
Molecular Mechanisms and the Significance of Synonymous Mutations


Abstract
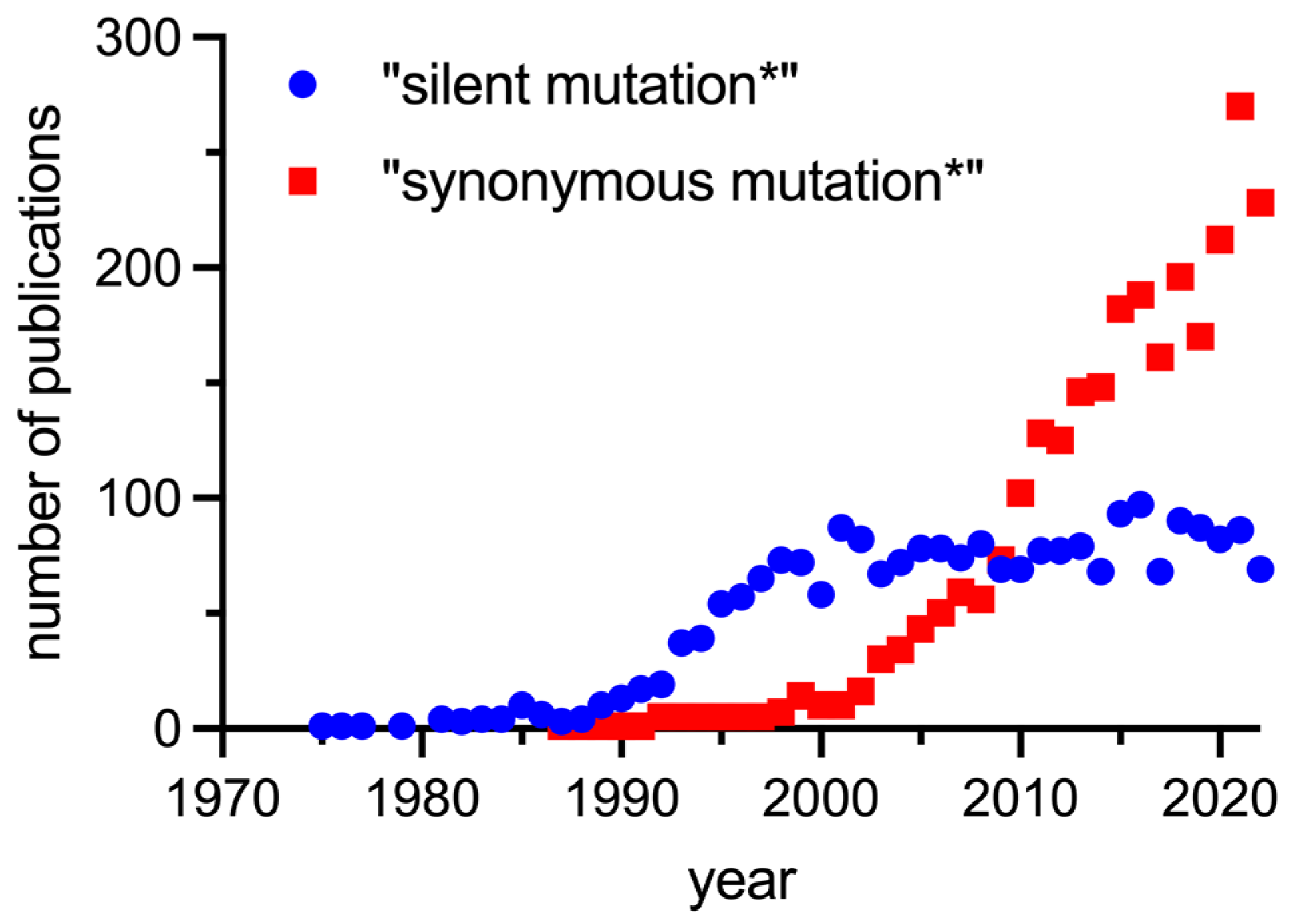
1. Introduction
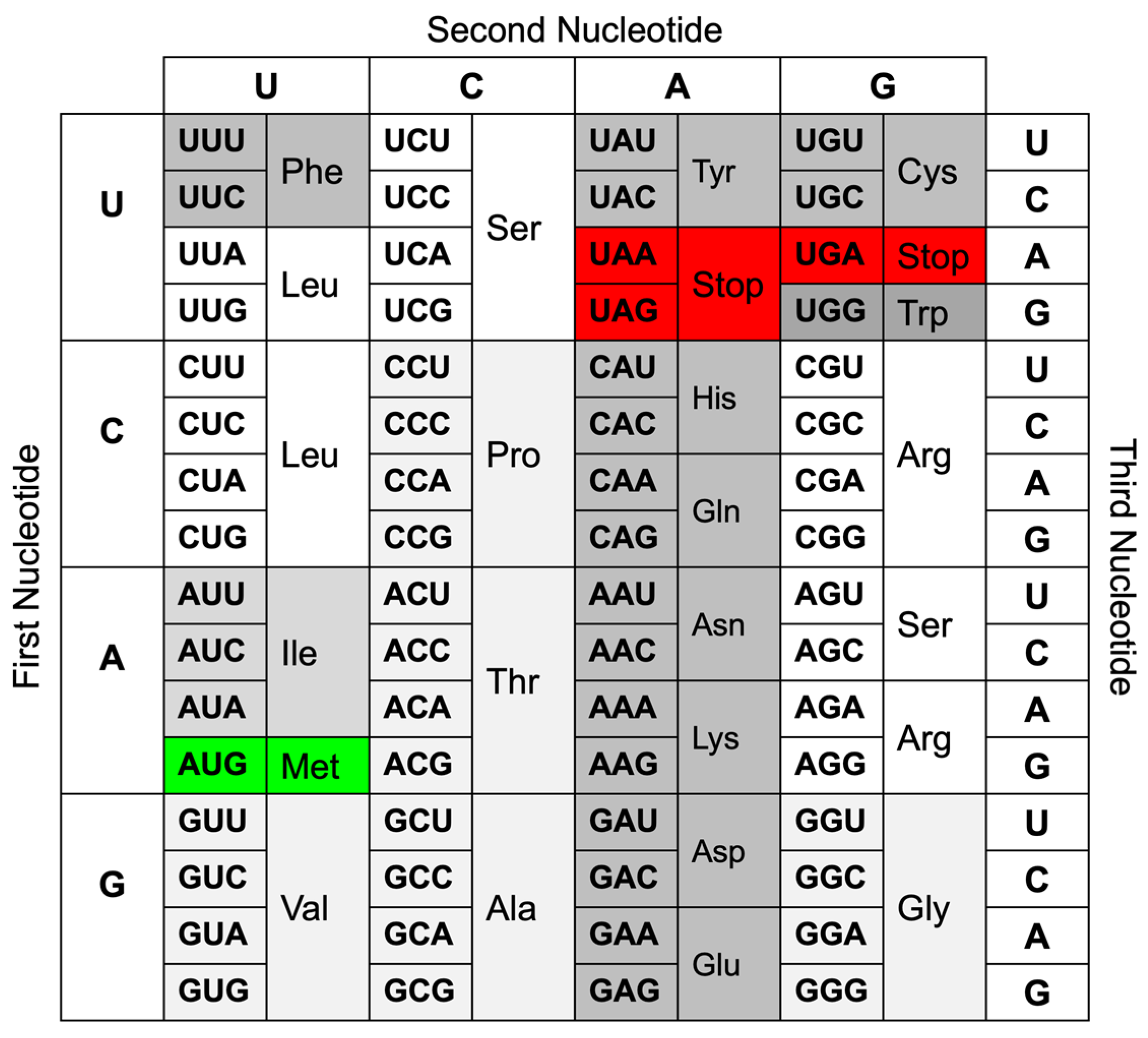
2. Definition of Single-Nucleotide Mutations
2.1. Single-Nucleotide Mutations Outside Coding Sequences
