
Potential Plasma Proteins (LGALS9, LAMP3, PRSS8 and AGRN) as Predictors of Hospitalisation Risk in COVID-19 Patients

 , , ,
, , ,  , , , , , , ,
, , , , , , ,  ,
,  , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Recruitment
2.2. Handling of Biological Samples
2.2.1. OLINK Plasma Protein Quantification
2.2.2. DNA Isolation
2.3. Genome Analysis
2.4. Statistical Testing
2.5. Differential Regulation Analysis
2.6. Proteomic Separation
2.7. Pathway Analysis
2.8. Machine Learning
2.9. Protein Association Analysis
3. Results
3.1. Demographics of COVID-19 Cohort
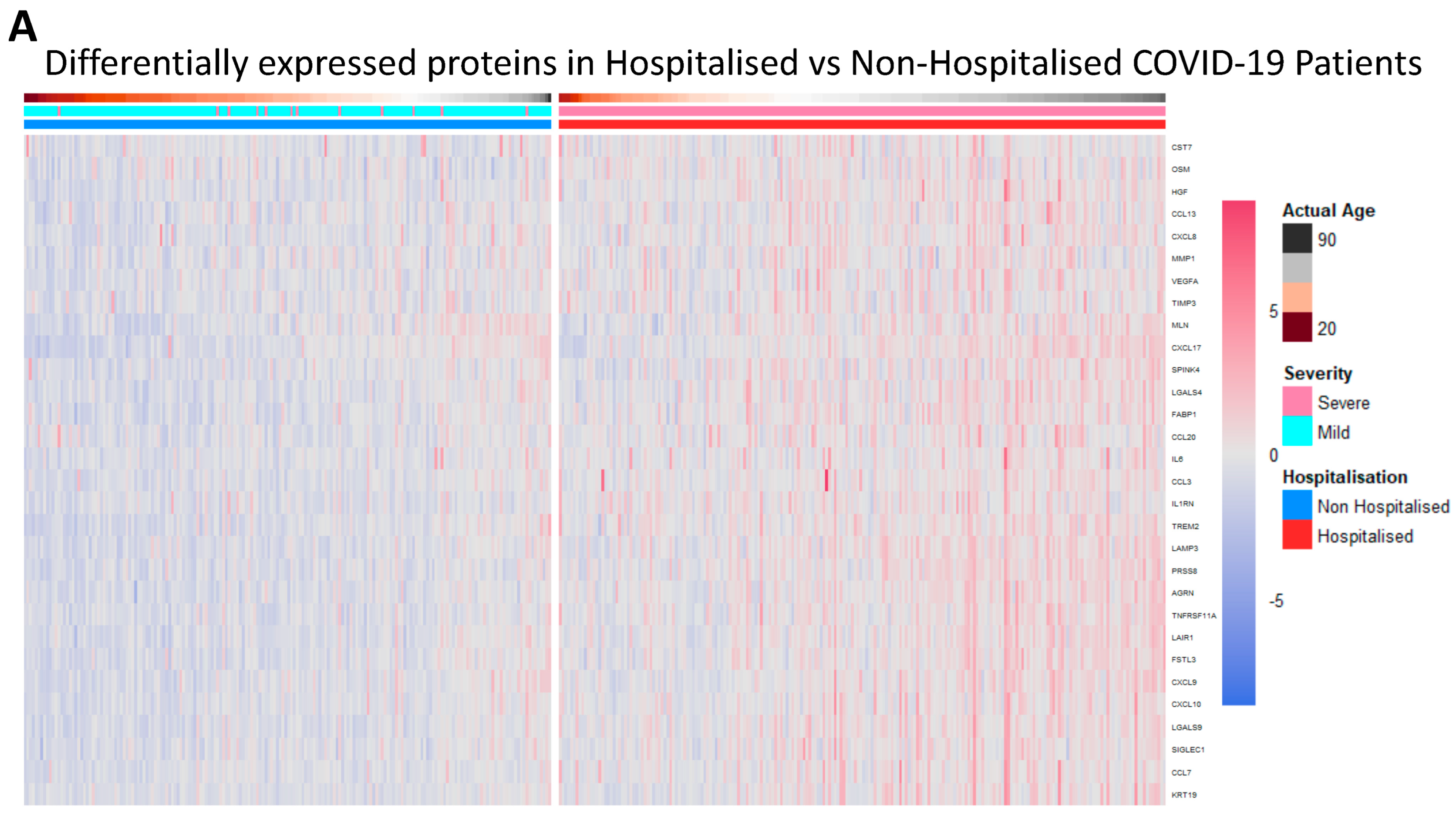
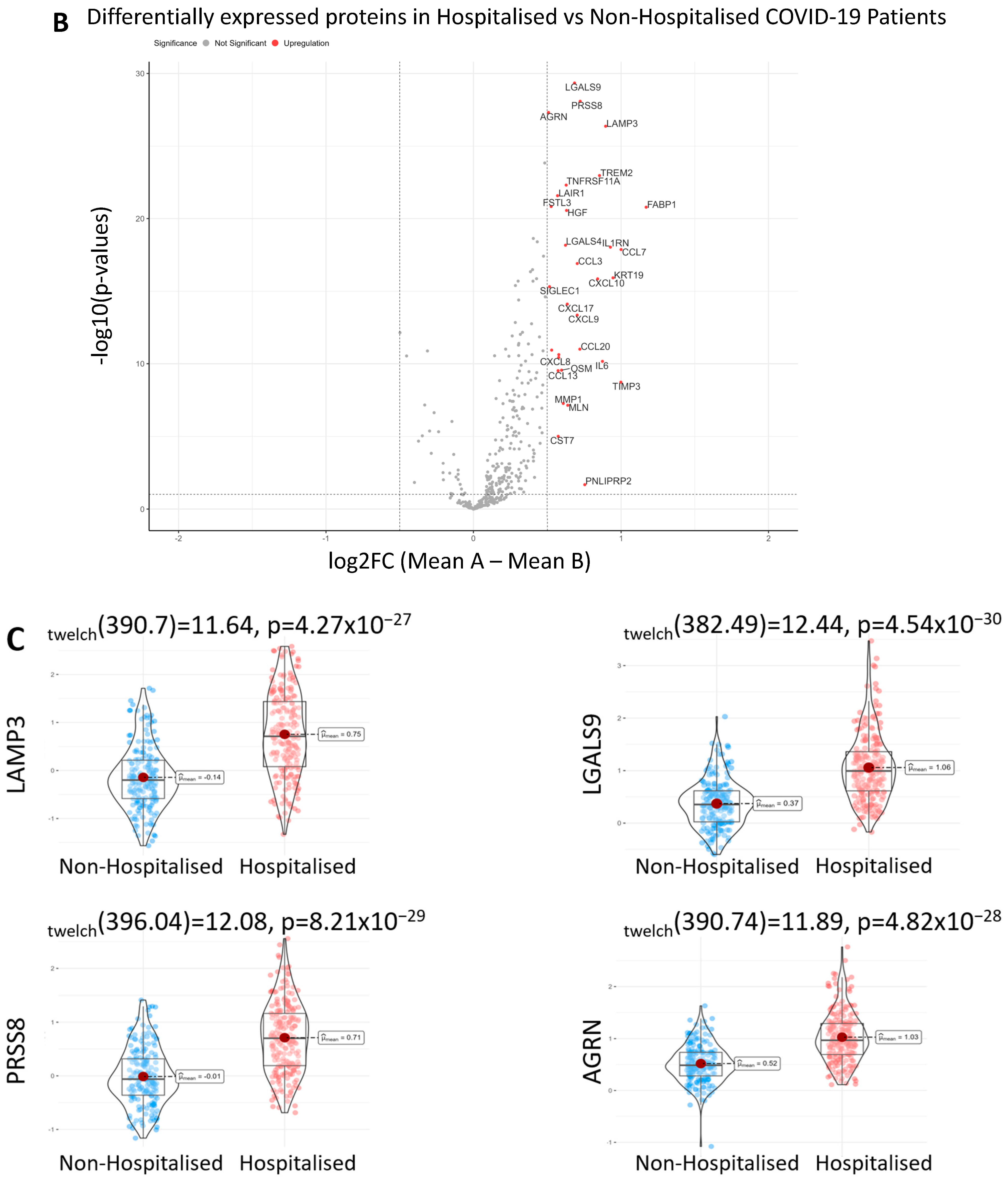
