
Mechanisms of Diseases Associated with Mutation in GJC2/Connexin 47


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Glial Connexins
3. Mouse Models for the Study of Glial Connexins
4. Manifestations of GJC2 Mutations
5. PMLD (HLD2)
6. Hereditary Spastic Paraparesis (SPG44)
7. Subclinical Leukodystrophy
8. Mechanisms of GJC2 Associated Diseases
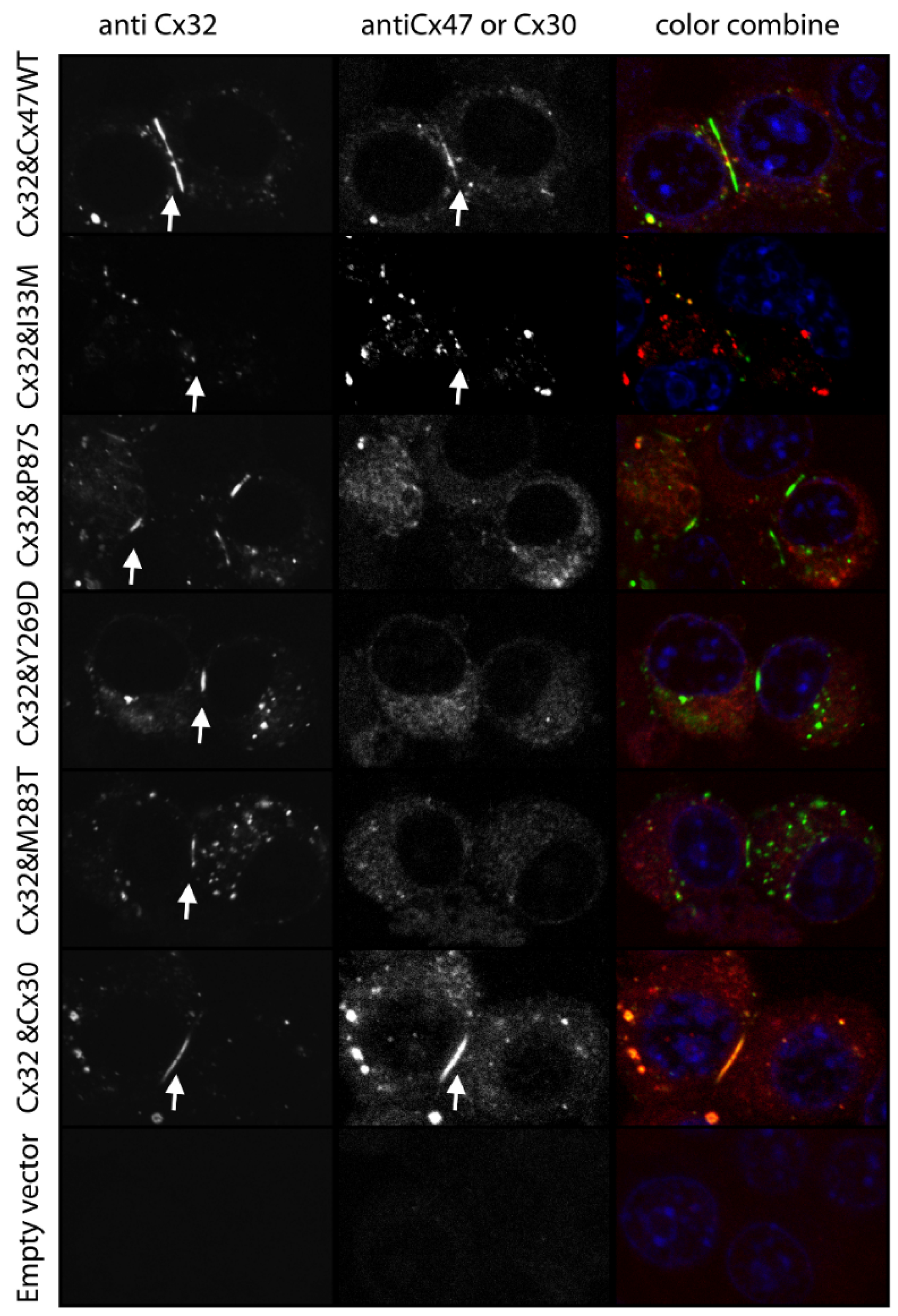
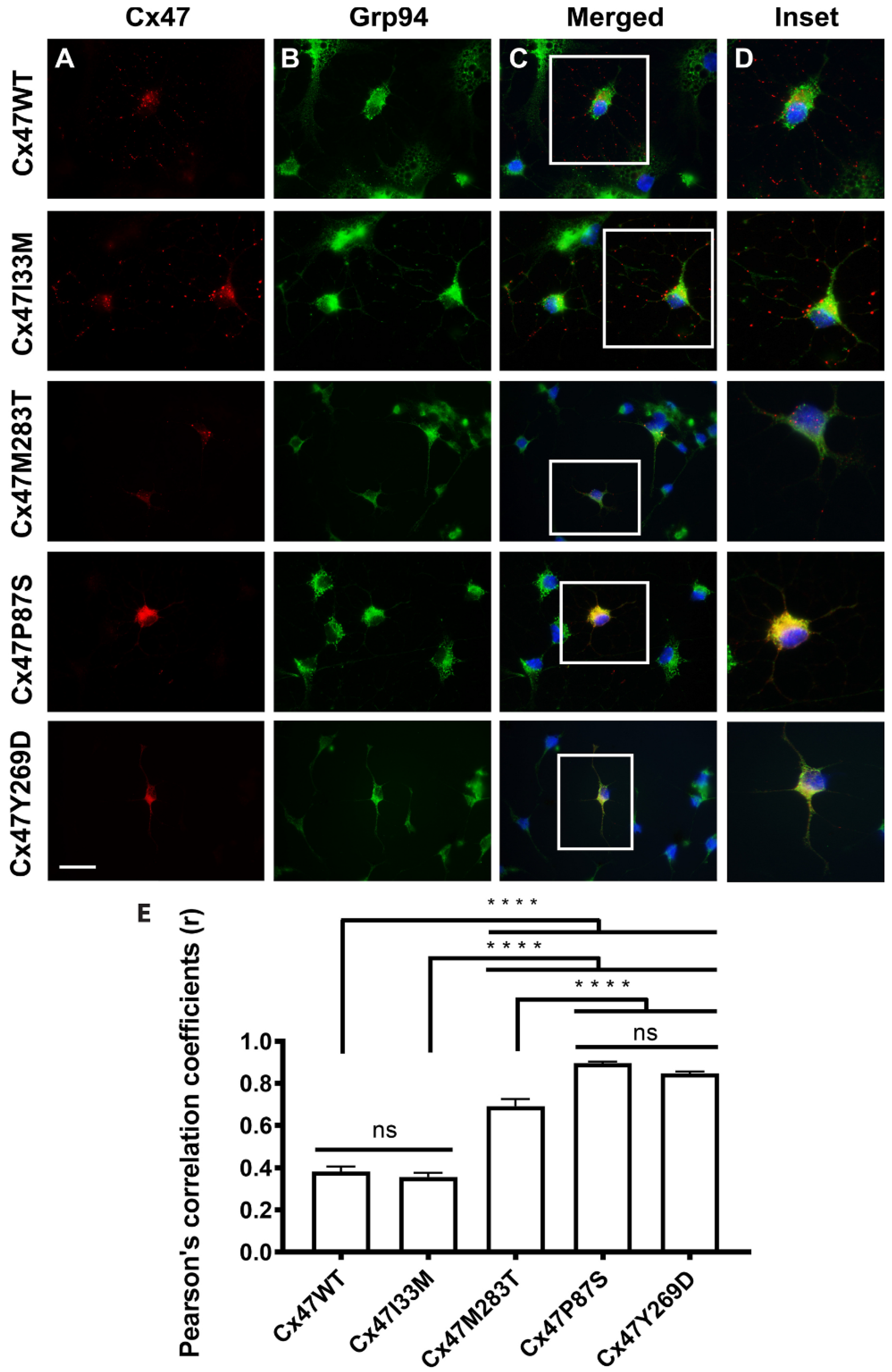
8.1. Mislocalization
8.2. Activation of the Unfolded Protein Response (UPR)
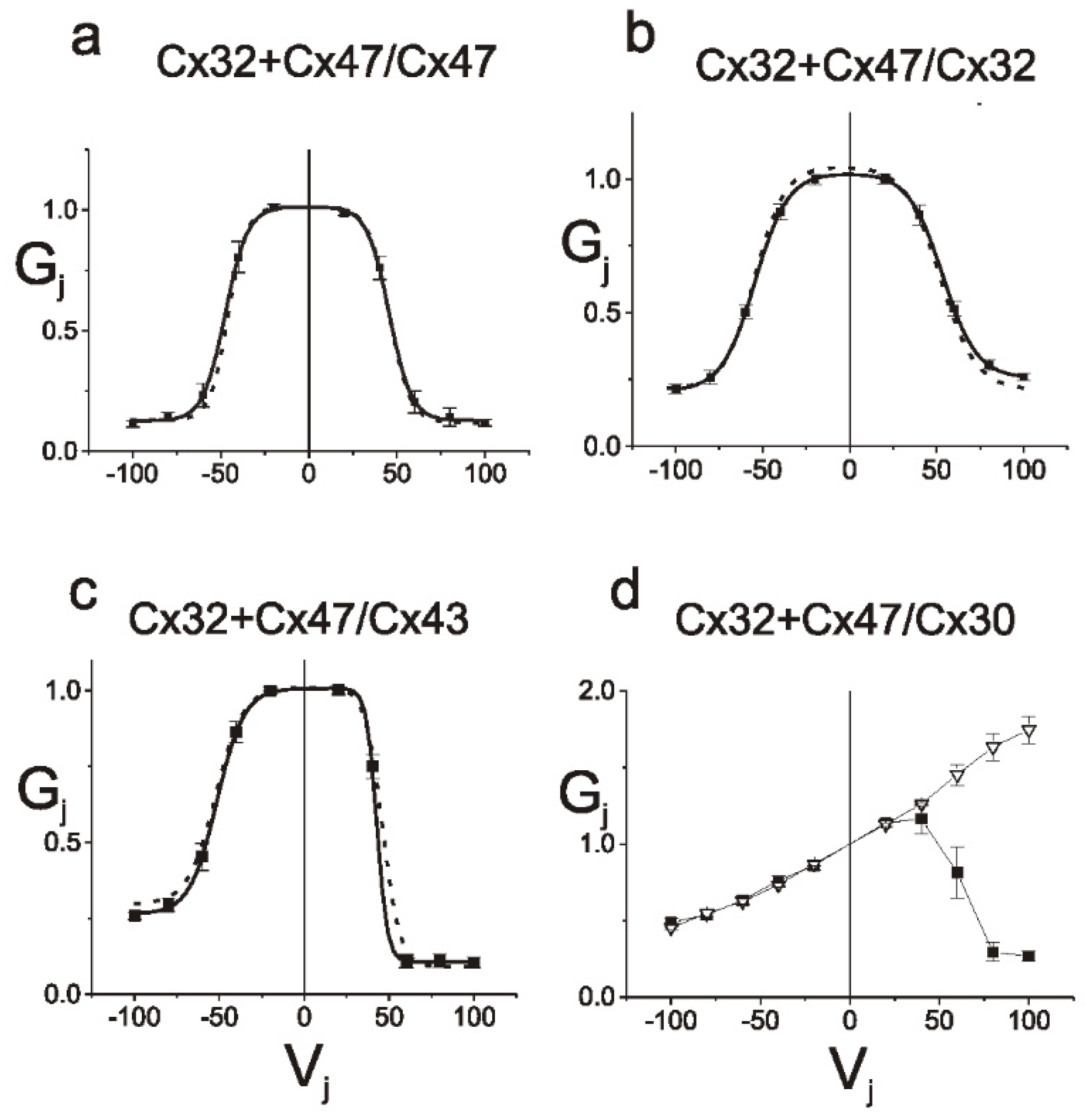
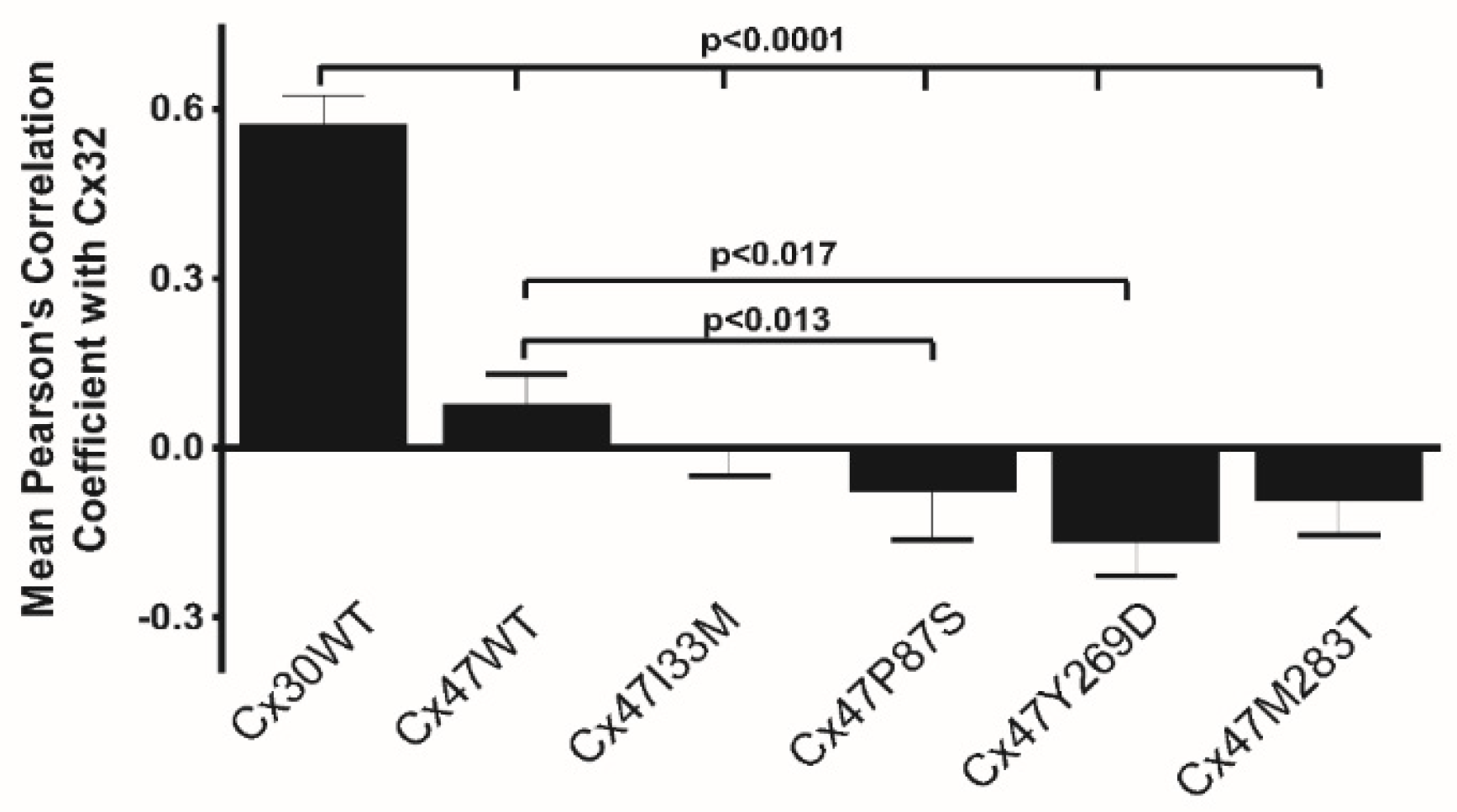
8.3. Differential Interactions with Other Connexin Isoforms
9. Conclusions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kumar, N.M.; Gilula, N.B. The gap junction communication channel. Cell 1996, 84, 381–388. [Google Scholar] [CrossRef]
- Mugnaini, E. Cell junctions of astrocytes, ependyma, and related cells in the mammalian central nervous system, with emphasis on the hypothesis of a generalized functional syncytium of supporting cells. In Astrocytes, Vol I; Fedoroff, S., Vernadakis, A., Eds.; Academic Press: New York, NY, USA, 1986; pp. 329–371. [Google Scholar]
- Scemes, E.; Giaume, C. Astrocyte calcium waves: What they are and what they do. Glia 2006, 54, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Kamasawa, N.; Sik, A.; Morita, M.; Yasumura, T.; Davidson, K.G.; Nagy, J.I.; Rash, J.E. Connexin-47 and connexin-32 in gap junctions of oligodendrocyte somata, myelin sheaths, paranodal loops and Schmidt-Lanterman incisures: Implications for ionic homeostasis and potassium siphoning. Neuroscience 2005, 136, 65–86. [Google Scholar] [CrossRef] [PubMed]