2.2. Single-Nucleotide Mutations in Coding Sequences
3. Impact of Synonymous Mutations at the Molecular Level
3.1. Transcription Efficiency
3.2. Translation Efficiency
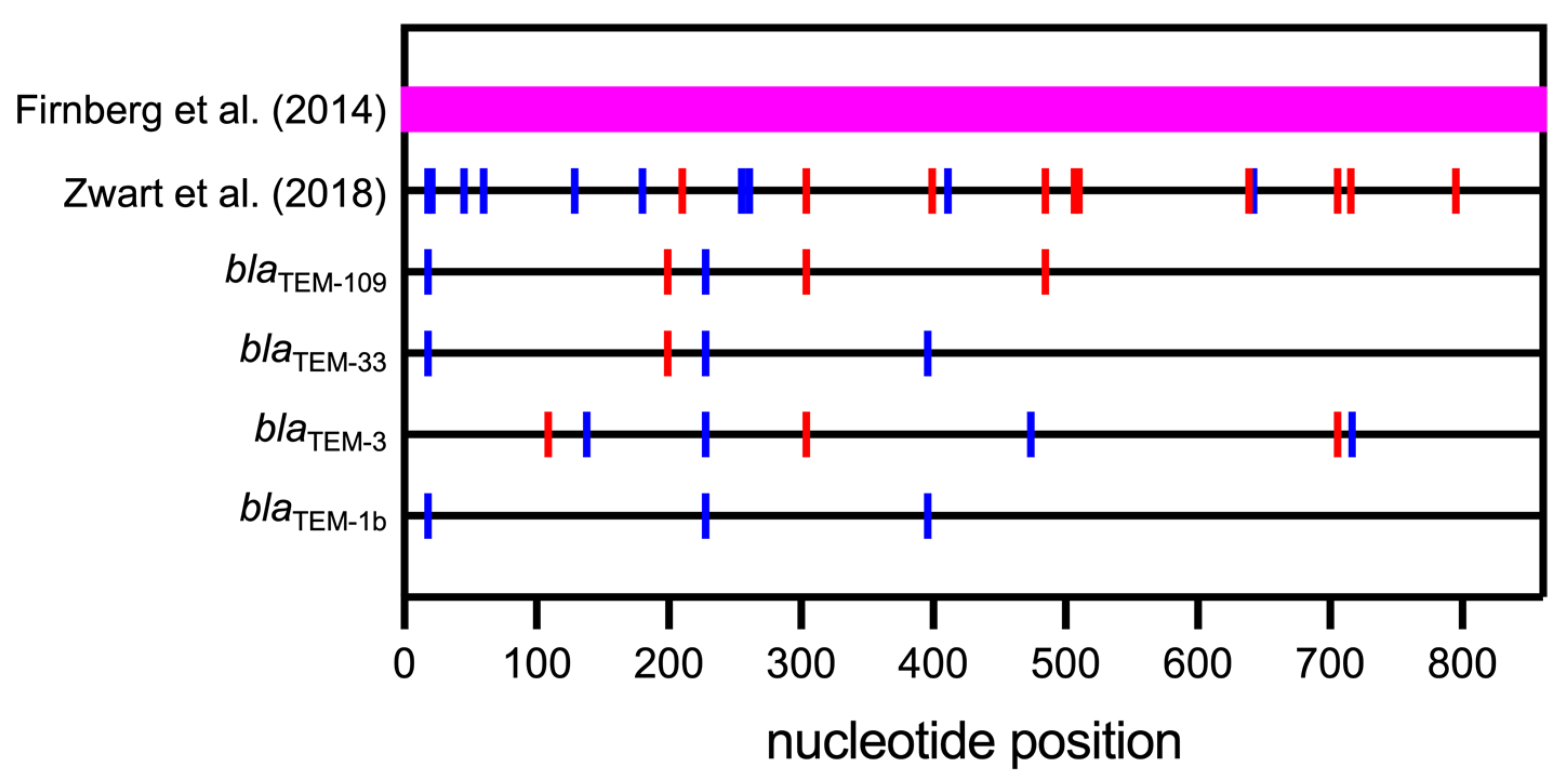
4. Synonymous Mutations in Prokaryotes
5. Synonymous Mutations in Eukaryotes
- Transcription and translation are decoupled both in space and time. Transcription occurs in the nucleus and translation in the cytoplasm, either at soluble or endoplasmic reticulum-bound ribosomes.