3.2. Differentially Expressed Proteins in Hospitalised Patients
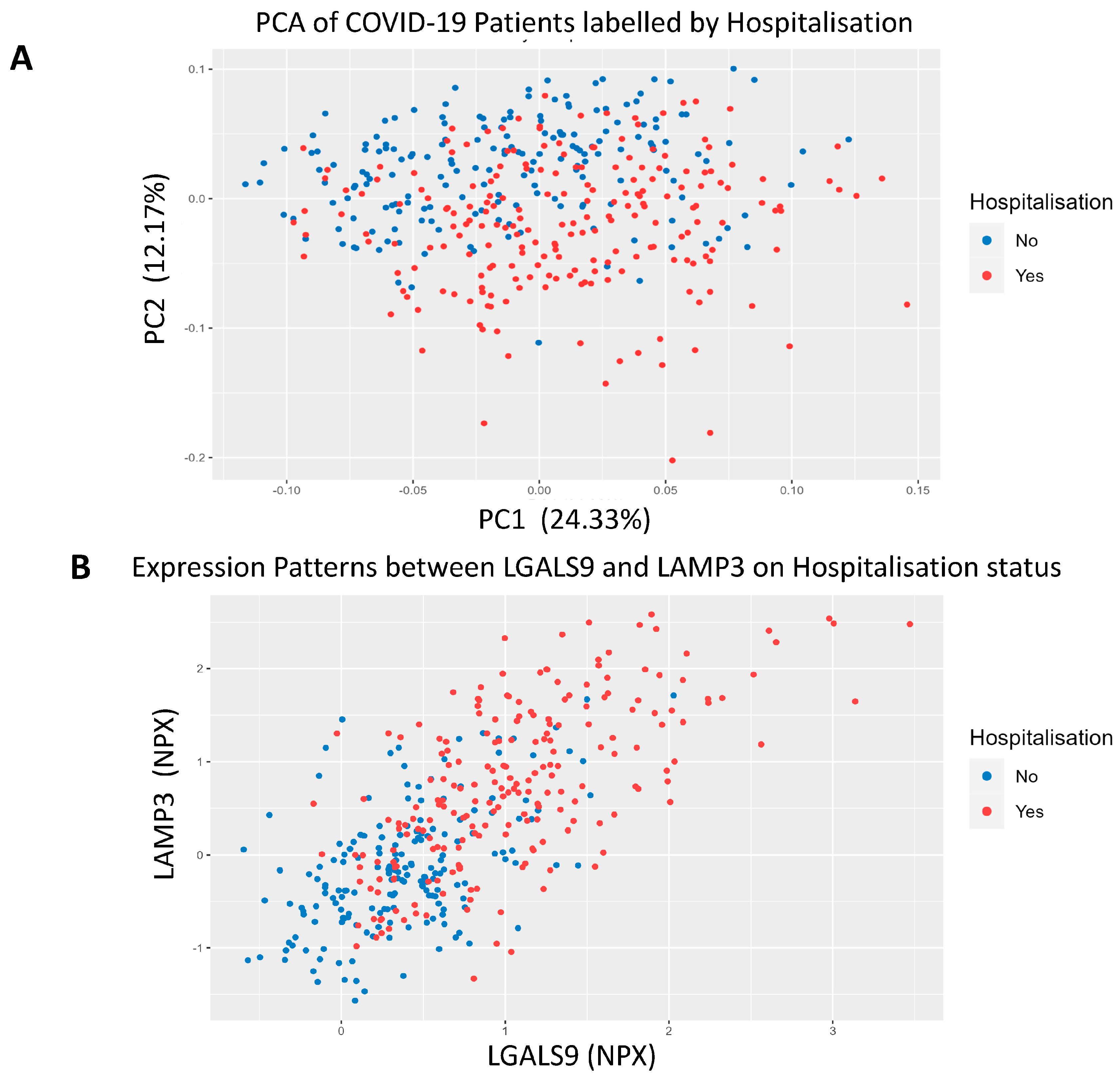
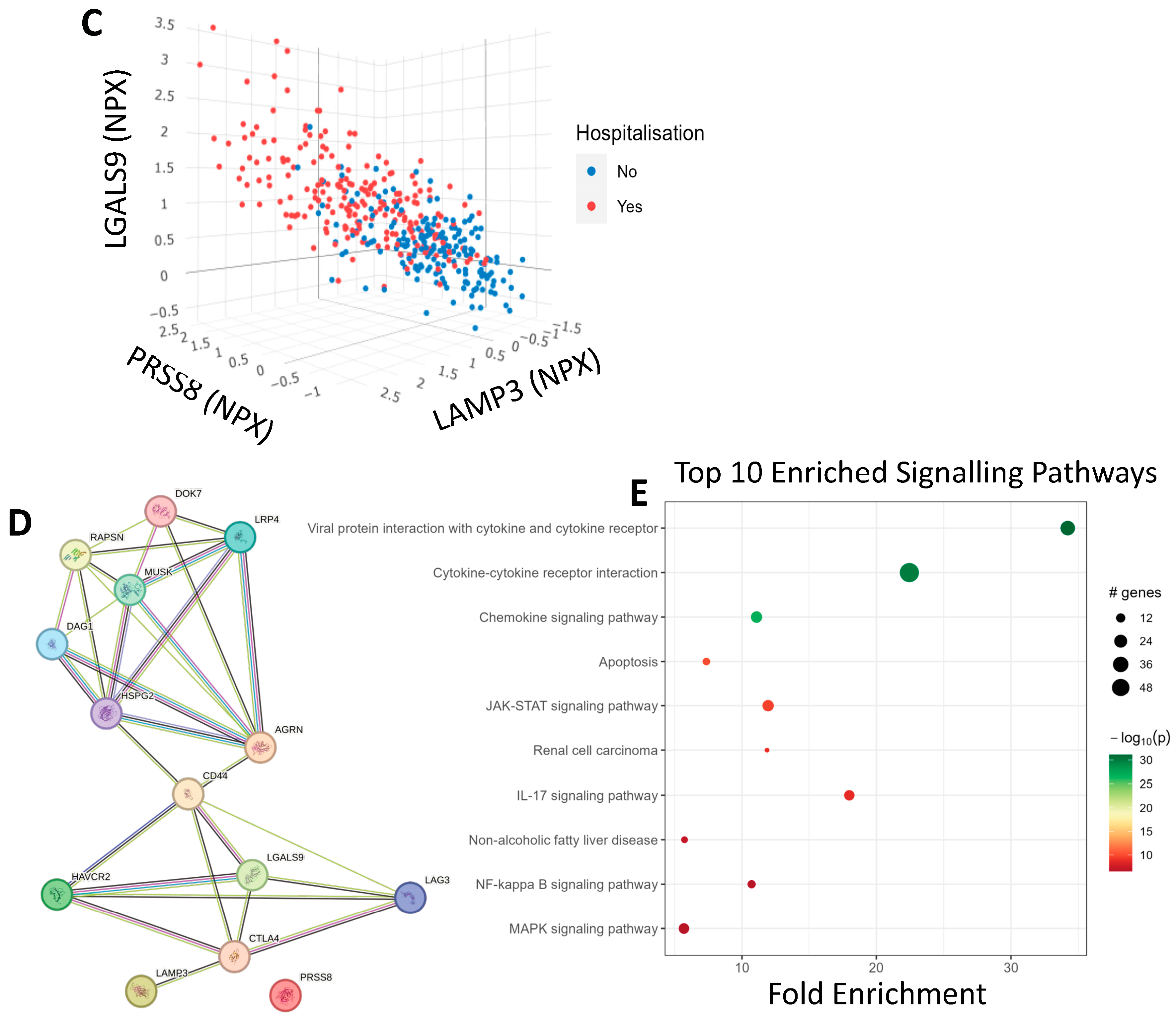
3.3. COVID-19 Patient Separation and Differential Signalling
