; McCuaig, R.; Hardy, K.; Donovan, T.; Tsimbalyuk, S.; Forwood, J.K.; Rao, S. Lysine-Specific Histone Demethylase 1A Regulates Macrophage Polarization and Checkpoint Molecules in the Tumor Microenvironment of Triple-Negative Breast Cancer. Front. Immunol. 2019, 10, 1351. [Google Scholar] [CrossRef]
- Boulding, T.; McCuaig, R.D.; Tan, A.; Hardy, K.; Wu, F.; Dunn, J.; Kalimutho, M.; Sutton, C.R.; Forwood, J.K.; Bert, A.G.; et al. LSD1 activation promotes inducible EMT programs and modulates the tumour microenvironment in breast cancer. Sci. Rep. 2018, 8, 73. [Google Scholar] [CrossRef]
- Zhuo, X.; Wu, Y.; Yang, Y.; Gao, L.; Qiao, X.; Chen, T. Knockdown of LSD1 meliorates Ox-LDL-stimulated NLRP3 activation and inflammation by promoting autophagy via SESN2-mesiated PI3K/Akt/mTOR signaling pathway. Life Sci. 2019, 233, 116696. [Google Scholar] [CrossRef]
- Doi, K.; Murata, K.; Ito, S.; Suzuki, A.; Terao, C.; Ishie, S.; Umemoto, A.; Murotani, Y.; Nishitani, K.; Yoshitomi, H.; et al. Role of Lysine-Specific Demethylase 1 in Metabolically Integrating Osteoclast Differentiation and Inflammatory Bone Resorption Through Hypoxia-Inducible Factor 1α and E2F1. Arthritis Rheumatol. 2022, 74, 948–960. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, H.; Kuranaga, Y.; Kumazaki, M.; Sugito, N.; Yoshikawa, Y.; Takai, T.; Taniguchi, K.; Ito, Y.; Akao, Y. Regulated Polarization of Tumor-Associated Macrophages by miR-145 via Colorectal Cancer-Derived Extracellular Vesicles. J. Immunol. 2017, 199, 1505–1515. [Google Scholar] [CrossRef]
- Wang, Y.C.; Wu, Y.S.; Hung, C.Y.; Wang, S.A.; Young, M.J.; Hsu, T.I.; Hung, J.J. USP24 induces IL-6 in tumor-associated microenvironment by stabilizing p300 and β-TrCP and promotes cancer malignancy. Nat. Commun. 2018, 9, 3996. [Google Scholar] [CrossRef]
- Shen, Y.; Wei, W.; Zhou, D.-X. Histone Acetylation Enzymes Coordinate Metabolism and Gene Expression. Trends Plant Sci. 2015, 20, 614–621. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Aksoylar, H.I.; Yu, J.; Snyder, N.W.; Worth, A.J.; Iyer, S.S.; Wang, J.; Ben-Sahra, I.; Byles, V.; Polynne-Stapornkul, T.; et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. eLife 2016, 5, e11612. [Google Scholar] [CrossRef] [PubMed]
- Noe, J.T.; Rendon, B.E.; Geller, A.E.; Conroy, L.R.; Morrissey, S.M.; Young, L.E.; Bruntz, R.C.; Kim, E.J.; Wise-Mitchell, A.; Rizzo, M.B.d.S.; et al. Lactate supports a metabolic-epigenetic link in macrophage polarization. Sci. Adv. 2021, 7, eabi8602. [Google Scholar] [CrossRef]
- Lauterbach, M.A.; Hanke, J.E.; Serefidou, M.; Mangan, M.S.; Kolbe, C.-C.; Hess, T.; Rothe, M.; Kaiser, R.; Hoss, F.; Gehlen, J.; et al. Toll-like Receptor Signaling Rewires Macrophage Metabolism and Promotes Histone Acetylation via ATP-Citrate Lyase. Immunity 2019, 51, 997–1011.e7. [Google Scholar] [CrossRef]