- Rash, J.E. Molecular disruptions of the panglial syncytium block potassium siphoning and axonal saltatory conduction: Pertinence to neuromyelitis optica and other demyelinating diseases of the central nervous system. Neuroscience 2010, 168, 982–1008. [Google Scholar] [CrossRef] [PubMed]
- Rash, J.E.; Vanderpool, K.G.; Yasumura, T.; Hickman, J.; Beatty, J.T.; Nagy, J.I. KV1 channels identified in rodent myelinated axons, linked to Cx29 in innermost myelin: Support for electrically active myelin in mammalian saltatory conduction. J. Neurophysiol. 2016, 115, 1836–1859. [Google Scholar] [CrossRef] [PubMed]
- Menichella, D.M.; Majdan, M.; Awatramani, R.; Goodenough, D.A.; Sirkowski, E.; Scherer, S.S.; Paul, D.L. Genetic and physiological evidence that oligodendrocyte gap junctions contribute to spatial buffering of potassium released during neuronal activity. J. Neurosci. 2006, 26, 10984–10991. [Google Scholar] [CrossRef]
- Ye, Z.C.; Wyeth, M.S.; Baltan-Tekkok, S.; Ransom, B.R. Functional hemichannels in astrocytes: A novel mechanism of glutamate release. J. Neurosci. 2003, 23, 3588–3596. [Google Scholar] [CrossRef]
- Quan, Y.; Du, Y.; Wu, C.; Gu, S.; Jiang, J.X. Connexin hemichannels regulate redox potential via metabolite exchange and protect lens against cellular oxidative damage. Redox Biol. 2021, 46, 102102. [Google Scholar] [CrossRef]
- Dale, N. Dynamic ATP signalling and neural development. J. Physiol. 2008, 586, 2429–2436. [Google Scholar] [CrossRef]
- Stout, C.E.; Costantin, J.L.; Naus, C.C.; Charles, A.C. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J. Biol. Chem. 2002, 277, 10482–10488. [Google Scholar] [CrossRef]
