
AAV-Mediated Targeting of the Activin A-ACVR1R206H Signaling in Fibrodysplasia Ossificans Progressiva



{kind=link}
{kind=link}
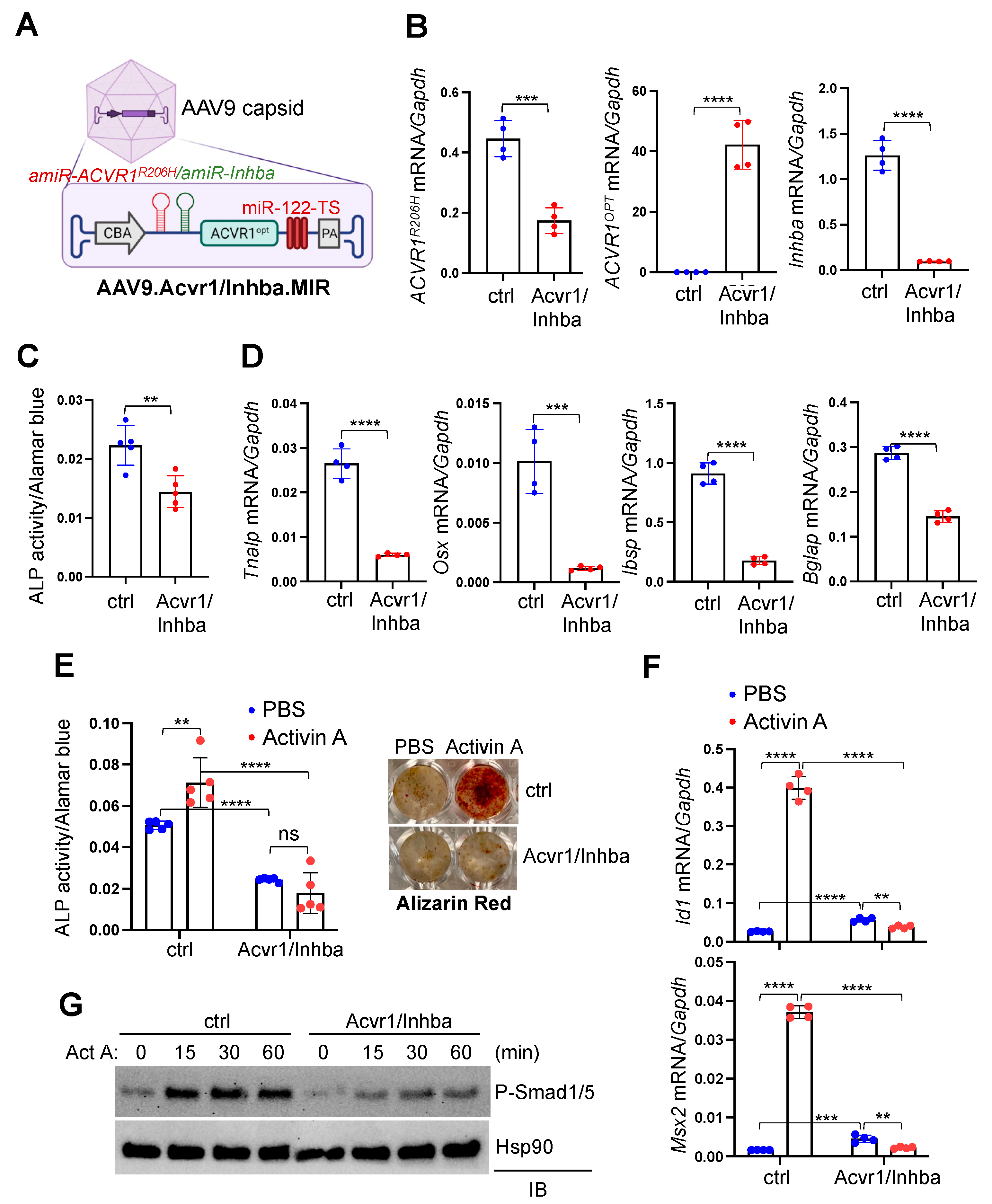
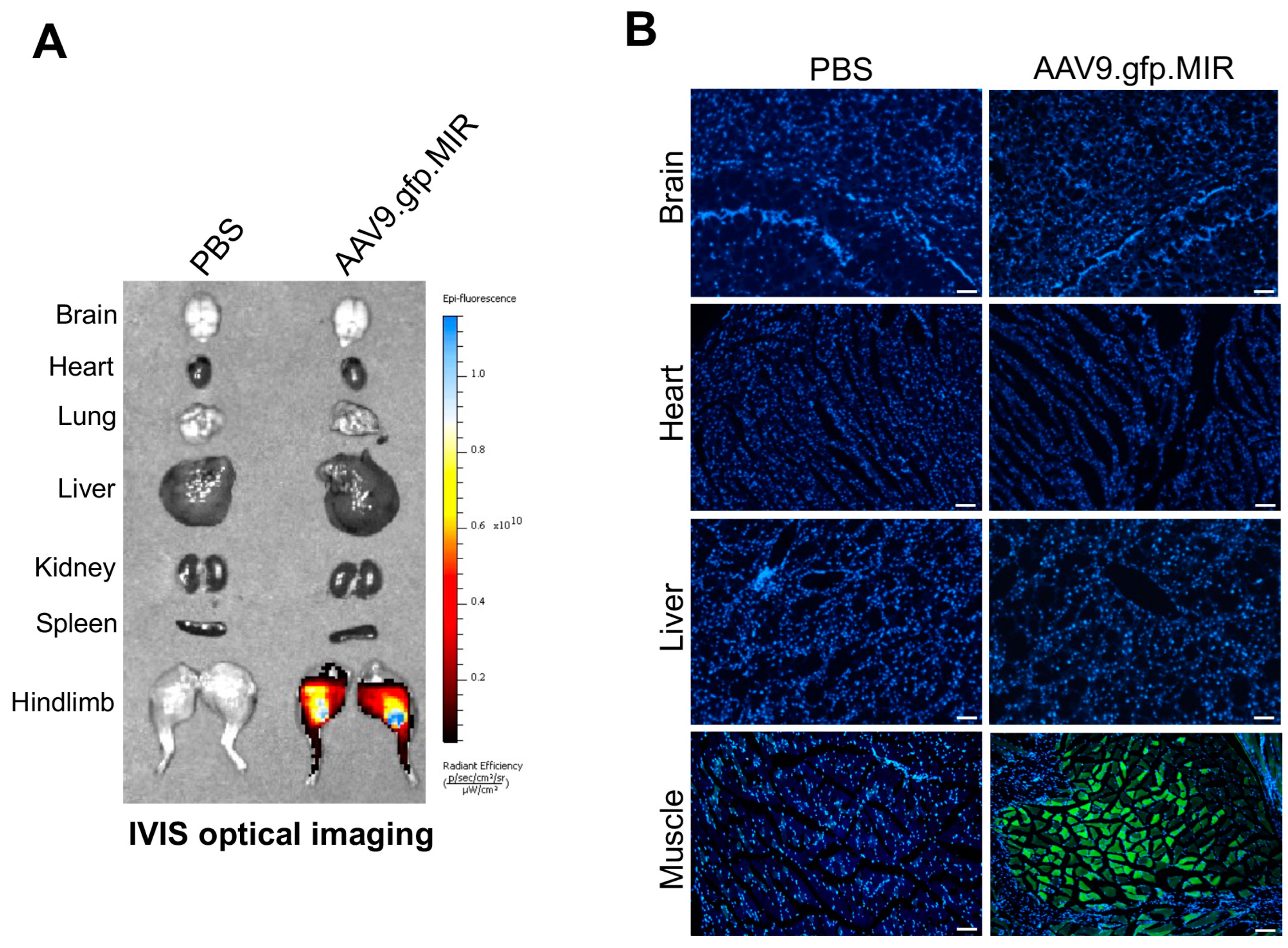{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Cell Culture and Reagents
2.3. rAAV Vector Design and Production
2.4. Quantitative RT-PCR, Immunoblotting, and ELISA Analyses
2.5. Trauma-Induced HO
2.6. Osteoblast Differentiation
2.7. MicroCT and Radiography
2.8. Histology
2.9. Statistical Analysis
3. Results
3.1. Upregulation of Activin A in HO-Tissues of FOP Mice
3.2. Generation of the AAV Gene Therapy Targeting Activin A
3.3. Generation of the AAV Gene Therapy with Liver-Specific Repression
3.4. AAV Gene Therapy Inhibits Aberrant ACVR1R206H Signaling and Osteogenic Differentiation of Acvr1R206H Skeletal Progenitors
3.5. AAV Gene Therapy Prevents Trauma-Induced HO in FOP Mice
3.6. Treatment of Traumatic HO in FOP Mice through AAV Gene Therapy
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pignolo, R.J.; Hsiao, E.C.; Baujat, G.; Lapidus, D.; Sherman, A.; Kaplan, F.S. Prevalence of fibrodysplasia ossificans progressiva (FOP) in the United States: Estimate from three treatment centers and a patient organization. Orphanet J. Rare Dis. 2021, 16, 350. [Google Scholar] [CrossRef] [PubMed]
- Pignolo, R.J.; Shore, E.M.; Kaplan, F.S. Fibrodysplasia ossificans progressiva: Diagnosis, management, and therapeutic horizons. Pediatr. Endocrinol. Rev. 2013, 10 (Suppl. 2), 437–448. [Google Scholar]
- Wentworth, K.L.; Masharani, U.; Hsiao, E.C. Therapeutic advances for blocking heterotopic ossification in fibrodysplasia ossificans progressiva. Br. J. Clin. Pharmacol. 2019, 85, 1180–1187. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.S.; Xie, J.; Wang, D.; Kim, J.M.; Tai, P.W.L.; Gravallese, E.; Gao, G.; Shim, J.H. Bone-targeting AAV-mediated silencing of Schnurri-3 prevents bone loss in osteoporosis. Nat. Commun. 2019, 10, 2958. [Google Scholar] [CrossRef] [PubMed]
- Herzog, R.W. Encouraging and Unsettling Findings in Long-Term Follow-up of AAV Gene Transfer. Mol. Ther. 2020, 28, 341–342. [Google Scholar] [CrossRef]
