2.1. Synthetic Procedures
General procedure A-reaction of substituted diphenylmethanol with thiophen-2-ylmethanethiol
An amount of 3.0–10.0 mmol of substituted diphenylmethanol and 3.0–10.0 mmol of thiophen-2-ylmethanethiol (molar ratio 1:1) were dissolved in glacial acetic acid. Then, 1.1 equivalents of 48% BF3·Et2O were added, and the reaction mixture was left under stirring overnight at room temperature. The reaction mixture was then poured over ice, a small amount of water was added, and the acetic acid was neutralized with the addition of solid NaHCO3. The product was then extracted (3×) with 50 mL of EtOAc. Organic layers were collected, combined, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure.
General procedure B-oxydation to sulfoxide with 30% H2O2
In total, 0.96–5.1 mmol of unoxidized products were dissolved in 5–12 mL of glacial acetic acid and 0.15–0.77 mL (0.15 mL per 1 mmol of unoxidized material) of 30% H2O2 and left to stir overnight (12–14 h) at room tem sperature. The reaction mixture was then neutralized with a cold 5% sodium bicarbonate solution. The reaction product was extracted (3 × 50 mL) with EtOAc and dried over anhydrous Na2SO4. The organic solution was concentrated under reduced pressure, and the residue was purified by flash column chromatography on silica gel (2.5 or 5% MeOH in DCM). The product was dried in a high vacuum.
- 1.
Synthesis of 2-((((2-fluorophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene (5a–8a)
Following general procedure A, 1.0 g (4.9 mmol) of (2-fluorophenyl)(phenyl)methanol, 0.64 g (4.9 mmol) of thiophen-2-ylmethanethiol, and (0.65 mL, 5.4 mmol) of 48% BF3·Et2O were reacted, affording 1.4 g of 3a as a yellow oily product (yield 91%).
Following general procedure B, 1.4 g (4.5 mmol) of 3a dissolved in 12 mL of glacial acetic acid was reacted with 30% H2O2 (0.54 mL) to give 1.17 g of 4a as a yellow solid (yield 79%).
Racemic 4a was further separated into two individual pairs of enantiomers by means of an MPLC system equipped with a glass column packed with silica gel. Toluol/EtOAc = 80/20 was used as the mobile phase, and the separation was carried out with a flow rate of 5 mL/min. Individual pairs of enantiomers were further separated into the individual enantiomers 5a–8a by means of semipreparative chiral HPLC. A semipreparative HPLC instrument was equipped with a CHIRALPAK IA column (10 mm Φ × 25 cm L) (Daicel Inc., Tokyo, Japan), and Hexane/EtOAc = 50/50 was used as the mobile phase in the cases of 5a and 6a and 100% EtOAc in the cases of 7a and 8a.
5a. (R,R)-2-((((2-fluorophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 331.0622 [M+H]
+ (calcd for C
18H
16FOS
2+, 331.0621, Δ = −0.3 ppm) (
Figure S1). HPLC R
t 24.25 min, purity > 99% (
Figure S1a). Chiral HPLC R
t 11.93 min, 95.4% ee (
Figure S1b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.67 (td, 1H, CH-6, aromatic), 7.47 (m, 2H, CH-2,6, phenyl), 7.39 (m, ov, 2H, CH-3,5, phenyl), 7.37 (ov, 1H, CH-4, phenyl), 7.34 (ov, 1H, CH-4, aromatic), 7.32 (dd, ov, 1H, CH-5, thiophene), 7.22 (td, 1H, CH-5, aromatic), 7.11 (m, 1H, CH-3, aromatic), 7.03 (dd, 1H, CH-4, thiophene), 6.96 (dd, 1H, CH-3, thiophene), 5.10 (s, 1H, CH), 4.12/3.97 (AB, 2H, CH
2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 159.91 (C
q-2, aromatic), 133.15 (C
q-1, phenyl), 130.3 (C
q, thiophene-2), 129.99 (CH-4, aromatic), 129.86 (CH-2,6, phenyl), 129.64 (CH-6, aromatic), 128.91 (CH-3, thiophene), 128.73 (CH-3,5, phenyl), 128.56 (CH-4, phenyl), 127.39 (CH-4, thiophene), 126.89 (CH-5, thiophene), 124.79 (CH-5, aromatic), 123.78 (C
q-1, aromatic), 116.1 (CH-3, aromatic), 61.39 (CH), 50.84 (CH
2) (
Figure S1c).
6a. (S,S)-2-((((2-fluorophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 331.0621 [M+H]
+ (calcd for C
18H
16FOS
2+, 331.0621, Δ = 0.1 ppm) (
Figure S2). HPLC R
t 24.23 min, purity > 99% (
Figure S2a). Chiral HPLC R
t 12.76 min, 95.4% ee (
Figure S2b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.67 (CH-6, aromatic), 7.47 (CH-2,6, phenyl), 7.39 (CH-3,5, phenyl), 7.37 (CH-4, phenyl), 7.34 (CH-4, aromatic), 7.32 (s, 1H, CH-5, thiophene), 7.22 (CH-5, aromatic), 7.11 (CH-3, aromatic), 7.03 (s, 1H, CH-4, thiophene), 6.96 (s, 1H, CH-3, thiophene), 5.10 (s, 1H, CH), 4.12/3.97 (AB, 2H, CH
2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 159.91 (C
q-2, aromatic), 133.15 (C
q-1, phenyl), 130.30 (C
q, thiophene-2), 129.99 (CH-4, aromatic), 129.87 (CH-2,6, phenyl), 129.64 (CH-6, aromatic), 128.92 (CH-3, thiophene), 128.74 (CH-3,5, phenyl), 128.56 (CH-4, phenyl), 127.39 (CH-4, thiophene), 126.89 (CH-5, thiophene), 124.79 (CH-5, aromatic), 123.78 (C
q-1, aromatic), 116.10 (CH-3, aromatic), 61.39 (CH), 50.84 (CH
2) (
Figure S2c).
7a. (R,S)-2-((((2-fluorophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 331.0623 [M+H]
+ (calcd for C
18H
16FOS
2+, 331.0621, Δ = −0.6 ppm) (
Figure S3). HPLC R
t 24.48 min, purity > 99% (
Figure S3a). Chiral HPLC R
t 6.96 min, >99% ee (
Figure S3b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.73 (CH-6, aromatic), 7.47 (CH-2,6, phenyl), 7.39 (CH-3,5, phenyl), 7.34 (CH-4, phenyl), 7.33 (CH-4, aromatic), 7.32 (s, 1H, CH-5, thiophene), 7.21 (CH-5, aromatic), 7.12 (CH-3, aromatic), 7.03 (s, 1H, CH-4, thiophene), 6.99 (s, 1H, CH-3, thiophene), 5.28 (s, 1H, CH), 4.16/3.91 (AB, 2H, CH
2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 160.84 (C
q-2, aromatic), 135.47 (C
q-1, phenyl), 130.49 (CH-6, aromatic), 130.40 (C
q, thiophene-2), 129.90 (CH-4, aromatic), 129.24 (CH-3,5, phenyl), 129.06 (CH-3, thiophene), 128.81 (CH-2,6, phenyl), 128.46 (CH-4, phenyl), 127.4 (CH-4, thiophene), 126.91 (CH-5, thiophene), 124.55 (CH-5, aromatic), 121.93 (C
q-1, aromatic), 115.68 (CH-3, aromatic), 61.31 (CH), 50.82 (CH
2) (
Figure S3c).
8a. (S,R)-2-((((2-fluorophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 331.0622 [M+H]
+ (calcd for C
18H
16FOS
2+, 331.0621, Δ = −0.2 ppm) (
Figure S4). HPLC R
t 24.48 min, purity 98.6% (
Figure S4a). Chiral HPLC R
t 10.94 min, >99% ee (
Figure S4b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.73 (CH-6, aromatic), 7.47 (CH-2,6, phenyl), 7.39 (CH-3,5, phenyl), 7.34 (CH-4, phenyl), 7.33 (CH-4, aromatic), 7.32 (s, 1H, CH-5, thiophene), 7.21 (CH-5, aromatic), 7.12 (CH-3, aromatic), 7.03 (s, 1H, CH-4, thiophene), 6.99 (s, 1H, CH-3, thiophene), 5.28 (s, 1H, CH), 4.16/3.91 (AB, 2H, CH
2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 160.84 (C
q-2, aromatic), 135.47 (C
q-1, phenyl), 130.48 (CH-6, aromatic), 130.39 (C
q, thiophene-2) 129.89 (CH-4, aromatic), 129.23 (CH-3,5, phenyl), 129.06 (CH-3, thiophene), 128.80 (CH-2,6, phenyl), 128.45 (CH-4, phenyl), 127.40 (CH-4, thiophene), 126.91 (CH-5, thiophene), 124.55 (CH-5, aromatic), 121.93 (C
q-1, aromatic), 115.67 (CH-3, aromatic), 61.30 (CH), 50.82 (CH
2) (
Figure S4c).
- 2.
Synthesis of 2-((((3-fluorophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene (5b–8b)
Following general procedure A, 1.0 g (4.9 mmol) of (3-fluorophenyl)(phenyl)methanol, 0.64 g (4.9 mmol) of thiophen-2-ylmethanethiol, and (0.65 mL, 5.4 mmol) of 48% BF3·Et2O were reacted, affording 0.8 g of 3b as a yellow oily product (yield 53%).
Following general procedure B, 0.8 g (5.2 mmol) of 3b dissolved in 12 mL of glacial acetic acid was reacted with 30% H2O2 (0.54 mL) to give 0.7 g of 4b as a yellow solid (yield 84%).
Racemic 4b was further separated into two individual pairs of enantiomers by means of an MPLC system equipped with a glass column packed with silica gel. Toluol/EtOAc = 90/10 was used as the mobile phase, and the separation was carried out with a flow rate of 5 mL/min. Individual pairs of enantiomers were further separated into the individual enantiomers 5b–8b by means of semipreparative chiral HPLC. A semipreparative HPLC instrument was equipped with a CHIRALPAK IA column (10 mm Φ × 25 cm L) (Daicel Inc., Osaka, Japan), and EtOAc was used as the mobile phase.
5b. HRESIMS
m/
z 331.0613 [M+H]
+ (calcd for C
18H
16FOS
2+, 331.0621, Δ = 2.6 ppm) (
Figure S5). HPLC R
t 24.52 min, purity 96.0% (
Figure S5a). Chiral HPLC R
t 6.24 min, >99% ee (but contains 2.4% 7b) (
Figure S5b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.44 (C
H-3,5, phenyl), 7.40 (C
H-2,6, phenyl), 7.40 (C
H-4, phenyl), 7.34 (C
H-5, thiophene), 7.33 (C
H, aromatic-5), 7.20 (C
H, aromatic-6), 7.15 (C
H, aromatic-2), 7.06 (C
H-4, thiophene), 7.03 (C
H, aromatic-4), 6.96 (C
H-3, thiophene), 4.73 (s, 1H, C
H), 4.16/3.89 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 162.71 (
C-3, aromatic), 136.70 (
C-1, aromatic), 134.91 (
C-1, phenyl), 130.08 (
C-5, aromatic), 129.85 (
C-2, thiophene), 129.44 (
CH-3,5, phenyl), 129.16 (
C-3, thiophene), 128.77 (
CH-2,6, phenyl), 128.71 (
CH-4, phenyl), 127.33 (
C-4, thiophene), 129.96 (
C-5, thiophene), 125.45 (
C-6, aromatic), 116.56 (
C-2, aromatic), 115.39 (
C-4, aromatic), 68.71 (
CH), 50.09 (
CH
2) (
Figure S5c).
6b. HRESIMS
m/
z 331.0616 [M+H]
+ (calcd for C
18H
16FOS
2+, 331.0621, Δ = 1.6 ppm) (
Figure S6). HPLC R
t 24.48 min, purity > 99% (
Figure S6a). Chiral HPLC R
t 9.21 min, >99% ee (
Figure S6b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.44 (C
H-3,5, phenyl), 7.40 (C
H-2,6, phenyl), 7.40 (C
H-4, phenyl), 7.34 (C
H-5, thiophene), 7.33 (C
H, aromatic-5), 7.20 (C
H, aromatic-6), 7.15 (C
H, aromatic-2), 7.06 (C
H-4, thiophene), 7.03 (C
H, aromatic-4), 6.96 (C
H-3, thiophene), 4.73 (s, 1H, C
H), 4.11/3.89 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 162.70 (
C-3, aromatic), 136.70 (
C-1, aromatic), 134.91 (
C-1, phenyl), 130.08 (
C-5, aromatic), 129.85 (
C-2, thiophene), 129.44 (
CH-3,5, phenyl), 129.16 (
C-3, thiophene), 128.77 (
CH-2,6, phenyl), 128.70 (
CH-4, phenyl), 127.32 (
C-4, thiophene), 129.95 (
C-5, thiophene), 125.45 (
C-6, aromatic), 116.56 (
C-2, aromatic), 115.39 (
C-4, aromatic), 68.71 (
CH), 50.09 (
CH
2) (
Figure S6c).
7b. HRESIMS
m/
z 331.0614 [M+H]
+ (calcd for C
18H
16FOS
2+, 331.0621, Δ = 2.3 ppm) (
Figure S7). HPLC R
t 24.27 min, purity 98.2% (
Figure S7a). Chiral HPLC R
t 6.72 min, >99% ee (but contains 2.9% 5b) (
Figure S7b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.43 (C
H-2,6, phenyl), 7.40 (C
H-3,5, phenyl), 7.39 (C
H-4, phenyl), 7.39 (C
H, aromatic-5), 7.34 (C
H-5, thiophene), 7.23 (C
H, aromatic-6), 7.14 (C
H, aromatic-2), 7.05 (C
H-4, thiophene), 7.05 (C
H, aromatic-4), 6.98 (C
H-3, thiophene), 4.72 (s, 1H, C
H), 4.09/3.90 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 162.97 (
C-3, aromatic), 138.12 (
C-1, aromatic), 133.33 (
C-1, phenyl), 130.79 (
C-5, aromatic), 130.09 (
C-2, thiophene), 129.72 (
CH-2,6, phenyl), 129.02 (
C-3, thiophene), 128.81 (
CH-3,5, phenyl), 128.67 (
CH-4, phenyl), 127.42 (
C-4, thiophene), 126.89 (
C-5, thiophene), 124.48 (
C-6, aromatic), 115.95 (
C-2, aromatic), 115.42 (
C-4, aromatic), 68.19 (
CH), 50.28 (
CH
2) (
Figure S7c).
8b. HRESIMS
m/
z 331.0615 [M+H]
+ (calcd for C
18H
16FOS
2+, 331.0621, Δ = 1.9 ppm) (
Figure S8). HPLC R
t 24.23 min, purity > 99% (
Figure S8a). Chiral HPLC R
t 10.60 min, >99% ee (
Figure S8b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.43 (C
H-2,6, phenyl), 7.40 (C
H-3,5, phenyl), 7.39 (C
H-4, phenyl), 7.39 (C
H, aromatic-5), 7.34 (C
H-5, thiophene), 7.23 (C
H, aromatic-6), 7.14 (C
H, aromatic-2), 7.05 (C
H-4, thiophene), 7.05 (C
H, aromatic-4), 6.98 (C
H-3, thiophene), 4.72 (s, 1H, C
H), 4.09/3.90 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 162.97 (
C-3, aromatic), 138.12 (
C-1, aromatic), 133.33 (
C-1, phenyl), 130.79 (
C-5, aromatic), 130.08 (
C-2, thiophene), 129.71 (
CH-2,6, phenyl), 129.02 (
C-3, thiophene), 128.81 (
CH-3,5, phenyl), 128.67 (
CH-4, phenyl), 127.42 (
C-4, thiophene), 126.89 (
C-5, thiophene), 124.48 (
C-6, aromatic), 115.94 (
C-2, aromatic), 115.41 (
C-4, aromatic), 68.18 (
CH), 50.28 (
CH
2) (
Figure S8c).
- 3.
Synthesis of 2-((((4-fluorophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene (5c–8c)
Following general procedure A, 0.6 g (3.0 mmol) of (4-fluorophenyl)(phenyl)methanol, 0.4 g (3.0 mmol) of thiophen-2-ylmethanethiol, and (0.41 mL, 3.3 mmol) of 48% BF3·Et2O were reacted, affording 0.8 g of 3c as a yellow oily product (yield 92%).
Following general procedure B, 0.8 g (2.7 mmol) of 3c dissolved in 10 mL of glacial acetic acid was reacted with 30% H2O2 (0.31 mL) to give 0.4 g of 4c as orange oil (yield 45%).
Racemic 4c was further separated into two individual pairs of enantiomers by means of an MPLC system equipped with a glass column packed with silica gel. Toluol/EtOAc = 80/20 was used as the mobile phase, and the separation was carried out with a flow rate of 5 mL/min. Individual pairs of enantiomers were further separated into the individual enantiomers 5c–8c by means of semipreparative chiral HPLC. A semipreparative HPLC instrument was equipped with a CHIRALPAK IA column (10 mm Φ × 25 cm L) (Daicel Inc.), and EtOAc was used as the mobile phase.
5c. HRESIMS
m/
z 353.0443 [M+Na]
+ (calcd for C
18H
15FOS
2Na
+, 353.0441, Δ = −0.7 ppm) (
Figure S9). HPLC R
t 23.03 min, purity 97.2% (
Figure S9a). Chiral HPLC R
t 6.52 min, >99% ee (but contains 1.1% 7c) (
Figure S9b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.44 (C
H-3,5, phenyl), 7.39 (C
H-2,6, phenyl), 7.39 (C
H-4, phenyl), 7.39 (C
H-2,6, aromatic), 7.34 (C
H-5, thiophene), 7.06 (C
H-3,5, aromatic), 7.05 (C
H-4, thiophene), 6.96 (C
H-3, thiophene), 4.73 (s, 1H, C
H), 4.09/3.88 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 162.82 (
C-4, aromatic), 135.29 (
C-1, phenyl), 131.38 (
CH-2,6, aromatic), 129.97 (
C-2, thiophene), 129.92 (
C-1, aromatic), 129.40 (
CH-3,5, phenyl), 129.08 (
C-3, thiophene), 128.77 (
CH-2,6, phenyl), 128.58 (
C-4, phenyl), 127.32 (
C-4, thiophene), 126.89 (
C-5, thiophene), 115.62 (
CH-3,5, aromatic), 68.20 (
CH), 50.01 (
CH
2) (
Figure S9c).
6c. HRESIMS
m/
z 353.0442 [M+Na]
+ (calcd for C
18H
15FOS
2Na
+, 353.0441, Δ = −0.5 ppm) (
Figure S10). HPLC R
t 23.07 min, purity 98.3% (
Figure S10a). Chiral HPLC R
t 10.18 min, >99% ee (
Figure S10b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.44 (C
H-3,5, phenyl), 7.39 (C
H-2,6, phenyl), 7.39 (C
H-4, phenyl), 7.39 (C
H-2,6, aromatic), 7.34 (C
H-5, thiophene), 7.06 (C
H-3,5, aromatic), 7.05 (C
H-4, thiophene), 6.96 (C
H-3, thiophene), 4.73 (s, 1H, C
H), 4.09/3.88 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 162.82 (
C-4, aromatic), 135.29 (
C-1, phenyl), 131.38 (
CH-2,6, aromatic), 129.97 (
C-2, thiophene), 129.92 (
C-1, aromatic), 129.40 (
CH-3,5, phenyl), 129.08 (
C-3, thiophene), 128.77 (
CH-2,6, phenyl), 128.58 (
C-4, phenyl), 127.32 (
C-4, thiophene), 126.89 (
C-5, thiophene), 115.62 (
CH-3,5, aromatic), 68.20 (
CH), 50.01 (
CH
2) (
Figure S10c).
