
Sequences of Alterations in Inflammation and Autophagy Processes in Rd1 Mice

 ,
,  , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
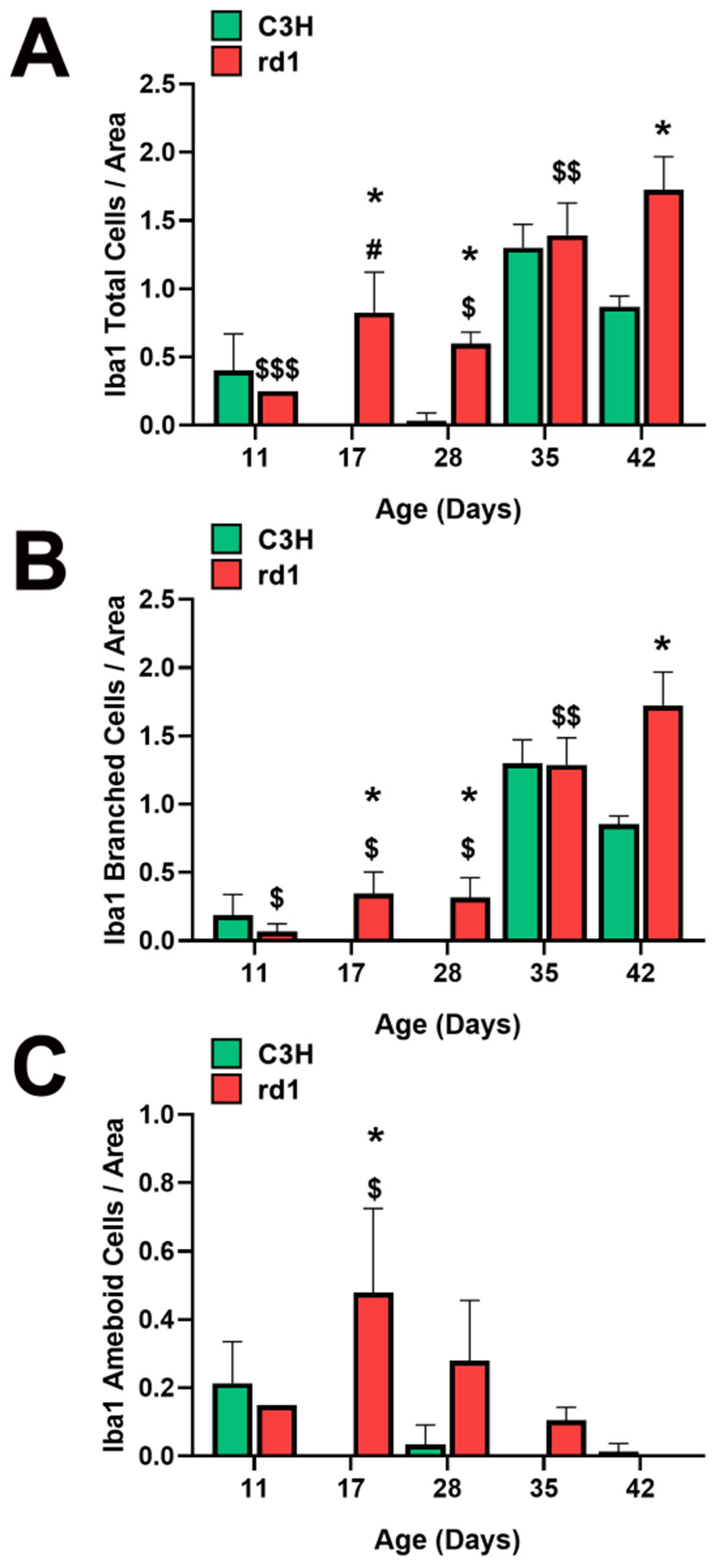
3.1. Microglia Expression in the Retina of the Rd1 Mice Model
3.2. Quantification of Macroautophagy and CMA Markers in the Retinas of Control and Rd1 Mice at Different Postnatal Ages
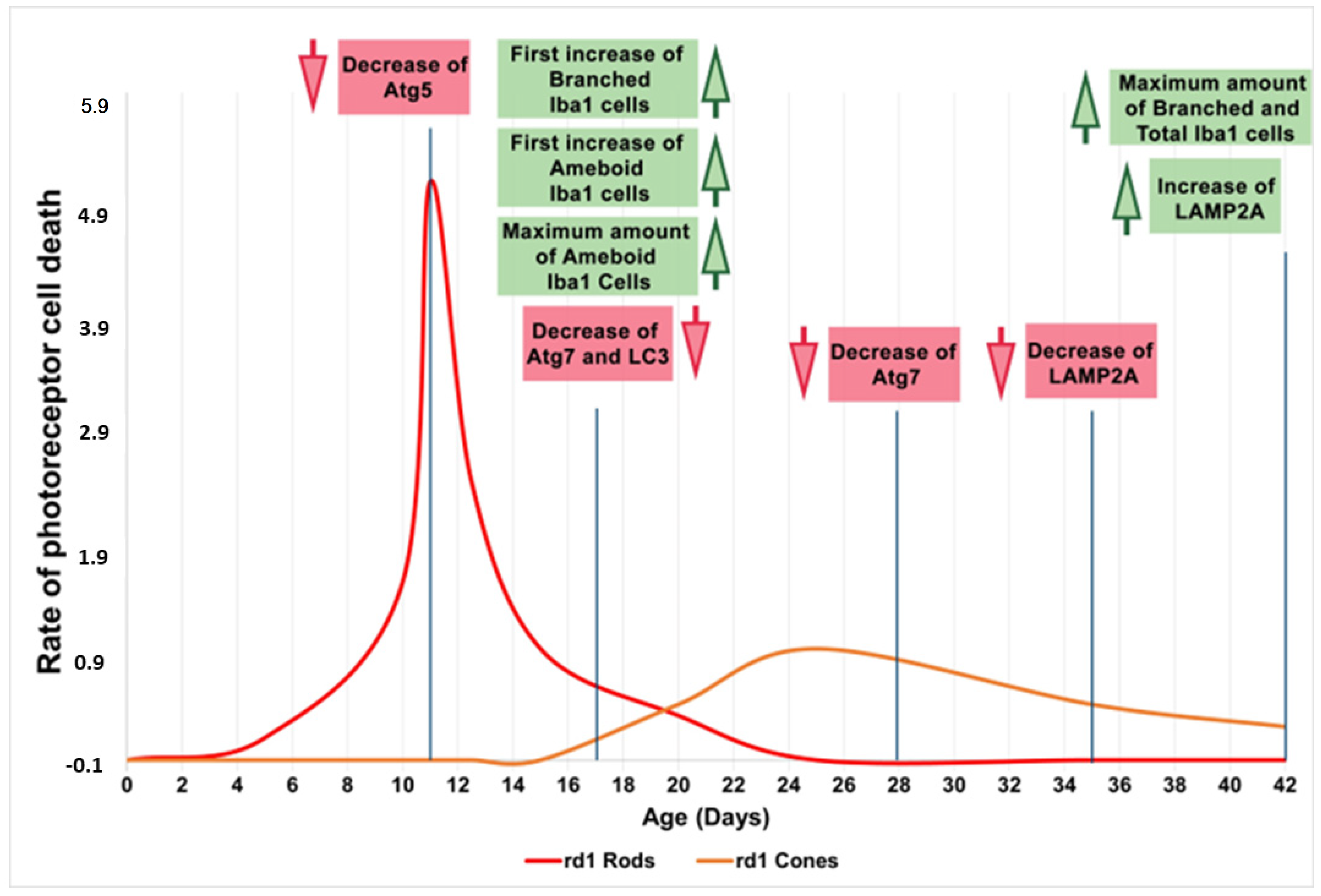
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hamel, C. Retinitis pigmentosa. Orphanet J. Rare Dis. 2006, 1, 40. [Google Scholar] [CrossRef]
- Tsujikawa, M.; Wada, Y.; Sukegawa, M.; Sawa, M.; Gomi, F.; Nishida, K.; Tano, Y. Age at onset curves of retinitis pigmentosa. Arch. Ophthalmol. 2008, 126, 337–340. [Google Scholar] [CrossRef]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar]
- Li, Z.Y.; Possin, D.E.; Milam, A.H. Histopathology of bone spicule pigmentation in retinitis pigmentosa. Ophthalmology 1995, 102, 805–816. [Google Scholar] [CrossRef]
- Olivares-González, L.; Velasco, S.; Campillo, I.; Rodrigo, R. Retinal Inflammation, Cell Death and Inherited Retinal Dystrophies. Int. J. Mol. Sci. 2021, 22, 2096. [Google Scholar] [CrossRef]
- McMurtrey, J.J.; Tso, M.O.M. A review of the immunologic findings observed in retinitis pigmentosa. Surv. Ophthalmol. 2018, 63, 769–781. [Google Scholar] [CrossRef]
- Massengill, M.T.; Ahmed, C.M.; Lewin, A.S.; Ildefonso, C.J. Neuroinflammation in Retinitis Pigmentosa, Diabetic Retinopathy, and Age-Related Macular Degeneration: A Minireview. Adv. Exp. Med. Biol. 2018, 1074, 185–191. [Google Scholar]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Reichenbach, A.; Bringmann, A. New functions of Müller cells. Glia 2013, 61, 651–678. [Google Scholar]
- Penn, J.S.; Madan, A.; Caldwell, R.B.; Bartoli, M.; Caldwell, R.W.; Hartnett, M.E. Vascular endothelial growth factor in eye disease. Prog. Retin. Eye Res. 2008, 27, 331–371. [Google Scholar]
- Wang, S.K.; Cepko, C.L. Targeting Microglia to Treat Degenerative Eye Diseases. Front. Immunol. 2022, 13, 843558. [Google Scholar] [CrossRef]
- Newton, F.; Megaw, R. Mechanisms of Photoreceptor Death in Retinitis Pigmentosa. Genes 2020, 11, 1120. [Google Scholar] [CrossRef]