- Shore, E.M.; Xu, M.; Feldman, G.J.; Fenstermacher, D.A.; Cho, T.J.; Choi, I.H.; Connor, J.M.; Delai, P.; Glaser, D.L.; LeMerrer, M.; et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 2006, 38, 525–527. [Google Scholar] [CrossRef]
- Lees-Shepard, J.B.; Yamamoto, M.; Biswas, A.A.; Stoessel, S.J.; Nicholas, S.E.; Cogswell, C.A.; Devarakonda, P.M.; Schneider, M.J., Jr.; Cummins, S.M.; Legendre, N.P.; et al. Activin-dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva. Nat. Commun. 2018, 9, 471. [Google Scholar] [CrossRef]
- Wosczyna, M.N.; Biswas, A.A.; Cogswell, C.A.; Goldhamer, D.J. Multipotent progenitors resident in the skeletal muscle interstitium exhibit robust BMP-dependent osteogenic activity and mediate heterotopic ossification. J. Bone Miner. Res. 2012, 27, 1004–1017. [Google Scholar] [CrossRef]
- Yang, Y.S.; Kim, J.M.; Xie, J.; Chaugule, S.; Lin, C.; Ma, H.; Hsiao, E.; Hong, J.; Chun, H.; Shore, E.M.; et al. Suppression of heterotopic ossification in fibrodysplasia ossificans progressiva using AAV gene delivery. Nat. Commun. 2022, 13, 6175. [Google Scholar] [CrossRef]
- Hino, K.; Ikeya, M.; Horigome, K.; Matsumoto, Y.; Ebise, H.; Nishio, M.; Sekiguchi, K.; Shibata, M.; Nagata, S.; Matsuda, S.; et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc. Natl. Acad. Sci. USA 2015, 112, 15438–15443. [Google Scholar] [CrossRef]
- Aykul, S.; Corpina, R.A.; Goebel, E.J.; Cunanan, C.J.; Dimitriou, A.; Kim, H.J.; Zhang, Q.; Rafique, A.; Leidich, R.; Wang, X.; et al. Activin A forms a non-signaling complex with ACVR1 and type II Activin/BMP receptors via its finger 2 tip loop. eLife 2020, 9, e54582. [Google Scholar] [CrossRef] [PubMed]
- Hatsell, S.J.; Idone, V.; Wolken, D.M.; Huang, L.; Kim, H.J.; Wang, L.; Wen, X.; Nannuru, K.C.; Jimenez, J.; Xie, L.; et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci. Transl. Med. 2015, 7, 303ra137. [Google Scholar] [CrossRef]
- Alessi Wolken, D.M.; Idone, V.; Hatsell, S.J.; Yu, P.B.; Economides, A.N. The obligatory role of Activin A in the formation of heterotopic bone in Fibrodysplasia Ossificans Progressiva. Bone 2018, 109, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Stoessel, S.J.; Yamamoto, S.; Goldhamer, D.J. Overexpression of Wild-Type ACVR1 in Fibrodysplasia Ossificans Progressiva Mice Rescues Perinatal Lethality and Inhibits Heterotopic Ossification. J. Bone Miner. Res. 2022, 37, 2077–2093. [Google Scholar] [CrossRef] [PubMed]
