

Application of Spatial Transcriptomics in Digestive System Tumors


Abstract
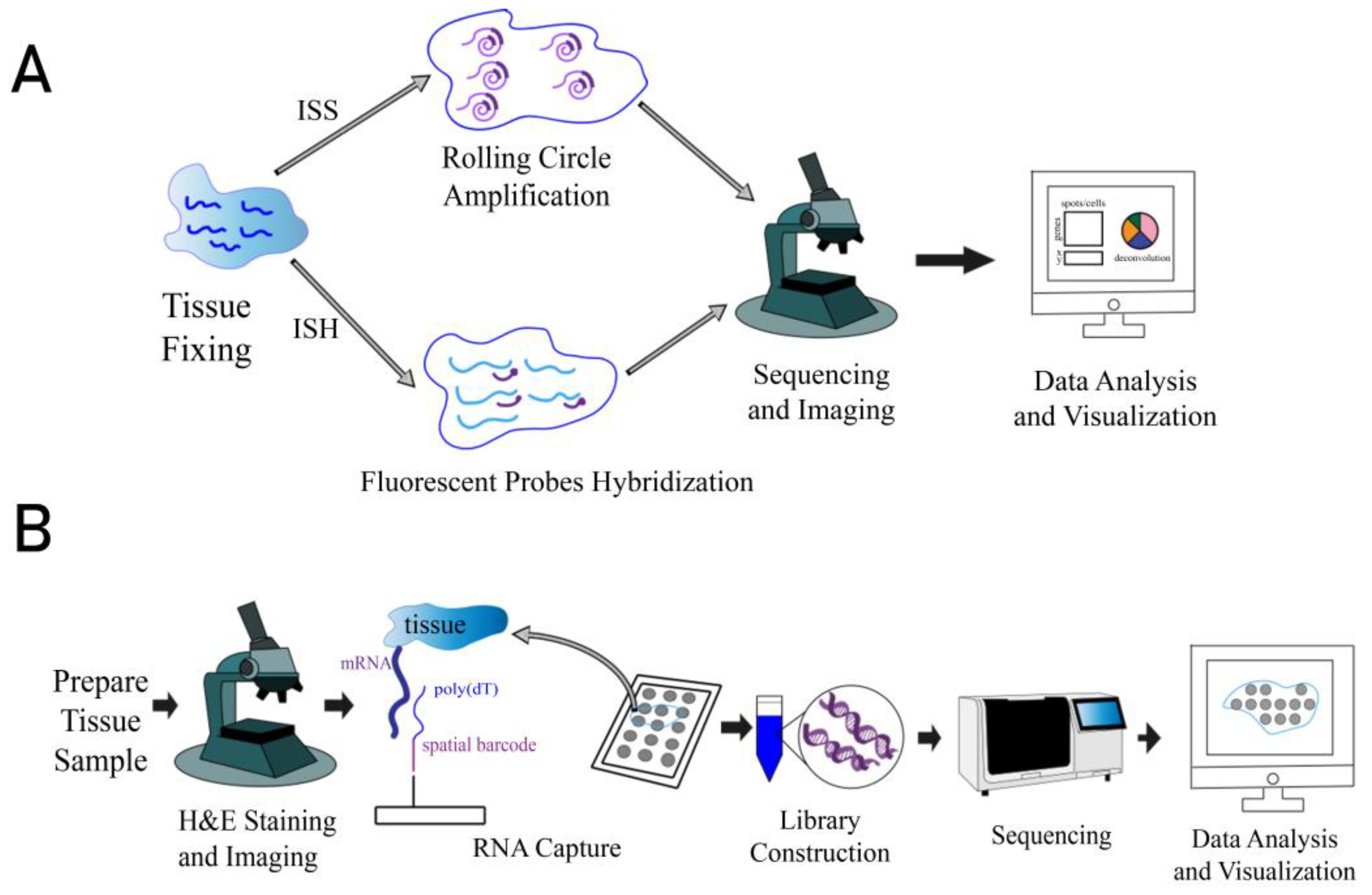
1. Introduction
2. Application of Spatial Transcriptomics in Digestive System Tumor Research
2.1. Application in Characterizing Tumor Heterogeneity and Uncovering Tumor Cell Subpopulations
2.2. Application in Studying Tumor Microenvironment
2.2.1. Application in Studying Spatial Distribution Preference of Non-Cancer Cells in Tumor Microenvironment
2.2.2. Application in Studying Function of Non-Cancer Cells and Their Interactions
2.3. Application in Studying Function of Tumor Heterogeneity in Treatment Responses
2.4. Application in Tracking Cellular Transitions in Cancer and Elucidating Cancer Evolution
2.5. Clinical Application
3. How to Select a Suitable Spatial Transcriptomics Method
4. Data Collection and Analysis in Spatial Transcriptomics Cancer Research
5. Challenges and Future Perspectives
5.1. Recent Advancements of Spatial Transcriptomics in Cancer Research
5.2. Discussion on Technical Challenges in Spatial Transcriptomics
5.3. Future Perspectives of Spatial Transcriptomics in Cancer Research
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Williams, C.G.; Lee, H.J.; Asatsuma, T.; Vento-Tormo, R.; Haque, A. An introduction to spatial transcriptomics for biomedical research. Genome Med. 2022, 14, 68. [Google Scholar] [CrossRef]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef]
- Wang, Q.; Zhi, Y.; Zi, M.; Mo, Y.; Wang, Y.; Liao, Q.; Zhang, S.; Gong, Z.; Wang, F.; Zeng, Z.; et al. Spatially resolved transcriptomics technology facilitates cancer research. Adv. Sci. 2023, 10, e2302558. [Google Scholar] [CrossRef]
- Park, H.E.; Jo, S.H.; Lee, R.H.; Macks, C.P.; Ku, T.; Park, J.; Lee, C.W.; Hur, J.K.; Sohn, C.H. Spatial transcriptomics: Technical aspects of recent developments and their applications in neuroscience and cancer research. Adv. Sci. 2023, 10, e2206939. [Google Scholar] [CrossRef]
- Rao, A.; Barkley, D.; França, G.S.; Yanai, I. Exploring tissue architecture using spatial transcriptomics. Nature 2021, 596, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Jiang, M.; Wu, L. Spatial transcriptomics technology in cancer research. Front. Oncol. 2022, 12, 1019111. [Google Scholar] [CrossRef] [PubMed]
- Lubeck, E.; Cai, L. Single-cell systems biology by super-resolution imaging and combinatorial labeling. Nat. Methods 2012, 9, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.L.; Lawson, M.; Zhu, Q.; Dries, R.; Koulena, N.; Takei, Y.; Yun, J.; Cronin, C.; Karp, C.; Yuan, G.C.; et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+. Nature 2019, 568, 235–239. [Google Scholar] [CrossRef]
- Moffitt, J.R.; Hao, J.; Wang, G.; Chen, K.H.; Babcock, H.P.; Zhuang, X. High-throughput single-cell gene-expression profiling with multiplexed error-robust fluorescence in situ hybridization. Proc. Natl. Acad. Sci. USA 2016, 113, 11046–11051. [Google Scholar] [CrossRef]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.M.; Chen, F.; Macosko, E.Z. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 2019, 363, 1463–1467. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Liao, S.; Cheng, M.; Ma, K.; Wu, L.; Lai, Y.; Qiu, X.; Yang, J.; Xu, J.; Hao, S.; et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell 2022, 185, 1777–1792. [Google Scholar] [CrossRef]