7c. HRESIMS
m/
z 353.0443 [M+Na]
+ (calcd for C
18H
15FOS
2Na
+, 353.0441, Δ = −0.6 ppm) (
Figure S11). HPLC R
t 22.93 min, purity > 99% (
Figure S11a). Chiral HPLC R
t 7.50 min, >99% ee (
Figure S11b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.42 (C
H-2,6, phenyl), 7.41 (C
H-2,6, aromatic), 7.39 (C
H-3,5, phenyl), 7.37 (C
H-4, phenyl), 7.34 (C
H-5, thiophene), 7.10 (C
H-3,5, aromatic), 7.05 (C
H-4, thiophene), 6.96 (C
H-3, thiophene), 4.73 (s, 1H, C
H), 4.08/3.89 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 162.53 (
C-4, aromatic), 135.79 (
C-1, phenyl), 131.52 (
C-1, aromatic), 130.55 (
CH-2,6, aromatic), 130.17 (
C-2, thiophene), 129.63 (
CH-2,6, phenyl), 128.98 (
C-3, thiophene), 128.76 (
CH-3,5, phenyl), 128.55 (
C-4, phenyl), 127.39 (
C-4, thiophene), 126.84 (
C-5, thiophene), 116.24 (
CH-3,5, aromatic), 67.87 (
CH), 50.20 (
CH
2) (
Figure S11c).
8c. HRESIMS
m/
z 353.0441 [M+Na]
+ (calcd for C
18H
15FOS
2Na
+, 353.0441, Δ = 0.1 ppm)% (
Figure S12). HPLC R
t 21.80 min, purity > 99% (
Figure S12a). Chiral HPLC R
t 11.61 min, >99% ee (
Figure S12b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.42 (C
H-2,6, phenyl), 7.41 (C
H-2,6, aromatic), 7.39 (C
H-3,5, phenyl), 7.37 (C
H-4, phenyl), 7.34 (C
H-5, thiophene), 7.10 (C
H-3,5, aromatic), 7.05 (C
H-4, thiophene), 6.96 (C
H-3, thiophene), 4.73 (s, 1H, C
H), 4.08/3.89 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 162.53 (
C-4, aromatic), 135.79 (
C-1, phenyl), 131.52 (
C-1, aromatic), 130.55 (
CH-2,6, aromatic), 130.17 (
C-2, thiophene), 129.63 (
CH-2,6, phenyl), 128.98 (
C-3, thiophene), 128.76 (
CH-3,5, phenyl), 128.55 (
C-4, phenyl), 127.39 (
C-4, thiophene), 126.84 (
C-5, thiophene), 116.24 (
CH-3,5, aromatic), 67.87 (
CH), 50.20 (
CH
2) (
Figure S12c).
- 4.
Synthesis of 2-((((2-chlorophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene (5d–8d)
Following general procedure A, 1.0 g (4.5 mmol) of (2-chlorophenyl)(phenyl)methanol, 0.59 g (4.5 mmol) of thiophen-2-ylmethanethiol, and (0.61 mL, 4.9 mmol) of 48% BF3·Et2O were reacted, affording 1.3 g of 3d as a yellow oily product (yield 88%).
Following general procedure B, 1.3 g (3.9 mmol) of 3d dissolved in 12 mL of glacial acetic acid was reacted with 30% H2O2 (0.5 mL) to give 1.0 g of 4d as a yellow solid (yield 78%).
Racemic 4d was further separated into the individual enantiomers 5d–8d by means of a semipreparative chiral HPLC method. A semipreparative HPLC instrument was equipped with a CHIRALPAK IA column (10 mm Φ × 25 cm L) (Daicel Inc.), and EtOAc was used as the mobile phase. During this process, 5d and 8d were fully separated, whereas the unresolved pair of 6d and 7d was further separated using Hexane/THF = 80/20 (v/v) as the mobile phase.
5d. (
R,
R)-
2-((((2-chlorophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 347.0323 [M+H]
+ (calcd for C
18H
16ClOS
2+, 347.0326, Δ = 0.7 ppm) (
Figure S13). HPLC R
t 25.60 min, purity 98.8% (
Figure S13a). Chiral HPLC R
t 4.98 min, 96.5% ee (
Figure S13b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.77 (C
H-6, aromatic), 7.48 (C
H-2,6, phenyl), 7.42 (C
H-3, aromatic), 7.39 (C
H-3,5, phenyl), 7.36 (C
H-4, phenyl), 7.35 (C
H-5, aromatic), 7.32 (s, 1H, C
H-5, thiophene), 7.28 (C
H-4, aromatic), 7.02 (s, 1H, C
H-4, thiophene), 6.95 (s, 1H, C
H-3, thiophene), 5.32 (s, 1H, C
H), 4.13/4.02 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 134.39 (
Cq-1, aromatic), 134.04 (
Cq-2, aromatic), 132.78 (
Cq-1, phenyl), 130.47 (
Cq, thiophene-2), 130.33 (
CH-3, aromatic), 130.09 (
CH-2,6, phenyl), 129.53 (
CH-6, aromatic), 129.39 (
CH-4, aromatic), 128.92 (
CH-3, thiophene), 128.68 (
CH-3,5, phenyl), 128.56 (
CH-4, phenyl), 127.48 (
CH-5, aromatic), 127.45 (
CH-4, thiophene), 126.88 (
CH-5, thiophene), 64.77 (
CH), 50.99 (
CH
2) (
Figure S13b).
6d. (
S,
S)-
2-((((2-chlorophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 347.0323 [M+H]
+ (calcd for C
18H
16ClOS
2+, 347.0326, Δ = 0.7 ppm) (
Figure S14). HPLC R
t 25.60 min, purity > 99% (
Figure S14a). Chiral HPLC R
t 21.95 min, >99% ee (
Figure S14b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.77 (C
H-6, aromatic), 7.48 (C
H-2,6, phenyl), 7.42 (C
H-3, aromatic), 7.39 (C
H-3,5, phenyl), 7.36 (C
H-4, phenyl), 7.35 (C
H-5, aromatic), 7.32 (s, 1H, C
H-5, thiophene), 7.28 (C
H-4, aromatic), 7.02 (s, 1H, C
H-4, thiophene), 6.95 (s, 1H, C
H-3, thiophene), 5.32 (s, 1H, C
H), 4.13/4.02 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 134.39 (
Cq-1, aromatic), 134.04 (
Cq-2, aromatic) 132.78 (
Cq-1, phenyl), 130.47 (
Cq, thiophene-2), 130.33 (
CH-3, aromatic), 130.09 (
CH-2,6, phenyl), 129.53 (
CH-6, aromatic), 129.39 (
CH-4, aromatic), 128.92 (
CH-3, thiophene), 128.68 (
CH-3,5, phenyl), 128.56 (
CH-4, phenyl), 127.48 (
CH-5, aromatic), 127.45 (
CH-4, thiophene), 126.88 (
CH-5, thiophene), 64.77 (
CH), 50.99 (
CH
2) (
Figure S14c).
7d. (
R,
S)-
2-((((2-chlorophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 347.0324 [M+H]
+ (calcd for C
18H
16ClOS
2+, 347.0326, Δ = 0.4 ppm) (
Figure S15). HPLC R
t 25.82 min, purity 95.9% (
Figure S15a). Chiral HPLC R
t 22.37 min, >99% ee (
Figure S15b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.95 (C
H-6, aromatic), 7.47 (C
H-2,6, phenyl), 7.43 (C
H-3, aromatic), 7.39 (C
H-5, aromatic), 7.39 (C
H-3,5, phenyl), 7.35 (C
H-4, phenyl), 7.32 (s, 1H, C
H-5, thiophene), 7.28 (C
H-4, aromatic), 7.03 (s, 1H, C
H-4, thiophene), 6.97 (s, 1H, C
H-3, thiophene), 5.53 (s, 1H, C
H), 4.19/3.92 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 135.21 (
Cq-1, phenyl), 134.91 (
Cq-2, aromatic), 132.96 (
Cq-1, aromatic), 130.43 (
Cq, thiophene-2), 130.15 (
CH-6, aromatic), 130.00 (
CH-3, aromatic), 129.28 (
CH-4, aromatic), 129.21 (
CH-3,5, phenyl), 129.13 (
CH-3, thiophene), 128.91 (
CH-2,6, phenyl), 128.47 (
CH-4, phenyl), 127.38 (
CH-4, thiophene), 127.32 (
CH-5, aromatic), 126.90 (
CH-5, thiophene), 64.75 (
CH), 50.54 (
CH
2) (
Figure S15c).
8d. (
S,
R)-
2-((((2-chlorophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 347.0325 [M+H]
+ (calcd for C
18H
16ClOS
2+, 347.0326, Δ = 0.3 ppm) (
Figure S16). HPLC R
t 25.82 min, purity 98.5% (
Figure S16a). Chiral HPLC R
t 8.32 min, >99% ee (
Figure S16b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.95 (C
H-6, aromatic), 7.47 (C
H-2,6, phenyl), 7.43 (C
H-3, aromatic), 7.39 (C
H-5, aromatic), 7.39 (C
H-3,5, phenyl), 7.35 (C
H-4, phenyl), 7.32 (s, 1H, C
H-5, thiophene), 7.28 (C
H-4, aromatic), 7.03 (s, 1H, C
H-4, thiophene), 6.97 (s, 1H, C
H-3, thiophene), 5.53 (s, 1H, C
H), 4.19/3.92 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 135.21 (
Cq-1, phenyl), 134.91 (
Cq-2, aromatic), 132.96 (
Cq-1, aromatic), 130.43 (
Cq, thiophene-2), 130.15 (
CH-6, aromatic), 130.00 (
CH-3, aromatic), 129.28 (
CH-4, aromatic), 129.21 (
CH-3,5, phenyl), 129.13 (
CH-3, thiophene), 128.91 (
CH-2,6, phenyl), 128.47 (
CH-4, phenyl), 127.38 (
CH-4, thiophene), 127.32 (
CH-5, aromatic), 126.90 (
CH-5, thiophene), 64.75 (
CH), 50.54 (
CH
2) (
Figure S16c).
- 5.
Synthesis of 2-((((3-chlorophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene (5e–8e)
Following general procedure A, 2.2 g (10.0 mmol) of (3-chlorophenyl)(phenyl)methanol, 1.3 g (10.0 mmol) of thiophen-2-ylmethanethiol, and (1.4 mL, 11 mmol) of 48% BF3·Et2O were reacted, affording 1.6 g of 3e as a yellow oily product (yield 50%).
Following general procedure B, 1.6 g (4.8 mmol) of 3e dissolved in 20 mL of glacial acetic acid was reacted with 30% H2O2 (0.5 mL) to give 0.5 g of 4e as a yellow-brown solid (yield 9%).
Racemic 4e was further separated into two individual pairs of enantiomers by means of an MPLC system equipped with a glass column packed with silica gel. Toluol/EtOAc = 80/20 was used as the mobile phase, and the separation was carried out with a flow rate of 5 mL/min. Individual pairs of enantiomers were further separated into the individual enantiomers 5e–8e by means of semipreparative chiral HPLC. A semipreparative HPLC instrument was equipped with a CHIRALPAK IA column (10 mm Φ × 25 cm L) (Daicel Inc.), and EtOAc was used as the mobile phase.
5e. (
S,
R)-
2-((((3-chlorophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 347.0322 [M+H]
+ (calcd for C
18H
16ClOS
2+, 347.0326, Δ = 1.1 ppm) (
Figure S17). HPLC R
t 24.65 min, purity 95.3% (
Figure S17a). Chiral HPLC R
t 6.68 min, >99% ee (but contains 4.7% 7e) (
Figure S17b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.44 (C
H-3,5, phenyl), 7.40 (C
H-2, aromatic), 7.40 (C
H-2,6, phenyl), 7.40 (C
H-4, phenyl), 7.34 (C
H-5, thiophene), 7.32 (C
H-6, aromatic), 7.31 (C
H-4, aromatic), 7.31 (C
H-5, aromatic), 7.06 (C
H-4, thiophene), 6.96 (C
H-3, thiophene), 4.71 (s, 1H, C
H), 4.10/3.89 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 136.33 (
C-1, aromatic), 134.53 (
C-3, aromatic), 134.53 (
C-4, phenyl), 134.53 (
C-2, thiophene), 129.85 (
C-5, aromatic), 129.55 (
C-2, aromatic), 129.48 (
CH-3,5, phenyl), 129.19 (
C-3, thiophene), 128.76 (
CH-2,6, phenyl), 128.73 (
C-4, phenyl), 128.59 (
C-4, aromatic), 127.89 (
C-6, aromatic), 127.34 (
C-4, thiophene), 126.98 (
C-4, thiophene), 68.72 (
CH), 50.08 (
CH
2) (
Figure S17c).
6e. (
R,
S)-
2-((((3-chlorophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 347.0322 [M+H]
+ (calcd for C
18H
16ClOS
2+, 347.0326, Δ = 1.0 ppm) (
Figure S18). HPLC R
t 26.03 min, purity 98.5% (
Figure S18a). Chiral HPLC R
t 9.04 min, >99% ee (
Figure S18b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.44 (C
H-3,5, phenyl), 7.40 (C
H-2, aromatic), 7.40 (C
H-2,6, phenyl), 7.40 (C
H-4, phenyl), 7.34 (C
H-5, thiophene), 7.32 (C
H-6, aromatic), 7.31 (C
H-4, aromatic), 7.31 (C
H-5, aromatic), 7.06 (C
H-4, thiophene), 6.96 (C
H-3, thiophene), 4.71 (s, 1H, C
H), 4.10/3.89 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 136.33 (
C-1, aromatic), 134.53 (
C-3, aromatic), 134.53 (
C-4, phenyl), 134.53 (
C-2, thiophene), 129.85 (
C-5, aromatic), 129.55 (
C-2, aromatic), 129.48 (
CH-3,5, phenyl), 129.19 (
C-3, thiophene), 128.76 (
CH-2,6, phenyl), 128.73 (
C-4, phenyl), 128.59 (
C-4, aromatic), 127.89 (
C-6, aromatic), 127.34 (
C-4, thiophene), 126.98 (
C-4, thiophene), 68.72 (
CH), 50.08 (
CH
2) (
Figure S18c).
7e. (
S,
S)-
2-((((3-chlorophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 347.0324 [M+H]
+ (calcd for C
18H
16ClOS
2+, 347.0326, Δ = 0.3 ppm) (
Figure S19). HPLC R
t 25.87 min, purity > 99% (
Figure S19a). Chiral HPLC R
t 6.95 min, >99% ee (but contains 5.0% 5e) (
Figure S19b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.43 (C
H-2,6, phenyl), 7.43 (C
H-2, aromatic), 7.40 (C
H-6, aromatic), 7.40 (C
H-3,5, phenyl), 7.39 (C
H-4, phenyl), 7.34 (C
H-5, thiophene), 7.34 (C
H-4, aromatic), 7.34 (C
H-5, aromatic), 7.06 (C
H-4, thiophene), 6.97 (C
H-3, thiophene), 4.70 (s, 1H, C
H), 4.10/3.90 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 137.77 (
C-1, aromatic), 135.06 (
C-3, aromatic), 133.23 (
C-1, phenyl), 130.43 (
C-5, aromatic), 130.05 (
C-2, thiophene), 129.71 (
C-2, aromatic), 129.71 (
CH-2,6, phenyl), 129.01 (
C-3, thiophene), 128.92 (
C-6, aromatic), 128.82 (
CH-3,5, phenyl), 128.68 (
C-4, phenyl), 127.43 (
C-4, phenyl), 126.90 (
C-5, phenyl), 68.08 (
CH), 50.31 (
CH
2) (
Figure S19c).
8e. (
R,
R)-
2-((((3-chlorophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 347.0321 [M+H]
+ (calcd for C
18H
16ClOS
2+, 347.0326, Δ = 1.3 ppm) (
Figure S20). HPLC R
t 25.87 min, purity > 99% (
Figure S20a). Chiral HPLC R
t 9.29 min, >99% ee (
Figure S20b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.43 (C
H-2,6, phenyl), 7.43 (C
H-2, aromatic), 7.40 (C
H-6, aromatic), 7.40 (C
H-3,5, phenyl), 7.39 (C
H-4, phenyl), 7.34 (C
H-5, thiophene), 7.34 (C
H-4, aromatic), 7.34 (C
H-5, aromatic), 7.06 (C
H-4, thiophene), 6.97 (C
H-3, thiophene), 4.70 (s, 1H, C
H), 4.10/3.90 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 137.77 (
C-1, aromatic), 135.06 (
C-3, aromatic), 133.23 (
C-1, phenyl), 130.43 (
C-5, aromatic), 130.05 (
C-2, thiophene), 129.71 (
C-2, aromatic), 129.71 (
CH-2,6, phenyl), 129.01 (
C-3, thiophene), 128.92 (
C-6, aromatic), 128.82 (
CH-3,5, phenyl), 128.68 (
C-4, phenyl), 127.43 (
C-4, phenyl), 126.90 (
C-5, phenyl), 68.08 (
CH), 50.31 (
CH
2) (
Figure S20c).
- 6.
Synthesis of 2-((((4-chlorophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene (5f–8f)
Following general procedure A, 1.68 g (7.7 mmol) of (4-chlorophenyl)(phenyl)methanol, 1.0 g (7.7 mmol) of thiophen-2-ylmethanethiol, and (1.0 mL, 8.5 mmol) of 48% BF3·Et2O were reacted, affording 1.7 g of 3f as a yellow oily product (yield 67%).
Following general procedure B, 1.7 g (5.1 mmol) of 3f dissolved in 15 mL of glacial acetic acid was reacted with 30% H2O2 (0.6 mL) to give 0.22 g of 4f as a pale-yellow solid (yield 11%).
Racemic 4f was further separated into two individual pairs of enantiomers by means of an MPLC system equipped with a glass column packed with silica gel. Toluol/EtOAc = 80/20 was used as the mobile phase, and the separation was carried out with a flow rate of 5 mL/min. Individual pairs of enantiomers were further separated into the individual enantiomers 5f–8f by means of semipreparative chiral HPLC. A semipreparative HPLC instrument was equipped with a CHIRALPAK IA column (10 mm Φ × 25 cm L) (Daicel Inc.), and EtOAc was used as the mobile phase.
5f. (
S,
R)-
2-((((4-chlorophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 369.0143 [M+Na]
+ (calcd for C
18H
15ClNaOS
2+, 369.0145, Δ = 0.6 ppm) (
Figure S21). HPLC Rt 24.67 min, purity > 99% (
Figure S21a). Chiral HPLC R
t 7.10 min, >99% ee (but contains 1.8% 7f) (
Figure S21b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.43 (C
H-2,6, phenyl), 7.39 (C
H-3,5, phenyl), 7.39 (C
H-4, phenyl), 7.35 (C
H-3,5, aromatic), 7.34 (C
H-2,6, aromatic), 7.34 (s, 1H, C
H-5, thiophene), 7.05 (s, 1H, C
H-4, thiophene), 6.95 (s, 1H, C
H-3, thiophene), 4.71 (s, 1H, C
H), 4.09/3.88 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 135.01 (
Cq-1, phenyl), 134.56 (
Cq-4, aromatic), 132.65 (
Cq-1, aromatic), 130.96 (
CH-2,6, aromatic), 130.08 (
Cq, thiophene-2), 129.64 (
CH-2,6, phenyl), 129.00 (
CH-3, thiophene), 128.81 (
CH-3,5, aromatic), 128.79 (
CH-3,5, phenyl), 128.61 (
CH-4, phenyl), 127.40 (
CH-4, thiophene), 126.87 (
CH-5, thiophene), 68.27 (
CH), 50.05 (
CH
2) (
Figure S21c).
6f. (
R,
S)-
2-((((4-chlorophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 369.0145 [M+Na]
+ (calcd for C
18H
15ClNaOS
2+, 369.0145, Δ = −0.1 ppm) (
Figure S22). HPLC R
t 24.67 min, purity 97.9% (
Figure S22a). Chiral HPLC R
t 12.94 min, >99% ee (
Figure S22b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.43 (C
H-2,6, phenyl), 7.39 (C
H-3,5, phenyl), 7.39 (C
H-4, phenyl), 7.35 (C
H-3,5, aromatic), 7.34 (C
H-2,6, aromatic), 7.34 (s, 1H, C
H-5, thiophene), 7.05 (s, 1H, C
H-4, thiophene), 6.95 (s, 1H, C
H-3, thiophene), 4.71 (s, 1H, C
H), 4.09/3.88 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 135.01 (
Cq-1, phenyl), 134.56 (
Cq-4, aromatic), 132.65 (
Cq-1, aromatic), 130.96 (
CH-2,6, aromatic), 130.08 (
Cq, thiophene-2), 129.64 (
CH-2,6, phenyl), 129.00 (
CH-3, thiophene), 128.81 (
CH-3,5, aromatic), 128.79 (
CH-3,5, phenyl), 128.61 (
CH-4, phenyl), 127.40 (
CH-4, thiophene), 126.87 (
CH-5, thiophene), 68.27 (
CH), 50.05 (
CH
2) (
Figure S22c).
