
Reactive Oxygen Species, a Potential Therapeutic Target for Vascular Dementia

 , , and
, , and 
Abstract
1. Introduction
2. Vascular Dementia and Oxidative Stress
3. Antioxidant Activity of Repurposed Treatments for the Management of Vascular Dementia
4. Importance of ROS in Alzheimer’s Diseases and Vascular Dementia
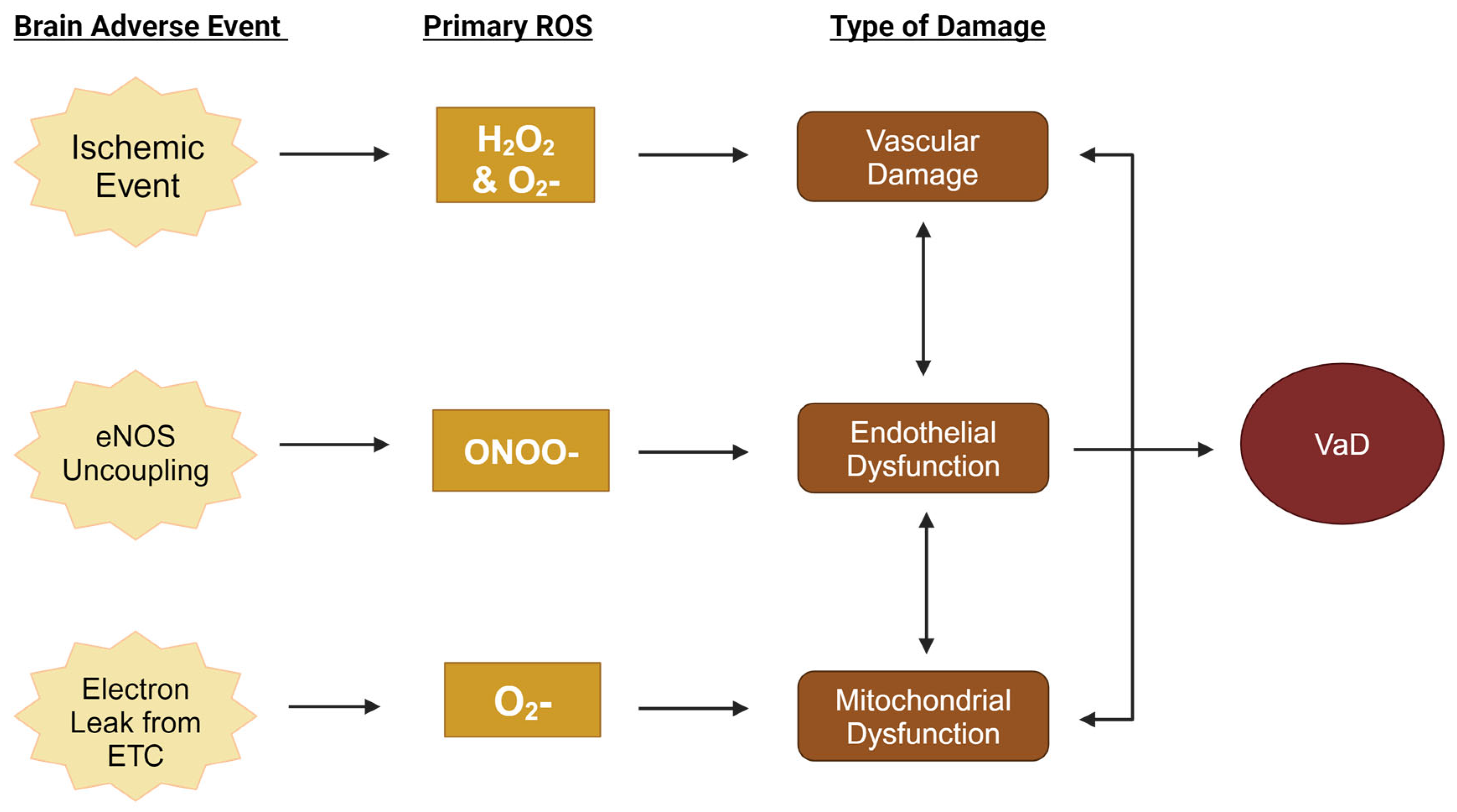
5. Primary Reactive Oxygen Species in the Pathogenesis of Vascular Dementia
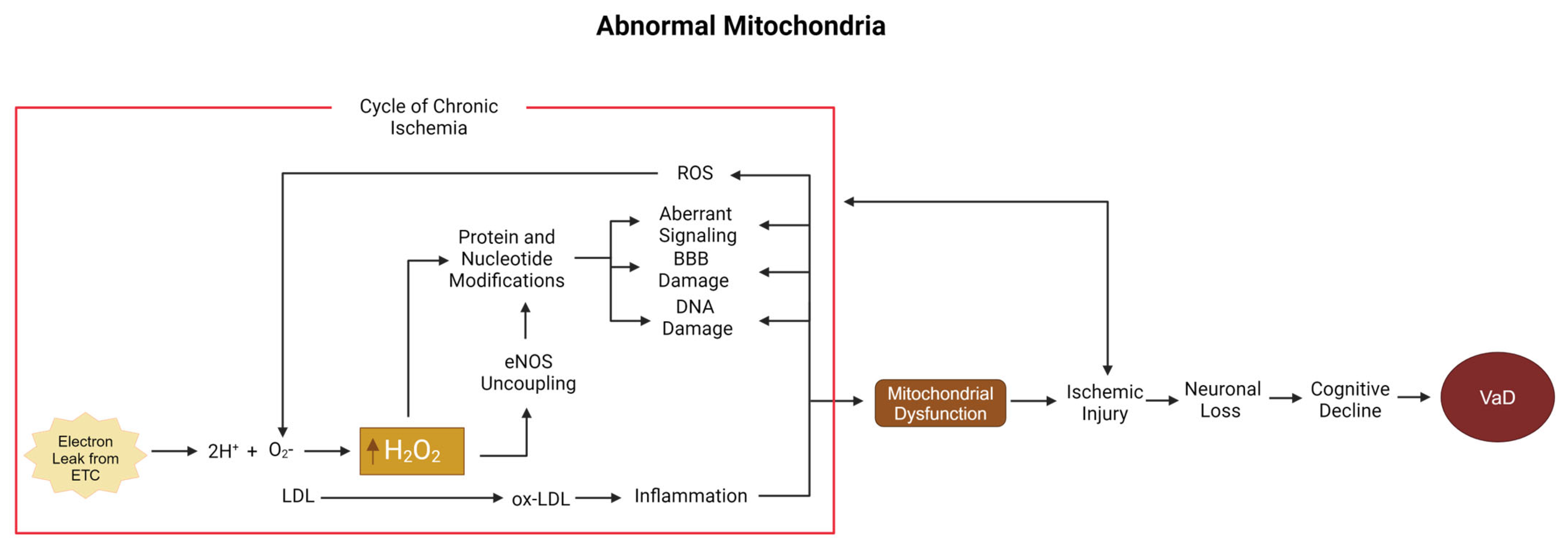
5.1. Low-Density Lipoprotein, Hydrogen Peroxide, and the Mitochondria
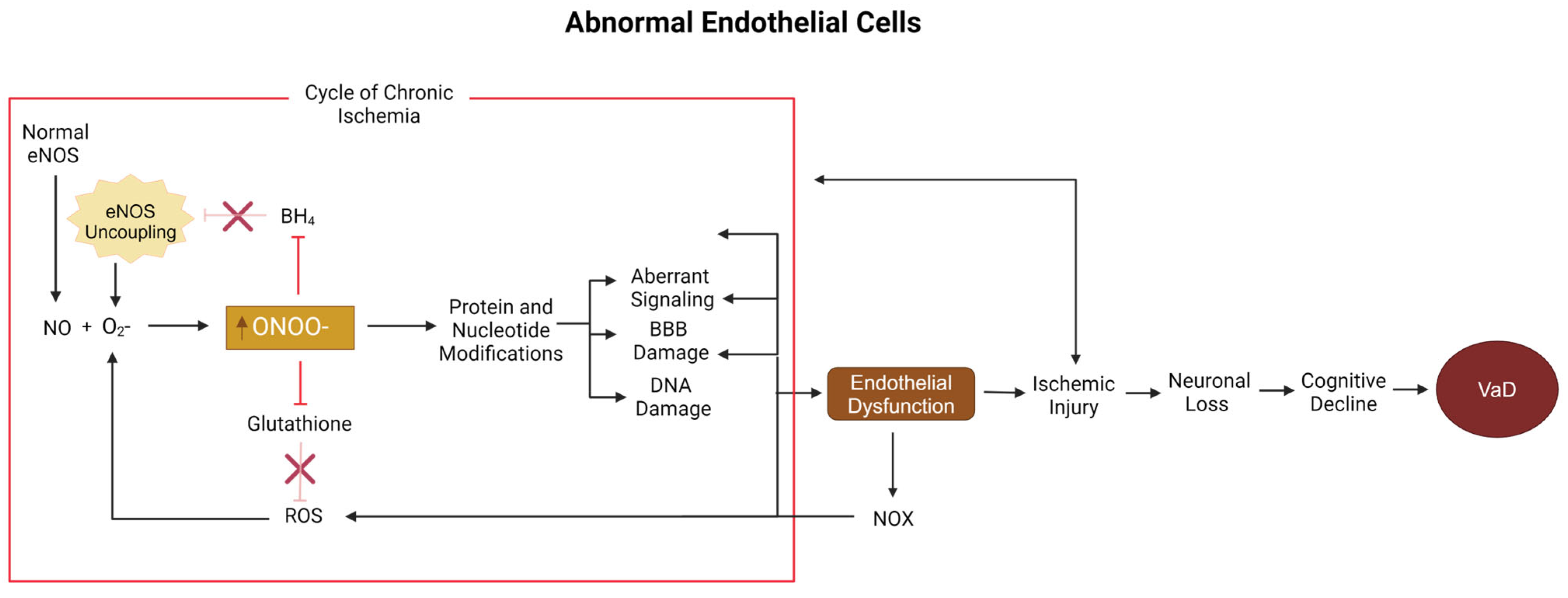
5.2. Peroxynitrite, Nitric Oxide, and Endothelial Cells
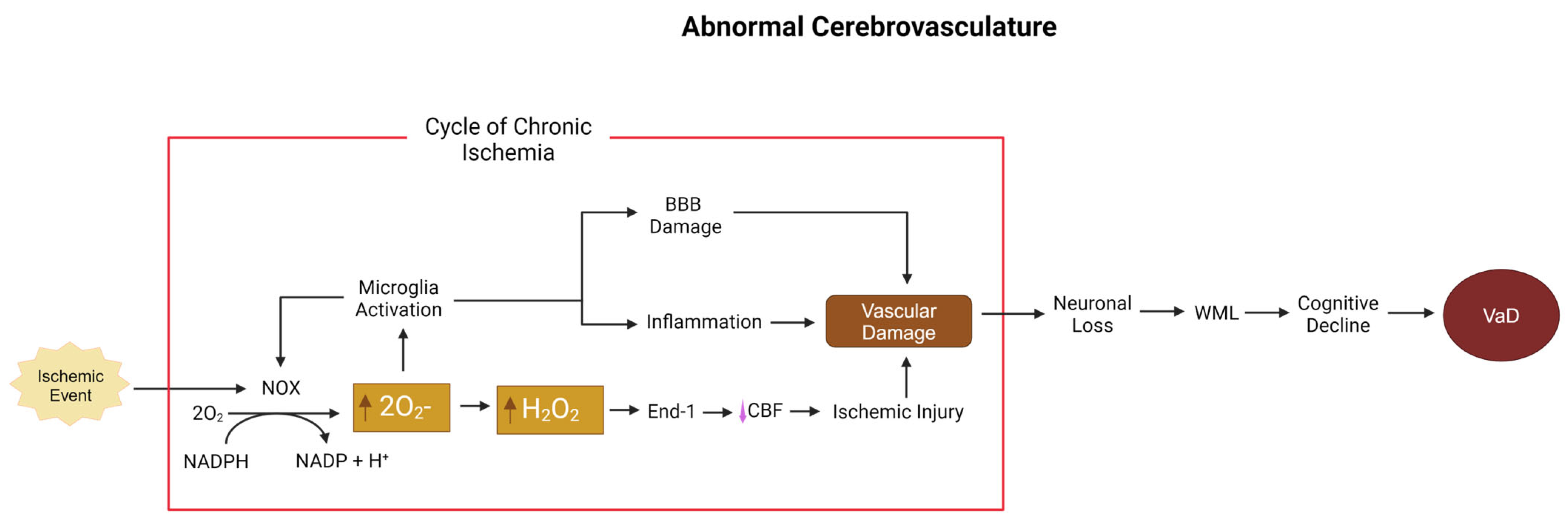
5.3. Superoxide and Hydrogen Peroxide, NOX, and the Cerebrovasculature
6. Potential Translational Antioxidant Therapeutics for Vascular Dementia
6.1. Carotenoids
6.2. Idebenone
6.3. Alpha-Lipoic Acid
6.4. Resveratrol
6.5. Selenium
6.6. Curcumin
6.7. Endothelial-Cell-Targted Antioxidants
6.8. Mitochondria-Targeted Antioxidants
6.9. Cerebrovasculature-Targeted Antioxidants
7. Clinical Trials Using Antioxidants for Vascular Dementia
8. Discussion and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MHPG | 3-methoxy-4-hydroxyphenylglycol |
| FeTPPs | 4-sulfonatophenyl porphyrinato iron chloride |
| ALA | Alpha-lipoic acid |
| AD | Alzheimer’s disease |
| Aβ | Amyloid beta |
| BCCAO | Bilateral common carotid artery occlusion |
| BBB | Blood–brain barrier |
| BDNF | Brain-derived neurotrophic factor |
| CSF | Cerebral spinal fluid |
| CADASIL | Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy |
| CARASIL | Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy |
| CSF | Cerebral spinal fluid |
| CoQ10 | Coenzyme Q10 |
| eNOS | Endothelial nitric oxide synthase |
| ERK | Extracellular signal-regulated kinase |
| GSH | Glutathione |
| CA1 | Hippocampal circuit 1 |
| HVA | Homovanillic acid |
| H2O2 | Hydrogen peroxide |
| HIF-1α | Hypoxia inducible factor-1 |
| ICAM | Intercellular adhesion molecule 1 |