- Chen, T.Y.; You, L.; Hardillo, J.; Chien, M.P. Spatial transcriptomic technologies. Cells 2023, 12, 2042. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer. J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, N.A.; Sanchez-Salas, R.; Peske, J.D.; Vano, Y.; Becht, E.; Petitprez, F.; Validire, P.; Ingels, A.; Cathelineau, X.; Fridman, W.H.; et al. The clinical role of the TME in solid cancer. Br. J. Cancer 2019, 120, 45–53. [Google Scholar] [CrossRef]
- Maniatis, S.; Petrescu, J.; Phatnani, H. Spatially resolved transcriptomics and its applications in cancer. Curr. Opin. Genet. Dev. 2021, 66, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Guo, W.; Qiu, X.; Wang, S.; Sui, C.; Lian, Q.; Wu, J.; Shan, Y.; Yang, Z.; Yang, S.; et al. Comprehensive analysis of spatial architecture in primary liver cancer. Sci. Adv. 2021, 7, eabg3750. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Li, N.; Liang, Y.; Li, R.; Tong, X.; Xiao, J.; Tang, H.; Jiang, D.; Xie, K.; Fang, C.; et al. Multi-omics analyses reveal spatial heterogeneity in primary and metastatic oesophageal squamous cell carcinoma. Clin. Transl. Med. 2023, 13, e1493. [Google Scholar] [CrossRef]
- Yang, W.; Chen, H.; Li, G.; Zhang, T.; Sui, Y.; Liu, L.; Hu, J.; Wang, G.; Chen, H.; Wang, Y.; et al. Caprin-1 influences autophagy-induced tumor growth and immune modulation in pancreatic cancer. J. Transl. Med. 2023, 21, 903. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, S.; Das, P.; Holliday, E.B.; Shen, L.; Lu, W.; Johnson, B.; Messick, C.A.; Taniguchi, C.M.; Skibber, J.; Ludmir, E.B.; et al. Differential spatial gene and protein expression associated with recurrence following chemoradiation for localized anal squamous cell cancer. Cancers 2023, 15, 1701. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Long, J.; Li, L.; Wu, Z.X.; Da, T.T.; Wang, X.Q.; Huang, C.; Jiang, Y.H.; Yao, X.Q.; Ma, H.Q.; et al. Single-cell and spatial transcriptome analysis reveals the cellular heterogeneity of liver metastatic colorectal cancer. Sci. Adv. 2023, 9, eadf5464. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Singh, S.; Cheng, C.; Natarajan, S.; Sheppard, H.; Abu-Zaid, A.; Durbin, A.D.; Lee, H.W.; Wu, Q.; Steele, J.; et al. Genome-wide mapping of cancer dependency genes and genetic modifiers of chemotherapy in high-risk hepatoblastoma. Nat. Commun. 2023, 14, 4003. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, N.P.; Webbink, L.; Haddad, T.S.; Rutgers, N.; van Vliet, S.; Wood, C.S.; Jansen, P.W.; Lafarge, M.W.; de Wilt, J.H.; Hugen, N.; et al. Transcriptomics and proteomics reveal distinct biology for lymph node metastases and tumour deposits in colorectal cancer. J. Pathol. 2023, 261, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, S.; Huang, Y.E.; Yuan, M.; Lei, W.; Chen, J.; Lin, K.; Jiang, W. Estimating metastatic risk of pancreatic ductal adenocarcinoma at single-cell resolution. Int. J. Mol. Sci. 2022, 23, 15020. [Google Scholar] [CrossRef] [PubMed]
- Moncada, R.; Barkley, D.; Wagner, F.; Chiodin, M.; Devlin, J.C.; Baron, M.; Hajdu, C.H.; Simeone, D.M.; Yanai, I. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat. Biotechnol. 2020, 38, 333–342. [Google Scholar] [CrossRef]
- Guo, W.; Zhou, B.; Yang, Z.; Liu, X.; Huai, Q.; Guo, L.; Xue, X.; Tan, F.; Li, Y.; Xue, Q.; et al. Integrating microarray-based spatial transcriptomics and single-cell RNA-sequencing reveals tissue architecture in esophageal squamous cell carcinoma. eBioMedicine 2022, 84, 104281. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Wang, S.; Wei, C.; Fang, Y.; Huang, S.; Yin, T.; Xiong, B.; Yang, C. Tumour microenvironment: A non-negligible driver for epithelial-mesenchymal transition in colorectal cancer. Expert. Rev. Mol. Med. 2021, 23, e16. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.Y.; Ham, I.H.; Lee, S.H.; Ryu, D.; Son, S.Y.; Han, S.U.; Kim, T.M.; Hur, H. Spatially distinct reprogramming of the tumor microenvironment based on tumor invasion in diffuse-type gastric cancers. Clin. Cancer Res. 2021, 27, 6529–6542. [Google Scholar] [CrossRef]