- Rodríguez-Ubreva, J.; Català-Moll, F.; Obermajer, N.; Álvarez-Errico, D.; Ramirez, R.N.; Company, C.; Vento-Tormo, R.; Moreno-Bueno, G.; Edwards, R.P.; Mortazavi, A.; et al. Prostaglandin E2 Leads to the Acquisition of DNMT3A-Dependent Tolerogenic Functions in Human Myeloid-Derived Suppressor Cells. Cell Rep. 2017, 21, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Luker, A.J.; Graham, L.J.; Smith, T.M.; Camarena, C.; Zellner, M.P.; Gilmer, J.-J.S.; Damle, S.R.; Conrad, D.H.; Bear, H.D.; Martin, R.K. The DNA methyltransferase inhibitor, guadecitabine, targets tumor-induced myelopoiesis and recovers T cell activity to slow tumor growth in combination with adoptive immunotherapy in a mouse model of breast cancer. BMC Immunol. 2020, 21, 8. [Google Scholar] [CrossRef]
- Sasidharan Nair, V.; Saleh, R.; Toor, S.M.; Taha, R.Z.; Ahmed, A.A.; Kurer, M.A.; Murshed, K.; Alajez, N.M.; Abu Nada, M.; Elkord, E. Transcriptomic profiling disclosed the role of DNA methylation and histone modifications in tumor-infiltrating myeloid-derived suppressor cell subsets in colorectal cancer. Clin. Epigenet. 2020, 12, 13. [Google Scholar] [CrossRef]
- Cartwright, A.N.; Suo, S.; Badrinath, S.; Kumar, S.; Melms, J.; Luoma, A.; Bagati, A.; Saadatpour, A.; Izar, B.; Yuan, G.C.; et al. Immunosuppressive Myeloid Cells Induce Nitric Oxide-Dependent DNA Damage and p53 Pathway Activation in CD8+ T Cells. Cancer Immunol. Res. 2021, 9, 470–485. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, P.; Parikh, F.; Lopez-Rivera, E.; Hailemichael, Y.; Clark, A.; Ma, G.; Cannan, D.; Ramacher, M.; Kato, M.; Overwijk, W.W.; et al. Tumor-expressed inducible nitric oxide synthase controls induction of functional myeloid-derived suppressor cells through modulation of vascular endothelial growth factor release. J. Immunol. 2012, 188, 5365–5376. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Liu, Z.; Klement, J.D.; Yang, D.; Merting, A.D.; Poschel, D.; Albers, T.; Waller, J.L.; Shi, H.; Liu, K. WDR5-H3K4me3 epigenetic axis regulates OPN expression to compensate PD-L1 function to promote pancreatic cancer immune escape. J. Immunother. Cancer 2021, 9, e002624. [Google Scholar] [CrossRef]
- Wysocka, J.; Swigut, T.; Milne, T.A.; Dou, Y.; Zhang, X.; Burlingame, A.L.; Roeder, R.G.; Brivanlou, A.H.; Allis, C.D. WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell 2005, 121, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Sahakian, E.; Powers, J.J.; Chen, J.; Deng, S.L.; Cheng, F.; Distler, A.; Woods, D.M.; Rock-Klotz, J.; Sodre, A.L.; Youn, J.-I.; et al. Histone deacetylase 11: A novel epigenetic regulator of myeloid derived suppressor cell expansion and function. Mol. Immunol. 2015, 63, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.-I.; Kumar, V.; Collazo, M.; Nefedova, Y.; Condamine, T.; Cheng, P.; Villagra, A.; Antonia, S.; McCaffrey, J.C.; Fishman, M.; et al. Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nat. Immunol. 2013, 14, 211–220. [Google Scholar] [CrossRef]
- de Almeida Nagata, D.E.; Chiang, E.Y.; Jhunjhunwala, S.; Caplazi, P.; Arumugam, V.; Modrusan, Z.; Chan, E.; Merchant, M.; Jin, L.; Arnott, D.; et al. Regulation of Tumor-Associated Myeloid Cell Activity by CBP/EP300 Bromodomain Modulation of H3K27 Acetylation. Cell Rep. 2019, 27, 269–281.e4. [Google Scholar] [CrossRef]
- Yang, R.; Cheng, S.; Luo, N.; Gao, R.; Yu, K.; Kang, B.; Wang, L.; Zhang, Q.; Fang, Q.; Zhang, L.; et al. Distinct epigenetic features of tumor-reactive CD8+ T cells in colorectal cancer patients revealed by genome-wide DNA methylation analysis. Genome Biol. 2019, 21, 2. [Google Scholar] [CrossRef]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017, 27, 109–118. [Google Scholar] [CrossRef]