- Orellana, J.A.; Froger, N.; Ezan, P.; Jiang, J.X.; Bennett, M.V.; Naus, C.C.; Giaume, C.; Saez, J.C. ATP and glutamate released via astroglial connexin 43 hemichannels mediate neuronal death through activation of pannexin 1 hemichannels. J. Neurochem. 2011, 118, 826–840. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Dringen, R. Gap junction hemichannel-mediated release of glutathione from cultured rat astrocytes. Neurosci. Lett. 2007, 415, 45–48. [Google Scholar] [CrossRef] [PubMed]
- O’Carroll, S.J.; Alkadhi, M.; Nicholson, L.F.; Green, C.R. Connexin 43 mimetic peptides reduce swelling, astrogliosis, and neuronal cell death after spinal cord injury. Cell Commun. Adhes 2008, 15, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Sohl, G.; Willecke, K. Gap junctions and the connexin protein family. Cardiovasc. Res. 2004, 62, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Paznekas, W.A.; Boyadjiev, S.A.; Shapiro, R.E.; Daniels, O.; Wollnik, B.; Keegan, C.E.; Innis, J.W.; Dinulos, M.B.; Christian, C.; Hannibal, M.C.; et al. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Amer. J. Hum. Genet. 2003, 72, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Gorlin, R.J.; Meskin, L.H.; St. Geme, J.W. Oculodentodigital dysplasia. J. Ped. 1963, 63, 69–75. [Google Scholar] [CrossRef]
- Loddenkemper, T.; Grote, K.; Evers, S.; Oelerich, M.; Stogbauer, F. Neurological manifestations of the oculodentodigital dysplasia syndrome. J. Neurol. 2002, 249, 584–595. [Google Scholar] [CrossRef]
- Stanislaw, C.L.; Narvaez, C.; Roger, R.G.; Woodard, C.S. Oculodentodigital dysplasia with cerebral white matter abnormalies: An additional case. Proc. Greenwood Genet. Center 1998, 17, 20–24. [Google Scholar]
- Schrander-Stumpel, C.T.; Franke, C.L. Central nervous system abnormalities in oculodentodigital dysplasia. Genet. Couns. 1996, 7, 233–235. [Google Scholar]
