1. Introduction
There is an increasing demand for artificial bone materials to heal massive bone defects in patients after revision surgery, malignancy resection, infection, or severe trauma [
1]. In recent years, there has been increased interest in porous scaffold materials. Scaffolds are a promising technique, acting as supportive structures while providing essential nutrients to facilitate cell attachment and growth on their surfaces [
2]. They also create space for vascularisation and bone formation [
3]. The long-term success of implanted ex vivo-generated tissue constructs depends significantly on rapid and sufficient integration with the host vasculature to ensure an adequate supply of oxygen and nutrients to the implant. However, the difficulty of inducing rapid vascular ingrowth during new bone tissue development is a major limitation of bone tissue engineering approaches for bone defect repair [
4].
Low-intensity pulsed ultrasound (LIPUS), as a specialised ultrasound modality, propagates with low intensity and is emitted in pulsed form, enabling the targeted delivery of acoustic energy to tissues while minimising thermal effects during treatment [
1]. LIPUS has been established as a non-invasive physical therapy modality with positive biological significance across all stages of fracture healing. In addition, it has been confirmed that LIPUS can promote osteoblast migration, proliferation, and differentiation and enhance bone ingrowth and bone formation in scaffolds [
5,
6]. However, traditional biomedical scaffolds are limited by the lack of vascularisation in synthetic bone structures, impacting implant survival and integration [
7]. Numerous investigations have highlighted the significance of direct cell-to-cell communication among osteoblasts, endothelial cells, and their precursors, along with the autocrine and paracrine factors they secrete, in fostering rapid vascularisation and effective bone formation [
4,
8,
9]. A prior study gathered conditioned media from LIPUS-treated human umbilical vein endothelial cells (HUVECs) co-cultured with osteoblasts, revealing an upregulation of osteogenic-related gene expression in osteoblasts compared to the control group. This finding confirms the influence of LIPUS on paracrine signalling and cell-to-cell communication in the context of angiogenesis–bone coupling. However, existing in vitro studies predominantly focus on osteoblasts or endothelial cells in isolation. As a result, the potential of osteoblasts within scaffold materials under LIPUS loading to influence vascularisation still requires further investigation.
Exosomes are extracellular bilayer lipid membrane vesicles between 30 and 150 nm in diameter, generated by endocytosis and budding as intraluminal vesicles in the luminal space of multivesicular bodies and secreted following the binding of multivesicular bodies with cell membranes [
10]. Cells can release exosomes in nearly all physiological and pathological states, carrying proteins, mRNA, non-coding RNA, DNA, lipids, and other nutrient-dense substances [
11]. Exosomes have the potential for multidirectional use. Exosomes can transmit differently expressed miRNAs between cells to impact intercellular communication and play an essential role in the paracrine coupling process of angiogenesis and osteogenesis, according to recent research [
12,
13]. Exosomes produced from a serum-free conditioned medium of human bone marrow mesenchymal stem cells stimulated early bone repair and angiogenesis in a rat model of femoral non-union while promoting cell migration and osteogenic and angiogenic gene expression in MSC cultured in vitro [
14]. Exosomes generated from human umbilical cord mesenchymal stem cells and released from a glass/hydrogel scaffold stimulated vascularised bone regeneration by transferring miR-23a-3p, as reported by Hongxing Hu et al. [
15]. Some research also indicates that LIPUS stimulation can enhance the release of exosomes from cells, potentially increasing cell-to-cell communication and the transfer of signalling molecules. Additionally, LIPUS may impact the cargo of exosomes, altering proteins, RNA, and other molecules they carry. By influencing the content of exosomes, LIPUS could potentially modulate the signalling pathways and biological functions mediated by these extracellular vesicles [
5,
6,
16]. Therefore, LIPUS may enhance osteogenesis–vascular coupling by modulating the exosomes secreted by osteoblasts on Ti-6Al-4V scaffold materials. This indicates that exosomes derived from 3D-cultured MC3T3-E1 cells under LIPUS stimulation could represent a promising therapeutic strategy for treating bone defects.