| LDL | Low-density lipoprotein |
| MDA | Malondialdehyde |
| MMP | Matrix metalloproteinase |
| MitoQ | Mitoquinone mesylate |
| MWM | Morris water maze |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NO | Nitric oxide |
| NFkB/NF-κB | Nuclear factor kappa light chain enhancer of activated B cells |
| Ox-LDL | Oxidized LDL |
| ONOO− | Peroxynitrite |
| ROS | Reactive oxygen species |
| RSVL | Reservatrol |
| 5-HIAA | Serotonin |
| SNP | Single-nucelotide polymorphism |
| SDH | Succinate dehydrogenase |
| O2− | Superoxide |
| SOD | Superoxide dismutase |
| BH4 | Tetrahydrobiopterin |
| UCCAO | Unilateral common carotid artery occlusion |
| VCAM-1 | Vascular cell adhesion molecules |
| VaD | Vascular dementia |
| RSVL | Resveratrol |
References
- Iemolo, F.; Duro, G.; Rizzo, C.; Castiglia, L.; Hachinski, V.; Caruso, C. Pathophysiology of vascular dementia. Immun. Ageing 2009, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Kuang, H.; Zhou, Z.F.; Zhu, Y.G.; Wan, Z.K.; Yang, M.W.; Hong, F.F.; Yang, S.L. Pharmacological Treatment of Vascular Dementia: A Molecular Mechanism Perspective. Aging Dis. 2021, 12, 308. [Google Scholar] [CrossRef] [PubMed]
- Luca, M.; Luca, A.; Calandra, C. The Role of Oxidative Damage in the Pathogenesis and Progression of Alzheimer’s Disease and Vascular Dementia. Oxidative Med. Cell. Longev. 2015, 2015, 504678. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.; Grant, M.M.; Aldred, S. Oxidative stress in vascular dementia and Alzheimer’s disease: A common pathology. J. Alzheimer’s Dis. 2009, 17, 245–257. [Google Scholar] [CrossRef]
- Sena, C.M.; Leandro, A.; Azul, L.; Seiça, R.; Perry, G. Vascular Oxidative Stress: Impact and Therapeutic Approaches. Front. Physiol. 2018, 9, 1668. [Google Scholar] [CrossRef]
- Definition of Reactive Oxygen Species—NCI Dictionary of Cancer Terms—NCI. Available online: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/reactive-oxygen-species (accessed on 19 November 2024).