- Gao, H.; Huang, X.; He, J.; Zou, T.; Chen, X.; Xu, H. The roles of microglia in neural remodeling during retinal degeneration. Histol. Histopathol. 2022, 37, 1–10. [Google Scholar]
- Di Pierdomenico, J.; García-Ayuso, D.; Pinilla, I.; Cuenca, N.; Vidal-Sanz, M.; Agudo-Barriuso, M.; Villegas-Pérez, M.P. Early Events in Retinal Degeneration Caused by Rhodopsin Mutation or Pigment Epithelium Malfunction: Differences and Similarities. Front. Neuroanat. 2017, 11, 14. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef]
- Platini, F.; Pérez-Tomás, R.; Ambrosio, S.; Tessitore, L. Understanding autophagy in cell death control. Curr. Pharm. Des. 2010, 16, 101–113. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy. Curr. Biol. 2005, 15, R282–R283. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef]
- Cuervo, A.M. Chaperone-mediated autophagy: Selectivity pays off. Trends Endocrinol. Metab. 2010, 21, 142–150. [Google Scholar] [CrossRef]
- Li, W.W.; Li, J.; Bao, J.K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef]
- Moreno, M.L.; Mérida, S.; Bosch-Morell, F.; Miranda, M.; Villar, V.M. Autophagy Dysfunction and Oxidative Stress, Two Related Mechanisms Implicated in Retinitis Pigmentosa. Front. Physiol. 2018, 9, 1008. [Google Scholar] [CrossRef]
- Chai, P.; Ni, H.; Zhang, H.; Fan, X. The Evolving Functions of Autophagy in Ocular Health: A Double-edged Sword. Int. J. Biol. Sci. 2016, 12, 1332–1340. [Google Scholar] [CrossRef]
- Pittler, S.J.; Baehr, W. Identification of a nonsense mutation in the rod photoreceptor cGMP phosphodiesterase beta-subunit gene of the rd mouse. Proc. Natl. Acad. Sci. USA 1991, 88, 8322–8326. [Google Scholar] [CrossRef]
- Bowes, C.; Li, T.; Frankel, W.N.; Danciger, M.; Coffin, J.M.; Applebury, M.L.; Farber, D.B. Localization of a retroviral element within the rd gene coding for the beta subunit of cGMP phosphodiesterase. Proc. Natl. Acad. Sci. USA 1993, 90, 2955–2959. [Google Scholar] [CrossRef]
- McLaughlin, M.E.; Ehrhart, T.L.; Berson, E.L.; Dryja, T.P. Mutation spectrum of the gene encoding the beta subunit of rod phosphodiesterase among patients with autosomal recessive retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 1995, 92, 3249–3253. [Google Scholar] [CrossRef]