3.4. Univariate Machine Learning Predictions for Hospitalisation Risk
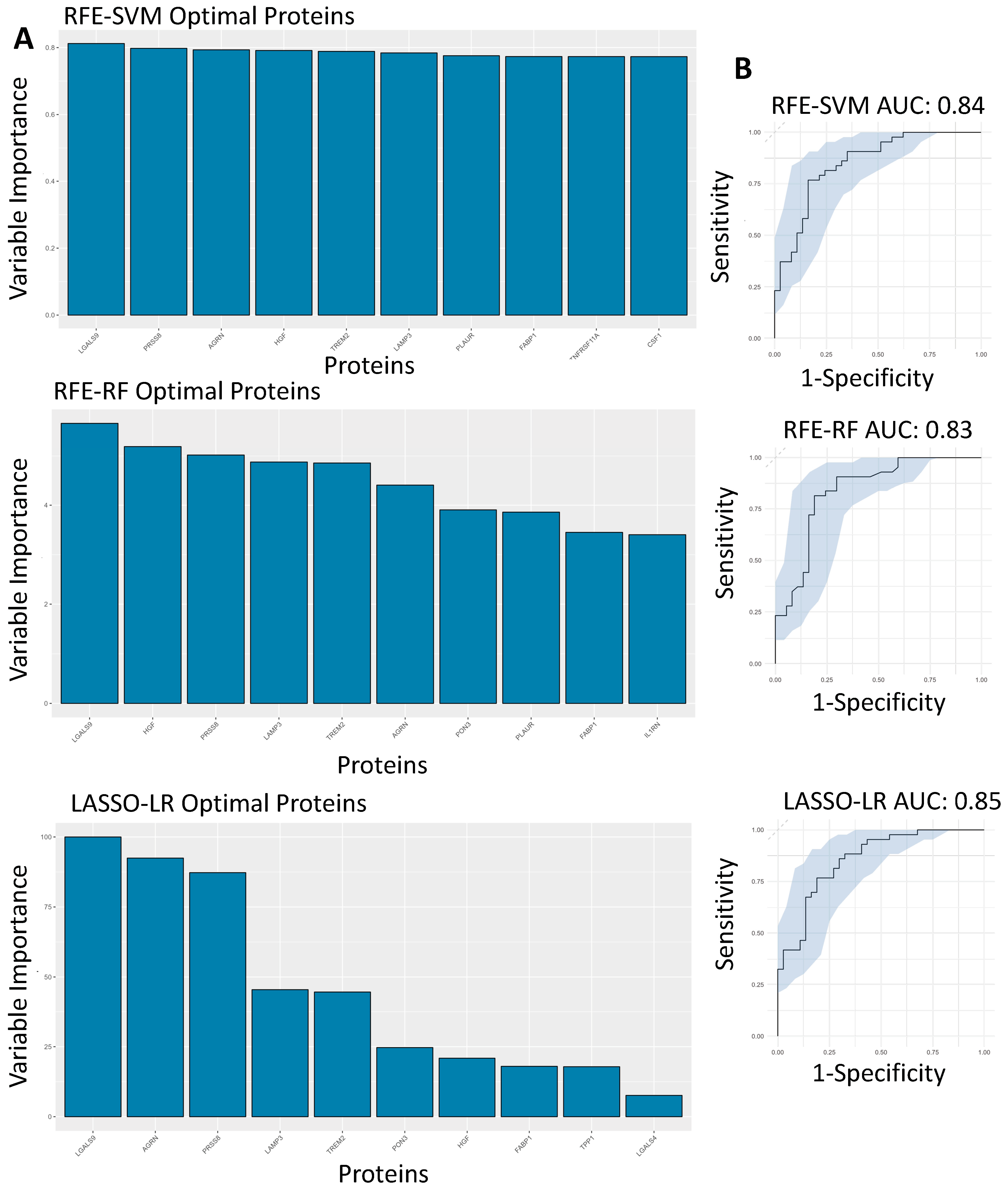
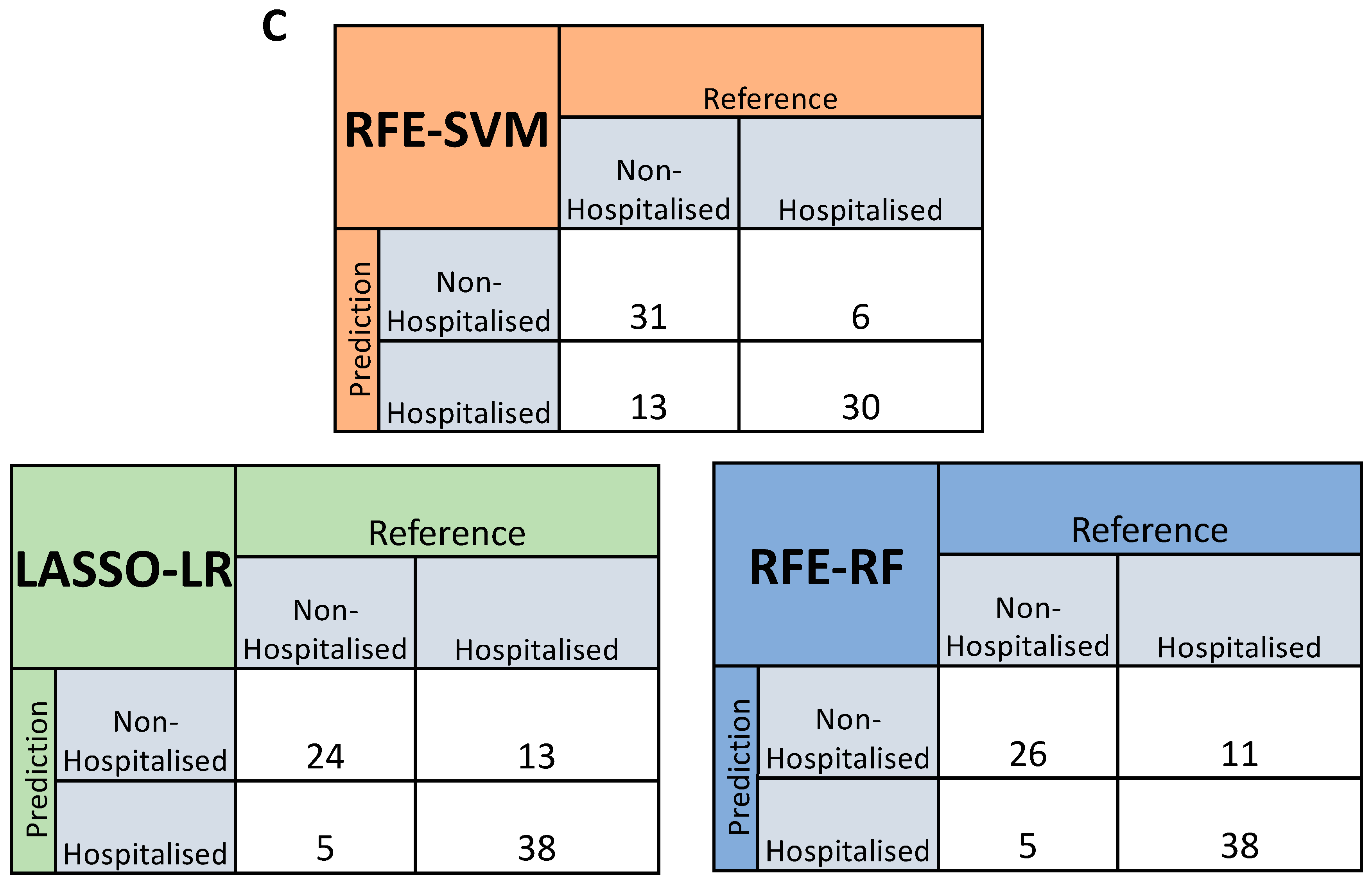
3.5. Feature Selected Machine Learning Predictions for Hospitalisation Risk
3.6. SNPs on Genes of Interest
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. WHO Coronavirus (COVID-19) Dashboard; World Health Organization: Geneva, Switzerland, 2023.
