

CYP24A1 DNA Methylation in Colorectal Cancer as Potential Prognostic and Predictive Markers


Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection for the Blood Cohort
2.2. Cell Culture and DAC Treatment
2.3. RNA Extraction, Reverse Transcription and qRT-PCR
2.4. Western Blot Analysis
2.5. DNA Extraction and Bisulfite Conversion
2.6. MethylTarget Sequencing and Methylation Data Processing
2.7. Data Sources for the Tissue Cohort
2.8. Differential Methylation Analysis
2.9. Differential Expression Analysis and In Vitro Validation
2.10. Functional Enrichment Analysis for CYP24A1 and CYP24A1-Related Genes
2.11. Correlation Analysis of DNA Methylation and Gene Expression and In Vitro Validation
2.12. Survival Analysis and Construction of Prognostic Risk Scores
2.13. Immune Infiltration Analysis
2.14. Statistical Analysis
3. Results
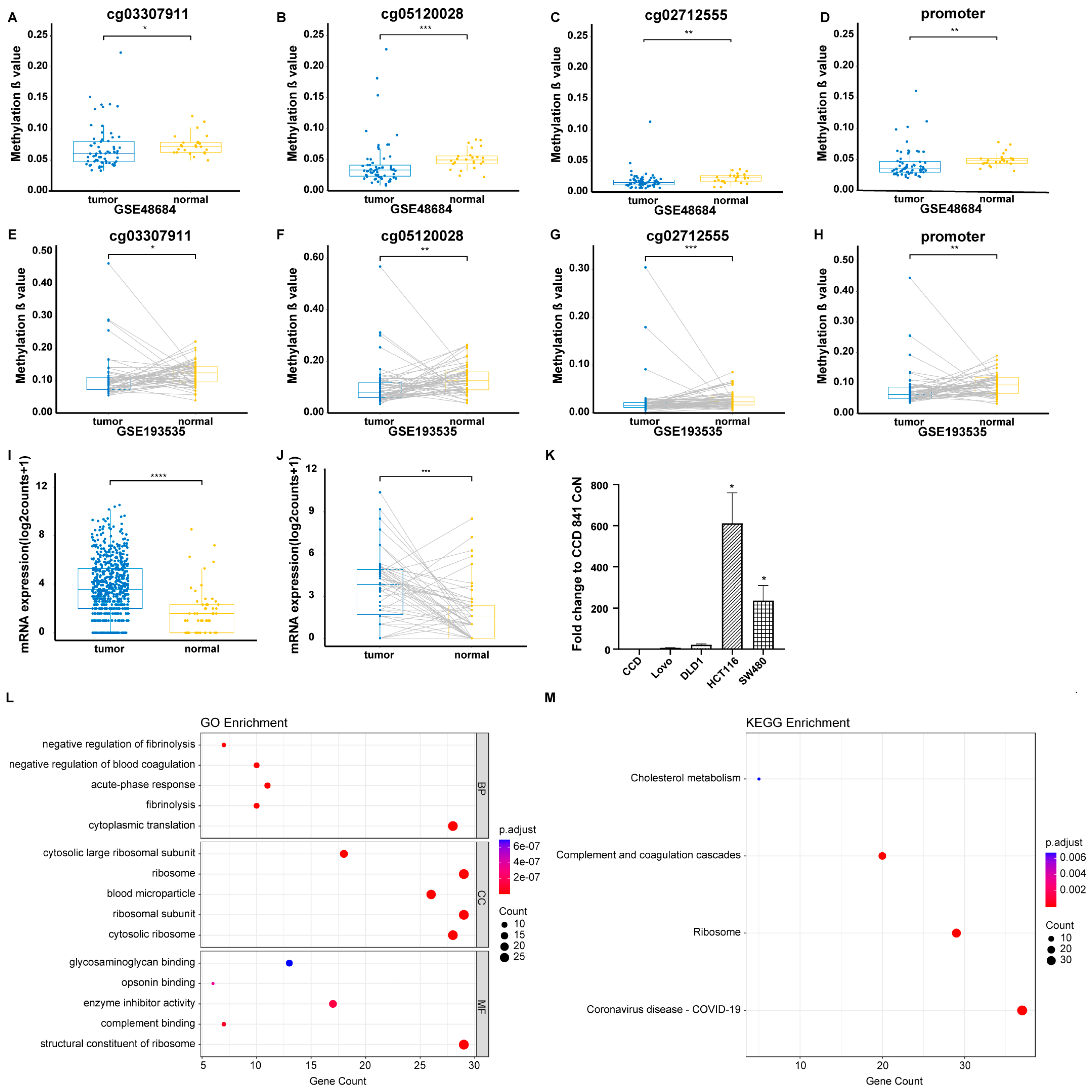
3.1. Identification of Significant Promoter CpG Sites in the Blood Cohort and GEO Tissue Cohort
3.2. Identification of Differential Expression of CYP24A1 in the TCGA Tissue Cohort and Validation Through In Vitro Experiments
3.3. GO and KEGG Enrichment Analyses of CYP24A1 and CYP24A1-Related Genes
3.4. Correlation Analysis of DNA Methylation and Gene Expression of CYP24A1 in the TCGA Tissue Cohort and Validation Through In Vitro Experiments
3.5. Clinical Characteristics of CRC Patients Included in the Survival Analysis
3.6. Prognostic Value of DNA Methylation of CYP24A1 in CRC
3.7. Predictive Performances of Prognostic Risk Scores for Overall Survival in CRC Patients
3.8. Correlation of CYP24A1 Methylation with Immune Infiltration in CRC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Zheng, R.; Zeng, H.; Wang, S.; Sun, K.; Chen, R.; Li, L.; Wei, W.; He, J. Cancer incidence and mortality in China, 2022. J. Natl. Cancer Cent. 2024, 4, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Chen, W.; Zheng, R.; Zhang, S.; Ji, J.S.; Zou, X.; Xia, C.; Sun, K.; Yang, Z.; Li, H.; et al. Changing cancer survival in China during 2003-15: A pooled analysis of 17 population-based cancer registries. Lancet Glob. Health 2018, 6, e555–e567. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J. Cellular and molecular effects of vitamin D on carcinogenesis. Arch. Biochem. Biophys. 2012, 523, 107–114. [Google Scholar] [CrossRef]
- Sung, V.; Feldman, D. 1,25-Dihydroxyvitamin D3 decreases human prostate cancer cell adhesion and migration. Mol. Cell Endocrinol. 2000, 164, 133–143. [Google Scholar] [CrossRef]
- González-Sancho, J.M.; Larriba, M.J.; Ordóñez-Morán, P.; Pálmer, H.G.; Muñoz, A. Effects of 1alpha,25-dihydroxyvitamin D3 in human colon cancer cells. Anticancer. Res. 2006, 26, 2669–2681. [Google Scholar]
- Colston, K.W.; Lowe, L.C.; Mansi, J.L.; Campbell, M.J. Vitamin D status and breast cancer risk. Anticancer. Res. 2006, 26, 2573–2580. [Google Scholar]