2. Materials and Methods
2.1. Scaffold Preparation
A cylindrical three-dimensional porous Ti-6Al-4V scaffold was supplied by the Institute of Metal Research, Chinese Academy of Sciences. The material exhibited compressive properties [
17]. A porous 3D Ti-6Al-4V scaffold with a diameter of 15 mm, height of 2 mm, pore size of 400–500 micrometres, and porosity of 65–70% was manufactured using an electron beam melting system (Arcam A1, Arcam, Mölndal, Sweden) as described in our previous work [
18].
Before experimentation, the materials were cleaned in an ultrasonic cleaning machine, dried with filter paper, and sterilised under high-temperature and -pressure conditions (0.21 MPa, 134 °C). Before use, the materials were wetted in the culture medium using a vacuum device.
2.2. Cell Culture
Mouse pre-osteoblast precursor cells MC3T3-E1 were provided by the Central Laboratory of China Medical University. MC3T3-E1 cells were cultured in alpha-minimum essential medium (α-MEM, HyClone Laboratories Inc., Logan, UT, USA) containing 10% foetal bovine serum (FBS, Clark, Richmond, VA, USA) and 1% penicillin–streptomycin dual antibiotics (HyClone Laboratories Inc., Logan, UT, USA). The medium was changed every two days, and the cells were passaged when 80% confluence was reached. The cells used in the experiments were in passages 5 to 15.
Human umbilical vein endothelial cells (HUVEC) were provided by the Central Laboratory of China Medical University. HUVECs were cultured in DMEM/F12 complete medium (HyClone Laboratories Inc., Logan, UT, USA) containing 10% FBS and 1% penicillin–streptomycin dual antibiotics. The cells were cultured in a CO2 incubator at 37 °C with a cell density of 80% for passaging. The cells used in the experiments were in passages 3 to 5.
2.3. Cell Seeding
Pre-treated porous Ti6Al4V materials were placed in a 6-well plate, with 3 materials placed in each well. A 20 µL suspension of MC3T3-E1 cells at a density of 1 × 106 cells/mL was seeded onto the materials and incubated in a cell culture incubator for 1 h to allow for cell adhesion. Subsequently, 3 mL of exosome-free complete culture medium was added to each well, and the plate was placed in a 37 °C, 5% CO2 cell culture incubator. After 24 h of culture, the samples were randomly assigned to the LIPUS group (loaded with ultrasound) and the control group (treated with sham irradiation under the same conditions).
2.4. LIPUS Ultrasonic Stimulation
LIPUS stimulation was applied using a Sonicator
® 740 (Mettler Electronics Corp, Anaheim, CA, USA) with a 5 cm
2 ultrasonic transducer (ME 7413). Based on preliminary experiments by our research group, the ultrasound parameters were set as follows: frequency at 1 MHz, intensity at 30 mW/cm
2, pulse width at 1 ms, pulse repetition frequency at 100 Hz; for 20 min per day, once daily, for a total of 7 days. During application, the ultrasonic transducer was placed in water, with the 6-well plates and the acoustic emission surface of the transducer being parallel and positioned above the water surface, with the bottom of the wells contacting the water surface at a vertical distance of 5 cm from the transducer’s emission surface, and the centre of the wells aligned with the centre of the transducer (
Figure 1A). The control group, i.e., the sham irradiation group, was placed on the water surface under the same conditions and exposed to a deactivated ultrasonic transducer for sham irradiation for 20 min per day, once daily, for a total of 7 days. Both the LIPUS and control groups had their medium changed after the fourth day of loading.
2.5. Exosome Extraction
Preparation of supernatant samples: On the fourth day of ultrasound application and within 24 h of the last application, the cell culture media from the LIPUS and control groups were collected and centrifuged at 300× g at 4 °C for 10 min, followed by centrifugation at 2000× g at 4 °C for 30 min to remove cells and cell debris. The supernatant was then transferred to new centrifuge tubes and stored at −80 °C.