- Tay, C.; Tanaka, A.; Sakaguchi, S. Tumor-infiltrating regulatory T cells as targets of cancer immunotherapy. Cancer Cell 2023, 41, 450–465. [Google Scholar] [CrossRef]
- Yang, R.; Qu, C.; Zhou, Y.; Konkel, J.E.; Shi, S.; Liu, Y.; Chen, C.; Liu, S.; Liu, D.; Chen, Y.; et al. Hydrogen Sulfide Promotes Tet1- and Tet2-Mediated Foxp3 Demethylation to Drive Regulatory T Cell Differentiation and Maintain Immune Homeostasis. Immunity 2015, 43, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Du, F.; Huang, W.; Ding, X.; Wang, Z.; Yan, F.; Wu, Z. Epigenetic control of Foxp3 in intratumoral T-cells regulates growth of hepatocellular carcinoma. Aging 2019, 11, 2343–2351. [Google Scholar] [CrossRef]
- Zou, Q.; Wang, X.; Ren, D.; Hu, B.; Tang, G.; Zhang, Y.; Huang, M.; Pai, R.K.; Buchanan, D.D.; Win, A.K.; et al. DNA methylation-based signature of CD8+ tumor-infiltrating lymphocytes enables evaluation of immune response and prognosis in colorectal cancer. J. Immunother. Cancer 2021, 9, e002671. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, J.; Bizet, M.; Desmedt, C.; Calonne, E.; Dedeurwaerder, S.; Garaud, S.; Koch, A.; Larsimont, D.; Salgado, R.; Van den Eynden, G.; et al. DNA methylation-based immune response signature improves patient diagnosis in multiple cancers. J. Clin. Investig. 2017, 127, 3090–3102. [Google Scholar] [CrossRef]
- Bian, Y.; Li, W.; Kremer, D.M.; Sajjakulnukit, P.; Li, S.; Crespo, J.; Nwosu, Z.C.; Zhang, L.; Czerwonka, A.; Pawłowska, A.; et al. Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature 2020, 585, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Nagarsheth, N.; Peng, D.; Kryczek, I.; Wu, K.; Li, W.; Zhao, E.; Zhao, L.; Wei, S.; Frankel, T.; Vatan, L.; et al. PRC2 Epigenetically Silences Th1-Type Chemokines to Suppress Effector T-Cell Trafficking in Colon Cancer. Cancer Res. 2016, 76, 275–282. [Google Scholar] [CrossRef]
- Tumes, D.J.; Onodera, A.; Suzuki, A.; Shinoda, K.; Endo, Y.; Iwamura, C.; Hosokawa, H.; Koseki, H.; Tokoyoda, K.; Suzuki, Y.; et al. The polycomb protein Ezh2 regulates differentiation and plasticity of CD4(+) T helper type 1 and type 2 cells. Immunity 2013, 39, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Allan, R.S.; Zueva, E.; Cammas, F.; Schreiber, H.A.; Masson, V.; Belz, G.T.; Roche, D.; Maison, C.; Quivy, J.-P.; Almouzni, G.; et al. An epigenetic silencing pathway controlling T helper 2 cell lineage commitment. Nature 2012, 487, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Adoue, V.; Binet, B.; Malbec, A.; Fourquet, J.; Romagnoli, P.; van Meerwijk, J.P.; Amigorena, S.; Joffre, O.P. The Histone Methyltransferase SETDB1 Controls T Helper Cell Lineage Integrity by Repressing Endogenous Retroviruses. Immunity 2019, 50, 629–644.e8. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, K.; Chen, W.; Wang, X.; Huang, Y.; Wang, W.; Wu, W.; Cai, Z.; Huang, W. Modulation of SRSF2 expression reverses the exhaustion of TILs via the epigenetic regulation of immune checkpoint molecules. Cell. Mol. Life Sci. 2020, 77, 3441–3452. [Google Scholar] [CrossRef]