- van Eijk, L.E.; Binkhorst, M.; Bourgonje, A.R.; Offringa, A.K.; Mulder, D.J.; Bos, E.M.; Kolundzic, N.; E Abdulle, A.; van der Voort, P.H.; Rikkert, M.G.O.; et al. COVID-19: Immunopathology, pathophysiological mechanisms, and treatment options. J. Pathol. 2021, 254, 307–331. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Liu, J.; Zhang, D.; Xu, Z.; Ji, J.; Wen, C. Cytokine Storm in COVID-19: The Current Evidence and Treatment Strategies. Front. Immunol. 2020, 11, 1708. [Google Scholar] [CrossRef]
- Guo, Y.-R.; Cao, Q.-D.; Hong, Z.-S.; Tan, Y.-Y.; Chen, S.-D.; Jin, H.-J.; Tan, K.-S.; Wang, D.-Y.; Yan, Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak—An update on the status. Mil. Med. Res. 2020, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Beaney, T.; Neves, A.L.; Alboksmaty, A.; Ashrafian, H.; Flott, K.; Fowler, A.; Benger, J.R.; Aylin, P.; Elkin, S.; Darzi, A.; et al. Trends and associated factors for Covid-19 hospitalisation and fatality risk in 2.3 million adults in England. Nat. Commun. 2022, 13, 2356. [Google Scholar] [CrossRef] [PubMed]
- Khedar, R.S.; Gupta, R.; Sharma, K.; Mittal, K.; Ambaliya, H.C.; Gupta, J.B.; Singh, S.; Sharma, S.; Singh, Y.; Mathur, A. Biomarkers and outcomes in hospitalised patients with COVID-19: A prospective registry. BMJ Open 2022, 12, e067430. [Google Scholar] [CrossRef] [PubMed]
- English, A.; McDaid, D.; Lynch, S.M.; McLaughlin, J.; Cooper, E.; Wingfield, B.; Kelly, M.; Bhavsar, M.; McGilligan, V.; Irwin, R.E.; et al. Genomic, Proteomic, and Phenotypic Biomarkers of COVID-19 Severity: Protocol for a Retrospective Observational Study. JMIR Res. Protoc. 2024, 13, e50733. [Google Scholar] [CrossRef] [PubMed]
- Yazici, D.; Cagan, E.; Tan, G.; Li, M.; Do, E.; Kucukkase, O.C.; Simsek, A.; Kizmaz, M.A.; Bozkurt, T.; Aydin, T.; et al. Disrupted epithelial permeability as a predictor of severe COVID-19 development. Allergy 2023, 78, 2644–2658. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Ma, X.; Ma, Y.; Du, Y.; Feng, J. A new emerging target in cancer immunotherapy: Galectin-9 (LGALS9). Genes Dis. 2023, 10, 2366–2382. [Google Scholar] [CrossRef] [PubMed]
- Teft, W.A.; Kirchhof, M.G.; Madrenas, J. A Molecular Perspective Of CTLA-4 Function. Annu. Rev. Immunol. 2006, 24, 65–97. [Google Scholar] [CrossRef] [PubMed]
- de Saint-Vis, B.; Vincent, J.; Vandenabeele, S.; Vanbervliet, B.; Pin, J.-J.; Aït-Yahia, S.; Patel, S.; Mattei, M.-G.; Banchereau, J.; Zurawski, S.; et al. A Novel Lysosome-Associated Membrane Glycoprotein, DC-LAMP, Induced upon DC Maturation, Is Transiently Expressed in MHC Class II Compartment. Immunity 1998, 9, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liang, J.; Lu, T.; Li, M.; Shan, G.; Bi, G.; Zhao, M.; Jin, X.; Wang, Q.; Chen, Z.; et al. AGRN promotes lung adenocarcinoma progression by activating Notch signaling pathway and acts as a therapeutic target. Pharmacol. Res. 2023, 194, 106819. [Google Scholar] [CrossRef] [PubMed]
- Klaric, L.; Gisby, J.S.; Papadaki, A.; Muckian, M.D.; Macdonald-Dunlop, E.; Zhao, J.H.; Peters, J.E. Mendelian randomisation identifies alternative splicing of the FAS death receptor as a mediator of severe COVID-19. MedRxiv 2021. [Google Scholar] [CrossRef]
- Groffen, A.J.A.; Buskens, C.A.F.; van Kuppevelt, T.H.; Veerkamp, J.H.; Monnens, L.A.H.; Heuvel, L.P.W.J.v.D. Primary structure and high expression of human agrin in basement membranes of adult lung and kidney. Eur. J. Biochem. 1998, 254, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.; Pachl, E.; Hellmuth, J.C.; Kneidinger, N.; Heydarian, M.; Frankenberger, M.; Stubbe, H.C.; Ryffel, B.; Petrera, A.; Hauck, S.M.; et al. Proteomics reveals antiviral host response and NETosis during acute COVID-19 in high-risk patients. Biochim. et Biophys. Acta (BBA)–Mol. Basis Dis. 2023, 1869, 166592. [Google Scholar] [CrossRef]