- mRNA must travel from the nucleus to the cytoplasm, and before that journey, it undergoes several modifications, including 5′-capping, 3′-polyadenylation, splicing, and binding to proteins and/or ribonucleoproteins.
5.1. Synonymous Mutations in Various Eukaryotic Organisms
5.2. Synonymous Mutations Associated with Human Pathophysiology
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Avery, O.T.; Macleod, C.M.; McCarty, M. Studies on the Chemical Nature of the Substance Inducing Transformation of Pneumococcal Types: Induction of Transformation by a Desoxyribonucleic Acid Fraction Isolated from Pneumococcus Type III. J. Exp. Med. 1944, 79, 137–158. [Google Scholar] [CrossRef]
- Franklin, R.E.; Gosling, R.G. Molecular configuration in sodium thymonucleate. Nature 1953, 171, 740–741. [Google Scholar] [CrossRef]
- Wilkins, M.H.; Stokes, A.R.; Wilson, H.R. Molecular structure of deoxypentose nucleic acids. Nature 1953, 171, 738–740. [Google Scholar] [CrossRef]
- Watson, J.D.; Crick, F.H. Molecular structure of nucleic acids: A structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef]
- Crick, F.H.; Watson, J.D. The compelemtary structure of deoxyribonucleic acid. Proc. R. Soc. A 1954, 223, 80–96. [Google Scholar]
- Crick, F.H. On protein synthesis. Symp. Soc. Exp. Biol. 1958, 12, 138–163. [Google Scholar]
- Crick, F.H.; Barnett, L.; Brenner, S.; Watts-Tobin, R.J. General nature of the genetic code for proteins. Nature 1961, 192, 1227–1232. [Google Scholar] [CrossRef]
- Nirenberg, M.W.; Matthaei, J.H.; Jones, O.W. An intermediate in the biosynthesis of polyphenylalanine directed by synthetic template RNA. Proc. Natl. Acad. Sci. USA 1962, 48, 104–109. [Google Scholar] [CrossRef]
- Crick, F.H. The genetic code: III. Sci. Am. 1966, 215, 55–60. [Google Scholar] [CrossRef]
- Casadesus, J.; Low, D. Epigenetic gene regulation in the bacterial world. Microbiol. Mol. Biol. Rev. 2006, 70, 830–856. [Google Scholar] [CrossRef]
- McClelland, M. Selection against dam methylation sites in the genomes of DNA of enterobacteriophages. J. Mol. Evol. 1984, 21, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Au, K.G.; Welsh, K.; Modrich, P. Initiation of methyl-directed mismatch repair. J. Biol. Chem. 1992, 267, 12142–12148. [Google Scholar] [CrossRef] [PubMed]
- Samy, M.D.; Yavorski, J.M.; Mauro, J.A.; Blanck, G. Impact of SNPs on CpG Islands in the MYC and HRAS oncogenes and in a wide variety of tumor suppressor genes: A multi-cancer approach. Cell Cycle 2016, 15, 1572–1578. [Google Scholar] [CrossRef] [PubMed]
- Shyamala, N.; Kongettira, C.L.; Puranam, K.; Kupsal, K.; Kummari, R.; Padala, C.; Hanumanth, S.R. In silico identification of single nucleotide variations at CpG sites regulating CpG island existence and size. Sci. Rep. 2022, 12, 3574. [Google Scholar] [CrossRef] [PubMed]
- Hunt, R.C.; Simhadri, V.L.; Iandoli, M.; Sauna, Z.E.; Kimchi-Sarfaty, C. Exposing synonymous mutations. Trend Genet. 2014, 30, 308–321. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.J.; Amode, M.R.; Aneja, A.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Becker, A.; Bennett, R.; Berry, A.; Bhai, J.; et al. Ensembl 2023. Nucleic Acids Res. 2023, 51, D933–D941. [Google Scholar] [CrossRef] [PubMed]
- DeLange, R.J.; Smith, E.L. Histones: Structure and function. Annu. Rev. Biochem. 1971, 40, 279–314. [Google Scholar] [CrossRef]