- Qiu, J.; Villa, M.; Sanin, D.E.; Buck, M.D.; O’Sullivan, D.; Ching, R.; Matsushita, M.; Grzes, K.M.; Winkler, F.; Chang, C.H.; et al. Acetate Promotes T Cell Effector Function during Glucose Restriction. Cell Rep. 2019, 27, 2063–2074.e5. [Google Scholar] [CrossRef]
- Vodnala, S.K.; Eil, R.; Kishton, R.J.; Sukumar, M.; Yamamoto, T.N.; Ha, N.H.; Lee, P.H.; Shin, M.; Patel, S.J.; Yu, Z.; et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science 2019, 363, eaau0135. [Google Scholar] [CrossRef] [PubMed]
- Luda, K.M.; Longo, J.; Kitchen-Goosen, S.M.; Duimstra, L.R.; Ma, E.H.; Watson, M.J.; Oswald, B.M.; Fu, Z.; Madaj, Z.; Kupai, A.; et al. Ketolysis drives CD8+ T cell effector function through effects on histone acetylation. Immunity 2023, 56, 2021–2035.e8. [Google Scholar] [CrossRef]
- Williams, C.J.; Naito, T.; Gomez-del Arco, P.; Seavitt, J.R.; Cashman, S.M.; De Souza, B.; Qi, X.; Keables, P.; Von Andrian, U.H.; Georgopoulos, K. The chromatin remodeler Mi-2beta is required for CD4 expression and T cell development. Immunity 2004, 20, 719–733. [Google Scholar] [CrossRef] [PubMed]
- Belk, J.A.; Yao, W.; Ly, N.; Freitas, K.A.; Chen, Y.-T.; Shi, Q.; Valencia, A.M.; Shifrut, E.; Kale, N.; Yost, K.E.; et al. Genome-wide CRISPR screens of T cell exhaustion identify chromatin remodeling factors that limit T cell persistence. Cancer Cell 2022, 40, 768–786.e7. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Zeng, S.; Su, C.; Li, J.; Xuan, Y.; Lin, Y.; Xu, E.; Fan, Q. The interaction between DNA methylation and tumor immune microenvironment: From the laboratory to clinical applications. Clin. Epigenet. 2024, 16, 24. [Google Scholar] [CrossRef] [PubMed]
- Sylvestre, M.; Tarte, K.; Roulois, D. Epigenetic mechanisms driving tumor supportive microenvironment differentiation and function: A role in cancer therapy? Epigenomics 2020, 12, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Lodewijk, I.; Nunes, S.P.; Henrique, R.; Jerónimo, C.; Dueñas, M.; Paramio, J.M. Tackling tumor microenvironment through epigenetic tools to improve cancer immunotherapy. Clin. Epigenet. 2021, 13, 63. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Ohtani, H.; Chakravarthy, A.; De Carvalho, D.D. Epigenetic therapy in immune-oncology. Nat. Rev. Cancer 2019, 19, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Qiao, X.; Fang, Y.; Guo, R.; Bai, P.; Liu, S.; Li, T.; Jiang, Y.; Wei, S.; Na, Z.; et al. Epigenetics-targeted drugs: Current paradigms and future challenges. Signal Transduct. Target. Ther. 2024, 9, 332. [Google Scholar] [PubMed]
- Wimalasena, V.K.; Wang, T.; Sigua, L.H.; Durbin, A.D.; Qi, J. Using Chemical Epigenetics to Target Cancer. Mol. Cell 2020, 78, 1086–1095. [Google Scholar] [CrossRef]
- Miranda Furtado, C.L.; Dos Santos Luciano, M.C.; Silva Santos RDa Furtado, G.P.; Moraes, M.O.; Pessoa, C. Epidrugs: Targeting epigenetic marks in cancer treatment. Epigenetics 2019, 14, 1164–1176. [Google Scholar] [CrossRef] [PubMed]
- Al-Kharashi, L.A.; Bakheet, T.; AlHarbi, W.A.; Al-Moghrabi, N.; Aboussekhra, A. Eugenol modulates genomic methylation and inactivates breast cancer-associated fibroblasts through E2F1-dependent downregulation of DNMT1/DNMT3A. Mol. Carcinog. 2021, 60, 784–795. [Google Scholar] [CrossRef]