- Farber, D.B.; Lolley, R.N. Enzymic basis for cyclic GMP accumulation in degenerative photoreceptor cells of mouse retina. J. Cycl. Nucleotide Res. 1976, 2, 139–148. [Google Scholar]
- Otani, A.; Dorrell, M.I.; Kinder, K.; Moreno, S.K.; Nusinowitz, S.; Banin, E.; Heckenlively, J.; Friedlander, M. Rescue of retinal degeneration by intravitreally injected adult bone marrow-derived lineage-negative hematopoietic stem cells. J. Clin. Investig. 2004, 114, 765–774. [Google Scholar] [CrossRef]
- Punzo, C.; Cepko, C. Cellular responses to photoreceptor death in the rd1 mouse model of retinal degeneration. Investig. Ophthalmol. Vis. Sci. 2007, 48, 849–857. [Google Scholar] [CrossRef]
- Pierce, E.A. Pathways to photoreceptor cell death in inherited retinal degenerations. BioEssays News Rev. Mol. Cell. Dev. Biol. 2001, 23, 605–618. [Google Scholar] [CrossRef]
- Bradford, M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Zhang, M.; Zhong, L.; Han, X.; Xiong, G.; Xu, D.; Zhang, S.; Cheng, H.; Chiu, K.; Xu, Y. Brain and Retinal Abnormalities in the 5xFAD Mouse Model of Alzheimer’s Disease at Early Stages. Front. Neurosci. 2021, 15, 681831. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Kim, H.P.; Jin, Y.; Choi, A.M.; Ryter, S.W. Beclin 1 deficiency is associated with increased hypoxia-induced angiogenesis. Autophagy 2011, 7, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Ross, B.X.; Jia, L.; Kong, D.; Wang, T.; Hager, H.M.; Abcouwer, S.F.; Zacks, D.N. Conditional Knock out of High-Mobility Group Box 1 (HMGB1) in Rods Reduces Autophagy Activation after Retinal Detachment. Cells 2021, 10, 2010. [Google Scholar] [CrossRef]
- Giansanti, V.; Rodriguez, G.E.; Savoldelli, M.; Gioia, R.; Forlino, A.; Mazzini, G.; Pennati, M.; Zaffaroni, N.; Scovassi, A.I.; Torriglia, A. Characterization of stress response in human retinal epithelial cells. J. Cell. Mol. Med. 2013, 17, 103–115. [Google Scholar] [CrossRef]