- Lyu, H.; Elkins, C.M.; Pierce, J.L.; Serezani, C.H.; Perrien, D.S. MyD88 Is Not Required for Muscle Injury-Induced Endochondral Heterotopic Ossification in a Mouse Model of Fibrodysplasia Ossificans Progressiva. Biomedicines 2021, 9, 630. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Tai, P.W.L.; Brown, A.; Gong, S.; Zhu, S.; Wang, Y.; Li, C.; Colpan, C.; Su, Q.; He, R.; et al. Effective and Accurate Gene Silencing by a Recombinant AAV-Compatible MicroRNA Scaffold. Mol. Ther. 2020, 28, 422–430. [Google Scholar] [CrossRef]
- Gregory, C.A.; Gunn, W.G.; Peister, A.; Prockop, D.J. An Alizarin red-based assay of mineralization by adherent cells in culture: Comparison with cetylpyridinium chloride extraction. Anal. Biochem. 2004, 329, 77–84. [Google Scholar] [CrossRef]
- Namwanje, M.; Brown, C.W. Activins and Inhibins: Roles in Development, Physiology, and Disease. Cold Spring Harb. Perspect. Biol. 2016, 8, a021881. [Google Scholar] [CrossRef]
- Convente, M.R.; Chakkalakal, S.A.; Yang, E.; Caron, R.J.; Zhang, D.; Kambayashi, T.; Kaplan, F.S.; Shore, E.M. Depletion of Mast Cells and Macrophages Impairs Heterotopic Ossification in an Acvr1(R206H) Mouse Model of Fibrodysplasia Ossificans Progressiva. J. Bone Miner. Res. 2018, 33, 269–282. [Google Scholar] [CrossRef]
- Mundy, C.; Yao, L.; Sinha, S.; Chung, J.; Rux, D.; Catheline, S.E.; Koyama, E.; Qin, L.; Pacifici, M. Activin A promotes the development of acquired heterotopic ossification and is an effective target for disease attenuation in mice. Sci. Signal 2021, 14, eabd0536. [Google Scholar] [CrossRef]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Nicolas, E.; Marks, D.; Sander, C.; Lerro, A.; Buendia, M.A.; Xu, C.; Mason, W.S.; Moloshok, T.; Bort, R.; et al. miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol. 2004, 1, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Lagos-Quintana, M.; Rauhut, R.; Yalcin, A.; Meyer, J.; Lendeckel, W.; Tuschl, T. Identification of tissue-specific microRNAs from mouse. Curr. Biol. 2002, 12, 735–739. [Google Scholar] [CrossRef]