- Cox, D.R.; DiSalvo, M.; Hall, B.D. Neurologic abnormalities in oculodentodigital dysplasia: A new finding. Clin. Res. 1978, 26, 193A. [Google Scholar]
- Uhlenberg, B.; Schuelke, M.; Ruschendorf, F.; Ruf, N.; Kaindl, A.M.; Henneke, M.; Thiele, H.; Stoltenburg-Didinger, G.; Aksu, F.; Topaloglu, H.; et al. Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. Am. J. Hum. Genet. 2004, 75, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Orthmann-Murphy, J.L.; Salsano, E.; Abrams, C.K.; Bizzi, A.; Uziel, G.; Freidin, M.M.; Lamantea, E.; Zeviani, M.; Scherer, S.S.; Pareyson, D. Hereditary spastic paraplegia is a novel phenotype for GJA12/GJC2 mutations. Brain 2009, 132, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Abrams, C.K.; Scherer, S.S.; Flores-Obando, R.; Freidin, M.M.; Wong, S.; Lamantea, E.; Farina, L.; Scaioli, V.; Pareyson, D.; Salsano, E. A new mutation in GJC2 associated with subclinical leukodystrophy. J. Neurol. 2014, 261, 1929–1938. [Google Scholar] [CrossRef] [PubMed]
- Taşdelen, E.; Durmaz, C.D.; Karabulut, H.G. Autosomal Recessive Oculodentodigital Dysplasia: A Case Report and Review of the Literature. Cytogenet. Genome Res. 2018, 154, 181–186. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, L.; Wang, P.; Chen, C.; Zhang, A.; Wang, W.; Ding, X. Novel ocular findings in oculodentodigital dysplasia (ODDD): A case report and literature review. Ophthalmic Genet. 2019, 40, 54–59. [Google Scholar] [CrossRef]
- Choi, J.; Yang, A.; Song, A.; Lim, M.; Kim, J.; Jang, J.H.; Park, K.T.; Cho, S.; Jin, D.K. Oculodentodigital Dysplasia with a Novel Mutation in GJA1 Diagnosed by Targeted Gene Panel Sequencing: A Case Report and Literature Review. Ann. Clin. Lab. Sci. 2018, 48, 776–781. [Google Scholar]