- Cohen, E.; Zafrir, Z.; Tuller, T. A code for transcription elongation speed. RNA Biol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Kudla, G.; Lipinski, L.; Caffin, F.; Helwak, A.; Zylicz, M. High guanine and cytosine content increases mRNA levels in mammalian cells. PLoS Biol. 2006, 4, e180. [Google Scholar] [CrossRef]
- Hia, F.; Yang, S.F.; Shichino, Y.; Yoshinaga, M.; Murakawa, Y.; Vandenbon, A.; Fukao, A.; Fujiwara, T.; Landthaler, M.; Natsume, T.; et al. Codon bias confers stability to human mRNAs. EMBO Rep. 2019, 20, e48220. [Google Scholar] [CrossRef]
- Zhou, Z.; Dang, Y.; Zhou, M.; Li, L.; Yu, C.H.; Fu, J.; Chen, S.; Liu, Y. Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proc. Natl. Acad. Sci. USA 2016, 113, E6117–E6125. [Google Scholar] [CrossRef] [PubMed]
- Landick, R. Transcriptional Pausing as a Mediator of Bacterial Gene Regulation. Annu. Rev. Microbiol. 2021, 75, 291–314. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Xu, J.; Chong, J.; Wang, D. Molecular basis of transcriptional pausing, stalling, and transcription-coupled repair initiation. Biochim. Biophys. Acta Gene Regul. Mech. 2021, 1864, 194659. [Google Scholar] [CrossRef] [PubMed]
- Hershberg, R.; Petrov, D.A. Selection on codon bias. Annu. Rev. Genet. 2008, 42, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, J.B.; Kudla, G. Synonymous but not the same: The causes and consequences of codon bias. Nat. Rev. Genet. 2011, 12, 32–42. [Google Scholar] [CrossRef]
- Supek, F. The Code of Silence: Widespread Associations Between Synonymous Codon Biases and Gene Function. J. Mol. Evol. 2016, 82, 65–73. [Google Scholar] [CrossRef]
- Ganesh, R.B.; Maerkl, S.J. Biochemistry of Aminoacyl tRNA Synthetase and tRNAs and Their Engineering for Cell-Free and Synthetic Cell Applications. Front. Bioeng. Biotechnol. 2022, 10, 918659. [Google Scholar] [CrossRef]
- Wen, J.D.; Lancaster, L.; Hodges, C.; Zeri, A.C.; Yoshimura, S.H.; Noller, H.F.; Bustamante, C.; Tinoco, I. Following translation by single ribosomes one codon at a time. Nature 2008, 452, 598–603. [Google Scholar] [CrossRef]
- Shine, J.; Dalgarno, L. Determinant of cistron specificity in bacterial ribosomes. Nature 1975, 254, 34–38. [Google Scholar] [CrossRef]
- Li, G.W.; Oh, E.; Weissman, J.S. The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature 2012, 484, 538–541. [Google Scholar] [CrossRef]
- Uemura, S.; Dorywalska, M.; Lee, T.H.; Kim, H.D.; Puglisi, J.D.; Chu, S. Peptide bond formation destabilizes Shine-Dalgarno interaction on the ribosome. Nature 2007, 446, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Tessier, L.H.; Sondermeyer, P.; Faure, T.; Dreyer, D.; Benavente, A.; Villeval, D.; Courtney, M.; Lecocq, J.P. The influence of mRNA primary and secondary structure on human IFN-gamma gene expression in E. coli. Nucleic Acids Res. 1984, 12, 7663–7675. [Google Scholar] [CrossRef] [PubMed]
- Kudla, G.; Murray, A.W.; Tollervey, D.; Plotkin, J.B. Coding-sequence determinants of gene expression in Escherichia coli. Science 2009, 324, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.N.; Gabay, J.; Debarbouille, M.; Schwartz, M. A role for mRNA secondary structure in the control of translation initiation. Nature 1982, 295, 616–618. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.; Roa, M.; Debarbouille, M. Mutations that affect lamB gene expression at a posttranscriptional level. Proc. Natl. Acad. Sci. USA 1981, 78, 2937–2941. [Google Scholar] [CrossRef] [PubMed]