- Carvalho, C.; Moreira, P.I. Oxidative Stress: A Major Player in Cerebrovascular Alterations Associated to Neurodegenerative Events. Front. Physiol. 2018, 9, 806. [Google Scholar] [CrossRef]
- Iadecola, C. The pathobiology of vascular dementia. Neuron 2013, 80, 844–866. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Tan, C.C.; Xu, W.; Hu, H.; Cao, X.P.; Dong, Q.; Tan, L.; Yu, J.T. The Prevalence of Dementia: A Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. 2020, 73, 1157–1166. [Google Scholar] [CrossRef] [PubMed]
- Román, G.C. Vascular dementia may be the most common form of dementia in the elderly. J. Neurol. Sci. 2002, 203–204, 7–10. [Google Scholar] [CrossRef]
- O’Brien, J.T.; Erkinjuntti, T.; Reisberg, B.; Roman, G.; Sawada, T.; Pantoni, L.; Bowler, J.V.; Ballard, C.; DeCarli, C.; Gorelick, P.B.; et al. Vascular cognitive impairment. Lancet Neurol. 2003, 2, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Pritam, P.; Deka, R.; Bhardwaj, A.; Srivastava, R.; Kumar, D.; Jha, A.K.; Jha, N.K.; Villa, C.; Jha, S.K. Antioxidants in Alzheimer’s Disease: Current Therapeutic Significance and Future Prospects. Biology 2022, 11, 212. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Grinberg, L.T.; Attems, J. Vascular dementia: Different forms of vessel disorders contribute to the development of dementia in the elderly brain. Exp. Gerontol. 2012, 47, 816–824. [Google Scholar] [CrossRef]
- Caruso, P.; Signori, R.; Moretti, R. Small vessel disease to subcortical dementia: A dynamic model, which interfaces aging, cholinergic dysregulation and the neurovascular unit. Vasc. Health Risk Manag. 2019, 15, 259. [Google Scholar] [CrossRef]
- Chen, X.; Chen, L.; Lin, G.; Wang, Z.; Kodali, M.C.; Li, M.; Chen, H.; Lebovitz, S.G.; Ortyl, T.C.; Li, L.; et al. White matter damage as a consequence of vascular dysfunction in a spontaneous mouse model of chronic mild chronic hypoperfusion with eNOS deficiency. Mol. Psychiatry 2022, 27, 4754. [Google Scholar] [CrossRef]
- Quick, S.; Moss, J.; Rajani, R.M.; Williams, A. A Vessel for Change: Endothelial Dysfunction in Cerebral Small Vessel Disease. Trends Neurosci. 2021, 44, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Madamanchi, N.R.; Runge, M.S. Mitochondrial dysfunction in atherosclerosis. Circ. Res. 2007, 100, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.Y.; Hong, F.F.; Yang, S.L. The Roles of Nitric Oxide Synthase/Nitric Oxide Pathway in the Pathology of Vascular Dementia and Related Therapeutic Approaches. Int. J. Mol. Sci. 2021, 22, 4540. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.B.; Holland, N.W. Vascular Dementia. In The 5-Minute Clinical Consult Standard 2016, 24th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2015. [Google Scholar] [CrossRef]
- Asiri, Y.A.; Mostafa, G.A.E. Donepezil. In Profiles of Drug Substances, Excipients and Related Methodology; Academic Press: Cambridge, MA, USA, 2023; Volume 35, pp. 117–150. [Google Scholar] [CrossRef]
- Kalola, U.K.; Patel, P.; Nguyen, H. Galantamine. In Naturally Occurring Chemicals against Alzheimer’s Disease; Academic Press: Cambridge, MA, USA, 2024; pp. 83–92. [Google Scholar] [CrossRef]
- Kuns, B.; Rosani, A.; Patel, P.; Varghese, D. Memantine. In The Essence of Analgesia and Analgesics; Cambridge University Press: Cambridge, UK, 2024; pp. 319–321. [Google Scholar] [CrossRef]
- Schneider, J.A.; Arvanitakis, Z.; Leurgans, S.E.; Bennett, D.A. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann. Neurol. 2009, 66, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Agrawal, N.; Goyal, A. Role of Alpha-7-Nicotinic Acetylcholine Receptor in Alzheimer’s Disease. CNS Neurol. Disord. Drug Targets 2024, 23, 384–394. [Google Scholar] [CrossRef]
- Tamagno, E.; Guglielmotto, M.; Vasciaveo, V.; Tabaton, M. Oxidative stress and beta amyloid in alzheimer’s disease. Which comes first: The chicken or the egg? Antioxidants 2021, 10, 1479. [Google Scholar] [CrossRef] [PubMed]
- Gustaw-Rothenberg, K.; Kowalczuk, K.; Stryjecka-Zimmer, M. Lipids’ peroxidation markers in Alzheimer’s disease and vascular dementia. Geriatr. Gerontol. Int. 2010, 10, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Kishi, T.; Matsunaga, S.; Oya, K.; Nomura, I.; Ikuta, T.; Iwata, N. Memantine for Alzheimer’s Disease: An Updated Systematic Review and Meta-analysis. J. Alzheimer’s Dis. 2017, 60, 401–425. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, Y.; Zhao, T.; Gong, M.; Wang, X.; Zhang, Y.; Xu, L.; Li, W.; Jia, J. The role of N-methyl-D-aspartate glutamate receptors in Alzheimer’s disease: From pathophysiology to therapeutic approaches. Prog. Neurobiol. 2023, 231, 102534. [Google Scholar] [CrossRef]
- Sharma, K. Cholinesterase inhibitors as Alzheimer’s therapeutics. Mol. Med. Rep. 2019, 20, 1479–1487. [Google Scholar] [CrossRef]
- Grossberg, G.T. Cholinesterase inhibitors for the treatment of Alzheimer’s disease: Getting on and staying on. Curr. Ther. Res. Clin. Exp. 2003, 64, 216–235. [Google Scholar] [CrossRef] [PubMed]
- Brennan, A.M.; Won Suh, S.; Joon Won, S.; Narasimhan, P.; Kauppinen, T.M.; Lee, H.; Edling, Y.; Chan, P.H.; Swanson, R.A. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat. Neurosci. 2009, 12, 857–863. [Google Scholar] [CrossRef]
- Munishamappa, V.; Seethalakshmi, V.A.E.; Rajathilagam, T. Evaluation of the antioxidant activity of donepezil-in vitro study. Natl. J. Physiol. Pharm. Pharmacol. 2019, 108, 108–110. [Google Scholar] [CrossRef]
- Barajas-Carrillo, V.W.; Estolano-Cobián, A.; Díaz-Rubio, L.; Ayllón-Gutiérrez, R.R.; Salazar-Aranda, R.; Díaz-Molina, R.; García-González, V.; Almanza-Reyes, H.; Rivero, I.A.; Marrero, J.G.; et al. Antioxidant and acetylcholinesterase inhibition activity of aliphatic and aromatic edaravone derivatives. Med. Chem. Res. 2021, 30, 610–623. [Google Scholar] [CrossRef]
- Stankova, I.G.; Stoilkova, A.I.; Chayrov, R.L.; Tsvetanova, E.R.; Georgieva, A.P.; Alexandrova, A.V. In Vitro Antioxidant Activity of Memantine Derivatives Containing Amino Acids. Pharm. Chem. J. 2020, 54, 268–272. [Google Scholar] [CrossRef]
- Yamauchi, M.; Kitamura, Y.; Nagano, H.; Kawatsu, J.; Gotoh, H. DPPH Measurements and Structure—Activity Relationship Studies on the Antioxidant Capacity of Phenols. Antioxidants 2024, 13, 309. [Google Scholar] [CrossRef]
- Kamaljeet Singh, S.; Gupta, G.D.; Aran, K.R. Emerging role of antioxidants in Alzheimer’s disease: Insight into physiological, pathological mechanisms and management. Pharm. Sci. Adv. 2024, 2, 100021. [Google Scholar] [CrossRef]
- Singh, N.; Ghosh, K.K. Recent advances in the antioxidant therapies for Alzheimer’s disease: Emphasis on natural antioxidants. In Pathology, Prevention and Therapeutics of Neurodegenerative Disease; Springer: Singapore, 2019; pp. 253–263. [Google Scholar] [CrossRef]
- Persson, T.; Popescu, B.O.; Cedazo-Minguez, A. Oxidative Stress in Alzheimer’s Disease: Why Did Antioxidant Therapy Fail? Oxidative Med. Cell. Longev. 2014, 2014, 427318. [Google Scholar] [CrossRef]
- Choi, D.-H.; Lee, J. A Mini-Review of the NADPH oxidases in Vascular Dementia: Correlation with NOXs and Risk Factors for VaD. Int. J. Mol. Sci. 2017, 18, 2500. [Google Scholar] [CrossRef]
- Romay, M.C.; Toro, C.; Iruela-Arispe, M.L. Emerging Molecular Mechanisms of Vascular Dementia. Curr. Opin. Hematol. 2019, 26, 199. [Google Scholar] [CrossRef]
- Smits, L.L.; Van Harten, A.C.; Pijnenburg, Y.A.L.; Koedam, E.L.G.E.; Bouwman, F.H.; Sistermans, N.; Reuling, I.E.W.; Prins, N.D.; Lemstra, A.W.; Scheltens, P.; et al. Trajectories of cognitive decline in different types of dementia. Psychol. Med. 2015, 45, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.K.; Storandt, M.; Morris, J.C.; Galvin, J.E. Longitudinal Study of the Transition from Healthy Aging to Alzheimer Disease. Arch. Neurol. 2009, 66, 1254. [Google Scholar] [CrossRef] [PubMed]
- Hugo, J.; Ganguli, M. Dementia and Cognitive Impairment: Epidemiology, Diagnosis, and Treatment. Clin. Geriatr. Med. 2014, 30, 421. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515. [Google Scholar] [CrossRef]
- Kalaria, R.N. Neuropathological diagnosis of vascular cognitive impairment and vascular dementia with implications for Alzheimer’s disease. Acta Neuropathol. 2016, 131, 659–685. [Google Scholar] [CrossRef] [PubMed]