- De Bock, M.; Kerrebrouck, M.; Wang, N.; Leybaert, L. Neurological manifestations of oculodentodigital dysplasia: A Cx43 channelopathy of the central nervous system? Front. Pharmacol. 2013, 4, 120. [Google Scholar] [CrossRef]
- Abrams, C.K.; Scherer, S.S. Gap junctions in inherited human disorders of the central nervous system. Biochim. Biophys. Acta. 2012, 1818, 2030–2047. [Google Scholar] [CrossRef]
- Abrams, C.K. GJB1 Disorders: Charcot-Marie-Tooth Neuropathy (CMT1X) and Central Nervous System Phenotypes. In GeneReviews(®); Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Abrams, C.K. Diseases of connexins expressed in myelinating glia. Neurosci. Lett. 2019, 695, 91–99. [Google Scholar] [CrossRef]
- Srinivas, M.; Verselis, V.K.; White, T.W. Human diseases associated with connexin mutations. Biochim. Biophys. Acta Biomembr. 2018, 1860, 192–201. [Google Scholar] [CrossRef]
- Delmar, M.; Laird, D.W.; Naus, C.C.; Nielsen, M.S.; Verselis, V.K.; White, T.W. Connexins and Disease. Cold Spring Harb. Perspect. Biol. 2018, 10, a029348. [Google Scholar] [CrossRef] [PubMed]
- Laird, D.W.; Lampe, P.D. Cellular mechanisms of connexin-based inherited diseases. Trends Cell Biol. 2022, 32, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Zheng, J.; Chen, S.; Sun, Y. Connexin Mutations and Hereditary Diseases. Int. J. Mol. Sci. 2022, 23, 4255. [Google Scholar] [CrossRef] [PubMed]
- Orthmann-Murphy, J.L.; Freidin, M.; Fischer, E.; Scherer, S.S.; Abrams, C.K. Two distinct heterotypic channels mediate gap junction coupling between astrocyte and oligodendrocyte connexins. J. Neurosci. 2007, 27, 13949–13957. [Google Scholar] [CrossRef] [PubMed]
- Magnotti, L.M.; Goodenough, D.A.; Paul, D.L. Functional heterotypic interactions between astrocyte and oligodendrocyte connexins. Glia 2011, 59, 26–34. [Google Scholar] [CrossRef]