7f. (
S,
S)-
2-((((4-chlorophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 369.0144 [M+Na]
+ (calcd for C
18H
15ClNaOS
2+, 369.0145, Δ = 0.3 ppm) (
Figure S23). HPLC R
t 24.70 min, purity > 99% (
Figure S23a). Chiral HPLC R
t 7.63 min, >99% ee (but contains 0.9% 5f) (
Figure S23b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.42 (C
H-2,6, aromatic), 7.42 (C
H-2,6, phenyl), 7.39 (C
H-3,5, phenyl), 7.38 (C
H-3,5, aromatic), 7.37 (C
H-4, phenyl), 7.34 (s, 1H, C
H-5, thiophene), 7.05 (s, 1H, C
H-4, thiophene), 6.96 (s, 1H, C
H-3, thiophene), 4.71 (s, 1H, C
H), 4.09/3.89 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 134.40 (
Cq-4, aromatic), 134.22 (
Cq-1, aromatic), 133.50 (
Cq-1, phenyl), 131.14 (
CH-2,6, aromatic), 129.88 (
Cq, thiophene-2), 129.64 (
CH-2,6, phenyl), 129.40 (
CH-3,5, aromatic), 129.11 (
CH-3, thiophene), 128.79 (
CH-3,5, phenyl), 128.64 (
CH-4, phenyl), 127.34 (
CH-4, thiophene), 126.92 (
CH-5, thiophene), 67.94 (
CH), 50.23 (
CH
2) (
Figure S23c).
8f. (
R,
R)-
2-((((4-chlorophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 369.0145 [M+Na]
+ (calcd for C
18H
15ClNaOS
2+, 369.0145, Δ = 0.1 ppm) (
Figure S24). HPLC R
t 24.72 min, purity > 99% (
Figure S24a). Chiral HPLC R
t 11.39 min, >99% ee (but contains 0.8% 6f) (
Figure S24b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.42 (C
H-2,6, aromatic), 7.42 (C
H-2,6, phenyl), 7.39 (C
H-3,5, phenyl), 7.38 (C
H-3,5, aromatic), 7.37 (C
H-4, phenyl), 7.34 (s, 1H, C
H-5, thiophene), 7.05 (s, 1H, C
H-4, thiophene), 6.96 (s, 1H, C
H-3, thiophene), 4.71 (s, 1H, C
H), 4.09/3.89 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 134.40 (
Cq-4, aromatic), 134.22 (
Cq-1, aromatic), 133.50 (
Cq-1, phenyl), 131.14 (
CH-2,6, aromatic), 129.88 (
Cq, thiophene-2), 129.64 (
CH-2,6, phenyl), 129.40 (
CH-3,5, aromatic), 129.11 (
CH-3, thiophene), 128.79 (
CH-3,5, phenyl), 128.64 (
CH-4, phenyl), 127.34 (
CH-4, thiophene), 126.92 (
CH-5, thiophene), 67.94 (
CH), 50.23 (
CH
2) (
Figure S24c).
- 7.
Synthesis of 2-((((2-bromophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene (5g–8g)
Following general procedure A, 1.0 g (3.8 mmol) of (2-bromophenyl)(phenyl)methanol, 0.49 g (3.8 mmol) of thiophen-2-ylmethanethiol, and (0.5 mL, 4.2 mmol) of 48% BF3·Et2O were reacted, affording 1.34 g of 3g as a white oily product (yield 94%).
Following general procedure B, 1.34 g (3.5 mmol) of 3g dissolved in 12 mL of glacial acetic acid was reacted with 30% H2O2 (0.37 mL) to give 0.6 g of 4g as a white solid (yield 43%).
Racemic 4g was further separated into two individual pairs of enantiomers by means of flash column chromatography on silica gel with Toluol/EtOAc = 80/20 as the mobile phase. Individual pairs of enantiomers were further separated into the individual enantiomers 5g–8g by means of semipreparative chiral HPLC. A semipreparative HPLC instrument was equipped with a CHIRALPAK IA column (10 mm Φ × 25 cm L) (Daicel Inc.), and Heptane/EtOAc = 70/30 was used as the mobile phase in the case of 5g/6g and 100% EtOAc in the case of 7g/8g.
5g. HRESIMS
m/
z 390.9820 [M+H]
+ (calcd for C
18H
16BrOS
2+, 390.9820, Δ = 0.1 ppm) (
Figure S25). HPLC R
t 26.00 min, purity > 99% (
Figure S25a). Chiral HPLC R
t 4.99 min, >99% ee (
Figure S25b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.77 (C
H-6, aromatic), 7.61 (C
H-3, aromatic), 7.49 (C
H-2,6, phenyl), 7.40 (C
H-5, aromatic), 7.40 (C
H-3,5, phenyl), 7.38 (C
H-4, phenyl), 7.32 (s, 1H, C
H-5, thiophene), 7.20 (C
H-4, aromatic), 7.02 (s, 1H, C
H-4, thiophene), 6.96 (s, 1H, C
H-3, thiophene), 5.33 (s, 1H, C
H), 4.14/4.04 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 136.02 (
Cq-1, aromatic), 133.69 (
CH-3, aromatic), 132.76 (
Cq-1, phenyl), 130.53 (
Cq, thiophene-2), 130.13 (
CH-2,6, phenyl), 129.71 (
CH-6, aromatic), 129.65 (
CH-4, aromatic), 128.94 (
CH-3, thiophene), 128.68 (
CH-3,5, phenyl), 128.57 (
CH-4, phenyl), 128.11 (
CH-5, aromatic), 127.48 (
CH-4, thiophene), 126.89 (
CH-5, thiophene), 124.90 (
Cq-2, aromatic) 67.28 (
CH), 51.04 (
CH
2) (
Figure S25c).
6g. HRESIMS
m/
z 390.9817 [M+H]
+ (calcd for C
18H
16BrOS
2+, 390.9820, Δ = 0.9 ppm) (
Figure S26). HPLC R
t 25.98 min, purity > 99% (
Figure S26a). Chiral HPLC R
t 25.78 min, 97.0% ee (
Figure S26b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.77 (C
H-6, aromatic), 7.61 (C
H-3, aromatic), 7.49 (C
H-2,6, phenyl), 7.39 (C
H-5, aromatic), 7.39 (C
H-3,5, phenyl), 7.37 (C
H-4, phenyl), 7.32 (s, 1H, C
H-5, thiophene), 7.20 (C
H-4, aromatic), 7.02 (s, 1H, C
H-4, thiophene), 6.96 (s, 1H, C
H-3, thiophene), 5.33 (s, 1H, C
H), 4.13/4.04 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 136.02 (
Cq-1, aromatic), 133.68 (
CH-3, aromatic), 132.75 (
Cq-1, phenyl), 130.53 (
Cq, thiophene-2), 130.13 (
CH-2,6, phenyl), 129.71 (
CH-6, aromatic), 129.65 (
CH-4, aromatic), 128.93 (
CH-3, thiophene), 128.67 (
CH-3,5, phenyl), 128.56 (
CH-4, phenyl), 128.10 (
CH-5, aromatic), 127.47 (
CH-4, thiophene), 126.87 (
CH-5, thiophene), 124.90 (
Cq-2, aromatic), 67.27 (
CH), 51.04 (
CH
2) (
Figure S26c).
7g. HRESIMS
m/
z 390.9819 [M+H]
+ (calcd for C
18H
16BrOS
2+, 390.9820, Δ = 0.3 ppm) (
Figure S27). HPLC R
t 26.20 min, purity 97.6% (
Figure S27a). Chiral HPLC R
t 27.22 min, 91.2% ee (
Figure S27b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.96 (C
H-6, aromatic), 7.61 (C
H-3, aromatic), 7.48 (C
H-2,6, phenyl), 7.42 (C
H-5, aromatic), 7.39 (C
H-3,5, phenyl), 7.34 (C
H-4, phenyl), 7.32 (s, 1H, C
H-5, thiophene), 7.20 (C
H-4, aromatic), 7.03 (s, 1H, C
H-4, thiophene), 6.97 (s, 1H, C
H-3, thiophene), 5.53 (s, 1H, C
H), 4.21/3.92 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 135.18 (
Cq-1, phenyl), 134.67 (
Cq-1, aromatic), 133.36 (
CH-3, aromatic), 130.40 (
Cq, thiophene-2), 130.27 (
CH-6, aromatic), 129.56 (
CH-4, aromatic), 129.20 (
CH-3,5, phenyl), 129.16 (
CH-3, thiophene), 128.91 (
CH-2,6, phenyl), 128.47 (
CH-4, phenyl), 127.96 (
CH-5, aromatic), 127.39 (
CH-4, thiophene), 126.91 (
CH-5, thiophene), 125.92 (
Cq-2, aromatic), 67.22 (
CH), 50.47 (
CH
2) (
Figure S27c).
8g. HRESIMS
m/
z 390.9819 [M+H]
+ (calcd for C
18H
16BrOS
2+, 390.9820, Δ = 0.3 ppm) (
Figure S28). HPLC R
t 26.20 min, purity 97.2% (
Figure S28a). Chiral HPLC R
t 8.51 min, >99% ee (
Figure S28b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.96 (C
H-6, aromatic), 7.61 (C
H-3, aromatic), 7.48 (C
H-2,6, phenyl), 7.42 (C
H-5, aromatic), 7.39 (C
H-3,5, phenyl), 7.34 (C
H-4, phenyl), 7.32 (s, 1H, C
H-5, thiophene), 7.19 (C
H-4, aromatic), 7.03 (s, 1H, C
H-4, thiophene), 6.97 (s, 1H, C
H-3, thiophene), 5.53 (s, 1H, C
H), 4.20/3.92 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 135.18 (
Cq-1, phenyl), 134.67 (
Cq-1, aromatic), 133.37 (
CH-3, aromatic), 130.40 (
Cq, thiophene-2), 130.27 (
CH-6, aromatic), 129.56 (
CH-4, aromatic), 129.21 (
CH-3,5, phenyl), 129.17 (
CH-3, thiophene), 128.92 (
CH-2,6, phenyl), 128.48 (
CH-4, phenyl), 127.96 (
CH-5, aromatic), 127.40 (
CH-4, thiophene), 126.91 (
CH-5, thiophene), 125.92 (
Cq-2, aromatic) 67.24 (
CH), 50.47 (
CH
2) (
Figure S28c).
- 8.
Synthesis of 2-((((3-bromophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene (5h–8h)
Following general procedure A, 1.0 g (3.8 mmol) of (3-bromophenyl)(phenyl)methanol, 0.49 g (3.8 mmol) of thiophen-2-ylmethanethiol, and (0.5 mL, 4.18 mmol) of 48% BF3·Et2O were reacted, affording 1.13 g of 3h as a yellow oily product (yield 79%).
Following general procedure B, 1.13 g (3.0 mmol) of 3h dissolved in 12 mL of glacial acetic acid was reacted with 30% H2O2 (0.3 mL) to give 0.71 g of 4h as a pale-yellow solid (yield 60%).
Racemic 4h was further separated into two individual pairs of enantiomers by means of an MPLC system equipped with a glass column packed with silica gel. Toluol/EtOAc = 80/20 was used as the mobile phase, and the separation was carried out with a flow rate of 5 mL/min. Individual pairs of enantiomers were further separated into the individual enantiomers 5h–8h by means of semipreparative chiral HPLC. A semipreparative HPLC instrument was equipped with a CHIRALPAK IA column (10 mm Φ × 25 cm L) (Daicel Inc.), and EtOAc was used as the mobile phase.
5h. (
S,
R)-
2-((((3-bromophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 390.9811 [M+H]
+ (calcd for C
18H
16BrOS
2+, 390.9820, Δ = 2.3 ppm) (
Figure S29). HPLC R
t 28.23 min, purity 98.4% (
Figure S29a). Chiral HPLC R
t 6.75 min, >99% ee (
Figure S29b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.54 (C
H-2, aromatic), 7.46 (C
H-4, aromatic), 7.44 (C
H-3,5, phenyl), 7.39 (C
H-6, aromatic), 7.39 (C
H-2,6, aromatic), 7.39 (C
H-4, phenyl), 7.34 (C
H-5, thiophene), 7.24 (C
H-5, aromatic), 7.06 (C
H-4, thiophene), 6.95 (C
H-3, thiophene), 4.69 (s, 1H, C
H), 4.10/3.89 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 136.60 (
C-1, aromatic), 134.79 (
C-1, phenyl), 132.40 (
C-2, aromatic), 131.50 (
C-4, aromatic), 130.12 (
C-5, aromatic), 129.78 (
C-2, thiophene), 129.48 (
CH-3,5, phenyl), 129.18 (
C-3, thiophene), 128.74 (
CH-2,6, phenyl), 128.74 (
C-4, phenyl), 128.33 (
C-6, aromatic), 126.98 (
C-5, thiophene), 127.33 (
C-4, thiophene), 122.70 (
C-3, aromatic), 68.67 (
CH), 50.09 (
CH
2) (
Figure S29c).
6h. (
R,
S)-
2-((((3-bromophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 390.9810 [M+H]
+ (calcd for C
18H
16BrOS
2+, 390.9820, Δ = 2.7 ppm) (
Figure S30). HPLC R
t 26.50 min, purity 96.9% (
Figure S30a). Chiral HPLC R
t 9.10 min, >99% ee (
Figure S30b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.54 (C
H-2, aromatic), 7.46 (C
H-4, aromatic), 7.44 (C
H-3,5, phenyl), 7.39 (C
H-6, aromatic), 7.39 (C
H-2,6, aromatic), 7.39 (C
H-4, phenyl), 7.34 (C
H-5, thiophene), 7.24 (C
H-5, aromatic), 7.06 (C
H-4, thiophene), 6.95 (C
H-3, thiophene), 4.69 (s, 1H, C
H), 4.10/3.89 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 136.60 (
C-1, aromatic), 134.79 (
C-1, phenyl), 132.40 (
C-2, aromatic), 131.50 (
C-4, aromatic), 130.12 (
C-5, aromatic), 129.78 (
C-2, thiophene), 129.48 (
CH-3,5, phenyl), 129.18 (
C-3, thiophene), 128.74 (
CH-2,6, phenyl), 128.74 (
C-4, phenyl), 128.33 (
C-6, aromatic), 126.98 (
C-5, thiophene), 127.33 (
C-4, thiophene), 122.70 (
C-3, aromatic), 68.67 (
CH), 50.09 (
CH
2) (
Figure S30c).
7h. (
S,
S)-
2-((((3-bromophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 390.9812 [M+H]
+ (calcd for C
18H
16BrOS
2+, 390.9820, Δ = 2.2 ppm) (
Figure S31). HPLC R
t 26.36 min, purity 96.8% (
Figure S31a). Chiral HPLC Rt 7.15 min, >99% ee (but contains 0.8% 5m) (
Figure S31b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.55 (C
H-2, aromatic), 7.49 (C
H-4, aromatic), 7.42 (C
H-2,6, aromatic), 7.41 (C
H-3,5, phenyl), 7.39 (C
H-6, aromatic), 7.38 (C
H-4, phenyl), 7.34 (C
H-5, thiophene), 7.28 (C
H-5, aromatic), 7.06 (C
H-4, thiophene), 6.97 (C
H-3, thiophene), 4.69 (s, 1H, C
H), 4.10/3.90 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 138.04 (
C-1, aromatic), 133.22 (
C-1, phenyl), 131.79 (
C-2, aromatic), 131.50 (
C-4, aromatic), 130.69 (
C-5, aromatic), 130.04 (
C-2, thiophene), 129.71 (
CH-2,6, phenyl), 129.02 (
C-3, thiophene), 128.83 (
CH-3,5, phenyl), 128.69 (
C-4, phenyl), 127.43 (
C-4, thiophene), 127.35 (
C-6, aromatic), 126.90 (
C-5, thiophene), 123.21 (
C-3, aromatic), 68.03 (
CH), 50.33 (
CH
2) (
Figure S31c).
8h. (
R,
R)-
2-((((3-bromophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 390.9810 [M+H]
+ (calcd for C
18H
16BrOS
2+, 390.9820, Δ = 2.6 ppm) (
Figure S32). HPLC R
t 26.36 min, purity 96.4% (
Figure S32a). Chiral HPLC R
t 10.28 min, >99% ee (
Figure S32b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.55 (C
H-2, aromatic), 7.49 (C
H-4, aromatic), 7.42 (C
H-2,6, aromatic), 7.41 (C
H-3,5, phenyl), 7.39 (C
H-6, aromatic), 7.38 (C
H-4, phenyl), 7.34 (C
H-5, thiophene), 7.28 (C
H-5, aromatic), 7.06 (C
H-4, thiophene), 6.97 (C
H-3, thiophene), 4.69 (s, 1H, C
H), 4.10/3.90 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 138.04 (
C-1, aromatic), 133.22 (
C-1, phenyl), 131.79 (
C-2, aromatic), 131.50 (
C-4, aromatic), 130.69 (
C-5, aromatic), 130.04 (
C-2, thiophene), 129.71 (
CH-2,6, phenyl), 129.02 (
C-3, thiophene), 128.83 (
CH-3,5, phenyl), 128.69 (
C-4, phenyl), 127.43 (
C-4, thiophene), 127.35 (
C-6, aromatic), 126.90 (
C-5, thiophene), 123.21 (
C-3, aromatic), 68.03 (
CH), 50.33 (
CH
2) (
Figure S32c).
- 9.
Synthesis of 2-((((2-iodophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene (5i–8i)
Following general procedure A, 0.5 g (1.61 mmol) of (2-iodophenyl)(phenyl)methanol, 0.21 g (1.61 mmol) of thiophen-2-ylmethanethiol, and 0.22 mL (1.8 mmol) of 48% BF3·Et2O were reacted, affording 0.49 g of 3i as a white oily product (yield 72%).
Following general procedure B, 0.49 g (1.17 mmol) of 3i dissolved in 6 mL of glacial acetic acid was reacted with 30% H2O2 (0.12 mL) to give 1.5 g of 4i as white oil (yield >95%).
Racemic 4i was further separated into the four individual diastereomers 5i–8i by means of semipreparative chiral HPLC. A semipreparative HPLC instrument was equipped with a CHIRALPAK IA column (10 mm Φ × 25 cm L) (Daicel Inc.), and EtOAc was used as the mobile phase.
5i. HRESIMS
m/
z 438.9682 [M+H]
+ (calcd for C
18H
16IOS
2+, 438.9682, Δ = −0.1 ppm) (
Figure S33). HPLC R
t 26.52 min, purity 95.9% (
Figure S33a). Chiral HPLC R
t 5.06 min, 93.9% ee (
Figure S33b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.90 (C
H-3, aromatic), 7.73 (C
H-6, aromatic), 7.51 (C
H-2,6, phenyl), 7.44 (C
H-5, aromatic), 7.40 (C
H-3,5, phenyl), 7.37 (C
H-4, phenyl), 7.32 (C
H-5, thiophene), 7.03 (C
H-4, thiophene), 7.03 (C
H-4, aromatic), 6.96 (C
H-3, thiophene), 5.22 (s, 1H, C
H), 4.14/4.06 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 140.46 (
CH-3, aromatic), 139.01 (
Cq-1, aromatic), 132.76 (
Cq-1, phenyl), 130.57 (
Cq, thiophene-2), 130.22 (
CH-2,6, phenyl), 129.83 (
CH-4, aromatic), 129.24 (
CH-6, aromatic), 128.99 (
CH-5, aromatic), 128.93 (
CH-3, thiophene), 128.66 (
CH-3,5, phenyl), 128.56 (
CH-4, phenyl), 127.55 (
CH-4, thiophene), 126.90 (
CH-5, thiophene), 101.75 (
Cq-2, aromatic), 71.95 (
CH), 51.02 (
CH
2) (
Figure S33c).