- Feldman, D.; Krishnan, A.V.; Swami, S.; Giovannucci, E.; Feldman, B.J. The role of vitamin D in reducing cancer risk and progression. Nat. Rev. Cancer 2014, 14, 342–357. [Google Scholar] [CrossRef]
- Prosser, D.E.; Jones, G. Enzymes involved in the activation and inactivation of vitamin D. Trends Biochem. Sci. 2004, 29, 664–673. [Google Scholar] [CrossRef]
- Zhalehjoo, N.; Shakiba, Y.; Panjehpour, M. Gene expression profiles of CYP24A1 and CYP27B1 in malignant and normal breast tissues. Mol. Med. Rep. 2017, 15, 467–473. [Google Scholar] [CrossRef]
- Tannour-Louet, M.; Lewis, S.K.; Louet, J.F.; Stewart, J.; Addai, J.B.; Sahin, A.; Vangapandu, H.V.; Lewis, A.L.; Dittmar, K.; Pautler, R.G.; et al. Increased expression of CYP24A1 correlates with advanced stages of prostate cancer and can cause resistance to vitamin D3-based therapies. Faseb J. 2014, 28, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Parise, R.A.; Egorin, M.J.; Kanterewicz, B.; Taimi, M.; Petkovich, M.; Lew, A.M.; Chuang, S.S.; Nichols, M.; El-Hefnawy, T.; Hershberger, P.A. CYP24, the enzyme that catabolizes the antiproliferative agent vitamin D, is increased in lung cancer. Int. J. Cancer 2006, 119, 1819–1828. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wang, C.; Hao, M.; Sun, R.; Wang, Y.; Liu, T.; Cong, X.; Liu, Y. CYP24A1 is a potential biomarker for the progression and prognosis of human colorectal cancer. Hum. Pathol. 2016, 50, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Fetahu, I.S.; Höbaus, J.; Kállay, E. Vitamin D and the epigenome. Front. Physiol. 2014, 5, 164. [Google Scholar] [CrossRef]
- McMahon, K.W.; Karunasena, E.; Ahuja, N. The Roles of DNA Methylation in the Stages of Cancer. Cancer J. 2017, 23, 257–261. [Google Scholar] [CrossRef]
- Dhar, G.A.; Saha, S.; Mitra, P.; Nag Chaudhuri, R. DNA methylation and regulation of gene expression: Guardian of our health. Nucleus 2021, 64, 259–270. [Google Scholar] [CrossRef]
- Werner, R.J.; Kelly, A.D.; Issa, J.J. Epigenetics and Precision Oncology. Cancer J. 2017, 23, 262–269. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, C.; Shi, C.; Zhang, J.; Qian, T.; Wang, Z.; Ma, R.; Wu, J.; Jiang, F.; Feng, J. Hypermethylation of heparanase 2 promotes colorectal cancer proliferation and is associated with poor prognosis. J. Transl. Med. 2021, 19, 98. [Google Scholar] [CrossRef]
- Li, X.; Cai, D.; Huang, Y.; Xie, Y.; Shen, D.; Yuan, Z.; Liu, X.; Huang, M.; Luo, Y.; Yu, H.; et al. Aberrant methylation in neurofunctional gene serves as a hallmark of tumorigenesis and progression in colorectal cancer. BMC Cancer 2023, 23, 315. [Google Scholar] [CrossRef]
- Wang, Y.F.; Li, L.; Deng, X.Q.; Fang, Y.J.; Zhang, C.X. Association of DNA methylation of vitamin D metabolic pathway related genes with colorectal cancer risk. Clin. Epigenet. 2023, 15, 140. [Google Scholar] [CrossRef]