Ultracentrifugation method for exosome extraction: The cell-free supernatant was collected in sufficient quantities from multiple experiments, centrifuged at 10,000×
g at 4 °C for 30 min, transferred to centrifuge tubes, filtered through a 0.22 µm filter, transferred to pre-sterilised ultracentrifuge tubes, and centrifuged at 100,000×
g at 4 °C for 70 min. The supernatant was discarded, the pellet was washed with PBS and centrifuged again at 100,000×
g at 4 °C for 70 min, the supernatant was discarded, and the pellet was resuspended in PBS and stored at −80 °C (
Figure 1A).
2.6. Exosome Characterisation
Transmission electron microscopy (TEM, HT7700, Hitachi, Tokyo, Japan) was used to observe and record the morphology of the exosomes. The exosome samples were diluted with PBS to an appropriate concentration, and 20 µL of the suspension was dropped onto a copper grid, allowed to adsorb naturally for 5–10 min, and then dried. A 20 µL drop of 2% phosphotungstic acid solution was added to the copper grid, left to stain for 3–5 min, and then dried and photographed.
The exosome samples were analysed using a ZetaView PMX 110 nanoparticle tracking analyser (Particle Metrix, Meerbusch, Germany) and ZetaView 8.04.02 software (Particle Metrix, Meerbusch, Germany), which recorded the particles’ Brownian motion and measured their size and concentration.
Exosomes were lysed with RIPA buffer (BiYunTian Biotechnology, Shanghai, China). Protein concentration was determined using a BCA Protein Assay Kit (BiYunTian Biotechnology, Shanghai, China). Proteins were transferred to polyvinylidene fluoride (PVDF, Millipore, Darmstadt, Germany) membranes and blocked with 5% non-fat milk (Yili Co., Ltd., Inner Mongolia, China) in TBST (25 mM Tris, 140 mM NaCl, 0.1% Tween 20, pH = 7.5) at room