6i. HRESIMS
m/
z 438.9678 [M+H]
+ (calcd for C
18H
16IOS
2+, 438.9682, Δ = 0.8 ppm) (
Figure S34). HPLC R
t 26.52 min, purity 98.6% (
Figure S34a). Chiral HPLC R
t 6.27 min, >99% ee (
Figure S34b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.90 (C
H-3, aromatic), 7.73 (C
H-6, aromatic), 7.51 (C
H-2,6, phenyl), 7.44 (C
H-5, aromatic), 7.40 (C
H-3,5, phenyl), 7.37 (C
H-4, phenyl), 7.32 (C
H-5, thiophene), 7.03 (C
H-4, thiophene), 7.03 (C
H-4, aromatic), 6.96 (C
H-3, thiophene), 5.22 (s, 1H, C
H), 4.14/4.05 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 140.46 (
CH-3, aromatic), 139.02 (
Cq-1, aromatic), 132.76 (
Cq-1, phenyl), 130.57 (
Cq, thiophene-2), 130.22 (
CH-2,6, phenyl), 129.83 (
CH-4, aromatic), 129.24 (
CH-6, aromatic), 128.99 (
CH-5, aromatic), 128.93 (
CH-3, thiophene), 128.66 (
CH-3,5, phenyl), 128.56 (
CH-4, phenyl), 127.55 (
CH-4, thiophene), 126.90 (
CH-5, thiophene), 101.76 (
Cq-2, aromatic), 71.95 (
CH), 51.02 (
CH
2) (
Figure S34c).
7i. HRESIMS
m/
z 438.9679 [M+H]
+ (calcd for C
18H
16IOS
2+, 438.9682, Δ = 2.0 ppm) (
Figure S35). HPLC R
t 26.67 min, purity 97.0% (
Figure S35a). Chiral HPLC R
t 6.82 min, 93.0% ee (
Figure S35b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.95 (C
H-6, aromatic), 7.89 (C
H-3, aromatic), 7.50 (C
H-2,6, phenyl), 7.45 (C
H-5, aromatic), 7.39 (C
H-3,5, phenyl), 7.34 (C
H-4, phenyl), 7.33 (C
H-5, thiophene), 7.04 (C
H-4, thiophene), 7.02 (C
H-4, aromatic), 6.97 (C
H-3, thiophene), 5.41 (s, 1H, C
H), 4.21/3.92 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 140.16 (
CH-3, aromatic), 137.89 (
Cq-1, aromatic), 135.12 (
Cq-1, phenyl), 130.26 (
Cq, thiophene-2), 129.77 (
CH-4, aromatic), 129.67 (
CH-6, aromatic), 129.23 (
CH-3, thiophene), 129.20 (
CH-3,5, phenyl), 128.94 (
CH-2,6, phenyl), 128.82 (
CH-5, aromatic), 128.50 (
CH-4, phenyl), 127.43 (
CH-4, thiophene), 126.96 (
CH-5, thiophene), 103.01 (
Cq-2, aromatic), 71.83 (
CH), 50.27 (
CH
2) (
Figure S35c).
8i. HRESIMS
m/
z 438.9679 [M+H]
+ (calcd for C
18H
16IOS
2+, 438.9682, Δ = 0.5 ppm) (
Figure S36). HPLC R
t 26.67 min, purity 97.0% (
Figure S36a). Chiral HPLC R
t 8.37 min, 97.9% ee (
Figure S36b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.95 (C
H-6, aromatic), 7.89 (C
H-3, aromatic), 7.50 (C
H-2,6, phenyl), 7.45 (C
H-5, aromatic), 7.39 (C
H-3,5, phenyl), 7.35 (C
H-4, phenyl), 7.33 (C
H-5, thiophene), 7.04 (C
H-4, thiophene), 7.02 (C
H-4, aromatic), 6.97 (C
H-3, thiophene), 5.41 (s, 1H, C
H), 4.21/3.92 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 140.17 (
CH-3, aromatic), 137.94 (
Cq-1, aromatic), 135.19 (
Cq-1, phenyl), 130.35 (
Cq, thiophene-2), 129.77 (
CH-4, aromatic), 129.71 (
CH-6, aromatic), 129.21 (
CH-3,5, phenyl), 129.21 (
CH-3, thiophene), 128.96 (
CH-2,6, phenyl), 128.82 (
CH-5, aromatic), 128.49 (
CH-4, phenyl), 127.44 (
CH-4, thiophene), 126.94 (
CH-5, thiophene), 103.02 (
Cq-2, aromatic), 71.89 (
CH), 50.36 (
CH
2) (
Figure S36c).
- 10.
Synthesis of 2-((((3-iodophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene (5j–8j)
Following general procedure A, 1.5 g (1.61 mmol) of (3-iodophenyl)(phenyl)methanol, 0.21 g (1.61 mmol) of thiophen-2-ylmethanethiol and 0.22 mL (1.8 mmol) of 48% BF3·Et2O were reacted, affording 0.58 g of 3j as an orange oily product (yield 86%).
Following general procedure B, 0.58 g (1.39 mmol) of 3j dissolved in 6 mL of glacial acetic acid was reacted with 30% H2O2 (0.14 mL) to give 0.38 g of 4j as a yellow solid (yield 63%).
Racemic 4j was further separated into two individual pairs of enantiomers by means of an MPLC system equipped with a glass column packed with silica gel. Toluol/EtOAc = 80/20 was used as the mobile phase, and the separation was carried out with a flow rate of 5 mL/min. Individual pairs of enantiomers were further separated into the individual enantiomers 5j–8j by means of semipreparative chiral HPLC. A semipreparative HPLC instrument was equipped with a CHIRALPAK IA column (10 mm Φ × 25 cm L) (Daicel Inc.), and EtOAc was used as the mobile phase.
5j. HRESIMS
m/
z 438.9681 [M+H]
+ (calcd for C
18H
16IOS
2+, 438.9682, Δ = 0.3 ppm) (
Figure S37). HPLC R
t 27.10 min, purity > 99% (
Figure S37a). Chiral HPLC R
t 6.59 min, >99% ee (
Figure S37b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.72 (C
H-2, aromatic), 7.66 (C
H-4, aromatic), 7.44 (C
H-3,5, phenyl), 7.43 (C
H-6, aromatic), 7.38 (C
H-2,6, phenyl), 7.38 (C
H-4, phenyl), 7.34 (s, 1H, C
H-5, thiophene), 7.11 (C
H-5, aromatic), 7.06 (s, 1H, C
H-4, thiophene), 6.95 (s, 1H, C
H-3, thiophene), 4.66 (s, 1H, C
H), 4.09/3.89 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 138.24 (
CH-2, aromatic), 137.44 (
CH-4, aromatic), 136.65 (
Cq-1, aromatic), 134.84 (
Cq-1, phenyl), 130.28 (
CH-5, aromatic), 129.83 (
Cq, thiophene-2), 129.46 (
CH-3,5, phenyl), 129.15 (
CH-3, thiophene), 128.88 (
CH-6, aromatic), 128.72 (
CH-4, phenyl), 128.72 (
CH-2,6, phenyl), 127.34 (
CH-4, thiophene), 126.96 (
CH-5, thiophene), 94.51 (
Cq-3, aromatic), 68.59 (
CH), 50.12 (
CH
2) (
Figure S37c).
6j. HRESIMS
m/
z 438.9683 [M+H]
+ (calcd for C
18H
16IOS
2+, 438.9682, Δ = −0.3 ppm) (
Figure S38). HPLC R
t 27.10 min, purity > 99% (
Figure S38a). Chiral HPLC R
t 8.69 min, 98.6% ee (
Figure S38b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.72 (C
H-2, aromatic), 7.66 (C
H-4, aromatic), 7.44 (C
H-3,5, phenyl), 7.43 (C
H-6, aromatic), 7.38 (C
H-2,6, phenyl), 7.38 (C
H-4, phenyl), 7.34 (s, 1H, C
H-5, thiophene), 7.11 (C
H-5, aromatic), 7.06 (s, 1H, C
H-4, thiophene), 6.95 (s, 1H, C
H-3, thiophene), 4.66 (s, 1H, C
H), 4.09/3.89 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 138.24 (
CH-2, aromatic), 137.44 (
CH-4, aromatic), 136.65 (
Cq-1, aromatic), 134.84 (
Cq-1, phenyl), 130.28 (
CH-5, aromatic), 129.83 (
Cq, thiophene-2), 129.46 (
CH-3,5, phenyl), 129.15 (
CH-3, thiophene), 128.88 (
CH-6, aromatic), 128.72 (
CH-4, phenyl), 128.72 (
CH-2,6, phenyl), 127.34 (
CH-4, thiophene), 126.96 (
CH-5, thiophene), 94.51 (
Cq-3, aromatic), 68.59 (
CH), 50.12 (
CH
2) (
Figure S38c).
7j. HRESIMS
m/
z 438.9681 [M+H]
+ (calcd for C
18H
16IOS
2+, 438.9682, Δ = 0.1 ppm) (
Figure S39). HPLC R
t 26.97 min, purity 98.9% (
Figure S39a). Chiral HPLC R
t 7.04 min, >99% ee (but contains 1.4% 5j) (
Figure S39b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.73 (C
H-2, aromatic), 7.69 (C
H-4, aromatic), 7.44 (C
H-6, aromatic), 7.42 (C
H-2,6, phenyl), 7.41 (C
H-3,5, phenyl), 7.38 (C
H-4, phenyl), 7.34 (s, 1H, C
H-5, thiophene), 7.14 (C
H-5, aromatic), 7.06 (s, 1H, C
H-4, thiophene), 6.97 (s, 1H, C
H-3, thiophene), 4.65 (s, 1H, C
H), 4.10/3.90 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 138.08 (
Cq-1, aromatic), 137.63 (
CH-2, aromatic), 137.44 (
CH-4, aromatic), 133.25 (
Cq-1, phenyl), 130.03 (
CH-5, aromatic), 130.03 (
Cq, thiophene-2), 129.68 (
CH-2,6, phenyl), 129.02 (
CH-3, thiophene), 128.82 (
CH-3,5, phenyl), 128.67 (
CH-4, phenyl), 127.92 (
CH-6, aromatic), 127.42 (
CH-4, thiophene), 126.91 (
CH-5, thiophene), 95.00 (
Cq-3, aromatic), 67.95 (
CH), 50.34 (
CH
2) (
Figure S39c).
8j. HRESIMS
m/
z 438.9683 [M+H]
+ (calcd for C
18H
16IOS
2+, 438.9682, Δ = −0.3 ppm) (
Figure S40). HPLC R
t 26.98 min, purity > 99% (
Figure S40a). Chiral HPLC R
t 9.33 min, >99% ee (but contains 1.2% 6j) (
Figure S40b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.72 (C
H-2, aromatic), 7.69 (C
H-4, aromatic), 7.44 (C
H-6, aromatic), 7.42 (C
H-2,6, phenyl), 7.41 (C
H-3,5, phenyl), 7.38 (C
H-4, phenyl), 7.34 (s, 1H, C
H-5, thiophene), 7.14 (C
H-5, aromatic), 7.06 (s, 1H, C
H-4, thiophene), 6.97 (s, 1H, C
H-3, thiophene), 4.65 (s, 1H, C
H), 4.09/3.90 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 138.08 (
Cq-1, aromatic), 137.63 (
CH-2, aromatic) 137.44 (
CH-4, aromatic), 133.25 (
Cq-1, phenyl), 130.79 (
CH-5, aromatic), 130.03 (
Cq, thiophene-2), 129.68 (
CH-2,6, phenyl), 129.02 (
CH-3, thiophene), 128.82 (
CH-3,5, phenyl), 128.67 (
CH-4, phenyl), 127.92 (
CH-6, aromatic), 127.42 (
CH-4, thiophene), 126.91 (
CH-5, thiophene), 95.00 (
Cq-3, aromatic), 67.95 (
CH), 50.34 (
CH
2) (
Figure S40c).
- 11.
Synthesis of 2-((((4-iodophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene (5k–8k)
Following general procedure A, 0.3 g (0.97 mmol) of (4-iodophenyl)(phenyl)methanol, 0.13 g (0.97 mmol) of thiophen-2-ylmethanethiol, and (0.13 mL, 1.1 mmol) of 48% BF3·Et2O were reacted, affording 0.4 g of 3k as a yellow oily product (yield >95%).
Following general procedure B, 0.4 g (0.96 mmol) of 3k dissolved in 6 mL of glacial acetic acid was reacted with 30% H2O2 (0.54 mL) to give 0.37 g of 4k as white oil (yield 88%).
Racemic 4k was further separated into two individual pairs of enantiomers by means of an MPLC system equipped with a glass column packed with silica gel. Toluol/EtOAc = 80/20 was used as the mobile phase, and the separation was carried out with a flow rate of 5 mL/min. Individual pairs of enantiomers were further separated into the individual enantiomers 5k–8k by means of semipreparative chiral HPLC. A semipreparative HPLC instrument was equipped with a CHIRALPAK IA column (10 mm Φ × 25 cm L) (Daicel Inc), and EtOAc was used as the mobile phase.
5k. (
S,
R)-
2-((((4-iodophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 438.9678 [M+H]
+ (calcd for C
18H
16IOS
2+, 438.9682, Δ = 0.9 ppm) (
Figure S41). HPLC R
t 27.28 min, purity 98.8% (
Figure S41a). Chiral HPLC R
t 7.38 min, >99% ee (
Figure S41b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.69 (C
H-3,5, aromatic), 7.43 (C
H-3,5, phenyl), 7.38 (C
H-2,6, phenyl), 7.38 (C
H-4, phenyl), 7.34 (s, 1H, C
H-5, thiophene), 7.15 (C
H-2,6, aromatic), 7.05 (s, 1H, C
H-4, thiophene), 6.95 (s, 1H, C
H-3, thiophene), 4.68 (s, 1H, C
H), 4.09/3.88 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 137.72 (
CH-3,5, aromatic), 134.92 (
Cq-1, phenyl), 133.85 (
Cq-1, aromatic), 131.48 (
CH-2,6, aromatic), 129.87 (
Cq, thiophene-2), 129.40 (
CH-3,5, phenyl), 129.12 (
CH-3, thiophene), 128.74 (
CH-2,6, phenyl), 128.64 (
CH-4, phenyl), 127.34 (
CH-4, thiophene), 126.93 (
CH-5, thiophene), 94.62 (
Cq-4, aromatic), 68.45 (
CH), 50.07 (
CH
2) (
Figure S41c).
6k. (
R,
S)-
2-((((4-iodophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 438.9679 [M+H]
+ (calcd for C
18H
16IOS
2+, 438.9682, Δ = 0.7 ppm) (
Figure S42). HPLC R
t 27.28 min, purity 98.9% (
Figure S42a). Chiral HPLC R
t 13.96 min, 98.8% ee (
Figure S42b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.69 (C
H-3,5, aromatic), 7.43 (C
H-3,5, phenyl), 7.38 (C
H-4, phenyl), 7.38 (C
H-2,6, phenyl), 7.34 (s, 1H, C
H-5, thiophene), 7.15 (C
H-2,6, aromatic), 7.05 (s, 1H, C
H-4, thiophene), 6.95 (s, 1H, C
H-3, thiophene), 4.68 (s, 1H, C
H), 4.09/3.88 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 137.72 (
CH-3,5, aromatic), 134.92 (
Cq-1, phenyl), 133.85 (
Cq-1, aromatic), 131.48 (
CH-2,6, aromatic), 129.87 (
Cq, thiophene-2), 129.40 (
CH-3,5, phenyl), 129.11 (
CH-3, thiophene), 128.74 (
CH-2,6, phenyl), 128.64 (
CH-4, phenyl), 127.34 (
CH-4, thiophene), 126.93 (
CH-5, thiophene), 94.62 (
Cq-4, aromatic), 68.45 (
CH), 50.07 (
CH
2) (
Figure S42c).
7k. (
S,
S)-
2-((((4-iodophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 438.9677 [M+H]
+ (calcd for C
18H
16IOS
2+, 438.9682, Δ = 1.2 ppm) (
Figure S43). HPLC R
t 27.33 min, purity 98.4% (
Figure S43a). Chiral HPLC R
t 7.56 min, >99% ee (
Figure S43b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.74 (C
H-3,5, aromatic), 7.40 (C
H-3,5, phenyl), 7.40 (C
H-2,6, phenyl), 7.37 (C
H-4, phenyl), 7.34 (s, 1H, C
H-5, thiophene), 7.17 (C
H-2,6, aromatic), 7.05 (s, 1H, C
H-4, thiophene), 6.96 (s, 1H, C
H-3, thiophene), 4.67 (s, 1H, C
H), 4.09/3.89 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 138.30 (
CH-3,5, aromatic), 135.39 (
Cq-1, aromatic), 133.41 (
Cq-1, phenyl), 130.62 (
CH-2,6, aromatic), 130.04 (
Cq, thiophene-2), 129.62 (
CH-2,6, phenyl), 129.01 (
CH-3, thiophene), 128.79 (
CH-3,5, phenyl), 128.61 (
CH-4, phenyl), 127.39 (
CH-4, thiophene), 126.87 (
CH-5, thiophene), 94.19 (
Cq-4, aromatic), 68.15 (
CH), 50.25 (
CH
2) (
Figure S43c).
8k. (
R,
R)-
2-((((4-iodophenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 438.9676 [M+H]
+ (calcd for C
18H
16IOS
2+, 438.9682, Δ = 1.2 ppm) (
Figure S44). HPLC R
t 27.35 min, purity > 99% (
Figure S44a). Chiral HPLC R
t 10.83 min, >99% ee (
Figure S44b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.74 (C
H-3,5, aromatic), 7.40 (C
H-3,5, phenyl), 7.40 (C
H-2,6, phenyl), 7.37 (C
H-4, phenyl), 7.34 (s, 1H, C
H-5, thiophene), 7.17 (C
H-2,6, aromatic), 7.05 (s, 1H, C
H-4, thiophene), 6.96 (s, 1H, C
H-3, thiophene), 4.67 (s, 1H, C
H), 4.09/3.89 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 138.30 (
CH-3,5, aromatic), 135.39 (
Cq-1, aromatic), 133.41 (
Cq-1, phenyl), 130.62 (
CH-2,6, aromatic), 130.04 (
Cq, thiophene-2), 129.62 (
CH-2,6, phenyl), 129.02 (
CH-3, thiophene), 128.79 (
CH-3,5, phenyl), 128.62 (
CH-4, phenyl), 127.39 (
CH-4, thiophene), 126.87 (
CH-5, thiophene), 94.19 (
Cq-4, aromatic), 68.15 (
CH), 50.25 (
CH
2) (
Figure S44c).
- 12.
Synthesis of 2-(((phenyl(o-tolyl)methyl)sulfinyl)methyl)thiophene (5l–8l)
Following general procedure A, 1.0 g (5.0 mmol) of phenyl(o-tolyl)methanol, 0.65 g (5.0 mmol) of thiophen-2-ylmethanethiol, and (0.7 mL, 5.5 mmol) of 48% BF3·Et2O were reacted, affording 1.53 g of 3l as a yellow oily product (yield > 95%).
Following general procedure B, 1.53 g (4.92 mmol) of 3l dissolved in 12 mL of glacial acetic acid was reacted with 30% H2O2 (0.54 mL) to give 1.3 g of 4l as pale-yellow solid (yield 81%).
Racemic 4l was further separated into two individual pairs of enantiomers by means of an MPLC system equipped with a glass column packed with silica gel. Toluol/EtOAc = 80/20 was used as the mobile phase, and the separation was carried out with a flow rate of 5 mL/min. Individual pairs of enantiomers were further separated into the individual enantiomers 5l–8l by means of semipreparative chiral HPLC. A semipreparative HPLC instrument was equipped with a CHIRALPAK IA column (10 mm Φ × 25 cm L) (Daicel Inc.), and Hexane/EtOAc = 50/50 was used as the mobile phase in the case of 5l/6l and 100% EtOAc in the case of 7l/8l.
5l. HRESIMS
m/
z 349.0688 [M+Na]
+ (calcd for C
19H
18NaOS
2+, 349.0691, Δ = 1.1 ppm) (
Figure S45). HPLC R
t 25.45 min, purity > 99% (
Figure S45a). Chiral HPLC R
t 12.77 min, 97.6% ee (
Figure S45b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.59 (C
H-6, aromatic), 7.37 (C
H-2,6, phenyl), 7.34 (C
H-3,5, phenyl), 7.33 (s, 1H, C
H-5, thiophene), 7.32 (C
H-5, aromatic), 7.32 (C
H-4, phenyl), 7.26 (C
H-4, aromatic), 7.21 (C
H-3, aromatic), 7.03 (s, 1H, C
H-4, thiophene), 6.85 (s, 1H, C
H-3, thiophene), 4.82 (s, 1H, C
H), 4.18/4.01 (AB, 2H, C
H2), 2.10 (C
H3).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 136.50 (
Cq-2, aromatic), 134.81 (
Cq-1, aromatic), 133.57 (
Cq-1, phenyl), 131.34 (
CH-3, aromatic), 130.33 (
Cq, thiophene-2), 130.03 (
CH-2,6, phenyl), 128.84 (
CH-3, thiophene), 128.58 (
CH-3,5, phenyl), 128.36 (
CH-4, phenyl), 128.12 (
CH-4, aromatic), 127.59 (
CH-6, aromatic), 127.22 (
CH-4, thiophene), 126.85 (
CH-5, thiophene), 126.61 (
CH-5, aromatic), 65.63 (
CH), 50.26 (
CH
2), 19.61 (
CH
3) (
Figure S45c).