- Clasadonte, J.; Scemes, E.; Wang, Z.; Boison, D.; Haydon, P.G. Connexin 43-Mediated Astroglial Metabolic Networks Contribute to the Regulation of the Sleep-Wake Cycle. Neuron 2017, 95, 1365–1380. [Google Scholar] [CrossRef]
- Wallraff, A.; Kohling, R.; Heinemann, U.; Theis, M.; Willecke, K.; Steinhauser, C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J. Neurosci. 2006, 26, 5438–5447. [Google Scholar] [CrossRef]
- Nagy, J.I.; Patel, D.; Ochalski, P.A.; Stelmack, G.L. Connexin30 in rodent, cat and human brain: Selective expression in gray matter astrocytes, co-localization with connexin43 at gap junctions and late developmental appearance. Neuroscience 1999, 88, 447–468. [Google Scholar] [CrossRef]
- Nagy, J.I.; Ionescu, A.V.; Lynn, B.D.; Rash, J.E. Coupling of astrocyte connexins Cx26, Cx30, Cx43 to oligodendrocyte Cx29, Cx32, Cx47: Implications from normal and connexin32 knockout mice. Glia 2003, 44, 205–218. [Google Scholar] [CrossRef]
- Rash, J.E.; Yasumura, T.; Dudek, F.E.; Nagy, J.I. Cell-specific expression of connexins and evidence of restricted gap junctional coupling between glial cells and between neurons. J. Neurosci. 2001, 21, 1983–2000. [Google Scholar] [CrossRef]
- Altevogt, B.M.; Paul, D.L. Four classes of intercellular channels between glial cells in the CNS. J. Neurosci. 2004, 24, 4313–4323. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.I.; Li, X.; Rempel, J.; Stelmack, G.; Patel, D.; Staines, W.A.; Yasumura, T.; Rash, J.E. Connexin26 in adult rodent central nervous system: Demonstration at astrocytic gap junctions and colocalization with connexin30 and connexin43. J. Comp. Neurol. 2001, 441, 302–323. [Google Scholar] [CrossRef] [PubMed]