- Höbaus, J.; Hummel, D.M.; Thiem, U.; Fetahu, I.S.; Aggarwal, A.; Müllauer, L.; Heller, G.; Egger, G.; Mesteri, I.; Baumgartner-Parzer, S.; et al. Increased copy-number and not DNA hypomethylation causes overexpression of the candidate proto-oncogene CYP24A1 in colorectal cancer. Int. J. Cancer 2013, 133, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.G.; Nakane, M.; Ruan, X.; Kroeger, P.E.; Wu-Wong, J.R. Expression of VDR and CYP24A1 mRNA in human tumors. Cancer Chemother. Pharmacol. 2006, 57, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Hershberger, P.A.; Trump, D.L.; Johnson, C.S. 24-Hydroxylase in cancer: Impact on vitamin D-based anticancer therapeutics. J. Steroid Biochem. Mol. Biol. 2013, 136, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Kósa, J.P.; Horváth, P.; Wölfling, J.; Kovács, D.; Balla, B.; Mátyus, P.; Horváth, E.; Speer, G.; Takács, I.; Nagy, Z.; et al. CYP24A1 inhibition facilitates the anti-tumor effect of vitamin D3 on colorectal cancer cells. World J. Gastroenterol. 2013, 19, 2621–2628. [Google Scholar] [CrossRef]
- Zhang, Q.; Kanterewicz, B.; Buch, S.; Petkovich, M.; Parise, R.; Beumer, J.; Lin, Y.; Diergaarde, B.; Hershberger, P.A. CYP24 inhibition preserves 1α,25-dihydroxyvitamin D(3) anti-proliferative signaling in lung cancer cells. Mol. Cell Endocrinol. 2012, 355, 153–161. [Google Scholar] [CrossRef]
- Mimori, K.; Tanaka, Y.; Yoshinaga, K.; Masuda, T.; Yamashita, K.; Okamoto, M.; Inoue, H.; Mori, M. Clinical significance of the overexpression of the candidate oncogene CYP24 in esophageal cancer. Ann. Oncol. 2004, 15, 236–241. [Google Scholar] [CrossRef]
- Chen, G.; Kim, S.H.; King, A.N.; Zhao, L.; Simpson, R.U.; Christensen, P.J.; Wang, Z.; Thomas, D.G.; Giordano, T.J.; Lin, L.; et al. CYP24A1 is an independent prognostic marker of survival in patients with lung adenocarcinoma. Clin. Cancer Res. 2011, 17, 817–826. [Google Scholar] [CrossRef]
- Turi, Z.; Lacey, M.; Mistrik, M.; Moudry, P. Impaired ribosome biogenesis: Mechanisms and relevance to cancer and aging. Aging 2019, 11, 2512–2540. [Google Scholar] [CrossRef]
- Nait Slimane, S.; Marcel, V.; Fenouil, T.; Catez, F.; Saurin, J.C.; Bouvet, P.; Diaz, J.J.; Mertani, H.C. Ribosome Biogenesis Alterations in Colorectal Cancer. Cells 2020, 9, 2361. [Google Scholar] [CrossRef]
- Jiao, L.; Liu, Y.; Yu, X.Y.; Pan, X.; Zhang, Y.; Tu, J.; Song, Y.H.; Li, Y. Ribosome biogenesis in disease: New players and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 15. [Google Scholar] [CrossRef]