- Wang, H.-C.; Chen, C.-W.; Yang, C.-L.; Tsai, I.-M.; Hou, Y.-C.; Chen, C.-J.; Shan, Y.-S. Tumor-Associated Macrophages Promote Epigenetic Silencing of Gelsolin through DNA Methyltransferase 1 in Gastric Cancer Cells. Cancer Immunol. Res. 2017, 5, 885–897. [Google Scholar] [CrossRef]
- DuPage, M.; Chopra, G.; Quiros, J.; Rosenthal, W.L.; Morar, M.M.; Holohan, D.; Zhang, R.; Turka, L.; Marson, A.; Bluestone, J.A. The Chromatin-Modifying Enzyme Ezh2 Is Critical for the Maintenance of Regulatory T Cell Identity after Activation. Immunity 2015, 42, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Quiros, J.; Mahuron, K.; Pai, C.-C.; Ranzani, V.; Young, A.; Silveria, S.; Harwin, T.; Abnousian, A.; Pagani, M.; et al. Targeting EZH2 Reprograms Intratumoral Regulatory T Cells to Enhance Cancer Immunity. Cell Rep. 2018, 23, 3262–3274. [Google Scholar] [CrossRef]
- Sun, W.; Chen, L.; Tang, J.; Zhang, C.; Wen, Y.; Wen, W. Targeting EZH2 depletes LMP1-induced activated regulatory T cells enhancing antitumor immunity in nasopharyngeal carcinoma. J. Cancer Res. Ther. 2020, 16, 309–319. [Google Scholar] [CrossRef]
- Huang, S.; Wang, Z.; Zhou, J.; Huang, J.; Zhou, L.; Luo, J.; Wan, Y.Y.; Long, H.; Zhu, B. EZH2 Inhibitor GSK126 Suppresses Antitumor Immunity by Driving Production of Myeloid-Derived Suppressor Cells. Cancer Res. 2019, 79, 2009–2020. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Dunleavey, J.M.; Xiao, L.; Ollila, D.W.; Troester, M.A.; Otey, C.A.; Li, W.; Barker, T.H.; Dudley, A.C. Suppression of TGFβ-mediated conversion of endothelial cells and fibroblasts into cancer associated (myo)fibroblasts via HDAC inhibition. Br. J. Cancer 2018, 118, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Pazolli, E.; Alspach, E.; Milczarek, A.; Prior, J.; Piwnica-Worms, D.; Stewart, S.A. Chromatin remodeling underlies the senescence-associated secretory phenotype of tumor stromal fibroblasts that supports cancer progression. Cancer Res. 2012, 72, 2251–2261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, Y.; Tu, T.; Schmull, S.; Han, Y.; Wang, W.; Li, H. Dual inhibition of HDAC and tyrosine kinase signaling pathways with CUDC-907 attenuates TGFβ1 induced lung and tumor fibrosis. Cell Death Dis. 2020, 11, 765. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Zou, J.; Li, S.; Topper, M.J.; Tao, Y.; Zhang, H.; Jiao, X.; Xie, W.; Kong, X.; Vaz, M.; et al. Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature 2020, 579, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Ikegami, T.; Ago, Y.; Okada, N.; Tachibana, M. Valproic acid attenuates CCR2-dependent tumor infiltration of monocytic myeloid-derived suppressor cells, limiting tumor progression. Oncoimmunology 2020, 9, 1734268. [Google Scholar] [CrossRef] [PubMed]
- Christmas, B.J.; Rafie, C.I.; Hopkins, A.C.; Scott, B.A.; Ma, H.S.; Cruz, K.A.; Woolman, S.; Armstrong, T.D.; Connolly, R.M.; Azad, N.A.; et al. Entinostat Converts Immune-Resistant Breast and Pancreatic Cancers into Checkpoint-Responsive Tumors by Reprogramming Tumor-Infiltrating MDSCs. Cancer Immunol. Res. 2018, 6, 1561–1577. [Google Scholar] [CrossRef]