- Gupta, U.; Ghosh, S.; Wallace, C.T.; Shang, P.; Xin, Y.; Nair, A.P.; Yazdankhah, M.; Strizhakova, A.; Ross, M.A.; Liu, H.; et al. Increased LCN2 (lipocalin 2) in the RPE decreases autophagy and activates inflammasome-ferroptosis processes in a mouse model of dry AMD. Autophagy 2023, 19, 92–111. [Google Scholar] [CrossRef]
- Trachsel-Moncho, L.; Benlloch-Navarro, S.; Fernández-Carbonell, Á.; Ramírez-Lamelas, D.T.; Olivar, T.; Silvestre, D.; Poch, E.; Miranda, M. Oxidative stress and autophagy-related changes during retinal degeneration and development. Cell Death Dis. 2018, 9, 812. [Google Scholar] [CrossRef]
- Menon, M.B.; Dhamija, S. Beclin 1 Phosphorylation—At the Center of Autophagy Regulation. Front. Cell Dev. Biol. 2018, 6, 137. [Google Scholar] [CrossRef]
- Bestebroer, J.; V’kovski, P.; Mauthe, M.; Reggiori, F. Hidden behind autophagy: The unconventional roles of ATG proteins. Traffic 2013, 14, 1029–1041. [Google Scholar] [CrossRef] [PubMed]
- Sancho-Pelluz, J.; Wunderlich, K.A.; Rauch, U.; Romero, F.J.; van Veen, T.; Limb, G.A.; Crocker, P.R.; Perez, M.T. Sialoadhesin expression in intact degenerating retinas and following transplantation. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5602–5610. [Google Scholar] [CrossRef]
- Zhou, T.; Huang, Z.; Zhu, X.; Sun, X.; Liu, Y.; Cheng, B.; Li, M.; Liu, Y.; He, C.; Liu, X. Alpha-1 Antitrypsin Attenuates M1 Microglia-Mediated Neuroinflammation in Retinal Degeneration. Front. Immunol. 2018, 9, 1202. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Huang, Z.; Sun, X.; Zhu, X.; Zhou, L.; Li, M.; Cheng, B.; Liu, X.; He, C. Microglia Polarization with M1/M2 Phenotype Changes in rd1 Mouse Model of Retinal Degeneration. Front. Neuroanat. 2017, 11, 77. [Google Scholar] [CrossRef]
- Harada, T.; Harada, C.; Kohsaka, S.; Wada, E.; Yoshida, K.; Ohno, S.; Mamada, H.; Tanaka, K.; Parada, L.F.; Wada, K. Microglia-Müller glia cell interactions control neurotrophic factor production during light-induced retinal degeneration. J. Neurosci. 2002, 22, 9228–9236. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhao, C.; Dai, J.; Weng, C.H.; Bian, B.S.J.; Gong, Y.; Ge, L.; Fang, Y.; Liu, H.; Xu, H.; et al. Microglia mediate mynaptic material mlearance at the early stage of rats with retinitis pigmentosa. Front. Immunol. 2019, 10, 912. [Google Scholar] [CrossRef]