- Perez, L.; Heim, L.; SHerzai, A.; JaceLdo-Siegl, K.; Sherzai, A. Nutrition and vascular dementia. J. Nutr. Health Aging 2012, 16, 319–324. [Google Scholar] [CrossRef]
- Villalpando-Rodriguez, G.E.; Gibson, S.B. Review Article Reactive Oxygen Species (ROS) Regulates Different Types of Cell Death by Acting as a Rheostat. Oxidative Med. Cell. Longev. 2021, 2021, 9912436. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Liu, S.; Sun, Y.; Chen, C.; Yang, S.; Lin, M.; Long, J.; Yao, J.; Lin, Y.; Yi, F.; et al. Targeting oxidative stress as a preventive and therapeutic approach for cardiovascular disease. J. Transl. Med. 2023, 21, 519. [Google Scholar] [CrossRef]
- Cai, H. Hydrogen peroxide regulation of endothelial function: Origins, mechanisms, and consequences. Cardiovasc. Res. 2005, 68, 26–36. [Google Scholar] [CrossRef]
- Schreurs, M.P.H.; Cipolla, M.J. Cerebrovascular dysfunction and blood-brain barrier permeability induced by oxidized LDL are prevented by apocynin and magnesium sulfate in female rats. J. Cardiovasc. Pharmacol. 2014, 63, 33. [Google Scholar] [CrossRef]
- Sieg, S.F.; Bazdar, D.A.; Zidar, D.; Freeman, M.; Lederman, M.M.; Funderburg, N.T. Highly oxidized low-density lipoprotein mediates activation of monocytes but does not confer interleukin-1β secretion nor interleukin-15 transpresentation function. Immunology 2020, 159, 221–230. [Google Scholar] [CrossRef]
- Gui, Y.; Zheng, H.; Cao, R.Y. Foam Cells in Atherosclerosis: Novel Insights into Its Origins, Consequences, and Molecular Mechanisms. Front. Cardiovasc. Med. 2022, 9, 845942. [Google Scholar] [CrossRef] [PubMed]
- Doyle, B.; Caplice, N. Plaque Neovascularization and Antiangiogenic Therapy for Atherosclerosis. J. Am. Coll. Cardiol. 2007, 49, 2073–2080. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Huang, R.; Zhang, R.; Xiao, C.; Wang, L.; Luo, M.; Song, N.; Zhang, J.; Yang, F.; Liu, X.; et al. Gastrodin and Gastrodigenin Improve Energy Metabolism Disorders and Mitochondrial Dysfunction to Antagonize Vascular Dementia. Molecules 2023, 28, 2598. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.X.; Yang, H.; Wang, D.S.; Chen, T.T.; Qi, X.L.; Tao, L.; Chen, Y.; Shen, X.C. Gastrodin alleviates mitochondrial dysfunction by regulating SIRT3-mediated TFAM acetylation in vascular dementia. Phytomedicine 2024, 128, 155369. [Google Scholar] [CrossRef]
- Li, L.; Willets, R.S.; Polidori, M.C.; Stahl, W.; Nelles, G.; Sies, H.; Griffiths, H.R. Oxidative LDL modification is increased in vascular dementia and is inversely associated with cognitive performance. Free Radic. Res. 2010, 44, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Cao, Y.; Ma, L.; Pei, H.; Rausch, W.D.; Li, H. Dysfunction of Cerebrovascular Endothelial Cells: Prelude to Vascular Dementia. Front. Aging Neurosci. 2018, 10, 401453. [Google Scholar] [CrossRef] [PubMed]
- Fabian, R.H.; Perez-Polo, J.R.; Kent, T.A. Perivascular nitric oxide and superoxide in neonatal cerebral hypoxia-ischemia. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1809–H1814. [Google Scholar] [CrossRef]
- Mahdi, A.; Tengbom, J.; Alvarsson, M.; Wernly, B.; Zhou, Z.; Pernow, J. Red blood cell peroxynitrite causes endothelial dysfunction in type 2 diabetes mellitus via arginase. Cells 2020, 9, 1712. [Google Scholar] [CrossRef]
- El-Remessy, A.B.; Tawfik, H.E.; Matragoon, S.; Pillai, B.; Caldwell, R.B.; Caldwell, R.W. Peroxynitrite mediates diabetes-induced endothelial dysfunction: Possible role of Rho kinase activation. Exp. Diabetes Res. 2010, 2010, 247861. [Google Scholar] [CrossRef]
- Hafez, S.; Abdelsaid, M.; Fagan, S.C.; Ergul, A. Peroxynitrite-Induced Tyrosine Nitration Contributes to Matrix Metalloprotease-3 Activation: Relevance to Hyperglycemic Ischemic Brain Injury and Tissue Plasminogen Activator. Neurochem. Res. 2018, 43, 259. [Google Scholar] [CrossRef] [PubMed]
- Szabó, C.; Ischiropoulos, H.; Radi, R. Peroxynitrite: Biochemistry, pathophysiology and development of Therapeutics. Nat. Rev. Drug Discov. 2007, 6, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Ebadi, M.; Sharma, S.K. Peroxynitrite and mitochondrial dysfunction in the pathogenesis of Parkinson’s disease. Antioxid. Redox Signal. 2003, 5, 319–335. [Google Scholar] [CrossRef]
- Tauffenberger, A.; Magistretti, P.J. Reactive Oxygen Species: Beyond Their Reactive Behavior. Neurochem. Res. 2021, 46, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Trigiani, L.J.; Hamel, E. An endothelial link between the benefits of physical exercise in dementia. J. Cereb. Blood Flow Metab. 2017, 37, 2649–2664. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.Y.; Degendorfer, G.; Hammer, A.; Whitelock, J.M.; Malle, E.; Davies, M.J. Oxidation modifies the structure and function of the extracellular matrix generated by human coronary artery endothelial cells. Biochem. J. 2014, 459, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Ivan, C.S. Dementia after stroke: The Framingham Study. Stroke 2004, 35, 1264–1268. [Google Scholar] [CrossRef]
- Choi, B.R. Characterization of White Matter Injury in a Rat Model of Chronic Cerebral Hypoperfusion. Stroke 2016, 47, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Bi, R.; Sun, S.; Chen, S.; Chen, J.; Hu, B.; Jin, H. The Role of Oxidative Stress in Acute Ischemic Stroke-Related Thrombosis. Oxidative Med. Cell Longev. 2022, 2022, 8418820. [Google Scholar] [CrossRef]
- Back, D.B. Chronic cerebral hypoperfusion induces post-stroke dementia following acute ischemic stroke in rats. J. Neuroinflamm. 2017, 14, 216. [Google Scholar] [CrossRef]
- Zekry, D.; Epperson, T.K.; Krause, K.H. A role for NOX NADPH oxidases in Alzheimer’s disease and other types of dementia? IUBMB Life 2003, 55, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Bhayadia, R. Senescence-Induced Oxidative Stress Causes Endothelial Dysfunction. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 71, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Drummond, G.R. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2020, 17, 170–194. [Google Scholar] [CrossRef] [PubMed]
- Leto, T.L. Targeting and regulation of reactive oxygen species generation by Nox family NADPH oxidases. Antioxid. Redox Signal. 2009, 11, 2607–2619. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.P.; Jandeleit-Dahm, K.A. The role of NADPH oxidase in vascular disease–hypertension, atherosclerosis & stroke. Curr. Pharm. Des. 2015, 21, 5933–5944. [Google Scholar]
- Miller, A.A. NADPH oxidase activity and function are profoundly greater in cerebral versus systemic arteries. Circ. Res. 2005, 97, 1055–1062. [Google Scholar] [CrossRef]
- Chamseddine, A.H.; Miller, F.J., Jr. Gp91phox contributes to NADPH oxidase activity in aortic fibroblasts but not smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, 2284–2289. [Google Scholar] [CrossRef]
- Begum, R. NADPH oxidase family proteins: Signaling dynamics to disease management. Cell. Mol. Immunol. 2022, 19, 660–686. [Google Scholar] [CrossRef] [PubMed]
- Haurani, M.J.; Pagano, P.J. Adventitial fibroblast reactive oxygen species as autacrine and paracrine mediators of remodeling: Bellwether for vascular disease? Cardiovasc. Res. 2007, 75, 679–689. [Google Scholar] [CrossRef]
- Lassègue, B.; Clempus, R.E. Vascular NAD(P)H oxidases: Specific features, expression, and regulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, 277–297. [Google Scholar] [CrossRef] [PubMed]
- Lassègue, B.; Griendling, K.K. NADPH oxidases: Functions and pathologies in the vasculature. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.R.; Dib, S.I.; Wright, C.B. Vascular Dementia. Curr. Transl. Geriatr. Exp. Gerontol. Rep. 2013, 2, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Pastori, D.; Pignatelli, P.; Carnevale, R.; Violi, F. Nox-2 up-regulation and platelet activation: Novel insights. Prostaglandins Other Lipid Mediat. 2015, 120, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Katsuyama, M.; Matsuno, K.; Yabe-Nishimura, C. Physiological roles of NOX/NADPH oxidase, the superoxide-generating enzyme. J. Clin. Biochem. Nutr. 2011, 50, 9–22. [Google Scholar] [CrossRef]