temperature for 2 h. The membrane was incubated overnight at 4 °C with primary antibodies against TSG101 (diluted 1:5000) (Abcam, Cambridge, MA, USA), HSP70 (diluted 1:1000) (Abcam, Cambridge, MA, USA), and calnexin (diluted 1:3000) (Abcam, Cambridge, MA, USA). The membranes were washed three times with TBST, each time for 10 min, and then incubated for 1 h at room temperature with HRP-conjugated goat anti-rabbit IgG secondary antibody (Santa Cruz Biotechnology, Dallas, TX, USA). After three washes with TBST, each lasting one hour, the membranes were treated with an ECL detection reagent (Tanon5200, Shanghai Tianneng Technology Co., Ltd., Shanghai, China) and exposed to obtain protein band images (
Figure 1A).
2.7. Exosome Uptake Experiment
Exosomes were resuspended in DMEM/F12 medium to a concentration of 1 × 10
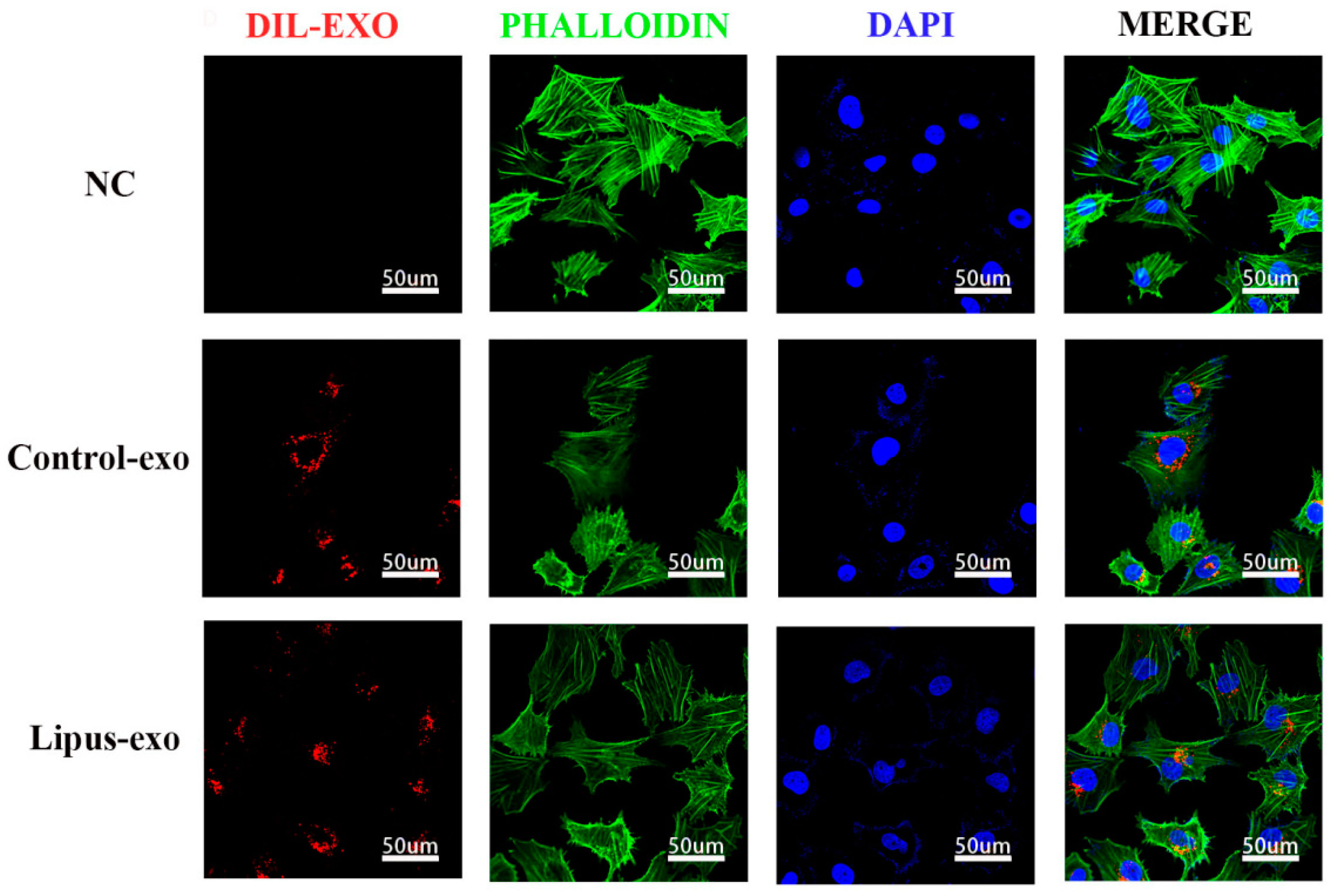
9 particles/well and added to a 24-well plate with 2 mL of complete Dil cell membrane red fluorescent staining solution (BiYunTian Biotechnology Co., Ltd., Shanghai, China) per well. Dil dye was co-cultured with exosomes for 12 h. Then, Dil-labelled exosomes were cultured with HUVEC cells for 24 h, followed by fixation with 4% paraformaldehyde for 10 minutes. Cells were washed three times with PBS for 3 min each, and then 400 μL/well of phalloidin working solution was added to stain the cytoskeleton (Boster Biological Engineering Co., Ltd., Wuhan, China) for 40–60 min. After three washes with PBS, the cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI, Boster Biological Engineering Co., Ltd., Wuhan, China) for 5 min and washed three more times with PBS. All procedures were performed in the dark, and the samples were observed and photographed under a laser confocal microscope (A1, Nikon, Tokyo, Japan) (
Figure 2).