- Chakkalakal, S.A.; Zhang, D.; Culbert, A.L.; Convente, M.R.; Caron, R.J.; Wright, A.C.; Maidment, A.D.; Kaplan, F.S.; Shore, E.M. An Acvr1 R206H knock-in mouse has fibrodysplasia ossificans progressiva. J. Bone Miner. Res. 2012, 27, 1746–1756. [Google Scholar] [CrossRef] [PubMed]
- Lounev, V.Y.; Ramachandran, R.; Wosczyna, M.N.; Yamamoto, M.; Maidment, A.D.; Shore, E.M.; Glaser, D.L.; Goldhamer, D.J.; Kaplan, F.S. Identification of progenitor cells that contribute to heterotopic skeletogenesis. J. Bone Jt. Surg. Am. 2009, 91, 652–663. [Google Scholar] [CrossRef]
- Carpentier, A.C.; Frisch, F.; Labbé, S.M.; Gagnon, R.; de Wal, J.; Greentree, S.; Petry, H.; Twisk, J.; Brisson, D.; Gaudet, D. Effect of alipogene tiparvovec (AAV1-LPL(S447X)) on postprandial chylomicron metabolism in lipoprotein lipase-deficient patients. J. Clin. Endocrinol. Metab. 2012, 97, 1635–1644. [Google Scholar] [CrossRef]
- Chiu, W.; Lin, T.Y.; Chang, Y.C.; Isahwan-Ahmad Mulyadi Lai, H.; Lin, S.C.; Ma, C.; Yarmishyn, A.A.; Lin, S.C.; Chang, K.J.; Chou, Y.B.; et al. An Update on Gene Therapy for Inherited Retinal Dystrophy: Experience in Leber Congenital Amaurosis Clinical Trials. Int. J. Mol. Sci. 2021, 22, 4534. [Google Scholar] [CrossRef]
- Mendell, J.R.; Al-Zaidy, S.A.; Lehman, K.J.; McColly, M.; Lowes, L.P.; Alfano, L.N.; Reash, N.F.; Iammarino, M.A.; Church, K.R.; Kleyn, A.; et al. Five-Year Extension Results of the Phase 1 START Trial of Onasemnogene Abeparvovec in Spinal Muscular Atrophy. JAMA Neurol. 2021, 78, 834–841. [Google Scholar] [CrossRef]
- Mullard, A. FDA approves first gene therapy for Duchenne muscular dystrophy, despite internal objections. Nat. Rev. Drug Discov. 2023. [Google Scholar] [CrossRef]
- Naddaf, M. Researchers welcome $3.5-million haemophilia gene therapy—But questions remain. Nature 2022, 612, 388–389. [Google Scholar] [CrossRef]
- Shore, E.M.; Kaplan, F.S. Insights from a rare genetic disorder of extra-skeletal bone formation, fibrodysplasia ossificans progressiva (FOP). Bone 2008, 43, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Heine, S.J.; Diaz-McNair, J.; Andar, A.U.; Drachenberg, C.B.; van de Verg, L.; Walker, R.; Picking, W.L.; Pasetti, M.F. Intradermal delivery of Shigella IpaB and IpaD type III secretion proteins: Kinetics of cell recruitment and antigen uptake, mucosal and systemic immunity, and protection across serotypes. J. Immunol. 2014, 192, 1630–1640. [Google Scholar] [CrossRef]