- Dotto, G.P.; Weinberg, R.A.; Ariza, A. Malignant transformation of mouse primary keratinocytes by harvey sarcoma virus and its modulation by surrounding normal cells. Proc. Natl. Acad. Sci. USA 1988, 85, 6389–6393. [Google Scholar] [CrossRef] [PubMed]
- Polanska, U.M.; Orimo, A. Carcinoma-associated fibroblasts: Non-neoplastic tumour-promoting mesenchymal cells. J. Cell. Physiol. 2013, 228, 1651–1657. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated sdf-1/cxcl12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Sun, H.; Zhang, Y.; Wang, Z.; Xun, Z.; Li, Z.; Ding, X.; Bao, R.; Hong, L.; Jia, W.; et al. Single-cell and spatial analysis reveal interaction of FAP+ fibroblasts and SPP1+ macrophages in colorectal cancer. Nat. Commun. 2022, 13, 1742. [Google Scholar] [CrossRef]
- Nieto, P.; Elosua-Bayes, M.; Trincado, J.L.; Marchese, D.; Massoni-Badosa, R.; Salvany, M.; Henriques, A.; Nieto, J.; Aguilar-Fernández, S.; Mereu, E.; et al. A single-cell tumor immune atlas for precision oncology. Genome Res. 2021, 31, 1913–1926. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Ye, M.; Ding, H.; Feng, Z.; Hu, K. Spatial transcriptomics atlas reveals the crosstalk between cancer-associated fibroblasts and tumor microenvironment components in colorectal cancer. J. Transl. Med. 2022, 20, 302. [Google Scholar] [CrossRef] [PubMed]
- Valdeolivas, A.; Amberg, B.; Giroud, N.; Richardson, M.; Gálvez, E.; Badillo, S.; Julien-Laferrière, A.; Túrós, D.; Voith, V.V.L.; Wells, I.; et al. Profiling the heterogeneity of colorectal cancer consensus molecular subtypes using spatial transcriptomics. NPJ Precis. Oncol. 2024, 8, 10. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, S.; Liu, T.; Zhao, X.; Xiang, T.; Hu, X.; Wu, C.; Lin, D. Aberrant epithelial cell interaction promotes esophageal squamous-cell carcinoma development and progression. Signal Transduct. Target. Ther. 2023, 8, 453. [Google Scholar] [CrossRef]
- Hunter, M.V.; Moncada, R.; Weiss, J.M.; Yanai, I.; White, R.M. Spatially resolved transcriptomics reveals the architecture of the tumor-microenvironment interface. Nat. Commun. 2021, 12, 6278. [Google Scholar] [CrossRef]
- Jang, E.; Shin, M.K.; Kim, H.; Lim, J.Y.; Lee, J.E.; Park, J.; Kim, J.; Kim, H.; Shin, Y.; Son, H.Y.; et al. Clinical molecular subtyping reveals intrinsic mesenchymal reprogramming in gastric cancer cells. Exp. Mol. Med. 2023, 55, 974–986. [Google Scholar] [CrossRef]
- Wu, Y.; Yang, S.; Ma, J.; Chen, Z.; Song, G.; Rao, D.; Cheng, Y.; Huang, S.; Liu, Y.; Jiang, S.; et al. Spatiotemporal immune landscape of colorectal cancer liver metastasis at single-cell level. Cancer Discov. 2022, 12, 134–153. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Deng, C.; Yang, C.; Yan, M.; Lu, H.; Zhang, Y.; Liu, H.; Tong, Z.; Ma, J.; Wang, J.; et al. Unraveling temporal and spatial biomarkers of epithelial-mesenchymal transition in colorectal cancer: Insights into the crucial role of immunosuppressive cells. J. Transl. Med. 2023, 21, 794. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, J.; Hirata, Y.; Otsuki, Y.; Suina, K.; Saito, Y.; Masuda, K.; Okazaki, S.; Ishimoto, T.; Saya, H.; Nagano, O. MEK inhibition suppresses metastatic progression of KRAS-mutated gastric cancer. Cancer Sci. 2022, 113, 916–925. [Google Scholar] [CrossRef]