- Tinoco, I., Jr.; Borer, P.N.; Dengler, B.; Levin, M.D.; Uhlenbeck, O.C.; Crothers, D.M.; Bralla, J. Improved estimation of secondary structure in ribonucleic acids. Nat. New Biol. 1973, 246, 40–41. [Google Scholar] [CrossRef] [PubMed]
- Oelschlaeger, P.; Lange, S.; Schmitt, J.; Siemann, M.; Reuss, M.; Schmid, R.D. Identification of factors impeding the production of a single-chain antibody fragment in Escherichia coli by comparing in vivo and in vitro expression. Appl. Microbiol. Biotechnol. 2003, 61, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Markham, N.R.; Zuker, M. DINAMelt web server for nucleic acid melting prediction. Nucleic Acids Res. 2005, 33, W577–W581. [Google Scholar] [CrossRef]
- Stent, G.S. The Operon: On Its Third Anniversary. Modulation of Transfer Rna Species Can Provide a Workable Model of an Operator-Less Operon. Science 1964, 144, 816–820. [Google Scholar] [CrossRef]
- Byrne, R.; Levin, J.G.; Bladen, H.A.; Nirenberg, M.W. The in Vitro Formation of a DNA-Ribosome Complex. Proc. Natl. Acad. Sci. USA 1964, 52, 140–148. [Google Scholar] [CrossRef]
- Mangiarotti, G.; Schlessinger, D. Polyribosome Metabolism in Escherichia coli II. Formation and Lifetime of Messenger RNA Molecules, Ribosomal Subunit Couples and Polyribosomes. J. Mol. Biol. 1967, 29, 395–418. [Google Scholar] [CrossRef]
- Miller, O.L., Jr.; Hamkalo, B.A.; Thomas, C.A., Jr. Visualization of bacterial genes in action. Science 1970, 169, 392–395. [Google Scholar] [CrossRef]
- Yanofsky, C. Attenuation in the control of expression of bacterial operons. Nature 1981, 289, 751–758. [Google Scholar] [CrossRef]
- Palzkill, T. Structural and Mechanistic Basis for Extended-Spectrum Drug-Resistance Mutations in Altering the Specificity of TEM, CTX-M, and KPC β-lactamases. Front. Mol. Biosci. 2018, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Stemmer, W.P. Rapid evolution of a protein in vitro by DNA shuffling. Nature 1994, 370, 389–391. [Google Scholar] [CrossRef]
- Zaccolo, M.; Gherardi, E. The effect of high-frequency random mutagenesis on in vitro protein evolution: A study on TEM-1 β-lactamase. J. Mol. Biol. 1999, 285, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Orencia, M.C.; Yoon, J.S.; Ness, J.E.; Stemmer, W.P.; Stevens, R.C. Predicting the emergence of antibiotic resistance by directed evolution and structural analysis. Nat. Struct. Biol. 2001, 8, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Schenk, M.F.; Szendro, I.G.; Krug, J.; de Visser, J.A. Quantifying the adaptive potential of an antibiotic resistance enzyme. PLoS Genet. 2012, 8, e1002783. [Google Scholar] [CrossRef]
- Zalucki, Y.M.; Gittins, K.L.; Jennings, M.P. Secretory signal sequence non-optimal codons are required for expression and export of β-lactamase. Biochem. Biophys. Res. Commun. 2008, 366, 135–141. [Google Scholar] [CrossRef]
- Palmer, I.; Wingfield, P.T. Preparation and extraction of insoluble (inclusion-body) proteins from Escherichia coli. Curr. Protoc. Protein Sci. 2004, 70, 6.3.1–6.3.18. [Google Scholar] [CrossRef]
- Tuller, T.; Carmi, A.; Vestsigian, K.; Navon, S.; Dorfan, Y.; Zaborske, J.; Pan, T.; Dahan, O.; Furman, I.; Pilpel, Y. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 2010, 141, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Choi, J.; Cottrell, K.A.; Lavagnino, Z.; Thomas, E.N.; Pavlovic-Djuranovic, S.; Szczesny, P.; Piston, D.W.; Zaher, H.S.; Puglisi, J.D.; et al. A short translational ramp determines the efficiency of protein synthesis. Nat. Commun. 2019, 10, 5774. [Google Scholar] [CrossRef] [PubMed]