- Kim, Y.-D.; Park, S.-M.; Ha, H.C.; Lee, A.R.; Won, H.; Cha, H.; Cho, S.; Cho, J.M. HDAC Inhibitor, CG-745, Enhances the Anti-Cancer Effect of Anti-PD-1 Immune Checkpoint Inhibitor by Modulation of the Immune Microenvironment. J. Cancer 2020, 11, 4059–4072. [Google Scholar] [CrossRef]
- Rosborough, B.R.; Castellaneta, A.; Natarajan, S.; Thomson, A.W.; Turnquist, H.R. Histone deacetylase inhibition facilitates GM-CSF-mediated expansion of myeloid-derived suppressor cells in vitro and in vivo. J. Leukoc. Biol. 2012, 91, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.H.; Elliott, I.A.; Wu, N.; Matsumura, C.; Vogelauer, M.; Attar, N.; Dann, A.; Ghukasyan, R.; Toste, P.A.; Patel, S.G.; et al. Histone deacetylase inhibitors provoke a tumor supportive phenotype in pancreatic cancer associated fibroblasts. Oncotarget 2017, 8, 19074–19088. [Google Scholar] [CrossRef]
- Adeegbe, D.O.; Liu, S.; Hattersley, M.M.; Bowden, M.; Zhou, C.W.; Li, S.; Vlahos, R.; Grondine, M.; Dolgalev, I.; Ivanova, E.V.; et al. BET Bromodomain Inhibition Cooperates with PD-1 Blockade to Facilitate Antitumor Response in Kras-Mutant Non–Small Cell Lung Cancer. Cancer Immunol. Res. 2018, 6, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Tateishi, K.; Kudo, Y.; Hoshikawa, M.; Tanaka, M.; Nakatsuka, T.; Fujiwara, H.; Miyabayashi, K.; Takahashi, R.; Tanaka, Y.; et al. Stromal remodeling by the BET bromodomain inhibitor JQ1 suppresses the progression of human pancreatic cancer. Oncotarget 2016, 7, 61469–61484. [Google Scholar] [CrossRef]
- Huang, Y.; Nahar, S.; Nakagawa, A.; Fernandez-Barrena, M.G.; Mertz, J.A.; Bryant, B.M.; Adams, C.E.; Mino-Kenudson, M.; Von Alt, K.N.; Chang, K.; et al. Regulation of GLI Underlies a Role for BET Bromodomains in Pancreatic Cancer Growth and the Tumor Microenvironment. Clin. Cancer Res. 2016, 22, 4259–4270. [Google Scholar] [CrossRef]
- Smith, A.D.; Lu, C.; Payne, D.; Paschall, A.V.; Klement, J.D.; Redd, P.S.; Ibrahim, M.L.; Yang, D.; Han, Q.; Liu, Z.; et al. Autocrine IL6-Mediated Activation of the STAT3–DNMT Axis Silences the TNFα–RIP1 Necroptosis Pathway to Sustain Survival and Accumulation of Myeloid-Derived Suppressor Cells. Cancer Res. 2020, 80, 3145–3156. [Google Scholar] [CrossRef]
- Chen, S.; Xie, P.; Cowan, M.; Huang, H.; Cardenas, H.; Keathley, R.; Tanner, E.J.; Fleming, G.F.; Moroney, J.W.; Pant, A.; et al. Epigenetic priming enhances antitumor immunity in platinum-resistant ovarian cancer. J. Clin. Investig. 2022, 132, e158800. [Google Scholar] [CrossRef] [PubMed]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G.; et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 2017, 170, 142–157.e19. [Google Scholar] [CrossRef] [PubMed]
- Stone, M.L.; Chiappinelli, K.B.; Li, H.; Murphy, L.M.; Travers, M.E.; Topper, M.J.; Mathios, D.; Lim, M.; Shih, I.-M.; Wang, T.-L.; et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc. Natl. Acad. Sci. USA 2017, 114, E10981–E10990. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Lee, J.H.; Zhang, Z.; Wu, Y.; Yang, M.; Liao, Y.; de la Rosa, R.; Scheirer, J.; Pechacek, D.; Zhang, N.; et al. PRC2-Mediated Epigenetic Suppression of Type I IFN-STAT2 Signaling Impairs Antitumor Immunity in Luminal Breast Cancer. Cancer Res. 2022, 82, 4624–4640. [Google Scholar] [CrossRef]