- Chaturvedi, A.K.; Udaltsova, N.; Engels, E.A.; Katzel, J.A.; Yanik, E.L.; Katki, H.A.; Lingen, M.W.; Silverberg, M.J. Oral leukoplakia and risk of progression to oral cancer: A population-based cohort study. J. Natl. Cancer. Inst. 2020, 112, 1047–1054. [Google Scholar] [CrossRef]
- Sun, L.; Kang, X.; Wang, C.; Wang, R.; Yang, G.; Jiang, W.; Wu, Q.; Wang, Y.; Wu, Y.; Gao, J.; et al. Single-cell and spatial dissection of precancerous lesions underlying the initiation process of oral squamous cell carcinoma. Cell Discov. 2023, 9, 28. [Google Scholar] [CrossRef]
- Croft, W.; Pearce, H.; Margielewska-Davies, S.; Lim, L.; Nicol, S.M.; Zayou, F.; Blakeway, D.; Marcon, F.; Powell-Brett, S.; Mahon, B.; et al. Spatial determination and prognostic impact of the fibroblast transcriptome in pancreatic ductal adenocarcinoma. eLife 2023, 12, e86125. [Google Scholar] [CrossRef]
- Wang, N.; Wang, R.; Li, X.; Song, Z.; Xia, L.; Wang, J.; Zhang, L.; Wu, A.; Ding, Z. Tumor microenvironment profiles reveal distinct therapy-oriented proteogenomic characteristics in colorectal cancer. Front. Bioeng. Biotechnol. 2021, 9, 757378. [Google Scholar] [CrossRef] [PubMed]
- Setayesh, T.; Hu, Y.; Vaziri, F.; Chen, X.; Lai, J.; Wei, D.; Yvonne, W.Y. Targeting stroma and tumor, silencing galectin 1 treats orthotopic mouse hepatocellular carcinoma. Acta Pharm. Sin. B 2024, 14, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.M.; Lin, D.C. Moving closer towards a comprehensive view of tumor biology and microarchitecture using spatial transcriptomics. Nat. Commun. 2023, 14, 7017. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Li, X.; Wang, R.; Ding, Z. Spatial transcriptomics and proteomics technologies for deconvoluting the tumor microenvironment. Biotechnol. J. 2021, 16, e2100041. [Google Scholar] [CrossRef] [PubMed]
- Elhanani, O.; Ben-Uri, R.; Keren, L. Spatial profiling technologies illuminate the tumor microenvironment. Cancer Cell 2023, 41, 404–420. [Google Scholar] [CrossRef]
- Matsubara, T.; Soh, J.; Morita, M.; Uwabo, T.; Tomida, S.; Fujiwara, T.; Kanazawa, S.; Toyooka, S.; Hirasawa, A. Dv200 index for assessing RNA integrity in next-generation sequencing. BioMed Res. Int. 2020, 2020, 9349132. [Google Scholar] [CrossRef] [PubMed]
- Asp, M.; Bergenstråhle, J.; Lundeberg, J. Spatially resolved transcriptomes-next generation tools for tissue exploration. BioEssays 2020, 42, e1900221. [Google Scholar] [CrossRef] [PubMed]
- Lein, E.; Borm, L.E.; Linnarsson, S. The promise of spatial transcriptomics for neuroscience in the era of molecular cell typing. Science 2017, 358, 64–69. [Google Scholar] [CrossRef]
- Xia, C.; Fan, J.; Emanuel, G.; Hao, J.; Zhuang, X. Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression. Proc. Natl. Acad. Sci. USA 2019, 116, 19490–19499. [Google Scholar] [CrossRef]
- Zheng, B.; Fang, L. Spatially resolved transcriptomics provide a new method for cancer research. J. Exp. Clin. Cancer Res. 2022, 41, 179. [Google Scholar] [CrossRef]
- Hafemeister, C.; Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019, 20, 296. [Google Scholar] [CrossRef]
- Zeng, Z.; Li, Y.; Li, Y.; Luo, Y. Statistical and machine learning methods for spatially resolved transcriptomics data analysis. Genome Biol. 2022, 23, 83. [Google Scholar] [CrossRef] [PubMed]
- Svensson, V.; Teichmann, S.A.; Stegle, O. SpatialDE: Identification of spatially variable genes. Nat. Methods 2018, 15, 343–346. [Google Scholar] [CrossRef]