- Ibi, M. Reactive oxygen species derived from NOX1/NADPH oxidase enhance inflammatory pain. J. Neurosci. 2008, 28, 9486–9494. [Google Scholar] [CrossRef] [PubMed]
- Camargo, L.L. Vascular Nox (NADPH Oxidase) Compartmentalization, Protein Hyperoxidation, and Endoplasmic Reticulum Stress Response in Hypertension. Hypertension 2018, 72, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Vermot, A. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef] [PubMed]
- Chay, K.O. NADPH Oxidase Mediates β-Amyloid Peptide-Induced Neuronal Death in Mouse Cortical Cultures. Chonnam Med. J. 2017, 53, 196–202. [Google Scholar] [CrossRef]
- Nortley, R. Amyloid β oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 2019, 365, eaav9518. [Google Scholar] [CrossRef]
- Rao, R. Oxidative stress-induced disruption of epithelial and endothelial tight junctions. Front. Biosci. 2008, 13, 7210–7226. [Google Scholar] [CrossRef] [PubMed]
- Diebold, I. The NADPH oxidase subunit NOX4 is a new target gene of the hypoxia-inducible factor-1. Mol. Biol. Cell 2010, 21, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Clempus, R.E. Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Nisimoto, Y. Nox4: A hydrogen peroxide-generating oxygen sensor. Biochemistry 2014, 53, 5111–5120. [Google Scholar] [CrossRef]
- Katsuyama, M. Sp3 transcription factor is crucial for transcriptional activation of the human NOX4 gene. FEBS J. 2011, 278, 964–972. [Google Scholar] [CrossRef]
- Mittal, M. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ. Res. 2007, 101, 258–267. [Google Scholar] [CrossRef]
- Frölich, L.; Götz, M.E.; Weinmüller, M.; Youdim, M.B.H.; Barth, N.; Dirr, A.; Gsell, W.; Jellinger, K.; Beckmann, H.; Riederer, P. (r)-, but not (s)-alpha lipoic acid stimulates deficient brain pyruvate dehydrogenase complex in vascular dementia, but not in Alzheimer dementia. J. Neural Transm. 2004, 111, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Gocmez, S.S.; Şahin, T.D.; Yazir, Y.; Duruksu, G.; Eraldemir, F.C.; Polat, S.; Utkan, T. Resveratrol prevents cognitive deficits by attenuating oxidative damage and inflammation in rat model of streptozotocin diabetes induced vascular dementia. Physiol. Behav. 2019, 201, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.R.; Xu, F.; Xu, X.C.; Tan, G.J.; Liu, L.M.; Wu, N.; Zhang, W.Z.; Liu, J.X. Effects of alpha-lipoic acid on spatial learning and memory, oxidative stress, and central cholinergic system in a rat model of vascular dementia. Neurosci. Lett. 2015, 587, 113–119. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.; Wang, Y.; Wang, G.; Mao, L.; Zhang, D.; Wang, J. Effects of resveratrol on learning and memory in rats with vascular dementia. Mol. Med. Rep. 2019, 20, 4587–4593. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.G.; Varatharajan, R.; Muthuraman, A. The Attenuating Effect of Beta-Carotene on Streptozotocin Induced Diabetic Vascular Dementia Symptoms in Rats. Molecules 2022, 27, 4293. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.W.; Yin, X.L.; Lin, R.; Fan, X.L.; Chen, S.J.; Zhu, Y.M.; Zhao, X.Z. Possible mechanisms of lycopene amelioration of learning and memory impairment in rats with vascular dementia. Neural Regen. Res. 2020, 15, 332. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, J.; Wang, J.; Li, Y.; Liu, W.; Xia, J. Lycopene supplementation protects vascular dementia gerbils against the impairment of learning and memory. Folia Neuropathol. 2021, 59, 161–173. [Google Scholar] [CrossRef]
- Asadi Nejad, H.; Yousefi Nejad, A.; Akbari, S.; Naseh, M.; Shid Moosavi, S.M.; Haghani, M. The low and high doses administration of lutein improves memory and synaptic plasticity impairment through different mechanisms in a rat model of vascular dementia. PLoS ONE 2024, 19, e0302850. [Google Scholar] [CrossRef]
- Zhu, N.; Liang, X.; Zhang, M.; Yin, X.; Yang, H.; Zhi, Y.; Ying, G.; Zou, J.; Chen, L.; Yao, X.; et al. Astaxanthin protects cognitive function of vascular dementia. Behav. Brain Funct. 2020, 16, 10. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Xu, Q.; Li, G.; Bu, Y.; Sun, F.; Zhang, J. Therapeutic Effect of Idebenone on Rats with Vascular Dementia via the MicroRNA-216a/RSK2/NF-κB Axis. Neuropsychiatr. Dis. Treat. 2021, 17, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.R.; Sun, Z.K.; Liu, Y.R.; Jia, Y.J.; Zhang, B.A.; Zhang, J.W. Resveratrol improves cognition and reduces oxidative stress in rats with vascular dementia. Neural Regen. Res. 2013, 8, 2050. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Sunkaria, A.; Singhal, N.; Sandhir, R. Resveratrol loaded solid lipid nanoparticles attenuate mitochondrial oxidative stress in vascular dementia by activating Nrf2/HO-1 pathway. Neurochem. Int. 2018, 112, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, J.; Wang, P.; Rao, Y.; Chen, L. Resveratrol Reverses the Synaptic Plasticity Deficits in a Chronic Cerebral Hypoperfusion Rat Model. J. Stroke Cerebrovasc. Dis. 2016, 25, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Ozacmak, V.H.; Sayan-Ozacmak, H.; Barut, F. Chronic treatment with resveratrol, a natural polyphenol found in grapes, alleviates oxidative stress and apoptotic cell death in ovariectomized female rats subjected to chronic cerebral hypoperfusion. Nutr. Neurosci. 2016, 19, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; He, J.; Pan, C.; Wang, J.; Ma, M.; Shi, X.; Xu, Z. Resveratrol Activates Autophagy via the AKT/mTOR Signaling Pathway to Improve Cognitive Dysfunction in Rats with Chronic Cerebral Hypoperfusion. Front. Neurosci. 2019, 13, 859. [Google Scholar] [CrossRef]
- Anastácio, J.R.; Netto, C.A.; Castro, C.C.; Sanches, E.F.; Ferreira, D.C.; Noschang, C.; Krolow, R.; Dalmaz, C.; Pagnussat, A. Resveratrol treatment has neuroprotective effects and prevents cognitive impairment after chronic cerebral hypoperfusion. Neurol. Res. 2014, 36, 627–633. [Google Scholar] [CrossRef]
- Shen, D.; Tian, X.; Sang, W.; Song, R. E-Mail Effect of Melatonin and Resveratrol against Memory Impairment and Hippocampal Damage in a Rat Model of Vascular Dementia. Neuroimmunomodulation 2017, 23, 318–331. [Google Scholar] [CrossRef]
- Zhu, M.L.; Zhang, J.; Guo, L.J.; Yue, R.Z.; Li, S.S.; Cui, B.Y.; Guo, S.; Niu, Q.Q.; Yu, Y.N.; Wang, H.H.; et al. Amorphous selenium inhibits oxidative stress injury of neurons in vascular dementia rats by activating NMDAR pathway. Eur. J. Pharmacol. 2023, 955, 175874. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhao, T.; Zheng, B.; Zhang, Y.; Li, X.; Zhang, F.; Cen, J.; Duan, S. Curcumin Derivative Cur20 Attenuated Cerebral Ischemic Injury by Antioxidant Effect and HIF-1α/VEGF/TFEB-Activated Angiogenesis. Front. Pharmacol. 2021, 12, 648107. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zhang, J.; Zhao, Y.; Zhang, Y.; Zhang, X.; Guan, J.; Liu, Y.; Fu, J. Curcumin protects against cognitive impairments in a rat model of chronic cerebral hypoperfusion combined with diabetes mellitus by suppressing neuroinflammation, apoptosis, and pyroptosis. Int. Immunopharmacol. 2021, 93, 107422. [Google Scholar] [CrossRef]
- Davinelli, S.; Ali, S.; Solfrizzi, V.; Scapagnini, G.; Corbi, G. Carotenoids and Cognitive Outcomes: A Meta-Analysis of Randomized Intervention Trials. Antioxidants 2021, 10, 223. [Google Scholar] [CrossRef]
- Polidori, M.C.; Stahl, W.; Griffiths, H.R. Nutritional cognitive neuroscience of aging: Focus on carotenoids and cognitive frailty. Redox Biol. 2021, 44, 101996. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Khan, S.T.; Aziz, A.; Gul, S.; Buvnariu, L.; Zia-Ul-Haq, M. Role of Carotenoids in Neurological Diseases. In Carotenoids: Structure and Function in the Human Body; Springer International Publishing: Cham, Switzerland, 2021; pp. 555–568. [Google Scholar]
- Foy, C.J.; Passmore, A.P.; Vahidassr, M.D.; Young, I.S.; Lawson, J.T. Plasma chain-breaking antioxidants in Alzheimer’s disease, vascular dementia and Parkinson’s disease. QJM Int. J. Med. 1999, 92, 39–45. [Google Scholar] [CrossRef]
- Polcz, M.E.; Barbul, A. The Role of Vitamin A in Wound Healing. Nutr. Clin. Pract. 2019, 34, 695–700. [Google Scholar] [CrossRef]