- Gupta, N.; Brown, K.E.; Milam, A.H. Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp. Eye Res. 2003, 76, 463–471. [Google Scholar] [CrossRef]
- Lin, W.; Xu, G. Autophagy: A Role in the Apoptosis, Survival, Inflammation, and Development of the Retina. Ophthalmic Res. 2019, 61, 65–72. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Gestwicki, J.E.; Murphy, L.O.; Klionsky, D.J. Potential therapeutic applications of autophagy. Nat. Rev. Drug Discov. 2007, 6, 304–312. [Google Scholar] [CrossRef]
- Williams, A.; Jahreiss, L.; Sarkar, S.; Saiki, S.; Menzies, F.M.; Ravikumar, B.; Rubinsztein, D.C. Aggregate-prone proteins are cleared from the cytosol by autophagy: Therapeutic implications. Curr. Top. Dev. Biol. 2006, 76, 89–101. [Google Scholar]
- Wang, T.; Lao, U.; Edgar, B.A. TOR-mediated autophagy regulates cell death in Drosophila neurodegenerative disease. J. Cell Biol. 2009, 186, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Jia, L.; Feathers, K.; Lin, C.; Khan, N.W.; Klionsky, D.J.; Ferguson, T.A.; Zacks, D.N. Autophagy-mediated catabolism of visual transduction proteins prevents retinal degeneration. Autophagy 2016, 12, 2439–2450. [Google Scholar] [CrossRef] [PubMed]
- Kunchithapautham, K.; Rohrer, B. Apoptosis and autophagy in photoreceptors exposed to oxidative stress. Autophagy 2007, 3, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Lohr, H.R.; Kuntchithapautham, K.; Sharma, A.K.; Rohrer, B. Multiple, parallel cellular suicide mechanisms participate in photoreceptor cell death. Exp. Eye Res. 2006, 83, 380–389. [Google Scholar] [CrossRef]
- Punzo, C.; Kornacker, K.; Cepko, C.L. Stimulation of the insulin/mTOR pathway delays cone death in a mouse model of retinitis pigmentosa. Nat. Neurosci. 2009, 12, 44–52. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Chaperones in autophagy. Pharmacol. Res. 2012, 66, 484–493. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rodríguez-Muela, N.; Koga, H.; García-Ledo, L.; de la Villa, P.; de la Rosa, E.J.; Cuervo, A.M.; Boya, P. Balance between autophagic pathways preserves retinal homeostasis. Aging Cell 2013, 12, 478–488. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Description | Concentration | Predicted Molecular Weight | Company | Catalog Number |