6l. HRESIMS
m/
z 349.0689 [M+Na]
+ (calcd for C
19H
18NaOS
2+, 349.0691, Δ = 0.6 ppm) (
Figure S46). HPLC R
t 25.43 min, purity > 99% (
Figure S46a). Chiral HPLC R
t 13.82 min, 96.5% ee (
Figure S46b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.59 (C
H-6, aromatic), 7.37 (C
H-2,6, phenyl), 7.34 (C
H-3,5, phenyl), 7.33 (s, 1H, C
H-5, thiophene), 7.32 (C
H-5, aromatic), 7.32 (C
H-4, phenyl), 7.26 (C
H-4, aromatic), 7.21 (C
H-3, aromatic), 7.03 (s, 1H, C
H-4, thiophene), 6.85 (s, 1H, C
H-3, thiophene), 4.82 (s, 1H, C
H), 4.18/4.01 (AB, 2H, C
H2), 2.10 (C
H3).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 136.50 (
Cq-2, aromatic), 134.81 (
Cq-1, aromatic), 133.57 (
Cq-1, phenyl), 131.34 (
CH-3, aromatic), 130.33 (
Cq, thiophene-2), 130.03 (
CH-2,6, phenyl), 128.84 (
CH-3, thiophene), 128.58 (
CH-3,5, phenyl), 128.36 (
CH-4, phenyl), 128.12 (
CH-4, aromatic), 127.59 (
CH-6, aromatic), 127.22 (
CH-4, thiophene), 126.85 (
CH-5, thiophene), 126.61 (
CH-5, aromatic), 65.63 (
CH), 50.26 (
CH
2), 19.61 (
CH
3) (
Figure S46c).
7l. HRESIMS
m/
z 349.0690 [M+Na]
+ (calcd for C
19H
18NaOS
2+, 349.0691, Δ = 0.3 ppm) (
Figure S47). HPLC R
t 25.22 min, purity 95.32% (
Figure S47a). Chiral HPLC R
t 6.66 min, >99% ee (
Figure S47b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.84 (C
H-6, aromatic), 7.40 (C
H-2,6, phenyl), 7.40 (C
H-3,5, phenyl), 7.33 (s, 1H, C
H-5, thiophene), 7.33 (C
H-4, phenyl), 7.28 (C
H-5, aromatic), 7.20 (C
H-4, aromatic), 7.16 (C
H-3, aromatic), 7.04 (s, 1H, C
H-4, thiophene), 6.92 (s, 1H, C
H-3, thiophene), 5.11 (s, 1H, C
H), 4.18/3.96 (AB, 2H, C
H2), 2.16 (C
H3).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 137.60 (
Cq-2, aromatic), 135.23 (
Cq-1, phenyl), 133.95 (
Cq-1, aromatic), 130.99 (
CH-3, aromatic), 130.16 (
Cq, thiophene-2), 129.24 (
CH-3,5, phenyl), 129.06 (
CH-3, thiophene), 129.04 (
CH-2,6, phenyl), 128.35 (
CH-4, phenyl), 127.95 (
CH-4, aromatic), 127.93 (
CH-6, aromatic), 127.22 (
CH-4, thiophene), 126.78 (
CH-5, thiophene), 126.52 (
CH-5, aromatic), 65.20 (
CH), 49.69 (
CH
2), 19.87 (
CH
3) (
Figure S47c).
8l. HRESIMS
m/
z 349.0690 [M+Na]
+ (calcd for C
19H
18NaOS
2+, 349.0691, Δ = 0.4 ppm) (
Figure S48). HPLC R
t 25.22 min, purity 95.68% (
Figure S48a). Chiral HPLC R
t 11.48 min, >96.1% ee (
Figure S48b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.84 (C
H-6, aromatic), 7.40 (C
H-2,6, phenyl), 7.40 (C
H-3,5, phenyl), 7.34 (s, 1H, C
H-5, thiophene), 7.33 (C
H-4, phenyl), 7.28 (C
H-5, aromatic), 7.20 (C
H-4, aromatic), 7.16 (C
H-3, aromatic), 7.04 (s, 1H, C
H-4, thiophene), 6.92 (s, 1H, C
H-3, thiophene), 5.11 (s, 1H, C
H), 4.18/3.96 (AB, 2H, C
H2), 2.16 (C
H3).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 137.60 (
Cq-2, aromatic), 135.23 (
Cq-1, phenyl), 133.95 (
Cq-1, aromatic), 130.99 (
CH-3, aromatic), 130.18 (
Cq, thiophene-2), 129.24 (
CH-3,5, phenyl), 129.06 (
CH-3, thiophene), 129.04 (
CH-2,6, phenyl), 128.35 (
CH-4, phenyl), 127.95 (
CH-4, aromatic), 127.93 (
CH-6, aromatic), 127.22 (
CH-4, thiophene), 126.79 (
CH-5, thiophene), 126.52 (
CH-5, aromatic), 65.20 (
CH), 49.69 (
CH
2), 19.87 (
CH
3) (
Figure S48c).
- 13.
Synthesis of 2-(((phenyl(m-tolyl)methyl)sulfinyl)methyl)thiophene (5m–8m)
Following general procedure A, 1.0 g (5.0 mmol) of phenyl(m-tolyl)methanol, 0.65 g (5.0 mmol) of thiophen-2-ylmethanethiol, and (0.7 mL, 5.5 mmol) of 48% BF3·Et2O were reacted, affording 1.49 g of 3m as a yellow oily product (yield 95%).
Following general procedure B, 1.49 g (4.8 mmol) of 3m dissolved in 10 mL of glacial acetic acid was reacted with 30% H2O2 (0.5 mL) to give 1.4 g of 4m as pale-yellow solid (yield 89%).
Racemic 4m was further separated into two individual pairs of enantiomers by means of an MPLC system equipped with a glass column packed with silica gel. Toluol/EtOAc = 80/20 was used as the mobile phase, and the separation was carried out with a flow rate of 5 mL/min. Individual pairs of enantiomers were further separated into the individual enantiomers 5m–8m by means of semipreparative chiral HPLC. A semipreparative HPLC instrument was equipped with a CHIRALPAK IA column (10 mm Φ × 25 cm L) (Daicel Inc.), and EtOAc was used as the mobile phase.
5m. (
S,
R)-
2-(((phenyl(m-tolyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 349.0685 [M+Na]
+ (calcd for C
19H
18NaOS
2+, 349.0691, Δ = 1.7 ppm) (
Figure S49). HPLC R
t 23.93 min, purity 98.4% (
Figure S49a). Chiral HPLC R
t 8.13 min, >99% ee (but contains 1.0% 7m) (
Figure S49b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.43 (C
H-2,6, phenyl), 7.42 (C
H-3,5, phenyl), 7.36 (C
H-4, phenyl), 7.32 (C
H-5, thiophene), 7.25 (C
H-5, aromatic), 7.25 (C
H-6, aromatic), 7.24 (C
H-2, aromatic), 7.14 (C
H-4, aromatic), 7.04 (C
H-4, thiophene), 6.96 (C
H-3, thiophene), 4.72 (s, 1H, C
H), 4.09/3.88 (AB, 2H, C
H2), 2.35 (s, 3H, C
H3).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 138.38 (
C-3, aromatic), 135.78 (
C-1, phenyl), 134.22 (
C-1, aromatic), 130.38 (
C-2, thiophene), 130.27 (
C-2, aromatic), 129.25 (
CH-3,5, phenyl), 129.20 (
C-4, aromatic), 128.99 (
C-3, thiophene), 128.74 (
CH-2,6, phenyl), 128.59 (
C-5, aromatic), 128.35 (
C-4, phenyl), 127.27 (
C-4, thiophene), 126.74 (
C-5, thiophene), 126.59 (
C-6, aromatic), 64.59 (
CH), 50.17 (
CH
2), 21.51 (
CH
3) (
Figure S49c).
6m. (
R,
S)-
2-(((phenyl(m-tolyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 349.0694 [M+Na]
+ (calcd for C
19H
18NaOS
2+, 349.0691, Δ = −0.7 ppm) (
Figure S50). HPLC R
t 23.95 min, purity 99.0% (
Figure S50a). Chiral HPLC R
t 14.67 min, >99% ee (
Figure S50b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.43 (C
H-2,6, phenyl), 7.42 (C
H-3,5, phenyl), 7.36 (C
H-4, phenyl), 7.32 (C
H-5, thiophene), 7.25 (C
H-5, aromatic), 7.25 (C
H-6, aromatic), 7.24 (C
H-2, aromatic), 7.14 (C
H-4, aromatic), 7.04 (C
H-4, thiophene), 6.96 (C
H-3, thiophene), 4.72 (s, 1H, C
H), 4.09/3.88 (AB, 2H, C
H2), 2.35 (s, 3H, C
H3).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 138.38 (
C-3, aromatic), 135.78 (
C-1, phenyl), 134.22 (
C-1, aromatic), 130.38 (
C-2, thiophene), 130.27 (
C-2, aromatic), 129.25 (
CH-3,5, phenyl), 129.20 (
C-4, aromatic), 128.99 (
C-3, thiophene), 128.74 (
CH-2,6, phenyl), 128.59 (
C-5, aromatic), 128.35 (
C-4, phenyl), 127.27 (
C-4, thiophene), 126.74 (
C-5, thiophene), 126.59 (
C-6, aromatic), 64.59 (
CH), 50.17 (
CH
2), 21.51 (
CH
3) (
Figure S50c).
7m. (
S,
S)-
2-(((phenyl(m-tolyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 349.0690 [M+Na]
+ (calcd for C
19H
18NaOS
2+, 349.0691, Δ = −0.4 ppm) (
Figure S51). HPLC R
t 24.02 min, purity > 99% (
Figure S51a). Chiral HPLC R
t 7.54 min, >99% ee (but contains 15.5% 5m) (
Figure S51b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.43 (C
H-2,6, phenyl), 7.37 (C
H-3,5, phenyl), 7.33 (C
H-4, phenyl), 7.33 (C
H-5, thiophene), 7.30 (C
H-5, aromatic), 7.22 (C
H-2, aromatic), 7.22 (C
H-6, aromatic), 7.17 (C
H-4, aromatic), 7.04 (C
H-4, thiophene), 6.96 (C
H-3, thiophene), 4.72 (s, 1H, C
H), 4.09/3.88 (AB, 2H, C
H2), 2.37 (s, 3H, C
H3).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 139.09 (
C-3, aromatic), 135.47 (
C-1, aromatic), 134.34 (
C-1, phenyl), 130.40 (
C-2, thiophene), 129.61 (
CH-2,6, phenyl), 129.42 (
C-2, aromatic), 129.18 (
C-4, aromatic), 128.99 (
C-3, thiophene), 128.99 (
C-5, aromatic), 128.65 (
CH-3,5, phenyl), 128.32 (
C-4, phenyl), 127.27 (
C-4, thiophene), 126.74 (
C-5, thiophene), 125.80 (
C-6, aromatic), 64.43 (
CH), 50.20 (
CH
2), 21.50 (
CH
3) (
Figure S51c).
8m. (
R,
R)-
2-(((phenyl(m-tolyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 349.0691 [M+Na]
+ (calcd for C
19H
18NaOS
2+, 349.0691, Δ = 0.1 ppm) (
Figure S52). HPLC R
t 24.02 min, purity > 99% (
Figure S52a). Chiral HPLC R
t 11.26 min, >99% ee (
Figure S52b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.43 (C
H-2,6, phenyl), 7.37 (C
H-3,5, phenyl), 7.33 (C
H-4, phenyl), 7.33 (C
H-5, thiophene), 7.30 (C
H-5, aromatic), 7.22 (C
H-2, aromatic), 7.22 (C
H-6, aromatic), 7.17 (C
H-4, aromatic), 7.04 (C
H-4, thiophene), 6.96 (C
H-3, thiophene), 4.72 (s, 1H, C
H), 4.09/3.88 (AB, 2H, C
H2), 2.37 (s, 3H, C
H3).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 139.09 (
C-3, aromatic), 135.47 (
C-1, aromatic), 134.34 (
C-1, phenyl), 130.40 (
C-2, thiophene), 129.61 (
CH-2,6, phenyl), 129.42 (
C-2, aromatic), 129.18 (
C-4, aromatic), 128.99 (
C-3, thiophene), 128.99 (
C-5, aromatic), 128.65 (
CH-3,5, phenyl), 128.32 (
C-4, phenyl), 127.27 (
C-4, thiophene), 126.74 (
C-5, thiophene), 125.80 (
C-6, aromatic), 64.43 (
CH), 50.20 (
CH
2), 21.50 (
CH
3) (
Figure S52c).
- 14.
Synthesis of 2-(((phenyl(p-tolyl)methyl)sulfinyl)methyl)thiophene (5n–8n)
Following general procedure A, 1.0 g (5.0 mmol) of phenyl(p-tolyl)methanol, 0.65 g (5.0 mmol) of thiophen-2-ylmethanethiol, and (0.7 mL, 5.5 mmol) of 48% BF3·Et2O were reacted, affording 1.5 g of 3n as a yellow oily product (yield >95%).
Following general procedure B, 1.5 g (4.8 mmol) of 3n dissolved in 12 mL of glacial acetic acid was reacted with 30% H2O2 (0.5 mL) to give 1.47 g of 4n as a pale-yellow solid (yield 94%).
Racemic 4n was further separated into two individual pairs of enantiomers by means of an MPLC system equipped with a glass column packed with silica gel. Toluol/EtOAc = 80/20 was used as the mobile phase, and the separation was carried out with a flow rate of 5 mL/min. Individual pairs of enantiomers were further separated into the individual enantiomers 5n–8n by means of semipreparative chiral HPLC. A semipreparative HPLC instrument was equipped with a CHIRALPAK IA column (10 mm Φ × 25 cm L) (Daicel Inc.), and EtOAc was used as the mobile phase.
5n. HRESIMS
m/
z 349.0694 [M+H]
+ (calcd for C
19H
18NaOS
2+, 349.0691, Δ = −0.6 ppm) (
Figure S53). HPLC R
t 24.08 min, purity > 99% (
Figure S53a). Chiral HPLC R
t 7.94 min, >99% ee (
Figure S53b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.42 (C
H-2,6, phenyl), 7.41 (C
H-3,5, phenyl), 7.35 (C
H-4, phenyl), 7.32 (C
H-5, thiophene), 7.32 (C
H-2,6, aromatic), 7.18 (C
H-3,5, phenyl), 7.04 (C
H-4, thiophene), 6.96 (C
H-3, thiophene), 4.72 (s, 1H, C
H), 4.08/3.87 (AB, 2H, C
H2), and 2.33 (s, 3H, C
H3).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 138.26 (
C-4, aromatic), 135.84 (
C-1, phenyl), 131.04 (
C-1, aromatic), 130.44 (
C-2, thiophene), 129.54 (
CH-2,6, aromatic), 129.40 (
CH-3,5, aromatic), 129.21 (
CH-3,5, phenyl), 128.94 (
C-3, thiophene), 128.76 (
CH-2,6, phenyl), 128.30 (
C-4, phenyl), 127.28 (
C-4, thiophene), 126.72 (
C-5, thiophene), 69.00 (
CH), 50.16 (
CH
2), 21.14 (
CH
3) (
Figure S53c).
6n. HRESIMS
m/
z 349.0689 [M+Na]
+ (calcd for C
19H
18NaOS
2+, 349.0691, Δ = 0.5 ppm) (
Figure S54). HPLC R
t 24.08 min, purity > 99% (
Figure S54a). Chiral HPLC R
t 14.87 min, >99% ee (
Figure S54b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.42 (C
H-2,6, phenyl), 7.41 (C
H-3,5, phenyl), 7.35 (C
H-4, phenyl), 7.32 (C
H-5, thiophene), 7.32 (C
H-2,6, aromatic), 7.18 (C
H-3,5, phenyl), 7.04 (C
H-4, thiophene), 6.96 (C
H-3, thiophene), 4.72 (s, 1H, C
H), 4.08/3.87 (AB, 2H, C
H2), and 2.33 (s, 3H, C
H3).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 138.26 (
C-4, aromatic), 135.85 (
C-1, phenyl), 131.04 (
C-1, aromatic), 130.44 (
C-2, thiophene), 129.54 (
CH-2,6, aromatic), 129.40 (
CH-3,5, aromatic), 129.21 (
CH-3,5, phenyl), 128.95 (
C-3, thiophene), 128.77 (
CH-2,6, phenyl), 128.30 (
C-4, phenyl), 127.29 (
C-4, thiophene), 126.72 (
C-5, thiophene), 69.00 (
CH), 50.16 (
CH
2), 21.14 (
CH
3) (
Figure S54c).
7n. (
S,
S)-
2-(((phenyl(p-tolyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 349.0690 [M+Na]
+ (calcd for C
19H
18NaOS
2+, 349.0691, Δ = 0.5 ppm) (
Figure S55). HPLC R
t 24.18 min, purity > 99% (
Figure S55a). Chiral HPLC R
t 7.85 min, >99% ee (
Figure S55b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.42 (C
H-2,6, phenyl), 7.36 (C
H-3,5, phenyl), 7.33 (C
H-4, phenyl), 7.33 (C
H-5, thiophene), 7.31 (C
H-2,6, aromatic), 7.22 (C
H-3,5, phenyl), 7.04 (C
H-4, thiophene), 6.97 (C
H-3, thiophene), 4.73 (s, 1H, C
H), 4.09/3.87 (AB, 2H, C
H2), 2.37 (s, 3H, C
H3).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 138.33 (
C-4, aromatic), 134.50 (
C-1, phenyl), 132.43 (
C-1, aromatic), 130.35 (
C-2, thiophene), 129.96 (
CH-3,5, aromatic), 129.56 (
CH-2,6, phenyl), 129.02 (
C-3, thiophene), 128.68 (
CH-2,6, aromatic), 128.64 (
CH-3,5, phenyl), 128.27 (
C-4, phenyl), 127.24 (
C-4, thiophene), 126.75 (
C-5, thiophene), 69.11 (
CH), 50.05 (
CH
2), 21.15 (
CH
3) (
Figure S55c).
8n. (
R,
R)-
2-(((phenyl(p-tolyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 349.0692 [M+Na]
+ (calcd for C
19H
18NaOS
2+, 349.0691, Δ = −0.3 ppm) (
Figure S56). HPLC R
t 24.20 min, purity > 99% (
Figure S56a). Chiral HPLC R
t 14.42 min, 9 8.8% (
Figure S56b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.42 (C
H-2,6, phenyl), 7.36 (C
H-3,5, phenyl), 7.33 (C
H-4, phenyl), 7.33 (C
H-5, thiophene), 7.31 (C
H-2,6, aromatic), 7.22 (C
H-3,5, phenyl), 7.05 (C
H-4, thiophene), 6.97 (C
H-3, thiophene), 4.73 (s, 1H, C
H), 4.09/3.87 (AB, 2H, C
H2), 2.37 (s, 3H, C
H3).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 138.32 (
C-4, aromatic), 134.49 (
C-1, phenyl), 132.42 (
C-1, aromatic), 130.35 (
C-2, thiophene), 129.96 (
CH-3,5, aromatic), 129.56 (
CH-2,6, phenyl), 129.01 (
C-3, thiophene), 128.67 (
CH-2,6, aromatic), 128.64 (
CH-3,5, phenyl), 128.27 (
C-4, phenyl), 127.24 (
C-4, thiophene), 126.74 (
C-5, thiophene), 69.11 (
CH), 50.04 (
CH
2), 21.15 (
CH
3) (
Figure S56c).
- 15.
Synthesis of 2-(((phenyl(2-(trifluoromethyl)phenyl)methyl)sulfinyl)methyl)thiophene (5o–8o)
Following general procedure A, 1.0 g (4.0 mmol) of phenyl(2-(trifluoromethyl)phenyl)methanol, 0.52 g (4.0 mmol) of thiophen-2-ylmethanethiol, and (0.54 mL, 4.4 mmol) of 48% BF3·Et2O were reacted, affording 1.37 g of 3o as a yellow oily product (yield 95%).