- Dias, I.H.K.; Polidori, M.C.; Li, L.; Weber, D.; Stahl, W.; Nelles, G.; Grune, T. Plasma levels of HDL and carotenoids are lower in dementia patients with vascular comorbidities. J. Alzheimer’s Dis. 2014, 40, 399–408. [Google Scholar] [CrossRef]
- Cole, G.M.; Frautschy, S.A. DHA may prevent age-related dementia. J. Nutr. 2010, 140, 869–874. [Google Scholar] [CrossRef]
- Jaber, S.M.; Ge, S.X.; Milstein, J.L.; VanRyzin, J.W.; Waddell, J.; Polster, B.M. Idebenone Has Distinct Effects on Mitochondrial Respiration in Cortical Astrocytes Compared to Cortical Neurons Due to Differential NQO1 Activity. J. Neurosci. 2020, 40, 4609–4619. [Google Scholar] [CrossRef] [PubMed]
- Dumont, M.; Beal, M.F. Neuroprotective strategies involving ROS in Alzheimer’s disease. Free Radic. Biol. Med. 2011, 51, 1014. [Google Scholar] [CrossRef] [PubMed]
- Chodari, L.; Sehati, F.; Hafazeh, L.; Nikbakhtzadeh, M.; Ataei, S.; Ranjbaran, M.; Ashabi, G.; Hosseindoost, S. Inhibition of histone methyltransferase promotes cognition and mitochondrial function in vascular dementia model. Behav. Brain Res. 2024, 473, 115194. [Google Scholar] [CrossRef] [PubMed]
- Marigliano, V.; Abate, G.; Barbagallo-Sangiorgi, G.; Bartorelli, L.; Capurso, A.; Cucinotta, D.; Cuzzupoli, M.; Senin, U.; Tammaro, A.E.; Fioravanti, M. Randomized, double-blind, placebo controlled, multicentre study of idebenone in patients suffering from multi-infarct dementia. Arch. Gerontol. Geriatr. 1992, 15, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.F.G.; Ruscica, M.; Banach, M. Resveratrol and cognitive decline: A clinician perspective. Arch. Med. Sci. 2019, 15, 936–943. [Google Scholar] [CrossRef] [PubMed]
- da Luz, P.L.; Fialdini, R.C.; Nishiyama, M. Red Wine, Resveratrol, and Vascular Aging: Implications for Dementia and Cognitive Decline. In Diet and Nutrition in Dementia and Cognitive Decline; Academic Press: Cambridge, MA, USA, 2015; pp. 943–953. [Google Scholar] [CrossRef]
- Morrison, C.D.; Pistell, P.J.; Ingram, D.K.; Johnson, W.D.; Liu, Y.; Fernandez-Kim, S.O.; White, C.L.; Purpera, M.N.; Uranga, R.M.; Bruce-Keller, A.J.; et al. High fat diet increases hippocampal oxidative stress and cognitive impairment in aged mice: Implications for decreased Nrf2 signaling. J. Neurochem. 2010, 114, 1581–1589. [Google Scholar] [CrossRef]
- Casaril, A.M.; Ignasiak, M.T.; Chuang, C.Y.; Vieira, B.; Padilha, N.B.; Carroll, L.; Lenardão, E.J.; Savegnago, L.; Davies, M.J. Selenium-containing indolyl compounds: Kinetics of reaction with inflammation-associated oxidants and protective effect against oxidation of extracellular matrix proteins. Free Radic. Biol. Med. 2017, 113, 395–405. [Google Scholar] [CrossRef]
- Storkey, C.; Pattison, D.I.; Ignasiak, M.T.; Schiesser, C.H.; Davies, M.J. Kinetics of reaction of peroxynitrite with selenium- and sulfur-containing compounds: Absolute rate constants and assessment of biological significance. Free Radic. Biol. Med. 2015, 89, 1049–1056. [Google Scholar] [CrossRef]
- Crow, J.P. Peroxynitrite scavenging by metalloporphyrins and thiolates. Free Radic. Biol. Med. 2000, 28, 1487–1494. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Clifton, P. Curcumin, Cardiometabolic Health and Dementia. Int. J. Environ. Res. Public Health 2018, 15, 2093. [Google Scholar] [CrossRef]
- Brondino, N.; Re, S.; Boldrini, A.; Cuccomarino, A.; Lanati, N.; Barale, F.; Politi, P. Curcumin as a therapeutic agent in dementia: A mini systematic review of human studies. Sci. World J. 2014, 2014, 174282. [Google Scholar] [CrossRef]
- Al Mheid, I.; Patel, R.; Murrow, J.; Morris, A.; Rahman, A.; Fike, L.; Kavtaradze, N.; Uphoff, I.; Hooper, C.; Tangpricha, V.; et al. Vitamin D status is associated with arterial stiffness and vascular dysfunction in healthy humans. J. Am. Coll. Cardiol. 2011, 58, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Gurses, K.M.; Tokgozoglu, L.; Yalcin, M.U.; Kocyigit, D.; Dural, M.; Canpinar, H.; Yorgun, H.; Sahiner, M.L.; Kaya, E.B.; Akin, S.; et al. Markers of subclinical atherosclerosis in premenopausal women with vitamin D deficiency and effect of vitamin D replacement. Atherosclerosis 2014, 237, 784–789. [Google Scholar] [CrossRef]
- Jablonski, K.L.; Chonchol, M.; Pierce, G.L.; Walker, A.E.; Seals, D.R. 25-Hydroxyvitamin D deficiency is associated with inflammation-linked vascular endothelial dysfunction in middle-aged and older adults. Hypertension 2011, 57, 63–69. [Google Scholar] [CrossRef]
- Chen, L.J.; Sha, S.; Stocker, H.; Brenner, H.; Schöttker, B. The associations of serum vitamin D status and vitamin D supplements use with all-cause dementia, Alzheimer’s disease, and vascular dementia: A UK Biobank based prospective cohort study. Am. J. Clin. Nutr. 2024, 119, 1052–1064. [Google Scholar] [CrossRef]
- Kim, D.H.; Meza, C.A.; Clarke, H.; Kim, J.S.; Hickner, R.C. Vitamin D and Endothelial Function. Nutrients 2020, 12, 575. [Google Scholar] [CrossRef]
- Janoušek, J.; Pilařová, V.; Macáková, K.; Nomura, A.; Veiga-Matos, J.; Silva DD da Remião, F.; Saso, L.; Malá-Ládová, K.; Malý, J.; Nováková, L.; et al. Vitamin D: Sources, physiological role, biokinetics, deficiency, therapeutic use, toxicity, and overview of analytical methods for detection of vitamin D and its metabolites. Crit. Rev. Clin. Lab. Sci. 2022, 59, 517–554. [Google Scholar] [CrossRef]
- Vauzour, D.; Vafeiadou, K.; Rodriguez-Mateos, A.; Rendeiro, C.; Spencer, J.P.E. The neuroprotective potential of flavonoids: A multiplicity of effects. Genes. Nutr. 2008, 3, 115. [Google Scholar] [CrossRef] [PubMed]
- Imperatrice, M.; Cuijpers, I.; Troost, F.J.; Sthijns, M.M.J.P.E. Hesperidin Functions as an Ergogenic Aid by Increasing Endothelial Function and Decreasing Exercise-Induced Oxidative Stress and Inflammation, Thereby Contributing to Improved Exercise Performance. Nutrients 2022, 14, 2955. [Google Scholar] [CrossRef]
- Williams, R.J.; Spencer, J.P.E. Flavonoids, cognition, and dementia: Actions, mechanisms, and potential therapeutic utility for Alzheimer disease. Free Radic. Biol. Med. 2012, 52, 35–45. [Google Scholar] [CrossRef]
- Valls, R.M.; Pedret, A.; Calderón-Pérez, L.; Llauradó, E.; Pla-Pagà, L.; Companys, J.; Moragas, A.; Martín-Luján, F.; Ortega, Y.; Giralt, M.; et al. Hesperidin in orange juice improves human endothelial function in subjects with elevated blood pressure and stage 1 hypertension: A randomized, controlled trial (Citrus study). J. Funct. Foods 2021, 85, 104646. [Google Scholar] [CrossRef]
- Chandra, R.; Tiwari, M.; Kaur, P.; Sharma, M.; Jain, R.; Dass, S. Metalloporphyrins-Applications and clinical significance. Indian J. Clin. Biochem. 2000, 15, 183–199. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.G.; Ji, Y.K.; Hae, Y.C.; Jeong, J.C. Zingerone as an antioxidant against peroxynitrite. J. Agric. Food Chem. 2005, 53, 7617–7622. [Google Scholar] [CrossRef]
- Gioscia-Ryan, R.A.; LaRocca, T.J.; Sindler, A.L.; Zigler, M.C.; Murphy, M.P.; Seals, D.R. Mitochondria-targeted antioxidant (MitoQ) ameliorates age-related arterial endothelial dysfunction in mice. J. Physiol. 2014, 592, 2549–2561. [Google Scholar] [CrossRef]
- Bucciantini, M.; Leri, M.; Nardiello, P.; Casamenti, F.; Stefani, M. Olive Polyphenols: Antioxidant and Anti-Inflammatory Properties. Antioxidants 2021, 10, 1044. [Google Scholar] [CrossRef]
- Abdallah, I.M.; Al-Shami, K.M.; Alkhalifa, A.E.; Al-Ghraiybah, N.F.; Guillaume, C.; Kaddoumi, A. Comparison of Oleocanthal-Low EVOO and Oleocanthal against Amyloid-β and Related Pathology in a Mouse Model of Alzheimer’s Disease. Molecules 2023, 28, 1249. [Google Scholar] [CrossRef] [PubMed]
- Batarseh, Y.S.; Kaddoumi, A. Oleocanthal-rich extra-virgin olive oil enhances donepezil effect by reducing amyloid-β load and related toxicity in a mouse model of Alzheimer’s disease. J. Nutr. Biochem. 2018, 55, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Grewal, R.; Reutzel, M.; Dilberger, B.; Hein, H.; Zotzel, J.; Marx, S.; Tretzel, J.; Sarafeddinov, A.; Fuchs, C.; Eckert, G.P. Purified oleocanthal and ligstroside protect against mitochondrial dysfunction in models of early Alzheimer’s disease and brain ageing. Exp. Neurol. 2020, 328, 113248. [Google Scholar] [CrossRef] [PubMed]