2.8. Cell Proliferation Experiment
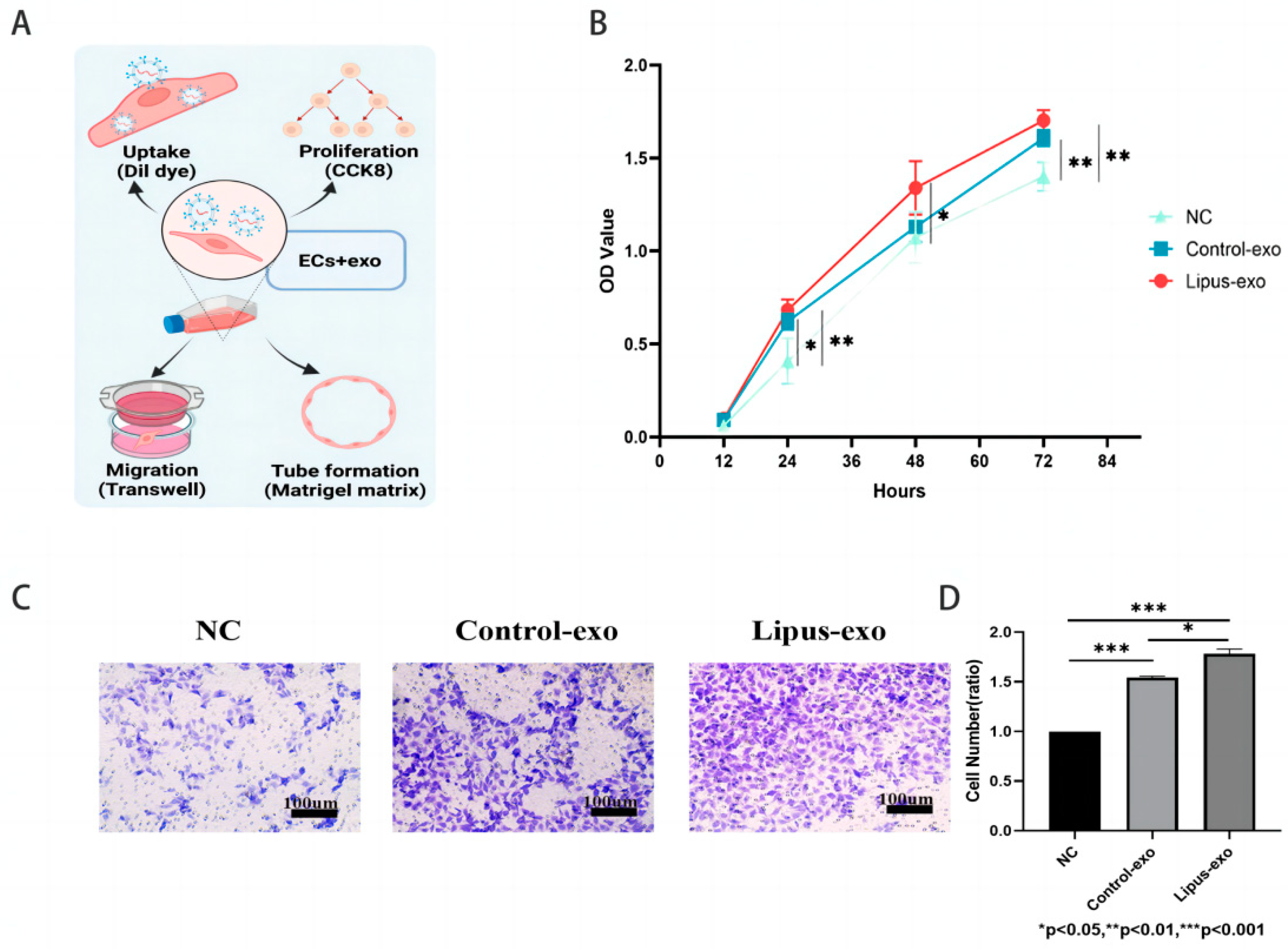
HUVEC cells were seeded at a density of 2000 cells/well in a 96-well plate. After cell adherence, the original medium was replaced with a complete medium containing 1 × 109 exosomes/mL. At 12, 24, 48, and 72 h of co-culture, 10 μL of CCK8 reagent (APExBIO, Houston, TX, USA) was added to each well and incubated for 1 h at 37 °C in a 5% CO2 incubator. The optical density at 450 nm was measured using an enzyme-linked immunosorbent assay reader (InfiniteF200, Tecan Trading AG, Männedorf, Switzerland), the values were recorded, and a cell proliferation curve was plotted.