- Zhou, L.; Mudianto, T.; Ma, X.; Riley, R.; Uppaluri, R. Targeting EZH2 Enhances Antigen Presentation, Antitumor Immunity, and Circumvents Anti-PD-1 Resistance in Head and Neck Cancer. Clin. Cancer Res. 2020, 26, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Tiffen, J.; Wilson, S.; Gallagher, S.J.; Hersey, P.; Filipp, F.V. Somatic Copy Number Amplification and Hyperactivating Somatic Mutations of EZH2 Correlate with DNA Methylation and Drive Epigenetic Silencing of Genes Involved in Tumor Suppression and Immune Responses in Melanoma. Neoplasia 2016, 18, 121–132. [Google Scholar] [CrossRef]
- Xu, T.; Dai, J.; Tang, L.; Yang, L.; Si, L.; Sheng, X.; Cui, C.; Chi, Z.; Kong, Y.; Guo, J. EZH2 Inhibitor Enhances the STING Agonist–Induced Antitumor Immunity in Melanoma. J. Investig. Dermatol. 2022, 142, 1158–1170.e8. [Google Scholar] [CrossRef] [PubMed]
- Zhao, E.; Maj, T.; Kryczek, I.; Li, W.; Wu, K.; Zhao, L.; Wei, S.; Crespo, J.; Wan, S.; Vatan, L.; et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat. Immunol. 2016, 17, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, Y.; Liu, S.; Adeegbe, D.O.; Christensen, C.L.; Quinn, M.M.; Dries, R.; Han, S.; Buczkowski, K.; Wang, X.; et al. NK Cells Mediate Synergistic Antitumor Effects of Combined Inhibition of HDAC6 and BET in a SCLC Preclinical Model. Cancer Res. 2018, 78, 3709–3717. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Zhao, K.; Wang, T.; Yuan, J.; Zhang, D.; Xiang, W.; Qian, J.; Luo, N.; Zhou, Y.; Tang, B.; et al. 5-aza-2′-deoxycytidine potentiates anti-tumor immunity in colorectal peritoneal metastasis by modulating ABC A9-mediated cholesterol accumulation in macrophages. Theranostics 2022, 12, 875–890. [Google Scholar] [CrossRef]
- Wang, Y.F.; Yu, L.; Hu, Z.L.; Fang, Y.F.; Shen, Y.Y.; Song, M.F.; Chen, Y. Regulation of CCL2 by EZH2 affects tumor-associated macrophages polarization and infiltration in breast cancer. Cell Death Dis. 2022, 13, 748. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Yuan, Y.; Yuan, H.; Wang, J.; Yun, H.; Geng, Y.; Zhao, M.; Li, L.; Weng, Y.; Liu, Z.; et al. Histone acetyltransferase 1 is a succinyltransferase for histones and non-histones and promotes tumorigenesis. EMBO Rep. 2021, 22, e50967. [Google Scholar] [CrossRef] [PubMed]
- Tihagam, R.D.; Bhatnagar, S. Detection Methods for Epigenetic Mechanisms in Breast Cancer. Adv. Exp. Med. Biol. 2024, 1465, 99–103. [Google Scholar] [PubMed]
- Blanco-Carmona, E.; Narayanan, A.; Hernandez, I.; Nieto, J.C.; Elosua-Bayes, M.; Sun, X.; Schmidt, C.; Pamir, N.; Özduman, K.; Herold-Mende, C.; et al. Tumor heterogeneity and tumor-microglia interactions in primary and recurrent IDH1-mutant gliomas. Cell Rep. Med. 2023, 4, 101249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, H.; Mo, H.; Hu, X.; Gao, R.; Zhao, Y.; Liu, B.; Niu, L.; Sun, X.; Yu, X.; et al. Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell 2021, 39, 1578–1593.e8. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Xu, D.; Wang, S.; Wong, C.K.; Martinez-Fundichely, A.; Lee, C.J.; Cohen, S.; Park, J.; Hill, C.E.; Eng, K.; et al. Chromatin profiles classify castration-resistant prostate cancers suggesting therapeutic targets. Science 2022, 376, eabe1505. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Cell Type | Major Markers | Function | Reference |