- Bozorgmehr, N.; Mashhouri, S.; Rosero, E.P.; Xu, L.; Shahbaz, S.; Sligl, W.; Osman, M.; Kutsogiannis, D.J.; MacIntyre, E.; O’neil, C.R.; et al. Galectin-9, a Player in Cytokine Release Syndrome and a Surrogate Diagnostic Biomarker in SARS-CoV-2 Infection. mBio 2021, 12, e00384-21. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Funaro, A.; Spagnoli, G.C.; Momo, M.; Knapp, W.; Malavasi, F. Stimulation of T cells via CD44 requires leukocyte-function-associated antigen interactions and interleukin-2 production. Hum. Immunol. 1994, 40, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Liu, S.; Liu, J.; Zhang, Z.; Wan, X.; Huang, B.; Chen, Y.; Zhang, Y. COVID-19: Immunopathogenesis and Immunotherapeutics. Signal Transduct. Target. Ther. 2020, 5, 128. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.D.; Kitchen, R.R.; Guseh, J.S.; McNeill, J.N.; Aid, M.; Martinot, A.J.; Yu, A.; Platt, C.; Rhee, J.; Weber, B.; et al. Plasma Proteomics of COVID-19–Associated Cardiovascular Complications. JACC Basic Transl. Sci. 2022, 7, 425–441. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| levels | Hospitalised | Non-Hospitalised | p-Value | |
|---|---|---|---|---|
| Age | Mean (SD) | 57.3 (13.1) | 45 (14.5) | <0.001 |
| Severity | Severe | 214 (100%) | 12 (6.5%) | <0.001 |
| Mild | 174 (93.5%) | |||
| (%) | ||||
| Gender | Female | 90 (42.1%) | 109 (58.6%) | <0.004 |
| Male | 120 (56.1%) | 75 (40.3%) | ||
| Other | 4 (1.9%) | 2 (1.1%) | ||
| (%) |
| Proteins | Log2FC | p-Value |
|---|---|---|
| LGALS9 | 0.686 | 4.537 × 10−30 |
| PRSS8 | 0.724 | 8.208 × 10−39 |
| AGRN | 0.509 | 4.820 × 10−29 |
| LAMP3 | 0.899 | 4.273 × 10−27 |
| PLAUR | 0.483 | 1.438 × 10−24 |
| TREM2 | 0.855 | 1.096 × 10−23 |
| TNFRSF11A | 0.629 | 4.978 × 10−23 |
| LAIR1 | 0.570 | 2.663 × 10−22 |
| FSTL3 | 0.527 | 1.482 × 10−21 |
| FABP1 | 1.166 | 1.648 × 10−21 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McLarnon, T.; McDaid, D.; Lynch, S.M.; Cooper, E.; McLaughlin, J.; McGilligan, V.E.; Watterson, S.; Shukla, P.; Zhang, S.-D.; Bucholc, M.; et al. Potential Plasma Proteins (LGALS9, LAMP3, PRSS8 and AGRN) as Predictors of Hospitalisation Risk in COVID-19 Patients. Biomolecules 2024, 14, 1163. https://doi.org/10.3390/biom14091163
McLarnon T, McDaid D, Lynch SM, Cooper E, McLaughlin J, McGilligan VE, Watterson S, Shukla P, Zhang S-D, Bucholc M, et al. Potential Plasma Proteins (LGALS9, LAMP3, PRSS8 and AGRN) as Predictors of Hospitalisation Risk in COVID-19 Patients. Biomolecules. 2024; 14(9):1163. https://doi.org/10.3390/biom14091163
Chicago/Turabian StyleMcLarnon, Thomas, Darren McDaid, Seodhna M. Lynch, Eamonn Cooper, Joseph McLaughlin, Victoria E. McGilligan, Steven Watterson, Priyank Shukla, Shu-Dong Zhang, Magda Bucholc, and et al. 2024. "Potential Plasma Proteins (LGALS9, LAMP3, PRSS8 and AGRN) as Predictors of Hospitalisation Risk in COVID-19 Patients" Biomolecules 14, no. 9: 1163. https://doi.org/10.3390/biom14091163
APA StyleMcLarnon, T., McDaid, D., Lynch, S. M., Cooper, E., McLaughlin, J., McGilligan, V. E., Watterson, S., Shukla, P., Zhang, S.-D., Bucholc, M., English, A., Peace, A., O’Kane, M., Kelly, M., Bhavsar, M., Murray, E. K., Gibson, D. S., Walsh, C. P., Bjourson, A. J., & Rai, T. S. (2024). Potential Plasma Proteins (LGALS9, LAMP3, PRSS8 and AGRN) as Predictors of Hospitalisation Risk in COVID-19 Patients. Biomolecules, 14(9), 1163. https://doi.org/10.3390/biom14091163