Following general procedure B, 1.37 g (4.4 mmol) of 3o dissolved in 12 mL of glacial acetic acid was reacted with 30% H2O2 (0.4 mL) to give 1.2 g of 4o as a white solid (yield 81%).
Racemic 4o was further separated into two individual pairs of enantiomers by means of a flash column chromatography system filled with silica gel and using Hexane/EtOAc = 2/1 as the mobile phase. Individual pairs of enantiomers were further separated into the individual enantiomers 5o–8o by means of semipreparative chiral HPLC. A semipreparative HPLC instrument was equipped with a CHIRALPAK IA column (10 mm Φ × 25 cm L) (Daicel Inc.), and EtOAc was used as the mobile phase.
5o. HRESIMS
m/
z 381.0592 [M+H]
+ (calcd for C
19H
16F
3OS
2+, 381.0589, Δ = −0.6 ppm) (
Figure S57). HPLC R
t 26.31 min, purity 98.7% (
Figure S57a). Chiral HPLC R
t 4.09 min, >99% ee (
Figure S57b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 8.00 (C
H-6, aromatic), 7.71 (C
H-3, aromatic), 7.65 (C
H-5, aromatic), 7.54 (C
H-2,6, phenyl), 7.46 (C
H-4, aromatic), 7.41 (C
H-3,5, phenyl), 7.38 (C
H-4, phenyl), 7.29 (s, 1H, C
H-5, thiophene), 6.99 (s, 1H, C
H-4, thiophene), 6.95 (s, 1H, C
H-3, thiophene), 5.22 (s, 1H, C
H), 4.04 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 135.06 (
Cq-1, aromatic), 133.21 (
Cq-1, phenyl), 132.52 (
CH-5, aromatic), 131.15 (
Cq, thiophene-2), 130.35 (
CH-6, aromatic), 129.73 (
CH-2,6, phenyl), 128.72 (
CH-3,5, phenyl), 128.67 (
CH-4, phenyl), 128.67 (
CH-3, thiophene), 128.41 (
Cq-2, aromatic), 128.25 (
CH-4, aromatic), 127.52 (
CH-4, thiophene), 126.79 (
CH-3, aromatic), 126.79 (
CH-5, thiophene), 123.98 (
CF
3), 64.58 (
CH), 52.02 (
CH
2) (
Figure S57c).
6o. HRESIMS
m/
z 381.0586 [M+H]
+ (calcd for C
19H
16F
3OS
2+, 381.0589, Δ = 0.8 ppm) (
Figure S58). HPLC R
t 26.31 min, purity > 99% (
Figure S58a). Chiral HPLC R
t 5.70 min, >99% ee (
Figure S58b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 8.00 (C
H-6, aromatic), 7.71 (C
H-3, aromatic), 7.65 (C
H-5, aromatic), 7.54 (C
H-2,6, phenyl), 7.46 (C
H-4, aromatic), 7.41 (C
H-3,5, phenyl), 7.38 (C
H-4, phenyl), 7.29 (s, 1H, C
H-5, thiophene), 6.99 (s, 1H, C
H-4, thiophene), 6.95 (s, 1H, C
H-3, thiophene), 5.22 (s, 1H, C
H), 4.04 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 135.06 (
Cq-1, aromatic), 133.21 (
Cq-1, phenyl), 132.52 (
CH-5, aromatic), 131.15 (
Cq, thiophene-2), 130.35 (
CH-6, aromatic), 129.73 (
CH-2,6, phenyl), 128.72 (
CH-3,5, phenyl), 128.67 (
CH-4, phenyl), 128.67 (
CH-3, thiophene), 128.41 (
Cq-2, aromatic), 128.25 (
CH-4, aromatic), 127.52 (
CH-4, thiophene), 126.79 (
CH-3, aromatic), 126.79 (
CH-5, thiophene), 123.98 (
CF
3), 64.58 (
CH), 52.02 (
CH
2) (
Figure S58c).
7o. HRESIMS
m/
z 381.0580 [M+H]
+ (calcd for C
19H
16F
3OS
2+, 381.0589, Δ = 2.4 ppm) (
Figure S59). HPLC R
t 26.28 min, purity > 99% (
Figure S59a). Chiral HPLC R
t 4.80 min, >99% ee (
Figure S59b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 8.16 (C
H-6, aromatic), 7.70 (C
H-3, aromatic), 7.67 (C
H-5, aromatic), 7.49 (C
H-2,6, phenyl), 7.44 (C
H-4, aromatic), 7.40 (C
H-3,5, phenyl), 7.34 (C
H-4, phenyl), 7.32 (s, 1H, C
H-5, thiophene), 7.03 (s, 1H, C
H-4, thiophene), 6.98 (s, 1H, C
H-3, thiophene), 5.35 (s, 1H, C
H), 4.16/3.83 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 135.27 (
Cq-1, phenyl), 134.25 (
Cq-1, aromatic), 132.37 (
CH-5, aromatic), 130.47 (
CH-6, aromatic), 130.12 (
Cq, thiophene-2), 129.62 (
Cq-2, aromatic), 129.30 (
CH-3,5, phenyl), 129.19 (
CH-3, thiophene), 128.63 (
CH-2,6, phenyl), 128.63 (
CH-4, phenyl), 128.05 (
CH-4, aromatic), 127.26 (
CH-4, thiophene), 126.94 (
CH-5, thiophene), 126.58 (
CH-3, aromatic), 124.08 (
CF
3), 64.85 (
CH), 50.43 (
CH
2) (
Figure S59c).
8o. HRESIMS
m/
z 381.0585 [M+H]
+ (calcd for C
19H
16F
3OS
2+, 381.0589, Δ = 1.0 ppm) (
Figure S60). HPLC R
t 26.28 min, purity > 99% (
Figure S60a). Chiral HPLC R
t 6.48 min, >99% ee (
Figure S60b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 8.16 (C
H-6, aromatic), 7.70 (C
H-3, aromatic), 7.67 (C
H-5, aromatic), 7.49 (C
H-2,6, phenyl), 7.44 (C
H-4, aromatic), 7.40 (C
H-3,5, phenyl), 7.34 (C
H-4, phenyl), 7.32 (s, 1H, C
H-5, thiophene), 7.03 (s, 1H, C
H-4, thiophene), 6.98 (s, 1H, C
H-3, thiophene), 5.35 (s, 1H, C
H), 4.16/3.83 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 135.27 (
Cq-1, phenyl), 134.25 (
Cq-1, aromatic), 132.37 (
CH-5, aromatic), 130.47 (
CH-6, aromatic), 130.12 (
Cq, thiophene-2), 129.62 (
Cq-2, aromatic), 129.30 (
CH-3,5, phenyl), 129.19 (
CH-3, thiophene), 128.63 (
CH-2,6, phenyl), 128.63 (
CH-4, phenyl), 128.05 (
CH-4, aromatic), 127.26 (
CH-4, thiophene), 126.94 (
CH-5, thiophene), 126.58 (
CH-3, aromatic), 124.08 (
CF
3), 64.85 (
CH), 50.43 (
CH
2) (
Figure S60c).
- 16.
Synthesis of 2-(((phenyl(3-(trifluoromethyl)phenyl)methyl)sulfinyl)methyl)thiophene (5p–8p)
Following general procedure A, 1.0 g (4.0 mmol) of phenyl(3-(trifluoromethyl)phenyl)methanol, 0.52 g (4.0 mmol) of thiophen-2-ylmethanethiol, and (0.54 mL, 4.4 mmol) of 48% BF3·Et2O were reacted, affording 1.3 g of 3p as a yellow oily product (yield 87%).
Following general procedure B, 1.3 g (3.6 mmol) of 3p dissolved in 12 mL of glacial acetic acid was reacted with 30% H2O2 (0.36 mL) to give 0.85 g of 4p as a pale-yellow solid (yield 63%).
Racemic 4p was further separated into two individual pairs of enantiomers by means of an MPLC system equipped with a glass column packed with silica gel. Toluol/EtOAc = 80/20 was used as the mobile phase, and the separation was carried out with a flow rate of 5 mL/min. Individual pairs of enantiomers were further separated into the individual enantiomers 5p–8p by means of semipreparative chiral HPLC. A semipreparative HPLC instrument was equipped with a CHIRALPAK IA column (10 mm Φ × 25 cm L) (Daicel Inc.), and EtOAc was used as the mobile phase.
5p. HRESIMS
m/
z 381.0588 [M+H]
+ (calcd for C
19H
16F
3OS
2+, 381.0589, Δ = 0.2 ppm) (
Figure S61). HPLC R
t 26.42 min, purity > 99% (
Figure S61a). Chiral HPLC R
t 4.81 min, >99% ee (
Figure S61b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.66 (C
H-6, aromatic), 7.63 (C
H-2, aromatic), 7.60 (C
H-4, aromatic), 7.50 (C
H-5, aromatic), 7.46 (C
H-3,5, phenyl), 7.46 (C
H-4, phenyl), 7.41 (C
H-2,6, phenyl), 7.35 (C
H-5, thiophene), 7.06 (C
H-4, thiophene), 6.96 (C
H-3, thiophene), 4.80 (s, 1H, C
H), 4.09/3.91 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 135.35 (
C-1, aromatic), 134.56 (
C-1, phenyl), 133.07 (
C-6, aromatic), 130.93 (
C-3, aromatic), 129.68 (
C-2, thiophene), 129.55 (
CH-3,5, phenyl), 129.20 (
C-3, thiophene), 129.10 (
C-5, aromatic), 128.84 (
C-4, phenyl), 128.76 (
CH-2,6, phenyl), 127.37 (
C-4, thiophene), 127.04 (
C-5, thiophene), 126.25 (
C-2, aromatic), 125.23 (
C-4, aromatic), 123.85 (
CF
3), 68.72 (
CH), 52.12 (
CH
2) (
Figure S61c).
6p. HRESIMS
m/
z 381.0587 [M+H]
+ (calcd for C
19H
16F
3OS
2+, 381.0589, Δ = 0.5 ppm) (
Figure S62). HPLC R
t 26.43 min, purity 98.3% (
Figure S62a). Chiral HPLC R
t 6.12 min, >99% ee (but contains 7.6%
7p) (
Figure S62b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.66 (C
H-6, aromatic), 7.63 (C
H-2, aromatic), 7.60 (C
H-4, aromatic), 7.50 (C
H-5, aromatic), 7.46 (C
H-3,5, phenyl), 7.46 (C
H-4, phenyl), 7.41 (C
H-2,6, phenyl), 7.35 (C
H-5, thiophene), 7.06 (C
H-4, thiophene), 6.96 (C
H-3, thiophene), 4.80 (s, 1H, C
H), 4.09/3.91 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 135.35 (
C-1, aromatic), 134.56 (
C-1, phenyl), 133.07 (
C-6, aromatic), 130.93 (
C-3, aromatic), 129.68 (
C-2, thiophene), 129.55 (
CH-3,5, phenyl), 129.20 (
C-3, thiophene), 129.10 (
C-5, aromatic), 128.84 (
C-4, phenyl), 128.76 (
CH-2,6, phenyl), 127.37 (
C-4, thiophene), 127.04 (
C-5, thiophene), 126.25 (
C-2, aromatic), 125.23 (
C-4, aromatic), 123.85 (
CF
3), 68.72 (
CH), 52.12 (
CH
2) (
Figure S62c).
7p. HRESIMS
m/
z 381.0588 [M+H]
+ (calcd for C
19H
16F
3OS
2+, 381.0589, Δ = 0.3 ppm) (
Figure S63). HPLC R
t 26.02 min, purity 97.7% (
Figure S63a). Chiral HPLC R
t 5.50 min, >99% ee (
Figure S63b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.68 (C
H-6, aromatic), 7.64 (C
H-2, aromatic), 7.62 (C
H-4, aromatic), 7.54 (C
H-5, aromatic), 7.44 (C
H-2,6, phenyl), 7.43 (C
H-3,5, phenyl), 7.40 (C
H-4, phenyl), 7.35 (C
H-5, thiophene), 7.05 (C
H-4, thiophene), 6.96 (C
H-3, thiophene), 4.77 (s, 1H, C
H), 4.09/3.90 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 136.98 (
C-1, aromatic), 132.89 (
C-1, phenyl), 132.08 (
C-6, aromatic), 131.50 (
C-3, aromatic), 129.95 (
CH-2,6, phenyl), 129.94 (
C-2, thiophene), 129.70 (
C-5, aromatic), 129.00 (
C-3, thiophene), 128.90 (
CH-3,5, phenyl), 128.82 (
C-4, phenyl), 127.48 (
C-4, thiophene), 126.95 (
C-5, thiophene), 125.68 (
C-2, aromatic), 125.17 (
C-4, aromatic), 123.71 (
CF
3), 67.85 (
CH), 50.41 (
CH
2) (
Figure S63c).
8p. HRESIMS
m/
z 381.0585 [M+H]
+ (calcd for C
19H
16F
3OS
2+, 381.0589, Δ = 1.2 ppm) (
Figure S64). HPLC R
t 26.03 min, purity > 99% (
Figure S64a). Chiral HPLC R
t 7.90 min, >99% ee (
Figure S64b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.68 (C
H-6, aromatic), 7.64 (C
H-2, aromatic), 7.62 (C
H-4, aromatic), 7.54 (C
H-5, aromatic), 7.44 (C
H-2,6, phenyl), 7.43 (C
H-3,5, phenyl), 7.40 (C
H-4, phenyl), 7.35 (C
H-5, thiophene), 7.05 (C
H-4, thiophene), 6.96 (C
H-3, thiophene), 4.77 (s, 1H, C
H), 4.09/3.90 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 136.98 (
C-1, aromatic), 132.89 (
C-1, phenyl), 132.08 (
C-6, aromatic), 131.50 (
C-3, aromatic), 129.95 (
CH-2,6, phenyl), 129.94 (
C-2, thiophene), 129.70 (
C-5, aromatic), 129.00 (
C-3, thiophene), 128.90 (
CH-3,5, phenyl), 128.82 (
C-4, phenyl), 127.48 (
C-4, thiophene), 126.95 (
C-5, thiophene), 125.68 (
C-2, aromatic), 125.17 (
C-4, aromatic), 123.71 (
CF
3), 67.85 (
CH), 50.41 (
CH
2) (
Figure S64c).
- 17.
Synthesis of 2-(((phenyl(4-(trifluoromethyl)phenyl)methyl)sulfinyl)methyl)thiophene (5q–8q)
Following general procedure A, 1.0 g (4.0 mmol) of phenyl(4-(trifluoromethyl)phenyl)methanol, 0.52 g (4.0 mmol) of thiophen-2-ylmethanethiol, and (0.54 mL, 4.4 mmol) of 48% BF3·Et2O were reacted, affording 0.95 g of 3q as a yellow oily product (yield 67%).
Following general procedure B, 1.95 g (2.6 mmol) of 3q dissolved in 10 mL of glacial acetic acid was reacted with 30% H2O2 (0.36 mL) to give 0.8 g of 4q as a yellow solid (yield 80%).
Racemic 4q was further separated into two individual pairs of enantiomers by means of an MPLC system equipped with a glass column packed with silica gel. Toluol/EtOAc = 80/20 was used as the mobile phase, and the separation was carried out with a flow rate of 5 mL/min. Individual pairs of enantiomers were further separated into the individual enantiomers 5q–8q by means of semipreparative chiral HPLC. A semipreparative HPLC instrument was equipped with a CHIRALPAK IA column (10 mm Φ × 25 cm L) (Daicel Inc.), and EtOAc was used as the mobile phase.
5q. HRESIMS
m/
z 381.0585 [M+H]
+ (calcd for C
19H
16F
3OS
2+, 381.0589, Δ = 1.2 ppm) (
Figure S65). HPLC R
t 26.65 min, purity > 99% (
Figure S65a). Chiral HPLC R
t 5.51 min, >99% ee (
Figure S65b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.62 (C
H-3,5, aromatic), 7.53 (C
H-2,6, aromatic), 7.45 (C
H-3,5, phenyl), 7.41 (C
H-2,6, phenyl), 7.39 (C
H-4, phenyl), 7.36 (C
H-5, thiophene), 7.07 (C
H-4, thiophene), 6.97 (C
H-3, thiophene), 4.80 (s, 1H, C
H), 4.12/3.91 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 138.28 (
C-1, aromatic), 134.56 (
C-1, phenyl), 130.46 (
C-4, aromatic), 129.99 (
CH-2,6, aromatic), 129.60 (
CH-3,5, phenyl), 129.60 (
C-2, thiophene), 129.27 (
C-3, thiophene), 128.91 (
C-4, phenyl), 128.79 (
CH-2,6, phenyl), 127.37 (
C-4, thiophene), 127.06 (
C-5, thiophene), 125.54 (
CH-3,5, aromatic), 123.94 (
CF
3), 68.59 (
CH), 50.05 (
CH
2) (
Figure S65a).
6q. HRESIMS
m/
z 381.0583 [M+H]
+ (calcd for C
19H
16F
3OS
2+, 381.0589, Δ = 1.7 ppm) (
Figure S66). HPLC R
t 26.63 min, purity > 99% (
Figure S66a). Chiral HPLC R
t 10.55 min, >99% ee (
Figure S66b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.62 (C
H-3,5, aromatic), 7.53 (C
H-2,6, aromatic), 7.45 (C
H-3,5, phenyl), 7.41 (C
H-2,6, phenyl), 7.39 (C
H-4, phenyl), 7.36 (C
H-5, thiophene), 7.07 (C
H-4, thiophene), 6.97 (C
H-3, thiophene), 4.80 (s, 1H, C
H), 4.12/3.91 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 138.28 (
C-1, aromatic), 134.56 (
C-1, phenyl), 130.46 (
C-4, aromatic), 129.99 (
CH-2,6, aromatic), 129.60 (
CH-3,5, phenyl), 129.60 (
C-2, thiophene), 129.27 (
C-3, thiophene), 128.91 (
C-4, phenyl), 128.79 (
CH-2,6, phenyl), 127.37 (
C-4, thiophene), 127.06 (
C-5, thiophene), 125.54 (
CH-3,5, aromatic), 123.94 (
CF
3), 68.59 (
CH), 50.05 (
CH
2) (
Figure S66c).
7q. HRESIMS
m/
z 381.0583 [M+H]
+ (calcd for C
19H
16F
3OS
2+, 381.0589, Δ = 1.6 ppm) (
Figure S67). HPLC R
t 26.40 min, purity > 99% (
Figure S67a). Chiral HPLC R
t 6.58 min, >99% ee (
Figure S67b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.66 (C
H-3,5, aromatic), 7.57 (C
H-2,6, aromatic), 7.44 (C
H-2,6, phenyl), 7.43 (C
H-3,5, phenyl), 7.40 (C
H-4, phenyl), 7.35 (C
H-5, thiophene), 7.06 (C
H-4, thiophene), 6.97 (C
H-3, thiophene), 4.78 (s, 1H, C
H), 4.10/3.92 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 139.95 (
C-1, aromatic), 132.93 (
C-1, phenyl), 130.49 (
C-4, aromatic), 129.22 (
CH-2,6, aromatic), 129.99 (
C-2, thiophene), 129.77 (
CH-2,6, phenyl), 129.00 (
C-3, thiophene), 128.89 (
CH-3,5, phenyl), 128.81 (
C-4, phenyl), 127.49 (
C-4, thiophene), 127.05 (
C-5, thiophene), 126.09 (
CH-3,5, aromatic), 123.79 (
CF
3), 67.95 (
CH), 50.41 (
CH
2) (
Figure S67c).
8q. HRESIMS
m/
z 381.0586 [M+H]
+ (calcd for C
19H
16F
3OS
2+, 381.0589, Δ = 0.7 ppm) (
Figure S68). HPLC R
t 26.38 min, purity 95.7% (
Figure S68a). Chiral HPLC R
t 9.28 min, >99% ee (
Figure S68b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.66 (C
H-3,5, aromatic), 7.57 (C
H-2,6, aromatic), 7.44 (C
H-2,6, phenyl), 7.43 (C
H-3,5, phenyl), 7.40 (C
H-4, phenyl), 7.35 (C
H-5, thiophene), 7.06 (C
H-4, thiophene), 6.97 (C
H-3, thiophene), 4.78 (s, 1H, C
H), 4.10/3.92 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 139.95 (
C-1, aromatic), 132.93 (
C-1, phenyl), 130.49 (
C-4, aromatic), 129.22 (
CH-2,6, aromatic), 129.99 (
C-2, thiophene), 129.77 (
CH-2,6, phenyl), 129.00 (
C-3, thiophene), 128.89 (
CH-3,5, phenyl), 128.81 (
C-4, phenyl), 127.49 (
C-4, thiophene), 127.05 (
C-5, thiophene), 126.09 (
CH-3,5, aromatic), 123.79 (
CF
3), 67.95 (
CH), 50.41 (
CH
2) (
Figure S68c).