- Ramnath, N.; Nadal, E.; Jeon, C.K.; Sandoval, J.; Colacino, J.; Rozek, L.S.; Christensen, P.J.; Esteller, M.; Beer, D.G.; Kim, S.H. Epigenetic regulation of vitamin D metabolism in human lung adenocarcinoma. J. Thorac. Oncol. 2014, 9, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Karpf, A.R.; Deeb, K.K.; Muindi, J.R.; Morrison, C.D.; Johnson, C.S.; Trump, D.L. Epigenetic regulation of vitamin D 24-hydroxylase/CYP24A1 in human prostate cancer. Cancer Res. 2010, 70, 5953–5962. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Jiang, C.; Cong, L.; Wu, N.; Wang, X.; Hao, M.; Liu, T.; Wang, L.; Liu, Y.; Cong, X. CYP24A1 Inhibition Facilitates the Antiproliferative Effect of 1,25(OH)(2)D(3) Through Downregulation of the WNT/β-Catenin Pathway and Methylation-Mediated Regulation of CYP24A1 in Colorectal Cancer Cells. DNA Cell Biol. 2018, 37, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Claus, R.; Lucas, D.M.; Stilgenbauer, S.; Ruppert, A.S.; Yu, L.; Zucknick, M.; Mertens, D.; Bühler, A.; Oakes, C.C.; Larson, R.A.; et al. Quantitative DNA methylation analysis identifies a single CpG dinucleotide important for ZAP-70 expression and predictive of prognosis in chronic lymphocytic leukemia. J. Clin. Oncol. 2012, 30, 2483–2491. [Google Scholar] [CrossRef]
- Koch, A.; Joosten, S.C.; Feng, Z.; De Ruijter, T.C.; Draht, M.X.; Melotte, V.; Smits, K.M.; Veeck, J.; Herman, J.G.; Van Neste, L.; et al. Analysis of DNA methylation in cancer: Location revisited. Nat. Rev. Clin. Oncol. 2018, 15, 459–466. [Google Scholar] [CrossRef]
- Xie, Y.; Li, P.; Sun, D.; Qi, Q.; Ma, S.; Zhao, Y.; Zhang, S.; Wang, T.; Wang, J.; Li, S.; et al. DNA Methylation-Based Testing in Peripheral Blood Mononuclear Cells Enables Accurate and Early Detection of Colorectal Cancer. Cancer Res. 2023, 83, 3636–3649. [Google Scholar] [CrossRef]
- Ganapathi, S.K.; Beggs, A.D.; Hodgson, S.V.; Kumar, D. Expression and DNA methylation of TNF, IFNG and FOXP3 in colorectal cancer and their prognostic significance. Br. J. Cancer 2014, 111, 1581–1589. [Google Scholar] [CrossRef]
- Yuasa, Y.; Nagasaki, H.; Oze, I.; Akiyama, Y.; Yoshida, S.; Shitara, K.; Ito, S.; Hosono, S.; Watanabe, M.; Ito, H.; et al. Insulin-like growth factor 2 hypomethylation of blood leukocyte DNA is associated with gastric cancer risk. Int. J. Cancer 2012, 131, 2596–2603. [Google Scholar] [CrossRef]
- Fridley, B.L.; Armasu, S.M.; Cicek, M.S.; Larson, M.C.; Wang, C.; Winham, S.J.; Kalli, K.R.; Koestler, D.C.; Rider, D.N.; Shridhar, V.; et al. Methylation of leukocyte DNA and ovarian cancer: Relationships with disease status and outcome. BMC Med. Genom. 2014, 7, 21. [Google Scholar] [CrossRef]
- Pufulete, M.; Al-Ghnaniem, R.; Leather, A.J.; Appleby, P.; Gout, S.; Terry, C.; Emery, P.W.; Sanders, T.A. Folate status, genomic DNA hypomethylation, and risk of colorectal adenoma and cancer: A case control study. Gastroenterology 2003, 124, 1240–1248. [Google Scholar] [CrossRef]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Galon, J.; Dieu-Nosjean, M.C.; Cremer, I.; Fisson, S.; Damotte, D.; Pagès, F.; Tartour, E.; Sautès-Fridman, C. Immune infiltration in human cancer: Prognostic significance and disease control. Curr. Top. Microbiol. Immunol. 2011, 344, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Diederichsen, A.C.; Hjelmborg, J.; Christensen, P.B.; Zeuthen, J.; Fenger, C. Prognostic value of the CD4+/CD8+ ratio of tumour infiltrating lymphocytes in colorectal cancer and HLA-DR expression on tumour cells. Cancer Immunol. Immunother. 2003, 52, 423–428. [Google Scholar] [CrossRef]