2.9. Cell Migration
A total of 5 × 104 HUVEC cells were seeded in the upper chamber of a Transwell 24-well plate (8 μm, Corning, NY, USA), and 600 μL of complete medium containing 1 × 109 exosomes/mL was added to the lower chamber. The chamber was incubated at 37 °C in a 5% CO2 incubator for 24 h, after which the inner layer cells of the chamber were gently removed with a moistened cotton swab. The chamber was then fixed in 4% paraformaldehyde for 20 min. After three washes with PBS, the chamber was stained with 1% crystal violet solution (BiYunTian Biotechnology Co., Ltd., Shanghai, China) for 30 min. The chamber was washed three more times with PBS to remove excess dye, and cells were observed and photographed under an inverted microscope.
2.10. Endothelial Tube Formation Assay
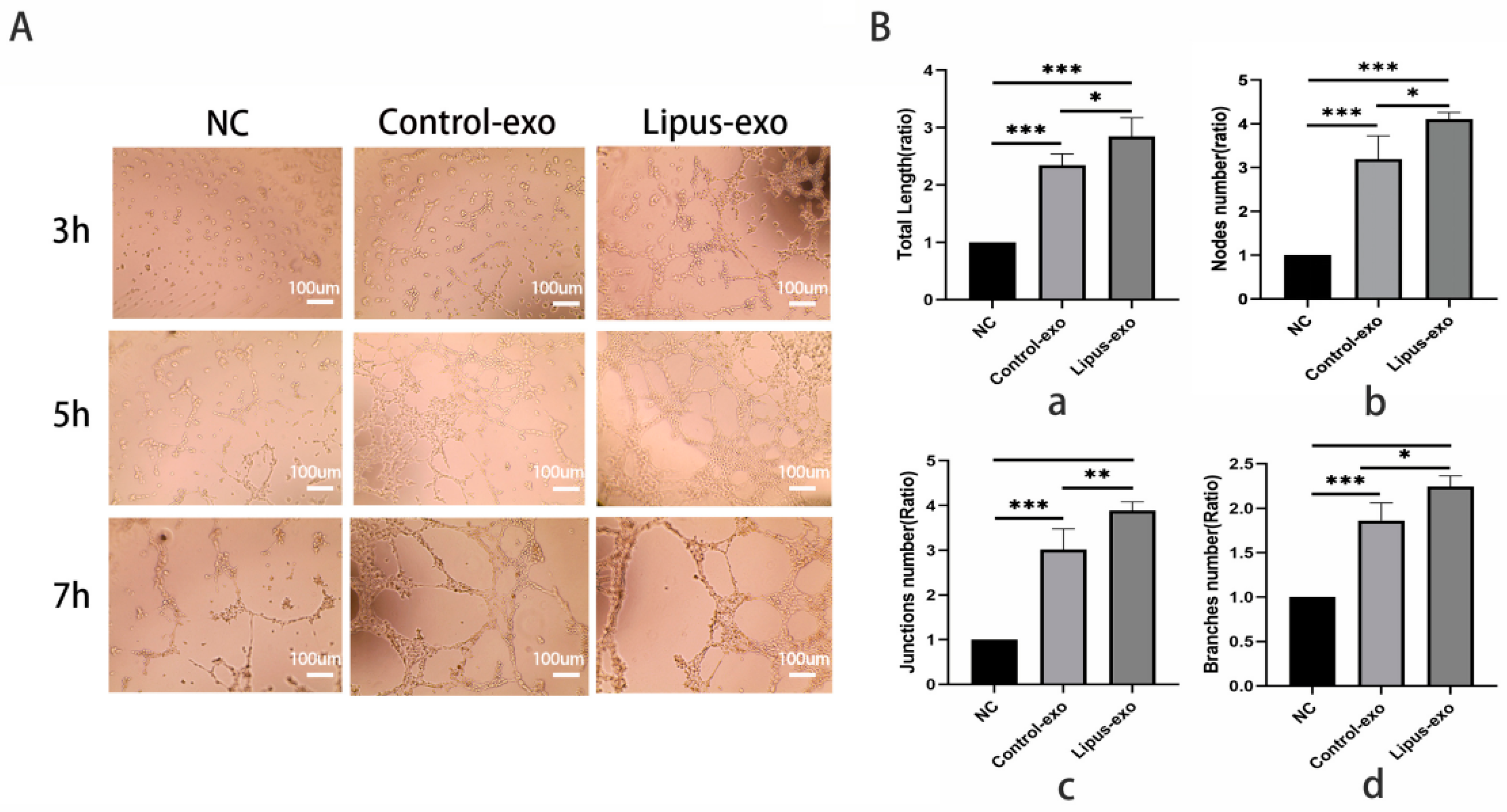
Matrigel basement membrane matrix (Corning, NY, USA) was coated at a concentration of 50 μL/well on a 96-well plate. 2 × 104 HUVEC cells/well were added, and an exosome solution was introduced to the experimental group at a concentration of 1 × 109/mL. A 100 μL cell suspension was seeded onto the solidified Matrigel gel and incubated at 37 °C in a 5% CO2 incubator. The observation began at 3 h post-seeding and continued every 1 h until tube-like structures formed, which were then quantified.
2.11. Statistical Analysis
All data are presented as mean ± standard error. Image analysis for the Transwell cell migration assay and tube formation assay was performed using Image J 1.54d software, and data processing was carried out with SPSS version 27.0.1. Comparisons between the two groups were made using an independent samples t-test, and for non-normally distributed data, the Mann–Whitney test was used. Comparisons among multiple groups were performed using one-way analysis of variance (one-way ANOVA), with post hoc comparisons using the LSD test. Graphical representations of statistical results were created using GraphPad Prism 8 software. A p-value < 0.05 was considered statistically significant, while a p-value > 0.05 was not considered statistically significant.
4. Discussion
Our previous results proved that LIPUS facilitated cellular ingrowth and enhanced the proliferation and early differentiation of osteoblasts and bone formation in Ti-6Al-4V scaffolds [
18,