- Hia, F.; Takeuchi, O. The effects of codon bias and optimality on mRNA and protein regulation. Cell. Mol. Life Sci. 2021, 78, 1909–1928. [Google Scholar] [CrossRef] [PubMed]
- Firnberg, E.; Labonte, J.W.; Gray, J.J.; Ostermeier, M. A comprehensive, high-resolution map of a gene’s fitness landscape. Mol. Biol. Evol. 2014, 31, 1581–1592. [Google Scholar] [CrossRef] [PubMed]
- Zwart, M.P.; Schenk, M.F.; Hwang, S.; Koopmanschap, B.; de Lange, N.; van de Pol, L.; Nga, T.T.T.; Szendro, I.G.; Krug, J.; de Visser, J. Unraveling the causes of adaptive benefits of synonymous mutations in TEM-1 β-lactamase. Heredity 2018, 121, 406–421. [Google Scholar] [CrossRef] [PubMed]
- Faheem, M.; Zhang, C.J.; Morris, M.N.; Pleiss, J.; Oelschlaeger, P. Role of Synonymous Mutations in the Evolution of TEM β-Lactamase Genes. Antimicrob. Agents Chemother. 2021, 65, e00018-21. [Google Scholar] [CrossRef]
- Boyd, D.A.; Tyler, S.; Christianson, S.; McGeer, A.; Muller, M.P.; Willey, B.M.; Bryce, E.; Gardam, M.; Nordmann, P.; Mulvey, M.R. Complete nucleotide sequence of a 92-kilobase plasmid harboring the CTX-M-15 extended-spectrum β-lactamase involved in an outbreak in long-term-care facilities in Toronto, Canada. Antimicrob. Agents Chemother. 2004, 48, 3758–3764. [Google Scholar] [CrossRef]
- Shen, X.; Song, S.; Li, C.; Zhang, J. Synonymous mutations in representative yeast genes are mostly strongly non-neutral. Nature 2022, 606, 725–731. [Google Scholar] [CrossRef]
- Dhindsa, R.S.; Wang, Q.; Vitsios, D.; Burren, O.S.; Hu, F.; DiCarlo, J.E.; Kruglyak, L.; MacArthur, D.G.; Hurles, M.E.; Petrovski, S. A minimal role for synonymous variation in human disease. Am. J. Hum. Genet. 2022, 109, 2105–2109. [Google Scholar] [CrossRef]
- Kruglyak, L.; Beyer, A.; Bloom, J.S.; Grossbach, J.; Lieberman, T.D.; Mancuso, C.P.; Rich, M.S.; Sherlock, G.; Kaplan, C.D. Insufficient evidence for non-neutrality of synonymous mutations. Nature 2023, 616, E8–E9. [Google Scholar] [CrossRef]
- Rahaman, S.; Faravelli, S.; Voegeli, S.; Becskei, A. Polysome propensity and tunable thresholds in coding sequence length enable differential mRNA stability. Sci. Adv. 2023, 9, eadh9545. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Averof, M.; Lloyd, A.T.; Matassi, G.; Peden, J.F. DNA sequence evolution: The sounds of silence. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1995, 349, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Akashi, H. Gene expression and molecular evolution. Curr. Opin. Genet. Dev. 2001, 11, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Akashi, H.; Kliman, R.M.; Eyre-Walker, A. Mutation pressure, natural selection, and the evolution of base composition in Drosophila. Genetica 1998, 102–103, 49–60. [Google Scholar] [CrossRef]