- Eekhoff, E.M.W.; de Ruiter, R.D.; Smilde, B.J.; Schoenmaker, T.; de Vries, T.J.; Netelenbos, C.; Hsiao, E.C.; Scott, C.; Haga, N.; Grunwald, Z.; et al. Gene therapy for Fibrodysplasia Ossificans Progressiva (FOP): Feasibility and obstacles. Hum. Gene Ther. 2022, 33, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.M.; Yeh, W.H.; Pendse, N.; Davis, J.R.; Hennessey, E.; Butcher, R.; Koblan, L.W.; Comander, J.; Liu, Q.; Liu, D.R. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat. Biomed. Eng. 2020, 4, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bamidele, N.; Liu, P.; Ojelabi, O.; Gao, X.D.; Rodriguez, T.; Cheng, H.; Kelly, K.; Watts, J.K.; Xie, J.; et al. Adenine Base Editing In Vivo with a Single Adeno-Associated Virus Vector. GEN Biotechnol. 2022, 1, 285–299. [Google Scholar] [CrossRef]
- Davis, J.R.; Wang, X.; Witte, I.P.; Huang, T.P.; Levy, J.M.; Raguram, A.; Banskota, S.; Seidah, N.G.; Musunuru, K.; Liu, D.R. Efficient in vivo base editing via single adeno-associated viruses with size-optimized genomes encoding compact adenine base editors. Nat. Biomed. Eng. 2022, 6, 1272–1283. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.-S.; Lin, C.; Ma, H.; Xie, J.; Kaplan, F.S.; Gao, G.; Shim, J.-H. AAV-Mediated Targeting of the Activin A-ACVR1R206H Signaling in Fibrodysplasia Ossificans Progressiva. Biomolecules 2023, 13, 1364. https://doi.org/10.3390/biom13091364
Yang Y-S, Lin C, Ma H, Xie J, Kaplan FS, Gao G, Shim J-H. AAV-Mediated Targeting of the Activin A-ACVR1R206H Signaling in Fibrodysplasia Ossificans Progressiva. Biomolecules. 2023; 13(9):1364. https://doi.org/10.3390/biom13091364
Chicago/Turabian StyleYang, Yeon-Suk, Chujiao Lin, Hong Ma, Jun Xie, Frederick S. Kaplan, Guangping Gao, and Jae-Hyuck Shim. 2023. "AAV-Mediated Targeting of the Activin A-ACVR1R206H Signaling in Fibrodysplasia Ossificans Progressiva" Biomolecules 13, no. 9: 1364. https://doi.org/10.3390/biom13091364
APA StyleYang, Y.-S., Lin, C., Ma, H., Xie, J., Kaplan, F. S., Gao, G., & Shim, J.-H. (2023). AAV-Mediated Targeting of the Activin A-ACVR1R206H Signaling in Fibrodysplasia Ossificans Progressiva. Biomolecules, 13(9), 1364. https://doi.org/10.3390/biom13091364