|---|---|---|---|
| CAFs | α-SMA, S100A4, FAP, PDGFRα/β | Enhance tumor cell survival and growth; In specific contexts, CAFs counteract tumor progression by promoting anticancer immunity and regulating tumor-inhibitory signaling. | [39,40,41,42,43] |
| TAMs | CD68, CD163, ARG1, CD11b | Promote angiogenesis, metastasis andimmune evasion: Phagocytosis of cancer cells exerts an anti-tumor effect. | [44,45,46,47,48,49] |
| MDSCs | CD11b, Lin-, CD33, Gr-1 | Suppress T-cell activity; Secrete immunosuppressive cytokines; Facilitate tumor progression. | [50,51,52,53,54,55,56] |
| CD8+ T | CD8, CD28, CD3, TCR, Tim 3, PD-1 | Release perforin and granzymes; Secrete cytokines like lFN-γ, TNF-α; Kill cancer cells. | [57,58] |
| CD4+ T | CD4, CD28, CD3, TCR | Th1 subtype promotes the anti-tumoral response; TH2 subtype exerts the pro-tumoral effect. | [59,60] |
| Treg cells | CD4, CD25, FOXP3 | Suppress the activity of effector T cells; Expediate tumor progression. | [61,62] |
| NK cells | CD56, CD16, KIRs, CD94 | Recognize and kill tumor cells directly. | [63,64] |
| B cells | CD19, CD20, CD22, CD27 | Drive the humoral immunity; Promote the anti-tumoral response. | [65,66] |
| Epigenetic Inhibitors | Target | Cell Type Within TME | Reference |
|---|---|---|---|
| DNMT inhibitors | |||
| Decitabine+eugenol | DNMT1/DNMT3A | CAFS | [78] |
| Guadecitabine | DNMT1 | MDSCs | [121] |
| Decitabine | DNMT1/DNMT3B | CAFS/MDSCs | [156] |
| 5-AZA | DNMTS | CAFS/TAMs | [157] |
| HKMT inhibitors | |||
| CPI-1205 | EZH2 | Tregs | [158,159] |
| Dzenp | EZH2 | Tregs | [160] |
| GSK126 | EZH2 | MDSCs | [161] |
| HDAC inhibitors | |||
| Scriptaid | HDAC1/3/8 | CAFS | [162] |
| CUDC-907 | Pan-HDAC | CAFS | [163,164] |
| Entinostat | Pan-HDAC | MDSCs/Tregs | [165] |
| SAHA | HDAC1/2/3 | CAFS/MDSCs | [166,167] |
| CG-745 | Pan-HDAC | MDSCS/TAMS | [168] |
| VPA | HDAC1 | MDSCs | [169,170] |
| BET inhibitors | |||
| JQ1 | BRD4 | CAFs/Tregs | [171,172] |
| CPI203 | BRD2/3 | Tregs | [173] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, K.; Li, Y.; Shen, M.; Xu, W.; Wu, S.; Yang, X.; Zhang, B.; Lin, N. Epigenetic Regulation of Stromal and Immune Cells and Therapeutic Targets in the Tumor Microenvironment. Biomolecules 2025, 15, 71. https://doi.org/10.3390/biom15010071
Liu K, Li Y, Shen M, Xu W, Wu S, Yang X, Zhang B, Lin N. Epigenetic Regulation of Stromal and Immune Cells and Therapeutic Targets in the Tumor Microenvironment. Biomolecules. 2025; 15(1):71. https://doi.org/10.3390/biom15010071
Chicago/Turabian StyleLiu, Kang, Yue Li, Minmin Shen, Wei Xu, Shanshan Wu, Xinxin Yang, Bo Zhang, and Nengming Lin. 2025. "Epigenetic Regulation of Stromal and Immune Cells and Therapeutic Targets in the Tumor Microenvironment" Biomolecules 15, no. 1: 71. https://doi.org/10.3390/biom15010071
APA StyleLiu, K., Li, Y., Shen, M., Xu, W., Wu, S., Yang, X., Zhang, B., & Lin, N. (2025). Epigenetic Regulation of Stromal and Immune Cells and Therapeutic Targets in the Tumor Microenvironment. Biomolecules, 15(1), 71. https://doi.org/10.3390/biom15010071