- 18.
Synthesis of 2-((((2-methoxyphenyl)(phenyl)methyl)sulfinyl)methyl)thiophene (5r–8r)
Following general procedure A, 0.4 g (1.8 mmol) of (2-methoxyphenyl)(phenyl)methanol, 0.24 g (1.8 mmol) of thiophen-2-ylmethanethiol, and (0.24 mL, 1.98 mmol) of 48% BF3·Et2O were reacted, affording 0.44 g of 3r as a yellow oily product (yield 75%).
Following general procedure B, 0.44 g (1.3 mmol) of 3r dissolved in 12 mL of glacial acetic acid was reacted with 30% H2O2 (0.15 mL) to give 0.21 g of 4r as a yellow solid (yield 46%).
Racemic 4r was further separated into two individual pairs of enantiomers by means of an MPLC system equipped with a glass column packed with silica gel. Toluol/EtOAc = 80/20 was used as the mobile phase, and the separation was carried out with a flow rate of 5 mL/min. Individual pairs of enantiomers were further separated into the individual enantiomers 5r–8r by means of semipreparative chiral HPLC. A semipreparative HPLC instrument was equipped with a CHIRALPAK IA column (10 mm Φ × 25 cm L) (Daicel Inc.), and EtOAc was used as the mobile phase in the case of 7r/8r and i-PrOH in the case of 5r/6r.
5r. (
R,
R)-
2-((((2-methoxyphenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 365.0643 [M+Na]
+ (calcd for C
19H
18NaO
2S
2+, 365.0640, Δ = −0.8 ppm) (
Figure S69). HPLC R
t 24.02 min, purity 96.6% (
Figure S69a). Chiral HPLC R
t 13.84 min, >90% ee (
Figure S69b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.54 (C
H-6, aromatic), 7.49 (C
H-2,6, phenyl), 7.36 (C
H-3,5, phenyl), 7.32 (C
H-4, phenyl), 7.32 (C
H-4, aromatic), 7.30 (C
H-5, thiophene), 7.02 (C
H-5, aromatic), 7.04 (C
H-4, thiophene), 6.91 (C
H-3, thiophene), 5.34 (s, 1H, C
H), 4.09/3.95 (AB, 2H, C
H2), 3.80 (s, 3H, OC
H3).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 156.47 (
C-2, aromatic), 134.39 (
C-1, phenyl), 131.25 (
C-2, thiophene), 129.92 (
CH-2,6, phenyl), 129.40 (
C-4, aromatic), 129.27 (
C-6, aromatic), 128.72 (
C-3, thiophene), 128.50 (
CH-3,5, phenyl), 128.11 (
C-4, phenyl), 127.17 (
C-4, thiophene), 126.56 (
C-5, thiophene), 124.52 (
C-1, aromatic), 120.97 (
C-5, aromatic), 111.12 (
C-3, aromatic), 62.95 (
CH), 55.49 (O
CH
3), 50.80 (
CH
2) (
Figure S69c).
6r. (
S,
S)-
2-((((2-methoxyphenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 365.0641 [M+Na]
+ (calcd for C
19H
18NaO
2S
2+, 365.0640, Δ = −0.1 ppm) (
Figure S70). HPLC R
t 24.00 min, purity 96.9% (
Figure S70a). Chiral HPLC R
t 14.36 min, ~75% ee (
Figure S70b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.54 (C
H-6, aromatic), 7.49 (C
H-2,6, phenyl), 7.36 (C
H-3,5, phenyl), 7.32 (C
H-4, phenyl), 7.32 (C
H-4, aromatic), 7.30 (C
H-5, thiophene), 7.02 (C
H-5, aromatic), 7.04 (C
H-4, thiophene), 6.92 (C
H-3, aromatic), 6.91 (C
H-3, thiophene), 5.34 (s, 1H, C
H), 4.09/3.95 (AB, 2H, C
H2), 3.80 (s, 3H, OC
H3).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 156.47 (
C-2, aromatic), 134.39 (
C-1, phenyl), 131.25 (
C-2, thiophene), 129.92 (
CH-2,6, phenyl), 129.40 (
C-4, aromatic), 129.27 (
C-6, aromatic), 128.72 (
C-3, thiophene), 128.50 (
CH-3,5, phenyl), 128.11 (
C-4, phenyl), 127.17 (
C-4, thiophene), 126.56 (
C-5, thiophene), 124.52 (
C-1, aromatic), 120.97 (
C-5, aromatic), 111.12 (
C-3, aromatic), 62.95 (
CH), 55.49 (O
CH
3), 50.80 (
CH
2) (
Figure S70c).
7r. (
S,
R)-
2-((((2-methoxyphenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 365.0641 [M+Na]
+ (calcd for C
19H
18NaO
2S
2+, 365.0640, Δ = −0.2 ppm) (
Figure S71). HPLC R
t 23.15 min, purity 97.9% (
Figure S71a). Chiral HPLC R
t 12.70 min, >99% ee (
Figure S71b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.54 (C
H-6, aromatic), 7.49 (C
H-2,6, phenyl), 7.36 (C
H-3,5, phenyl), 7.32 (C
H-4, aromatic), 7.30 (C
H-5, thiophene), 7.29 (C
H-4, phenyl), 7.04 (C
H-5, aromatic), 7.01 (C
H-4, thiophene), 6.97 (C
H-3, thiophene), 6.94 (C
H-3, aromatic), 5.53 (s, 1H, C
H), 4.14/3.84 (AB, 2H, C
H2), 3.84 (s, 3H, OC
H3).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 157.19 (
C-2, aromatic), 136.67 (
C-1, phenyl), 131.42 (
C-2, thiophene), 130.25 (
C-6, aromatic), 129.28 (
C-4, aromatic), 128.94 (
CH-3,5, phenyl), 128.88 (
CH-2,6, phenyl), 128.78 (
C-3, thiophene), 127.95 (
C-4, phenyl), 127.27 (
C-4, thiophene), 126.52 (
C-5, thiophene), 122.91 (
C-1, aromatic), 120.95 (
C-5, aromatic), 110.88 (
C-3, aromatic), 62.96 (
CH), 55.60 (O
CH
3), 50.85 (
CH
2) (
Figure S71c).
8r. (
S,
S)-
2-((((2-methoxyphenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 365.0642 [M+Na]
+ (calcd for C
19H
18NaO
2S
2+, 365.0640, Δ = −0.5 ppm) (
Figure S72). HPLC R
t 23.15 min, purity 97.0% (
Figure S72a). Chiral HPLC R
t 15.24 min, >99% ee (
Figure S72b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.54 (C
H-6, aromatic), 7.49 (C
H-2,6, phenyl), 7.36 (C
H-3,5, phenyl), 7.32 (C
H-4, aromatic), 7.30 (C
H-5, thiophene), 7.29 (C
H-4, phenyl), 7.04 (C
H-5, aromatic), 7.01 (C
H-4, thiophene), 6.97 (C
H-3, thiophene), 6.94 (C
H-3, aromatic), 5.53 (s, 1H, C
H), 4.14/3.84 (AB, 2H, C
H2), 3.84 (s, 3H, OC
H3).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 157.19 (
C-2, aromatic), 136.67 (
C-1, phenyl), 131.42 (
C-2, thiophene), 130.25 (
C-6, aromatic), 129.28 (
C-4, aromatic), 128.94 (
CH-3,5, phenyl), 128.88 (
CH-2,6, phenyl), 128.78 (
C-3, thiophene), 127.95 (
C-4, phenyl), 127.27 (
C-4, thiophene), 126.52 (
C-5, thiophene), 122.91 (
C-1, aromatic), 120.95 (
C-5, aromatic), 110.88 (
C-3, aromatic), 62.96 (
CH), 55.60 (O
CH
3), 50.85 (
CH
2) (
Figure S72c).
- 19.
Synthesis of 2-((((3-methoxyphenyl)(phenyl)methyl)sulfinyl)methyl)thiophene (5s–8s)
Following general procedure A, 1.0 g (4.6 mmol) of (3-methoxyphenyl)(phenyl)methanol, 0.6 g (4.6 mmol) of thiophen-2-ylmethanethiol, and 0.62 mL (5.1 mmol) of 48% BF3·Et2O were reacted, affording 1.1 g of 3s as a red-yellow oily product (yield 73%).
Following general procedure B, 1.1 g (3.4 mmol) of 3s dissolved in 12 mL of glacial acetic acid was reacted with 30% H2O2 (0.35 mL) to give 1.1 g of 4s as a yellow solid (yield 94%).
Racemic 4s was further separated into two individual pairs of enantiomers by means of an MPLC system equipped with a glass column packed with silica gel. Toluol/EtOAc = 80/20 was used as the mobile phase, and the separation was carried out with a flow rate of 5 mL/min. Individual pairs of enantiomers were further separated into the individual enantiomers 5s–8s by means of semipreparative chiral HPLC. A semipreparative HPLC instrument was equipped with a CHIRALPAK IA column (10 mm Φ × 25 cm L) (Daicel Inc.), and EtOAc was used as the mobile phase.
5s. HRESIMS
m/
z 365.0652 [M+Na]
+ (calcd for C
19H
18NaO
2S
2+, 365.0640, Δ = −3.2 ppm) (
Figure S73). HPLC R
t 23.83 min, purity 98.2% (
Figure S73a). Chiral HPLC R
t 7.05 min, >99% ee (
Figure S73b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.43 (C
H-2,6, phenyl), 7.37 (C
H-3,5, phenyl), 7.34 (C
H-4, phenyl), 7.34 (C
H-5, aromatic), 7.33 (C
H-5, thiophene), 7.05 (C
H-4, thiophene), 7.02 (C
H-6, aromatic), 6.98 (C
H-3, thiophene), 6.95 (C
H-2, aromatic), 6.89 (C
H-4, aromatic), 4.72 (s, 1H, C
H), 4.11/3.89 (AB, 2H, C
H2), 3.82 (s, 3H, OC
H3).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 160.09 (
C-3, aromatic), 136.92 (
C-1, aromatic), 134.16 (
C-1, phenyl), 130.34 (
C-5, aromatic), 130.27 (
C-2, thiophene), 129.60 (
CH-2,6, phenyl), 129.08 (
C-3, thiophene), 128.68 (
CH-3,5, phenyl), 128.39 (
C-4, phenyl), 127.28 (
C-4, thiophene), 126.79 (
C-5, thiophene), 120.97 (
C-6, aromatic), 114.72 (
C-2, aromatic), 113.57 (
C-4, aromatic), 69.36 (
CH), 55.31 (O
CH
3), 50.14 (
CH
2) (
Figure S73a).
6s. HRESIMS
m/
z 365.0645 [M+Na]
+ (calcd for C
19H
18NaO
2S
2+, 365.0640, Δ = −1.3 ppm) (
Figure S74). HPLC R
t 22.57 min, purity > 99% (
Figure S74a). Chiral HPLC R
t 10.55 min, 98.7% ee (
Figure S74b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.43 (C
H-2,6, phenyl), 7.37 (C
H-3,5, phenyl), 7.34 (C
H-4, phenyl), 7.34 (C
H-5, aromatic), 7.33 (C
H-5, thiophene), 7.05 (C
H-4, thiophene), 7.02 (C
H-6, aromatic), 6.98 (C
H-3, thiophene), 6.95 (C
H-2, aromatic), 6.89 (C
H-4, aromatic), 4.72 (s, 1H, C
H), 4.11/3.89 (AB, 2H, C
H2), 3.82 (s, 3H, OC
H3).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 160.09 (
C-3, aromatic), 136.92 (
C-1, aromatic), 134.16 (
C-1, phenyl), 130.34 (
C-5, aromatic), 130.27 (
C-2, thiophene), 129.60 (
CH-2,6, phenyl), 129.08 (
C-3, thiophene), 128.68 (
CH-3,5, phenyl), 128.39 (
C-4, phenyl), 127.28 (
C-4, thiophene), 126.79 (
C-5, thiophene), 120.97 (
C-6, aromatic), 114.72 (
C-2, aromatic), 113.57 (
C-4, aromatic), 69.36 (
CH), 55.31 (O
CH
3), 50.14 (
CH
2) (
Figure S74c).
7s. HRESIMS
m/
z 365.0644 [M+Na]
+ (calcd for C
19H
18NaO
2S
2+, 365.0640, Δ = −0.8 ppm) (
Figure S75). HPLC R
t 24.05 min, purity 96.7% (
Figure S75a). Chiral HPLC R
t 7.04 min, >99% ee (
Figure S75b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.43 (C
H-2,6, phenyl), 7.41 (C
H-3,5, phenyl), 7.36 (C
H-4, phenyl), 7.32 (C
H-5, thiophene), 7.29 (C
H-5, aromatic), 7.04 (C
H-4, thiophene), 7.03 (C
H-6, aromatic), 7.00 (C
H-2, aromatic), 6.96 (C
H-3, thiophene), 6.87 (C
H-4, aromatic), 4.72 (s, 1H, C
H), 4.09/3.89 (AB, 2H, C
H2), 3.79 (s, 3H, OC
H3).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 159.66 (
C-3, aromatic), 135.75 (
C-1, aromatic), 133.57 (
C-1, phenyl), 130.37 (
C-2, thiophene), 129.66 (
C-5, aromatic), 129.27 (
CH-3,5, phenyl), 129.01 (
C-3, thiophene), 128.75 (
CH-2,6, phenyl), 128.43 (
C-4, phenyl), 127.30 (
C-4, thiophene), 126.77 (
C-5, thiophene), 121.98 (
C-6, aromatic), 115.42 (
C-2, aromatic), 113.69 (
C-4, aromatic), 69.37 (
CH), 55.25 (O
CH
3), 50.25 (
CH
2) (
Figure S75c).
8s. HRESIMS
m/
z 365.0642 [M+Na]
+ (calcd for C
19H
18NaO
2S
2+, 365.0640, Δ = −0.4 ppm) (
Figure S76). HPLC R
t 22.80 min, purity > 99% (
Figure S76a). Chiral HPLC R
t 13.05 min, >99% ee (
Figure S76b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.43 (C
H-2,6, phenyl), 7.41 (C
H-3,5, phenyl), 7.36 (C
H-4, phenyl), 7.32 (C
H-5, thiophene), 7.29 (C
H-5, aromatic), 7.04 (C
H-4, thiophene), 7.03 (C
H-6, aromatic), 7.00 (C
H-2, aromatic), 6.96 (C
H-3, thiophene), 6.87 (C
H-4, aromatic), 4.72 (s, 1H, C
H), 4.09/3.89 (AB, 2H, C
H2), 3.79 (s, 3H, OC
H3).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 159.66 (
C-3, aromatic), 135.75 (
C-1, aromatic), 133.57 (
C-1, phenyl), 130.37 (
C-2, thiophene), 129.66 (
C-5, aromatic), 129.27 (
CH-3,5, phenyl), 129.01 (
C-3, thiophene), 128.75 (
CH-2,6, phenyl), 128.43 (
C-4, phenyl), 127.30 (
C-4, thiophene), 126.77 (
C-5, thiophene), 121.98 (
C-6, aromatic), 115.42 (
C-2, aromatic), 113.69 (
C-4, aromatic), 69.37 (
CH), 55.25 (O
CH
3), 50.25 (
CH
2) (
Figure S76c).
- 20.
Synthesis of 2-((((4-methoxyphenyl)(phenyl)methyl)sulfinyl)methyl)thiophene (5t–8t)
Following general procedure A, 1.64 g (7.7 mmol) of (2-fluorophenyl)(phenyl)methanol, 1.0 g (7.7 mmol) of thiophen-2-ylmethanethiol, and 1.0 mL (8.5 mmol) of 48% BF3·Et2O were reacted, affording 0.8 g of 3t as a yellow oily product (yield 30%).
Following general procedure B, 0.8 g (2.3 mmol) of 3t dissolved in 12 mL of glacial acetic acid was reacted with 30% H2O2 (0.27 mL) to give 0.4 g of 4t as dark orange-brown oil (yield 44%).
Racemic 4t was further separated into two individual pairs of enantiomers by means of an MPLC system equipped with a glass column packed with silica gel. Toluol/EtOAc = 80/20 was used as the mobile phase, and the separation was carried out with a flow rate of 5 mL/min. Individual pairs of enantiomers were further separated into the individual enantiomers 5t–8t by means of semipreparative chiral HPLC. A semipreparative HPLC instrument was equipped with a CHIRALPAK IA column (10 mm Φ × 25 cm L) (Daicel Inc.), and EtOAc was used as the mobile phase.
5t. (
S,
S)-
2-((((4-methoxyphenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 365.0639 [M+Na]
+ (calcd for C
19H
18NaO
2S
2+, 365.0640, Δ = 0.4 ppm) (
Figure S77). HPLC R
t 22.37 min, purity 91.7% (
Figure S77a). Chiral HPLC R
t 8.72 min, >99% ee (but contains 2.8%
7t) (
Figure S77b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.42 (C
H-2,6, phenyl), 7.37 (C
H-3,5, phenyl), 7.34 (C
H-2,6, aromatic), 7.33 (C
H-4, phenyl), 7.32 (C
H-5, thiophene), 7.04 (C
H-4, thiophene), 6.97 (C
H-3, thiophene), 6.94 (C
H-3,5, aromatic), 4.72 (s, 1H, C
H), 4.09/3.87 (AB, 2H, C
H2), 3.82 (s, 3H, OC
H3).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 159.55 (
C-4, aromatic), 134.60 (
C-1, phenyl), 130.37 (
C-2, thiophene), 130.02 (
CH-2,6, aromatic), 129.52 (
CH-2,6, phenyl), 128.99 (
C-3, thiophene), 128.64 (
CH-3,5, phenyl), 128.26 (
C-4, phenyl), 127.32 (
C-1, aromatic), 127.26 (
C-4, thiophene), 126.74 (
C-5, thiophene), 114.64 (
CH-3,5, aromatic), 68.61 (
CH), 55.32 (O
CH
3), 50.02 (
CH
2) (
Figure S77c).
6t. (
R,
R)-
2-((((4-methoxyphenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 365.0639 [M+Na]
+ (calcd for C
19H
18NaO
2S
2+, 365.0640, Δ = 0.3 ppm) (
Figure S78). HPLC R
t 23.63 min, purity > 99% (
Figure S78a). Chiral HPLC R
t 14.51 min, >99% ee (
Figure S78b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.42 (C
H-2,6, phenyl), 7.37 (C
H-3,5, phenyl), 7.34 (C
H-2,6, aromatic), 7.33 (C
H-4, phenyl), 7.32 (C
H-5, thiophene), 7.04 (C
H-4, thiophene), 6.97 (C
H-3, thiophene), 6.94 (C
H-3,5, aromatic), 4.72 (s, 1H, C
H), 4.09/3.87 (AB, 2H, C
H2), 3.82 (s, 3H, OC
H3).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 159.55 (
C-4, aromatic), 134.60 (
C-1, phenyl), 130.37 (
C-2, thiophene), 130.02 (
CH-2,6, aromatic), 129.52 (
CH-2,6, phenyl), 128.99 (
C-3, thiophene), 128.64 (
CH-3,5, phenyl), 128.26 (
C-4, phenyl), 127.32 (
C-1, aromatic), 127.26 (
C-4, thiophene), 126.74 (
C-5, thiophene), 114.64 (
CH-3,5, aromatic), 68.61 (
CH), 55.32 (O
CH
3), 50.02 (
CH
2) (
Figure S78c).