- Sun, S.; Zhu, J.; Zhou, X. Statistical analysis of spatial expression patterns for spatially resolved transcriptomic studies. Nat. Methods 2020, 17, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Edsgärd, D.; Johnsson, P.; Sandberg, R. Identification of spatial expression trends in single-cell gene expression data. Nat. Methods 2018, 15, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Sun, S.; Zhou, X. SPARK-X: Non-parametric modeling enables scalable and robust detection of spatial expression patterns for large spatial transcriptomic studies. Genome Biol. 2021, 22, 184. [Google Scholar] [CrossRef] [PubMed]
- Tsoucas, D.; Dong, R.; Chen, H.; Zhu, Q.; Guo, G.; Yuan, G.C. Accurate estimation of cell-type composition from gene expression data. Nat. Commun. 2019, 10, 2975. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Yuan, G.C. SpatialDWLS: Accurate deconvolution of spatial transcriptomic data. Genome Biol. 2021, 22, 145. [Google Scholar] [CrossRef]
- Elosua-Bayes, M.; Nieto, P.; Mereu, E.; Gut, I.; Heyn, H. SPOTlight: Seeded NMF regression to deconvolute spatial transcriptomics spots with single-cell transcriptomes. Nucleic Acids Res. 2021, 49, e50. [Google Scholar] [CrossRef] [PubMed]
- Cable, D.M.; Murray, E.; Zou, L.S.; Goeva, A.; Macosko, E.Z.; Chen, F.; Irizarry, R.A. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat. Biotechnol. 2022, 40, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.; Bergenstråhle, J.; Asp, M.; Bergenstråhle, L.; Jurek, A.; Fernández, N.J.; Lundeberg, J. Single-cell and spatial transcriptomics enables probabilistic inference of cell type topography. Commun. Biol. 2020, 3, 565. [Google Scholar] [CrossRef] [PubMed]
- Biancalani, T.; Scalia, G.; Buffoni, L.; Avasthi, R.; Lu, Z.; Sanger, A.; Tokcan, N.; Vanderburg, C.R.; Segerstolpe, Å.; Zhang, M.; et al. Deep learning and alignment of spatially resolved single-cell transcriptomes with tangram. Nat. Methods 2021, 18, 1352–1362. [Google Scholar] [CrossRef]
- Song, Q.; Su, J. DSTG: Deconvoluting spatial transcriptomics data through graph-based artificial intelligence. Brief. Bioinform. 2021, 22, bbaa414. [Google Scholar] [CrossRef] [PubMed]
- Arnol, D.; Schapiro, D.; Bodenmiller, B.; Saez-Rodriguez, J.; Stegle, O. Modeling cell-cell interactions from spatial molecular data with spatial variance component analysis. Cell Rep. 2019, 29, 202–211. [Google Scholar] [CrossRef]
- Boisset, J.C.; Vivié, J.; Grün, D.; Muraro, M.J.; Lyubimova, A.; van Oudenaarden, A. Mapping the physical network of cellular interactions. Nat. Methods 2018, 15, 547–553. [Google Scholar] [CrossRef]
- Yuan, Y.; Bar-Joseph, Z. GCNG: Graph convolutional networks for inferring gene interaction from spatial transcriptomics data. Genome Biol. 2020, 21, 300. [Google Scholar] [CrossRef]
- Palla, G.; Spitzer, H.; Klein, M.; Fischer, D.; Schaar, A.C.; Kuemmerle, L.B.; Rybakov, S.; Ibarra, I.L.; Holmberg, O.; Virshup, I.; et al. Squidpy: A scalable framework for spatial omics analysis. Nat. Methods 2022, 19, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Shengquan, C.; Boheng, Z.; Xiaoyang, C.; Xuegong, Z.; Rui, J. stPlus: A reference-based method for the accurate enhancement of spatial transcriptomics. Bioinformatics 2021, 37, i299–i307. [Google Scholar] [CrossRef] [PubMed]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.R.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Michailidis, G. A non-negative matrix factorization method for detecting modules in heterogeneous omics multi-modal data. Bioinformatics 2016, 32, 1–8. [Google Scholar] [CrossRef]