19,
20]. In the process of bone formation, angiogenesis plays a pivotal role because blood vessels not only act as a source of oxygen and nutrients but also facilitate the transportation of bone progenitor cells, supply essential minerals like calcium and phosphate for mineralisation, and contribute to the establishment of a niche for bone marrow stromal cells and hematopoietic stem cells. Therefore, studying how osteoblasts on Ti-6Al-4V scaffolds under LIPUS stimulation affect angiogenesis is essential to confirm their potential for repairing bone defects.
Contrary to the traditional belief that angiogenesis solely influences bone remodelling, recent research has underscored the intricate spatial and temporal correlation between bone formation and angiogenesis, termed “angiogenesis–bone coupling”. This coupled process has been proven to be related to paracrine mechanisms, with cytokines playing a significant role. For example, osteoblast-derived VEGF is essential for the early vascularisation response and macrophage infiltration in the initial stages of inflammation and later stimulates angiogenesis and osteoblast differentiation [
21]. MicroRNAs (miRNAs) have also been shown to control angiogenesis at multiple levels, expressed within vascular constituents, and play key roles in development and disease [
22]. This paracrine-mediated interplay offers insight into the potential function of exosomes in this process. Exosomes derived from bone marrow mesenchymal stem cells have been demonstrated to promote angiogenesis–osteogenesis coupling both in vitro and in vivo [
14].
The interaction between LIPUS and exosomes has become an intriguing area of study, with several studies suggesting that LIPUS can modulate the release, content, and uptake of exosomes, thereby affecting cellular communication and tissue regeneration. Both in vitro and in vivo experimental results demonstrate that extracellular vesicles secreted by LIPUS-treated BMSCs possess stronger anti-inflammatory properties compared to extracellular vesicles secreted by unstimulated BMSCs [
23]. However, to date, there have been no reports on the role of LIPUS in influencing communication between osteoblasts and endothelial cells through the effect on exosomes secreted by MC3T3-E1 cells on porous Ti-6Al-4V scaffolds.