- Iida, K.; Akashi, H. A test of translational selection at ‘silent’ sites in the human genome: Base composition comparisons in alternatively spliced genes. Gene 2000, 261, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Sauna, Z.E.; Kimchi-Sarfaty, C. Understanding the contribution of synonymous mutations to human disease. Nat. Rev. Genet. 2011, 12, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Wainwright, M.S.; Comeron, J.M.; Saitou, N.; Sanders, A.R.; Gelernter, J.; Gejman, P.V. Synonymous mutations in the human dopamine receptor D2 (DRD2) affect mRNA stability and synthesis of the receptor. Hum. Mol. Genet. 2003, 12, 205–216. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Bartoszewski, R.A.; Jablonsky, M.; Bartoszewska, S.; Stevenson, L.; Dai, Q.; Kappes, J.; Collawn, J.F.; Bebok, Z. A synonymous single nucleotide polymorphism in ΔF508 CFTR alters the secondary structure of the mRNA and the expression of the mutant protein. J. Biol. Chem. 2010, 285, 28741–28748. [Google Scholar] [CrossRef]
- Sato, S.; Ward, C.L.; Kopito, R.R. Cotranslational ubiquitination of cystic fibrosis transmembrane conductance regulator in vitro. J. Biol. Chem. 1998, 273, 7189–7192. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Kroliczewski, J.; Piotrowski, A.; Jasiecka, A.J.; Bartoszewska, S.; Vecchio-Pagan, B.; Fu, L.; Sobolewska, A.; Matalon, S.; Cutting, G.R.; et al. Codon bias and the folding dynamics of the cystic fibrosis transmembrane conductance regulator. Cell Mol. Biol. Lett. 2016, 21, 23. [Google Scholar] [CrossRef] [PubMed]
- Lazrak, A.; Fu, L.; Bali, V.; Bartoszewski, R.; Rab, A.; Havasi, V.; Keiles, S.; Kappes, J.; Kumar, R.; Lefkowitz, E.; et al. The silent codon change I507-ATC->ATT contributes to the severity of the ΔF508 CFTR channel dysfunction. FASEB J. 2013, 27, 4630–4645. [Google Scholar] [CrossRef] [PubMed]
- Bali, V.; Lazrak, A.; Guroji, P.; Fu, L.; Matalon, S.; Bebok, Z. A synonymous codon change alters the drug sensitivity of ΔF508 cystic fibrosis transmembrane conductance regulator. FASEB J. 2016, 30, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Brest, P.; Lapaquette, P.; Souidi, M.; Lebrigand, K.; Cesaro, A.; Vouret-Craviari, V.; Mari, B.; Barbry, P.; Mosnier, J.F.; Hebuterne, X.; et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat. Genet. 2011, 43, 242–245. [Google Scholar] [CrossRef]
- Gartner, J.J.; Parker, S.C.; Prickett, T.D.; Dutton-Regester, K.; Stitzel, M.L.; Lin, J.C.; Davis, S.; Simhadri, V.L.; Jha, S.; Katagiri, N.; et al. Whole-genome sequencing identifies a recurrent functional synonymous mutation in melanoma. Proc. Natl. Acad. Sci. USA 2013, 110, 13481–13486. [Google Scholar] [CrossRef]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Name Used in This Review | Outcome | Alternative Names |
|---|---|---|
| Synonymous mutation (SM) | No change in amino acid (AA) or Stop codon | Silent |
| Nonsynonymous mutation (NM) | Change from one AA to another AA | Missense |
| Change from an AA to a Stop codon | Nonsense, Stop | |
| Change from a Stop codon to an AA | Nonstop |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oelschlaeger, P. Molecular Mechanisms and the Significance of Synonymous Mutations. Biomolecules 2024, 14, 132. https://doi.org/10.3390/biom14010132
Oelschlaeger P. Molecular Mechanisms and the Significance of Synonymous Mutations. Biomolecules. 2024; 14(1):132. https://doi.org/10.3390/biom14010132
Chicago/Turabian StyleOelschlaeger, Peter. 2024. "Molecular Mechanisms and the Significance of Synonymous Mutations" Biomolecules 14, no. 1: 132. https://doi.org/10.3390/biom14010132
APA StyleOelschlaeger, P. (2024). Molecular Mechanisms and the Significance of Synonymous Mutations. Biomolecules, 14(1), 132. https://doi.org/10.3390/biom14010132