- Welch, J.D.; Kozareva, V.; Ferreira, A.; Vanderburg, C.; Martin, C.; Macosko, E.Z. Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell 2019, 177, 1873–1887. [Google Scholar] [CrossRef] [PubMed]
- Mourragui, S.; Loog, M.; van de Wiel, M.A.; Reinders, M.; Wessels, L. PRECISE: A domain adaptation approach to transfer predictors of drug response from pre-clinical models to tumors. Bioinformatics 2019, 35, i510–i519. [Google Scholar] [CrossRef]
- Abdelaal, T.; Mourragui, S.; Mahfouz, A.; Reinders, M. SpaGE: Spatial gene enhancement using scRNA-seq. Nucleic Acids Res. 2020, 48, e107. [Google Scholar] [CrossRef]
- Ogbeide, S.; Giannese, F.; Mincarelli, L.; Macaulay, I.C. Into the multiverse: Advances in single-cell multiomic profiling. Trends Genet. 2022, 38, 831–843. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, D.; Song, D.; Liu, X.; Zhang, Y.; Xu, X.; Wang, X. Clinical and translational values of spatial transcriptomics. Signal Transduct. Target. Ther. 2022, 7, 111. [Google Scholar] [CrossRef] [PubMed]
- Nerurkar, S.N.; Goh, D.; Cheung, C.; Nga, P.; Lim, J.; Yeong, J. Transcriptional spatial profiling of cancer tissues in the era of immunotherapy: The potential and promise. Cancers 2020, 12, 2572. [Google Scholar] [CrossRef]
- Stickels, R.R.; Murray, E.; Kumar, P.; Li, J.; Marshall, J.L.; Di Bella, D.J.; Arlotta, P.; Macosko, E.Z.; Chen, F. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat. Biotechnol. 2021, 39, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Boettiger, A.N.; Moffitt, J.R.; Wang, S.; Zhuang, X. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 2015, 348, aaa6090. [Google Scholar] [CrossRef]
- Mccart, R.A.; Bennett, J.; Kutasovic, J.R.; Kalaw, E.; Ferguson, K.; Yeong, J.; Simpson, P.T.; Lakhani, S.R. Digital spatial profiling application in breast cancer: A user’s perspective. Virchows Arch. 2020, 477, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Toki, M.I.; Merritt, C.R.; Wong, P.F.; Smithy, J.W.; Kluger, H.M.; Syrigos, K.N.; Ong, G.T.; Warren, S.E.; Beechem, J.M.; Rimm, D.L. High-plex predictive marker discovery for melanoma immunotherapy-treated patients using digital spatial profiling. Clin. Cancer Res. 2019, 25, 5503–5512. [Google Scholar] [CrossRef]
- Meylan, M.; Petitprez, F.; Becht, E.; Bougoüin, A.; Pupier, G.; Calvez, A.; Giglioli, I.; Verkarre, V.; Lacroix, G.; Verneau, J.; et al. Tertiary lymphoid structures generate and propagate anti-tumor antibody-producing plasma cells in renal cell cancer. Immunity 2022, 55, 527–541. [Google Scholar] [CrossRef]
- Lebrigand, K.; Bergenstråhle, J.; Thrane, K.; Mollbrink, A.; Meletis, K.; Barbry, P.; Waldmann, R.; Lundeberg, J. The spatial landscape of gene expression isoforms in tissue sections. Nucleic Acids Res. 2023, 51, e47. [Google Scholar] [CrossRef] [PubMed]
- Li, P.H.; Kong, X.Y.; He, Y.Z.; Liu, Y.; Peng, X.; Li, Z.H.; Xu, H.; Luo, H.; Park, J. Recent developments in application of single-cell RNA sequencing in the tumour immune microenvironment and cancer therapy. Mil. Med. Res. 2022, 9, 52. [Google Scholar] [CrossRef]
- Erickson, A.; He, M.; Berglund, E.; Marklund, M.; Mirzazadeh, R.; Schultz, N.; Kvastad, L.; Andersson, A.; Bergenstråhle, L.; Bergenstråhle, J.; et al. Spatially resolved clonal copy number alterations in benign and malignant tissue. Nature 2022, 608, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Chelebian, E.; Avenel, C.; Kartasalo, K.; Marklund, M.; Tanoglidi, A.; Mirtti, T.; Colling, R.; Erickson, A.; Lamb, A.D.; Lundeberg, J.; et al. Morphological features extracted by ai associated with spatial transcriptomics in prostate cancer. Cancers 2021, 13, 4837. [Google Scholar] [CrossRef]