In our experiments, following co-culture with Dil red fluorescent cell membrane dye, internalisation of exosomes by HUVECs was indicated by the presence of red fluorescent signals within the cells. The intensity of these signals increased in correlation with the concentration of exosomes and the duration of co-culture, suggesting that HUVECs are capable of taking up exosomes from the solution in a sustained manner, demonstrating a time-dependent and concentration-dependent uptake effect.
After internalisation by cells, exosomes can exert certain biological functions. This study found that the exosomes from both the LIPUS and control groups promoted proliferation, migration, and tube formation of HUVECs, which may be related to the paracrine effects of MC3T3-E1 cells on endothelial cell biological functions. Studies have confirmed that MC3T3-E1 cells can promote the recruitment and proliferation of endothelial cells at fracture sites by secreting vascular endothelial growth factor (VEGF) and angiogenic factors [
24]. Co-culture of HUVECs with conditioned medium from osteoblasts revealed that it could enhance endothelial cell proliferation, migration, and differentiation capabilities [
25]. Exosomes derived from senescent osteoblasts mediated by miR-139-5p can promote aging and apoptosis by targeting the TBX1 gene, thereby inhibiting the proliferation and invasion of endothelial cells [
11]. Building upon this foundation, our experiments confirmed that exosomes from MC3T3-E1 cells could play a positive role in the coupling process of angiogenesis and osteogenesis.
Endothelial cell migration, an early step in the angiogenesis cascade and a hallmark of angiogenesis, occurs autonomously but can acquire collective migration characteristics upon stimulation, when a group of cells migrate towards the chemotactic stimulus and the remaining cells follow in the same direction. This process is crucial for stimulating angiogenesis in tissue regeneration [
26]. Transwell assays in various experiments involving the co-culture of exosomes and HUVECs were employed to assess the migratory impact of exosomes from the lower chamber on HUVECs in the upper chamber [
27,
28,
29]. Our study utilised two sets of exosomes in the lower chamber of the transwell system to investigate their chemotactic effect on HUVECs in the upper chamber, and the results demonstrated that the LIPUS group was more effective in driving the migration of HUVECs towards the lower chamber compared to the control group (
p < 0.01), thereby enhancing the migratory capacity of HUVECs.
The basement membrane is a thin, highly specialised extracellular matrix that serves as a foundation for endothelial cells and maintains vascular morphology in vivo. In vitro, when endothelial cells are seeded on recombinant basement membrane matrices, they rapidly organise into tubular structures. This principle has been adapted into a matrix gel, which is a convenient and quantifiable method for assessing the tube-forming ability of endothelial cells [
30]. Our experiments found that the LIPUS group formed microtubes with complete luminal structures more rapidly compared to the control group and exosome-free control group, with a greater density of microtubes. Quantitative analysis using Image J 1.54d software revealed that the total length of microtubes, number of branching microtubes, number of nodes, and number of connecting points were all significantly higher in the LIPUS group than in the control group, which proves that exosomes from MC3T3-E1 cells under ultrasound can promote endothelial cell tube formation. These experimental outcomes corroborate the role of ultrasound-modulated exosomes secreted by MC3T3-E1 cells in enhancing the biological functions of endothelial cells.
5. Conclusions
In this study, MC3T3-E1 cells produced exosomes on porous Ti-6Al-4V scaffold materials under LIPUS loading. Compared to the non-ultrasound group, the LIPUS group enhanced endothelial cell proliferation and angiogenesis through exosomes secreted by MC3T3-E1 cells. The LIPUS group formed microtubes with complete luminal structures more rapidly, exhibiting greater density, increased total length, and a higher number of branching microtubes, nodes, and connecting points. Therefore, LIPUS may be a promising strategy to boost the efficacy of osteoblast-derived exosomes in porous Ti-6Al-4V scaffold materials to enhance angiogenesis. However, the effectiveness of osteoblast-derived exosomes in promoting angiogenesis within these scaffold materials remains unclear and requires further investigation.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}