- Masaki, K.H.; Losonczy, K.G.; Izmirlian, G.; Foley, D.J.; Ross, G.W.; Petrovitch, H.; Havlik, R.; White, L.R. Association of vitamin E and C supplement use with cognitive function and dementia in elderly men. Neurology 2000, 54, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Ryglewicz, D.; Rodo, M.; Kunicki, P.K.; Bednarska-Makaruk, M.; Graban, A.; Lojkowska, W.; Wehr, H. Plasma antioxidant activity and vascular dementia. J. Neurol. Sci. 2002, 203–204, 195–197. [Google Scholar] [CrossRef]
- Sinclair, A.J.; Bayer, A.J.; Johnston, J.O.; Warner, C.; Maxwell, S.R.J. Altered Plasma Antioxidant Status in Subjects with Alzheimer’s Disease and Vascular Dementia. Int. J. Geriatr. Psychiatry 1998, 13, 840–845. [Google Scholar] [CrossRef]
- Gautam, J. Dual Inhibition of NOX2 and Receptor Tyrosine Kinase by BJ-1301 Enhances Anticancer Therapy Efficacy via Suppression of Autocrine-Stimulatory Factors in Lung Cancer. Mol. Cancer Ther. 2017, 16, 2144–2156. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y. Rosuvastatin Alleviates Coronary Microembolization-Induced Cardiac Injury by Suppressing Nox2-Induced ROS Overproduction and Myocardial Apoptosis. Cardiovasc. Toxicol. 2022, 22, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, P. Rosuvastatin reduces platelet recruitment by inhibiting NADPH oxidase activation. Biochem. Pharmacol. 2012, 84, 1635–1642. [Google Scholar] [CrossRef]
- Chen, X. Dexmedetomidine Alleviates Hypoxia-Induced Synaptic Loss and Cognitive Impairment via Inhibition of Microglial NOX2 Activation in the Hippocampus of Neonatal Rats. Oxidative Med. Cell. Longev. 2021, 2021, 6643171. [Google Scholar] [CrossRef]
- Barua, S. The role of NOX inhibitors in neurodegenerative diseases. IBRO Rep. 2019, 7, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Chocry, M.; Leloup, L. The NADPH Oxidase Family and Its Inhibitors. Antioxid. Redox Signal. 2012, 33, 332–353. [Google Scholar] [CrossRef]
- Mariani, E.; Polidori, M.C.; Cherubini, A.; Mecocci, P. Oxidative stress in brain aging, neurodegenerative and vascular diseases: An overview. J. Chromatogr. B Analyt Technol. Biomed. Life Sci. 2005, 827, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Ernesto, C.; Thomas, R.G.; Klauber, M.R.; Schafer, K.; Grundman, M.; Woodbury, P.; Growdon, J.; Cotman, C.W.; Pfeiffer, E.; et al. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. N. Engl. J. Med. 1997, 336, 1216–1222. [Google Scholar] [CrossRef] [PubMed]
- Marder, K. Vitamin E and donepezil for the treatment of mild cognitive impairment. Curr. Neurol. Neurosci. Rep. 2005, 5, 337–338. [Google Scholar] [CrossRef] [PubMed]
- Baum, L.; Lam, C.W.K.; Cheung, S.K.-K.; Kwok, T.; Lui, V.; Tsoh, J.; Lam, L.; Leung, V.; Hui, E.; Ng, C.; et al. Six-month randomized, placebo-controlled, double-blind, pilot clinical trial of curcumin in patients with Alzheimer disease. J. Clin. Psychopharmacol. 2008, 28, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Commenges, D.; Scotet, V.; Renaud, S.; Jacqmin-Gadda, H.; Barberger-Gateau, P.; Dartigues, J.F. Intake of flavonoids and risk of dementia. Eur. J. Epidemiol. 2000, 16, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Shukla, D.; Tripathi, M.; Ersland, L. Cognitive Improvement with Glutathione Supplement in Alzheimer’s Disease: A Way Forward. J. Alzheimer’s Dis. 2019, 68, 531–535. [Google Scholar] [CrossRef]
- Hager, K.; Kenklies, M.; McAfoose, J.; Engel, J.; Münch, G. Alpha-lipoic acid as a new treatment option for Alzheimer’s disease—A 48 months follow-up analysis. J. Neural Transm. 2007, 72, 189–193. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, H.; Qi, X.; Wu, F.; Zhang, D. Effect of butylphthalide combined with idebenone on vascular dementia: A retrospective observational analysis. Medicine 2024, 103, e37495. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.X.; Hu, Y.; Kang, L.J.; Li, P.; Gao, T.C.; Zhang, X. Effects of Butyphthalide Combined with Idebenone on Inflammatory Cytokines and Vascular Endothelial Functions of Patients with Vascular Dementia. J. Coll. Physicians Surg. Pak. 2020, 30, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.Q.; Liang, X.M.; Juan-Wu Zhang, Y.F.; Zhu, C.X.; Jiang, X.J. Treatment with Huperzine A improves cognition in vascular dementia patients. Cell Biochem. Biophys. 2012, 62, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Nappi, G.; Bono, G.; Merlo, P.; Denaro, A.; Proietti, R.; Martucci, N.; Fioravanti, M. Long-term idebenone treatment of vascular and degenerative brain disorders of the elderly. Arch. Gerontol. Geriatr. 1992, 15, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Alshial, E.E.; Abdulghaney, M.I.; Wadan, A.-H.S.; Abdellatif, M.A.; Ramadan, N.E.; Suleiman, A.M.; Waheed, N.; Abdellatif, M.; Mohammed, H.S. Mitochondrial dysfunction and neurological disorders: A narrative review and treatment overview. Life Sci. 2023, 334, 122257. [Google Scholar] [CrossRef] [PubMed]
- Zs-Nagy, I. Chemistry, toxicology, pharmacology and pharmacokinetics of idebenone: A review. Arch. Gerontol. Geriatr. 1990, 11, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.A.; Perry, E.K.; Tomlinson, B.E. Acetylcholine and choline levels in post-mortem human brain tissue: Preliminary observations in Alzheimer’s disease. Life Sci. 1980, 26, 1683–1689. [Google Scholar] [CrossRef]
- Chen, D.-P.; Hou, S.-H.; Chen, Y.-G.; Chen, M.-S.; Hu, Z.-Z.; Zhang, Z.-J. L-butyl phthalein improves neural function of vascular dementia mice by regulating the PI3K/AKT signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 5377–5384. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.-S.; Wang, H.; Wei, Z.-H.; Song, Y.-Y.; Zhang, L.; Chen, H.-Z. Efficacy and safety of natural acetylcholinesterase inhibitor huperzine A in the treatment of Alzheimer’s disease: An updated meta-analysis. J. Neural Transm. 2009, 116, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Zheng, C.Y.; Yan, H.; Wang, Z.F.; Tang, L.L.; Gao, X.; Tang, X.C. Potential therapeutic targets of huperzine A for Alzheimer’s disease and vascular dementia. Chem. Biol. Interact. 2008, 175, 396–402. [Google Scholar] [CrossRef]
- Meyer, J.S.; Stoica, E.; Pascu, I.; Shimazu, K.; Hartmann, A. Catecholamine concentrations in csf and plasma of patients with cerebral infarction and hæmorrhage. Brain 1973, 96, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, M.; Itoh, T. Effects of idebenone on monoamine metabolites in cerebrospinal fluid of patients with cerebrovascular dementia. Arch. Gerontol. Geriatr. 1989, 8, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Linh, T.T.D.; Hsieh, Y.C.; Huang, L.K.; Hu, C.J. Clinical Trials of New Drugs for Vascular Cognitive Impairment and Vascular Dementia. Int. J. Mol. Sci. 2022, 23, 11067. [Google Scholar] [CrossRef]
- Browne, D.; McGuinness, B.; Woodside, J.V.; McKay, G.J. Vitamin E and Alzheimer’s disease: What do we know so far? Clin. Interv. Aging 2019, 14, 1303. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N.; Akinyemi, R.; Ihara, M. Stroke injury, cognitive impairment and vascular dementia. Biochim. Biophys. Acta 2016, 1862, 915–925. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Mechanism of Action | Effect | Side Effects | Inhibition % (DPPH) | Concentration Range (µg/mL) | Bioavailability | References | |
|---|---|---|---|---|---|---|---|---|
| Repurposed Medications | Donepezil | AChEi | Mental clarity | Nausea, vomiting, and QT prolongation | 33.5–42.3% | 10–1000 | Moderate (40–100%) | Vinay Munishamappa et al., 2018 [32] |
| Galantamine | AChEi | Mental clarity | Dry mouth, constipation, and QT prolongation | 20–35% | 50–200 | High (90%) | Victor Wagner Barajas-Carrillo et al., 2020 [33] | |
| Memantine | NMDAr antagonist | Neuronal protection | Dizziness, headache | 1–4% | 100–1000 | High (100%) | I.G. Stankova et al., 2020 [34] | |
| Antioxidant Vitamins and Supplements * | Curcumin * | Antioxidant/Anti-inflammatory | Anti-inflammatory | Gastrointestinal upset | 65–75% | 100–500 | Very low (<1%) | Moeka Yamauchi et al., 2024 [35] |
| Vitamin C * | Antioxidant | Immune support | Diarrhea (high doses) | 90% | 50–100 | High (70–90%) | Moeka Yamauchi et al., 2024 [35] | |
| Vitamin E * | Antioxidant | Skin health | Bleeding risk (high doses) | 70–80% | 100–200 | Low (20–40%) | Moeka Yamauchi et al., 2024 [35] | |