- He, B.; Bergenstråhle, L.; Stenbeck, L.; Abid, A.; Andersson, A.; Borg, Å.; Maaskola, J.; Lundeberg, J.; Zou, J. Integrating spatial gene expression and breast tumour morphology via deep learning. Nat. Biomed. Eng. 2020, 4, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Levy-Jurgenson, A.; Tekpli, X.; Kristensen, V.N.; Yakhini, Z. Spatial transcriptomics inferred from pathology whole-slide images links tumor heterogeneity to survival in breast and lung cancer. Sci. Rep. 2020, 10, 18802. [Google Scholar] [CrossRef] [PubMed]
- Yoosuf, N.; Navarro, J.F.; Salmén, F.; Ståhl, P.L.; Daub, C.O. Identification and transfer of spatial transcriptomics signatures for cancer diagnosis. Breast Cancer Res. 2020, 22, 6. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Common Downstream Analyses | Representative Methods | Characteristics | References |
|---|---|---|---|
| Cell-type deconvolution | spatialDWLS | Utilizes cell type features derived from scRNA-seq data to perform gene signature enrichment and uses DWLS to infer proportions of cell types at each spot. | [58,63,64] |
| SPOTlight | Based on a seeded NMF regression algorithm, utilizes cell-type signatures from scRNA-seq data to initialize basis and coefficient matrices, uses NNLS to populate coefficient matrices, and derives proportions of cell types at each spot. | [65] | |
| RCTD | Uses gene expression profiles for each cell type from scRNA-seq data as input and transcript counts as output to fit a statistical model and leverages maximum-likelihood estimation to infer proportions of cell types at each spot. | [66] | |
| stereoscope | Based on negative binomial distribution and leverages gene expression profiles from scRNA-seq to infer proportions of cell types at each spot probabilistically. | [67] | |
| Tangram | Aligns scRNA-seq and snRNA-seq data to spatial transcriptomics data through deep learning, thereby deconvolving low-resolution data into single cells and drawing location maps for different cell types. | [68] | |
| DSTG | First, generates synthetic pseudo-ST data from scRNA-seq data. Next, performs CCA to incorporate pseudo-ST and real-ST data into a common graph. Then, performs KNN to identify mutual nearest neighbors and construct a link graph of spot mapping. Finally, utilizes a semi-supervised GCN to explain the compositions of cell types at each spot based on the link graph. | [69] | |
| Cell–cell interactions and gene–gene interactions | MISTy | Dissects contribution of different predictor markers to prediction of target marker expression in a specific view and identifies potential interactions between target marker and predictor markers. | [58] |
| SVCA | Decomposes variation of gene expression into intrinsic effects, environmental effects, and cell–cell interaction effects and utilizes a gradient-based optimizer to calculate proportion of variance attributable to cell–cell interaction components through maximum likelihood. | [70] | |
| ProximID | Based on physical cell interaction and scRNA-seq and used to infer cell–cell interactions. | [71] | |
| GCNG | Encodes position of cells and expression of gene pairs in these cells into two matrices separately as inputs and leverages two graph convolutional layers and a sigmoid function output layer to infer gene–gene interactions. | [72] | |