|---|---|---|---|---|---|
| Anti-Iba1 | Monoclonal rabbit | 1:500 | 17 kDa | Wako | Z0334 |
| Anti-Beclin-1 (H-300) | Polyclonal rabbit | 1:1000 | 50 kDa | Santa Cruz Biotechnology | sc-11427 |
| Anti-Atg5 | Polyclonal rabbit | 1:500 | 35 kDa | Novus Biologicals | NB110-53818 |
| Anti-Atg7 | Polyclonal rabbit | 1:1000 | 78 kDa | Cell Signaling | 2631S |
| Anti-LC3 | Polyclonal rabbit | 1:1000 | 17 kDa | Cell Signaling | 2775S |
| Anti-LAMP-2A | Polyclonal rabbit | 1:1000 | 120 kDa | Invitrogen | 51-2200 |
| Anti-β-Actin-Peroxidase | Monoclonal rabbit | 1:30,000 | 35 kDa | Sigma-Aldrich | A3854 |
| Antibody | Description | Concentration | Company | Catalog Number |
|---|---|---|---|---|
| Alexa Fluor® 488 | Goat anti-rabbit | 1:200 | Life Technologies | A11034 |
| IGG F(AB′)2-HRP | F(ab′)2 goat | 1:5000 | Santa Cruz Biotechnology | sc-3837 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-González, J.; Fernández-Carbonell, Á.; Cantó, A.; Gimeno-Hernández, R.; Almansa, I.; Bosch-Morell, F.; Miranda, M.; Olivar, T. Sequences of Alterations in Inflammation and Autophagy Processes in Rd1 Mice. Biomolecules 2023, 13, 1277. https://doi.org/10.3390/biom13091277
Martínez-González J, Fernández-Carbonell Á, Cantó A, Gimeno-Hernández R, Almansa I, Bosch-Morell F, Miranda M, Olivar T. Sequences of Alterations in Inflammation and Autophagy Processes in Rd1 Mice. Biomolecules. 2023; 13(9):1277. https://doi.org/10.3390/biom13091277
Chicago/Turabian StyleMartínez-González, Javier, Ángel Fernández-Carbonell, Antolin Cantó, Roberto Gimeno-Hernández, Inmaculada Almansa, Francisco Bosch-Morell, María Miranda, and Teresa Olivar. 2023. "Sequences of Alterations in Inflammation and Autophagy Processes in Rd1 Mice" Biomolecules 13, no. 9: 1277. https://doi.org/10.3390/biom13091277
APA StyleMartínez-González, J., Fernández-Carbonell, Á., Cantó, A., Gimeno-Hernández, R., Almansa, I., Bosch-Morell, F., Miranda, M., & Olivar, T. (2023). Sequences of Alterations in Inflammation and Autophagy Processes in Rd1 Mice. Biomolecules, 13(9), 1277. https://doi.org/10.3390/biom13091277