- Deng, G.; Chen, A.; Hong, J.; Chae, H.S.; Kim, Y.S. Methylation of CpG in a small region of the hMLH1 promoter invariably correlates with the absence of gene expression. Cancer Res. 1999, 59, 2029–2033. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CpG Site/Promoter | Genome Position (GRCh38/hg38) | Distance to the TSS 1 | Patients (n = 101) Mean ± SD | Controls (n = 101) Mean ± SD | p |
|---|---|---|---|---|---|
| 102 | chr20:54174368 | −382 | 0.018 ± 0.011 | 0.021 ± 0.011 | 0.029 |
| 200 | chr20:54174270 | −284 | 0.011 ± 0.009 | 0.014 ± 0.009 | 0.007 |
| 41 | chr20:54173555 | 431 | 0.021 ± 0.008 | 0.023 ± 0.007 | 0.046 |
| 53 | chr20:54172987 | 999 | 0.050 ± 0.024 | 0.044 ± 0.011 | 0.025 |
| Promoter 1 | - | - | 0.017 ± 0.006 | 0.019 ± 0.005 | <0.001 |
| Promoter 2 | - | - | 0.025 ± 0.008 | 0.025 ± 0.006 | 0.322 |
| Characteristics | Blood Cohort Number (%) | TCGA Cohort Number (%) | ||
|---|---|---|---|---|
| K-M Survival Analysis | Cox Regression Analysis 1 | K-M Survival Analysis | Cox Regression Analysis 2 | |
| Overall | 101 | 99 | 380 | 362 |
| Age (years) (Mean ± SD) | 58 ± 10 | 58 ± 10 | 65 ± 13 | 65 ± 13 |
| Sex | ||||
| Male | 58 (57) | 58 (59) | 205 (54) | 194 (54) |
| Female | 43 (43) | 41 (41) | 175 (46) | 168 (46) |
| TNM stage | ||||
| Stage I/II | 41 (41) | 41 (41) | 194 (51) | 194 (54) |
| Stage III/IV | 58 (57) | 58 (59) | 168 (44) | 168 (46) |
| Not reported | 2 (2) | / | 18 (5) | / |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, R.-H.; Li, L.; Ou, Q.-J.; Wang, Y.-F.; Fang, Y.-J.; Zhang, C.-X. CYP24A1 DNA Methylation in Colorectal Cancer as Potential Prognostic and Predictive Markers. Biomolecules 2025, 15, 104. https://doi.org/10.3390/biom15010104
Zhou R-H, Li L, Ou Q-J, Wang Y-F, Fang Y-J, Zhang C-X. CYP24A1 DNA Methylation in Colorectal Cancer as Potential Prognostic and Predictive Markers. Biomolecules. 2025; 15(1):104. https://doi.org/10.3390/biom15010104
Chicago/Turabian StyleZhou, Ru-Hua, Lei Li, Qing-Jian Ou, Yi-Fan Wang, Yu-Jing Fang, and Cai-Xia Zhang. 2025. "CYP24A1 DNA Methylation in Colorectal Cancer as Potential Prognostic and Predictive Markers" Biomolecules 15, no. 1: 104. https://doi.org/10.3390/biom15010104
APA StyleZhou, R.-H., Li, L., Ou, Q.-J., Wang, Y.-F., Fang, Y.-J., & Zhang, C.-X. (2025). CYP24A1 DNA Methylation in Colorectal Cancer as Potential Prognostic and Predictive Markers. Biomolecules, 15(1), 104. https://doi.org/10.3390/biom15010104