7t. (
S,
R)-
2-((((4-methoxyphenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 365.0639 [M+Na]
+ (calcd for C
19H
18NaO
2S
2+, 365.0640, Δ = 0.4 ppm) (
Figure S79). HPLC R
t 23.88 min, purity > 99% (
Figure S79a). Chiral HPLC R
t 8.13 min, >99% ee (but contains 7.6%
5t) (
Figure S79b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.41 (C
H-2,6, phenyl), 7.41 (C
H-3,5, phenyl), 7.35 (C
H-2,6, aromatic), 7.35 (C
H-4, phenyl), 7.32 (C
H-5, thiophene), 7.04 (C
H-4, thiophene), 6.96 (C
H-3, thiophene), 6.91 (C
H-3,5, aromatic), 4.71 (s, 1H, C
H), 4.07/3.87 (AB, 2H, C
H2), 3.79 (s, 3H, OC
H3).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 159.69 (
C-4, aromatic), 135.89 (
C-1, phenyl), 130.89 (
CH-2,6, aromatic), 130.42 (
C-2, thiophene), 129.23 (
CH-3,5, phenyl), 128.93 (
C-3, thiophene), 128.76 (
CH-2,6, phenyl), 128.30 (
C-4, phenyl), 127.29 (
C-4, thiophene), 126.73 (
C-5, thiophene), 125.91 (
C-1, aromatic), 114.09 (
CH-3,5, aromatic), 68.52 (
CH), 55.32 (O
CH
3), 50.10 (
CH
2) (
Figure S79c).
8t. (
R,
S)-
2-((((4-methoxyphenyl)(phenyl)methyl)sulfinyl)methyl)thiophene. HRESIMS
m/
z 365.0641 [M+Na]
+ (calcd for C
19H
18NaO
2S
2+, 365.0640, Δ = −0.2 ppm) (
Figure S80). HPLC R
t 22.65 min, purity 98.4% (
Figure S80a). Chiral HPLC R
t 14.38 min, >99% ee (
Figure S80b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.41 (C
H-2,6, phenyl), 7.41 (C
H-3,5, phenyl), 7.35 (C
H-2,6, aromatic), 7.35 (C
H-4, phenyl), 7.32 (C
H-5, thiophene), 7.04 (C
H-4, thiophene), 6.96 (C
H-3, thiophene), 6.91 (C
H-3,5, aromatic), 4.71 (s, 1H, C
H), 4.07/3.87 (AB, 2H, C
H2), 3.79 (s, 3H, OC
H3).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 159.69 (
C-4, aromatic), 135.89 (
C-1, phenyl), 130.89 (
CH-2,6, aromatic), 130.42 (
C-2, thiophene), 129.23 (
CH-3,5, phenyl), 128.93 (
C-3, thiophene), 128.76 (
CH-2,6, phenyl), 128.30 (
C-4, phenyl), 127.29 (
C-4, thiophene), 126.73 (
C-5, thiophene), 125.91 (
C-1, aromatic), 114.09 (
CH-3,5, aromatic), 68.52 (
CH), 55.32 (O
CH
3), 50.10 (
CH
2) (
Figure S80c).
- 21.
Synthesis of 2-((((4-chlorophenyl)(4-fluorophenyl)methyl)sulfinyl)methyl)thiophene (5u–8u)
Following general procedure A, 1.0 g (3.9 mmol) of (4-chlorophenyl)(4-fluorophenyl)methanol, 0.52 g (3.9 mmol) of thiophen-2-ylmethanethiol, and 0.54 mL (4.3 mmol) of 48% BF3·Et2O were reacted, affording 0.6 g of 3u as a yellow oily product (yield 44%).
Following general procedure B, 0.6 g (1.72 mmol) of 3u dissolved in 12 mL of glacial acetic acid was reacted with 30% H2O2 (0.18 mL) to give 0.52 g of 4u as a yellow solid (yield 83%).
Racemic 4u was further separated into two individual pairs of enantiomers by means of an MPLC system equipped with a glass column packed with silica gel. Toluol/EtOAc = 80/20 was used as the mobile phase, and the separation was carried out with a flow rate of 5 mL/min. Individual pairs of enantiomers were further separated into the individual enantiomers 5u–8u by means of semipreparative chiral HPLC. A semipreparative HPLC instrument was equipped with a CHIRALPAK IA column (10 mm Φ × 25 cm L) (Daicel Inc.), and EtOAc was used as the mobile phase.
5u. HRESIMS
m/
z 387.0052 [M+Na]
+ (calcd for C
18H
14ClFNaOS
2+, 387.0051, Δ = −0.2 ppm) (
Figure S81). HPLC R
t 26.13 min, purity > 99% (
Figure S81a). Chiral HPLC R
t 7.22 min, >99% ee (but contains 15.5%
5m) (
Figure S81b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.37 (C
H-2’,6’, phenyl), 7.35 (C
H-3,5, phenyl), 7.34 (C
H-2,6, phenyl), 7.34 (C
H-5, thiophene), 7.12 (C
H-3’,5’, phenyl), 7.06 (C
H-4, thiophene), 6.96 (C
H-3, thiophene), 4.69 (s, 1H, C
H), 4.08/3.89 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 162.62 (
C-4, aromatic), 134.75 (
C-4, phenyl), 132.26 (
C-1, phenyl), 130.97 (
C-1, aromatic), 130.91 (
CH-2,6, phenyl), 130.51 (
CH-2,6, aromatic), 129.77 (
C-2, thiophene), 129.97 (
C-3, thiophene), 128.92 (
CH-3,5, phenyl), 127.44 (
C-4, thiophene), 126.97 (
C-5, thiophene), 116.41 (
CH-3,5, aromatic), 66.93 (
CH), 50.11 (
CH
2) (
Figure S81c).
6u. HRESIMS
m/
z 387.0053 [M+Na]
+ (calcd for C
18H
14ClFNaOS
2+, 387.0051, Δ = −0.5 ppm) (
Figure S82). HPLC R
t 26.17 min, purity > 99% (
Figure S82a). Chiral HPLC R
t 12.64 min, >99% ee (
Figure S82b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.37 (C
H-2’,6’, phenyl), 7.35 (C
H-3,5, phenyl), 7.34 (C
H-2,6, phenyl), 7.34 (C
H-5, thiophene), 7.12 (C
H-3’,5’, phenyl), 7.06 (C
H-4, thiophene), 6.96 (C
H-3, thiophene), 4.69 (s, 1H, C
H), 4.08/3.89 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 162.62 (
C-4, aromatic), 134.75 (
C-4, phenyl), 132.26 (
C-1, phenyl), 130.97 (
C-1, aromatic), 130.93 (
CH-2,6, phenyl), 130.51 (
CH-2,6, aromatic), 129.76 (
C-2, thiophene), 129.07 (
C-3, thiophene), 128.92 (
CH-3,5, phenyl), 127.44 (
C-4, thiophene), 126.97 (
C-5, thiophene), 116.41 (
CH-3,5, aromatic), 66.92 (
CH), 50.11 (
CH
2) (
Figure S82c).
7u. HRESIMS
m/
z 387.0052 [M+Na]
+ (calcd for C
18H
14ClFNaOS
2+, 387.0051, Δ = −0.3 ppm) (
Figure S83). HPLC R
t 26.27 min, purity 97.4% (
Figure S83a). Chiral HPLC R
t 6.38 min, >99% ee (
Figure S83b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.40 (C
H-2,6, phenyl), 7.37 (C
H-2’,6’, phenyl), 7.35 (C
H-5, thiophene), 7.33 (C
H-3,5, phenyl), 7.08 (C
H-3’,5’, phenyl), 7.06 (C
H-4, thiophene), 6.96 (C
H-3, thiophene), 4.68 (s, 1H, C
H), 4.08/3.89 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 162.62 (
C-4, aromatic), 134.59 (
C-4, phenyl), 133.94 (
C-1, phenyl), 131.38 (
CH-2,6, aromatic), 130.08 (
CH-2,6, phenyl), 129.77 (
C-2, thiophene), 129.53 (
CH-3,5, phenyl), 129.26 (
C-1, aromatic), 129.06 (
C-3, thiophene), 127.44 (
C-4, thiophene), 126.96 (
C-5, thiophene), 115.77 (
CH-3,5, aromatic), 66.93 (
CH), 50.11 (
CH
2) (
Figure S83c).
8u. HRESIMS
m/
z 387.0050 [M+Na]
+ (calcd for C
18H
14ClFNaOS
2+, 387.0051, Δ = 0.1 ppm) (
Figure S84). HPLC R
t 26.27 min, purity > 99% (
Figure S84a). Chiral HPLC R
t 10.81 min, >99% ee (
Figure S84b).
1H NMR (500 MHz, CDCl
3, 23 °C): δ = 7.40 (C
H-3,5, phenyl), 7.37 (C
H-2’,6’, phenyl), 7.35 (C
H-5, thiophene), 7.33 (C
H-2,6, phenyl), 7.08 (C
H-3’,5’, phenyl), 7.06 (C
H-4, thiophene), 6.96 (C
H-3, thiophene), 4.68 (s, 1H, C
H), 4.08/3.89 (AB, 2H, C
H2).
13C NMR (125.75 MHz, CDCl
3, 23 °C): δ = 162.62 (
C-4, aromatic), 134.59 (
C-4, phenyl), 133.93 (
C-1, phenyl), 131.38 (
CH-2,6, aromatic), 130.08 (
CH-2,6, phenyl), 129.77 (
C-2, thiophene), 129.53 (
CH-3,5, phenyl), 129.26 (
C-1, aromatic), 129.06 (
C-3, thiophene), 127.44 (
C-4, thiophene), 126.96 (
C-5, thiophene), 115.77 (
CH-3,5, aromatic), 66.93 (
CH), 50.11 (
CH
2) (
Figure S84c).



 ,
, 
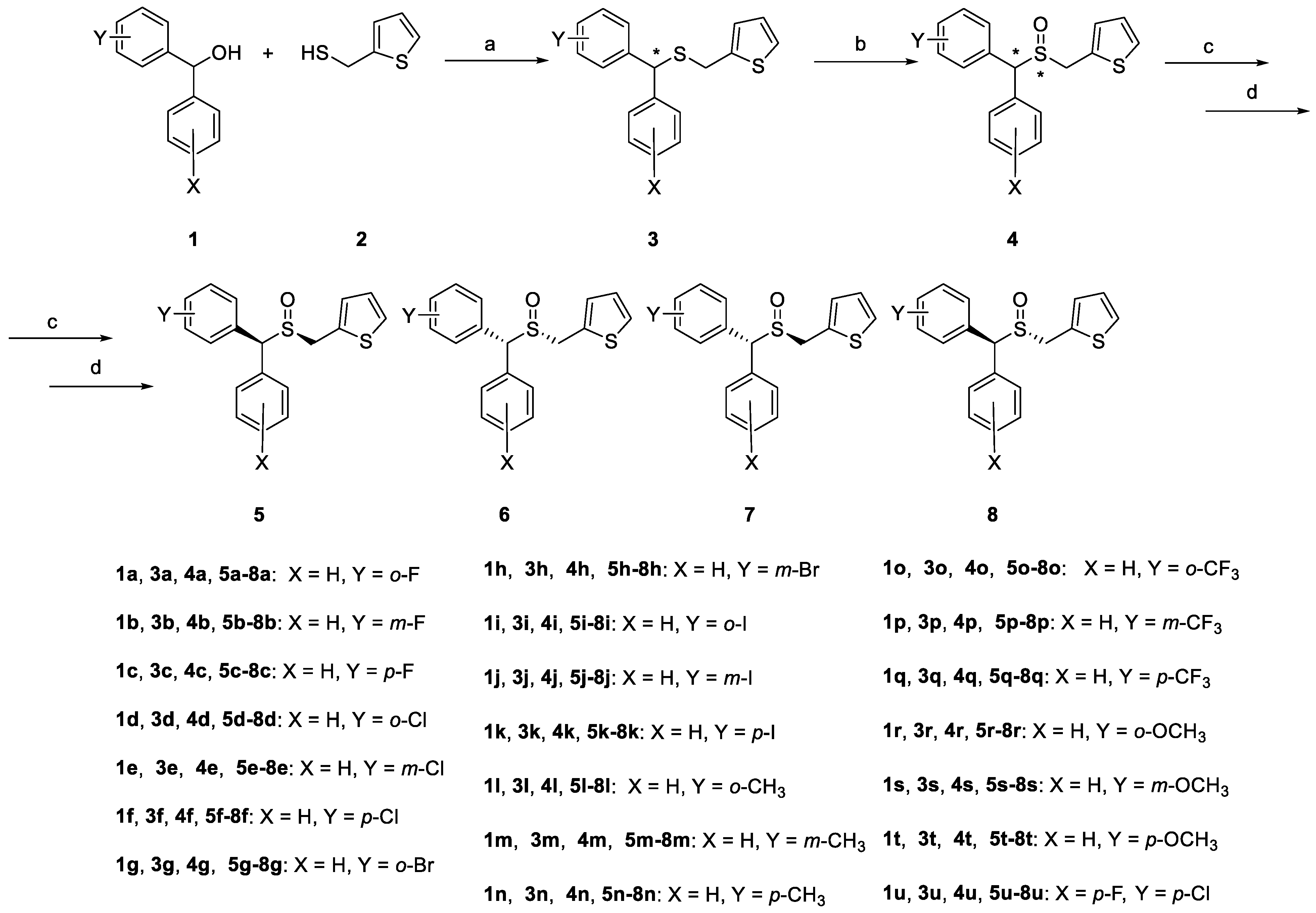
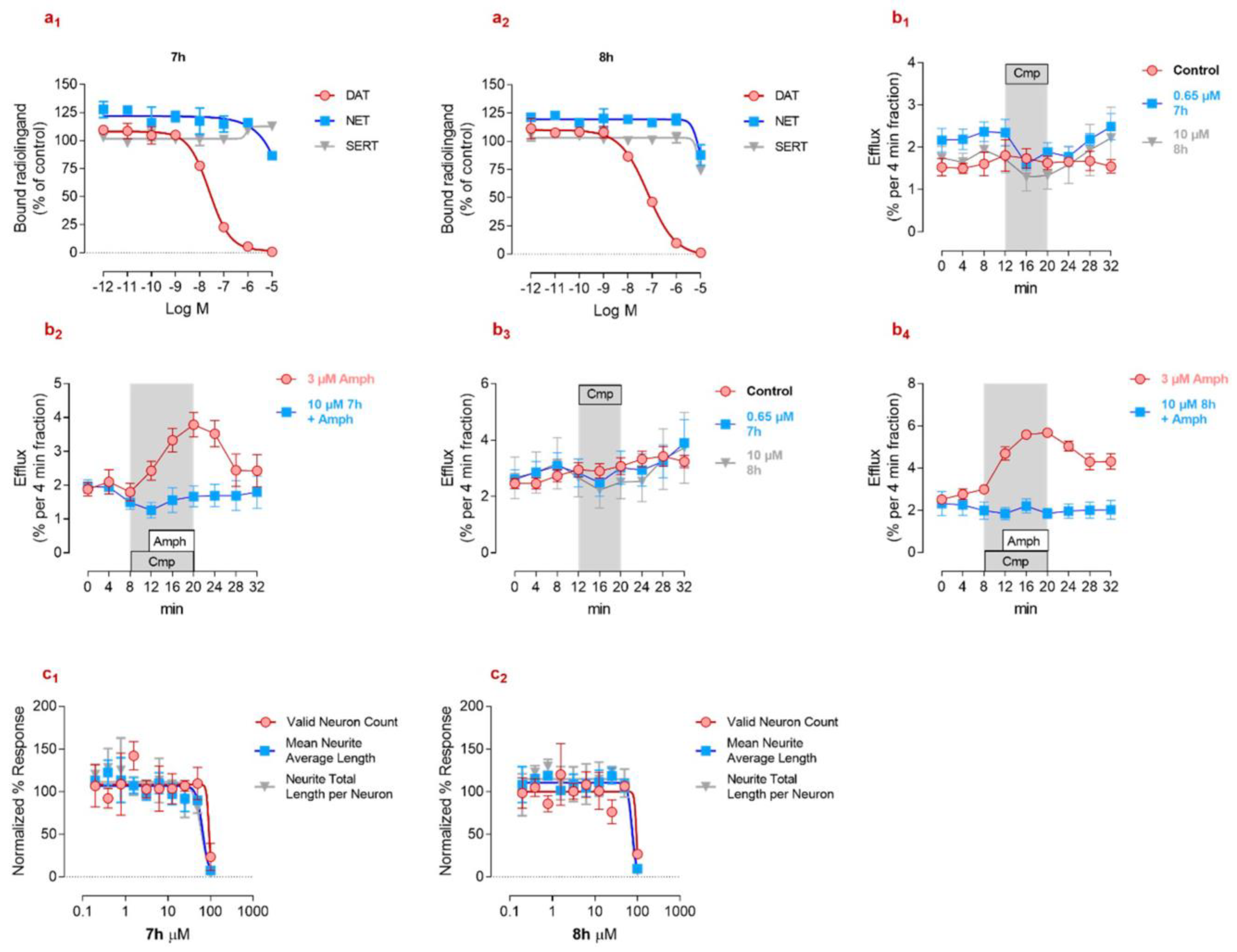


{kind=link}
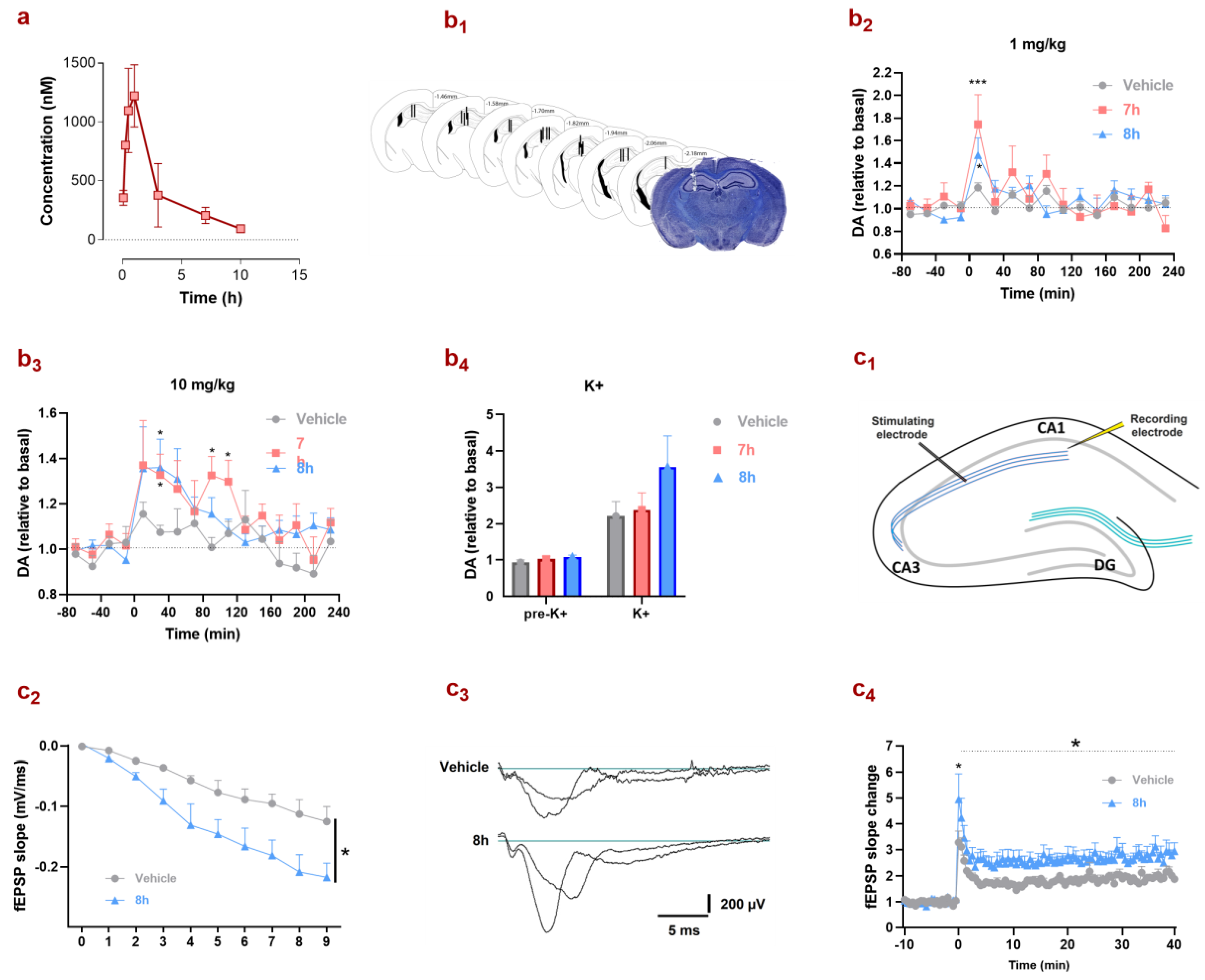{kind=link}
{kind=link}