| Glutathione * | Antioxidant/Detoxifier | Detoxification | Rare (allergic reactions) | 50–60% | 100–500 | Low (poor oral bioavailability) | Moeka Yamauchi et al., 2024 [35] | |
| Resveratrol * | Antioxidant | Cardiovascular health | Gastrointestinal upset | 60–70% | 100–200 | Very low (<1%) | Moeka Yamauchi et al., 2024 [35] |
| Species | VaD Model | Antioxidant | Dose | Results | Reference | |
|---|---|---|---|---|---|---|
| Increase | Decrease | |||||
| Human | VaD brain tissue | (r)-ALA | 1 µM, 10 µM, 100 µM, 1 mM, and 10 mM | PDHc activity (10 µm) SDH activity (1 mM) | PDHc activity (10 mM) SDH activity (10 mM) | L. Frolich et al., 2004 [97] |
| Human | VaD brain tissue | (s)-ALA | 1 µM, 10 µM, 100 µM, 1 mM, and 10 mM | SDH activity (1 mM) | PDHc activity (10 mM) SDH activity (10 mM) | L. Frolich et al., 2004 [97] |
| Male wistar rat adult | Induced diabetic VaD | RSVL | 20 mg/kg i.p. daily for 4 weeks | Cognition, body weight, blood glucose, carbachol, antioxidant levels, BDNF, and eNOS | Inflammation, HO-1, and NOX | Semil Selcen Gocmez et al., 2019 [98] |
| Male wistar rat adult | BCCAO | ALA | 50 mg/kg i.p. daily for 28 days | Cognition, GSH levels, ACh, and ChAT | ROS and AChE levels | Ran-ran Zhao et al., 2015 [99] |
| Male SD rat 2 mo | BCCAO | RSVL | 20 and 10 mL/kg i.p. daily for 4 weeks | Cognition and SOD levels | MDA and apoptosis | Yeqing Zhang et al., 2019 [100] |
| Male SD rat | Induced diabetic VaD | Beta-carotene | 50 mg/kg oral daily for 15 days | Cognition and GSH levels | AChE and thiobarbituric acid reactive substance levels | Khian Giap Lim et al., 2022 [101] |
| Male SD rat | Induced diabetic VaD | Beta-carotene | 100 mg/kg oral daily for 15 days | Cognition and GSH levels | AChE and Thiobarbituric acid reactive substances levels | Khian Giap Lim et al., 2022 [101] |
| Female SD rat 2 mo | BCCAO | Lycopene | 50 mg/kg i.g. every other day for 2 mo | ROS in CA1, CA3, and DG | −−− | Ning-Wei Zhu et al., 2020 [102] |
| Female SD rat 2 mo | BCCAO | Lycopene | 100 mg/kg i.g. every other day for 2 mo | Cognition and SOD activity | ROS in CA1, CA3, and DG | Ning-Wei Zhu et al., 2020 [102] |
| Female SD rat 2 mo | BCCAO | Lycopene | 200 mg/kg i.g. every other day for 2 mo | ROS in CA1, CA3, and DG | −−− | Ning-Wei Zhu et al., 2020 [102] |
| Gerbil | BCCAO | Lycopene | 20 mg/kg twice a day for 28 days | Cognition, neurons in CA1, and antioxidant activity | Inflammation, apoptosis, and GFAP in CA1 | Wei Chen et al., 2021 [103] |
| Male SD rat 7–10 weeks | BCCAO | Lutein | 0.5 mg/kg daily for 30 days | Cognition, pyramidal neuronal cells, and conduction in CA1 | MDA levels | Hamideh Asadi Nejad et al., 2024 [104] |
| Male SD rat 7–10 weeks | BCCAO | Lutein | 5 mg/kg | Cognition (NOR), pyramidal neuronal cells, and plasticity in CA1 | MDA levels | Hamideh Asadi Nejad et al., 2024 [104] |
| Male mice | UCCAO | Astaxanthin | 50 mg/kg daily for 30 days | Cognition (NOR), SOD levels, and IL-4 levels | IL1β levels | Ningwei Zhu et al., 2020 [105] |
| Male mice | UCCAO | Astaxanthin | 100 mg/kg daily for 30 days | Cognition (NOR), IL4, and SOD levels | IL1β and MDA levels | Ningwei Zhu et al., 2020 [105] |
| Male mice | UCCAO | Astaxanthin | 200 mg/kg daily for 30 days | Cognition (NOR and MWM), IL4, and SOD levels | IL1β and MDA levels | Ningwei Zhu et al., 2020 [105] |
| Male SD rat adult | BCCAO | Idebenone | 100 mg/kg oral daily for 3 weeks | Cognition and neuronal levels in CA1 | TNFα | Xudong Qian et al., 2021 [106] |
| Wistar rat 12–14 mo | BCCAO | RSVL | 25 mg/kg oral for 4 weeks | Cognition, neurons, and antioxidant activity | Inflammation and MDA levels | Xingrong Ma et al., 2013 [107] |
| Male SD rat | BCCAO | RSVL | 10 mg/kg oral daily for 4 weeks | Cognition, redox ratio, and SOD, HO1, and Nrf2 levels | ROS, GSSG, Hif1α, LPO, and protein carbonylation | Aarti Yadav et al., 2018 [108] |
| Male wistar rat | BCCAS | RSVL | 40 mg/kg i.p. for 4 weeks | Cognition, synaptic spines, and synapse-associated proteins | −−− | Huagang Li et al., 2016 [109] |
| Female wistar rat 8–10 mo | mBCCAO | RSVL | Daily 10 mg/kg oral for 15 days | GSH and pyramidal neurons | MDA and GFAP levels | Veysel Haktan Ozacmak et al., 2016 [110] |
| Male SD rat | BCCAO | RSVL | 50 mg/kg i.g. daily for 9 weeks | Cognition and antioxidant activity | AKT/mTOR signaling pathway, autophagy, apoptosis, and MDA levels | Nan Wang et al., 2019 [111] |
| Male wistar rat 3 mo | m2VO | RSVL | 20 mg/kg i.p. daily for 7 days | Cognition, neuronal levels, and NGF | −−− | Janine R Anastacio et al., 2014 [112] |
| Male wistar rat | BCCAO | RSVL | 10 mg/kg for 4 weeks | −−− | MDA, TNFα, and nitrite levels | Dongfang Shen et al., 2017 [113] |
| Male wistar rat | BCCAO | RSVL | 20 mg/kg for 4 weeks | Cognition and GSH | MDA, IL1β,TNFα, AChE activity, and nitrite levels | Dongfang Shenet al., 2017 [113] |
| Male SD rat | BCCAO | Selenium | 0.1 mg/kg p.o. daily for 4 mo | Cognition, posterior cerebral blood flow, neurons, synaptic plasticity, and GSH, NO, and SOD levels | ROS, NOX, and MDA level | Mo-li Zhu et al., 2023 [114] |
| Male ICR mice | UCCAO | Curcumin | 20 µmol/kg i.g. daily for 2 weeks | Cognition (MWM) and GSH | ROS, SOD, and MDA levels | Runfang Zhang et al., 2021 [115] |
| Male ICR mice | UCCAO | Cur20 | 20 µmol/kg i.g. daily for 2 weeks | Cognition (MWM), GSH, and Hif1α | ROS, SOD, and MDA levels | Runfang Zhang et al., 2021 [115] |
| Male SD rats | Induced diabetic VaD | Curcumin | 50 mg/kg i.p. every other day for 8 weeks | Cognition (MWM), NeuN, IL10, and IL4 | Inflammation and apoptosis | Yaling Zheng et al., 2021 [116] |
| Antioxidant | Treatment | Study details | Inference | Reference |
|---|---|---|---|---|
| Idebenone | 45 mg/day b.i.d., for 120 days of oral administration | Randomized double-blind, placebo-controlled, and multicenter study on 108 elderly patients with mild-to-moderate mental deterioration of vascular origin |
Improved acquisition and retention of verbal stimuli; improvements in memory, attention, and cognitive function | Vicenzo Marigliano et al., 1992 [127] |
| Idebenone and butyphthalide | Butyphthalide (0.2 g/time; 3 times/day) combined with idebenone (30 mg/time; 3 times/day) for 12 weeks | Retrospective clinical study of 126 patients with vascular dementia, average age of 67.3 ± 7.0 years | Improved daily living activities and cognitive function of patients | Hongxia Zhang et al., 2024 [169] |
| Idebenone and butyphthalide | −−− | Randomized observational study on 88 VaD patients | Combination of Butyphthalide with idebenone reduced serum inflammatory mediators levels in VaD patients, improved vascular endothelial functions and cognitive function | Fan Xing Qi et al., 2020 [170] |
| Huperzine A | Huperzine A (0.1 mg b.i.d.), 12 consecutive weeks | Randomized, double-blinded, and placebo-controlled study with 78 patients with mild-to-moderate VaD | Improved the cognitive function in patients with VaD | Zhi Qiang Xu et al., 2012 [171] |
| Idebenone | 45 mg b.i.d. orally (90 mg over 6 mo) | Open multicenter study | Improved cognitive function in patients with VaD (MID and CCVD) | Giuseppe Nappi et al., 1992 [172] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
White, A.L.; Talkington, G.M.; Ouvrier, B.; Ismael, S.; Solch-Ottaiano, R.J.; Bix, G. Reactive Oxygen Species, a Potential Therapeutic Target for Vascular Dementia. Biomolecules 2025, 15, 6. https://doi.org/10.3390/biom15010006
White AL, Talkington GM, Ouvrier B, Ismael S, Solch-Ottaiano RJ, Bix G. Reactive Oxygen Species, a Potential Therapeutic Target for Vascular Dementia. Biomolecules. 2025; 15(1):6. https://doi.org/10.3390/biom15010006
Chicago/Turabian StyleWhite, Amanda Louise, Grant M. Talkington, Blake Ouvrier, Saifudeen Ismael, Rebecca J. Solch-Ottaiano, and Gregory Bix. 2025. "Reactive Oxygen Species, a Potential Therapeutic Target for Vascular Dementia" Biomolecules 15, no. 1: 6. https://doi.org/10.3390/biom15010006
APA StyleWhite, A. L., Talkington, G. M., Ouvrier, B., Ismael, S., Solch-Ottaiano, R. J., & Bix, G. (2025). Reactive Oxygen Species, a Potential Therapeutic Target for Vascular Dementia. Biomolecules, 15(1), 6. https://doi.org/10.3390/biom15010006