| Squidpy | Based on Python, dissects spatial omics data and detects cell–cell interactions mediated by ligand–receptor interactions between identified cell clusters using CellPhoneDB. | [73] | |
| Gene imputation | gimVI | Integrates scRNA-seq and spatial transcriptomic data to impute missing genes. | [58] |
| Tangram | Takes scRNA-seq, snRNA-seq, and spatial transcriptomics data as inputs, rearranges scRNA-seq and snRNA-seq profiles in space, and obtains new spatial data containing all genes at single-cell resolution and with spatial position. | [68] | |
| stPlus | Based on an auto-encoder with a loss function, conducts weighted KNN to perform joint embedding projection and leverages scRNA-seq profiles to achieve accurate prediction for expression of unmeasured genes and effective imputation for measured genes. | [74] | |
| Harmony | Leverages iterations of maximum diversity clustering and mixture-model-based linear batch correction to project cells into a shared embedding with reduced dimension, thereby embedding scRNA-seq and spatial transcriptomics data into a common latent space and using KNN imputation to infer spatial expression and localization of unmeasured genes. | [75] | |
| LIGER | Utilizes iNMF to learn a low-dimensional space, integrates scRNA-seq and spatial transcriptomics data to assign spatial positions to cell clusters, and improves resolution for detecting cell clusters from in situ data. | [76,77] | |
| SpaGE | Based on domain adaptation using PRECISE, integrates scRNA-seq and spatial transcriptomics datasets, corrects differences in sensitivity of transcript detection, and performs KNN to predict spatial expression of unmeasured genes. | [78,79] | |
| SVGs identification | trendseek | Based on marked point theory, conducts pairwise analyses on points as a function of distance between points to evaluate whether there is a significant dependency between spatial distributions of points (represent spatial positions of cells or regions) and their related marks (represent expression levels), and identifies genes with significant spatial expression trends. | [61] |
| SpatialDE | Based on Gaussian process regression, decomposes expression variability of each gene into a spatial component (modeled as a spatial variance term that parametrizes gene expression covariance by pairwise spatial distances among locations) and a non-spatial component (modeled as a noise term) and compares full model to a model without spatial variance component to identify significant SVGs. | [59] | |
| SPARK | Based on a generalized linear spatial model; enables analyzing tens of thousands of genes across tens of thousands of spatial positions. | [60] | |
| SPARK-X | Based on a robust covariance test framework; enables including various spatial kernels for non-parametric spatial modeling of large-scale sparse spatial transcriptomic data. | [62] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, B.; Chen, Y.; Yuan, S. Application of Spatial Transcriptomics in Digestive System Tumors. Biomolecules 2025, 15, 21. https://doi.org/10.3390/biom15010021
Huang B, Chen Y, Yuan S. Application of Spatial Transcriptomics in Digestive System Tumors. Biomolecules. 2025; 15(1):21. https://doi.org/10.3390/biom15010021
Chicago/Turabian StyleHuang, Bowen, Yingjia Chen, and Shuqiang Yuan. 2025. "Application of Spatial Transcriptomics in Digestive System Tumors" Biomolecules 15, no. 1: 21. https://doi.org/10.3390/biom15010021
APA StyleHuang, B., Chen, Y., & Yuan, S. (2025). Application of Spatial Transcriptomics in Digestive System Tumors. Biomolecules, 15(1), 21. https://doi.org/10.3390/biom15010021