- Filippov, M.A.; Hormuzdi, S.G.; Fuchs, E.C.; Monyer, H. A reporter allele for investigating connexin 26 gene expression in the mouse brain. Eur. J. Neurosci. 2003, 18, 3183–3192. [Google Scholar] [CrossRef]
- Wingard, J.C.; Zhao, H.B. Cellular and Deafness Mechanisms Underlying Connexin Mutation-Induced Hearing Loss - A Common Hereditary Deafness. Front. Cell. Neurosci. 2015, 9, 202. [Google Scholar] [CrossRef]
- Lilly, E.; Sellitto, C.; Milstone, L.M.; White, T.W. Connexin channels in congenital skin disorders. Semin. Cell Dev. Biol. 2016, 50, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Odermatt, B.; Wellershaus, K.; Wallraff, A.; Seifert, G.; Degen, J.; Euwens, C.; Fuss, B.; Bussow, H.; Schilling, K.; Steinhauser, C.; et al. Connexin 47 (Cx47)-deficient mice with enhanced green fluorescent protein reporter gene reveal predominant oligodendrocytic expression of Cx47 and display vacuolized myelin in the CNS. J. Neurosci. 2003, 23, 4549–4559. [Google Scholar] [CrossRef] [PubMed]
- Menichella, D.M.; Goodenough, D.A.; Sirkowski, E.; Scherer, S.S.; Paul, D.L. Connexins are critical for normal myelination in the CNS. J. Neurosci. 2003, 23, 5963–5973. [Google Scholar] [CrossRef]
- Kleopa, K.A.; Orthmann, J.L.; Enriquez, A.; Paul, D.L.; Scherer, S.S. Unique distributions of the gap junction proteins connexin29, connexin32, and connexin47 in oligodendrocytes. Glia 2004, 47, 346–357. [Google Scholar] [CrossRef]
- Barrio, L.C.; Suchyna, T.; Bargiello, T.; Xu, L.X.; Roginski, R.S.; Bennett, M.V.; Nicholson, B.J. Gap junctions formed by connexins 26 and 32 alone and in combination are differently affected by applied voltage. Proc. Natl. Acad. Sci. USA 1991, 88, 8410–8414, erratum in Proc. Natl. Acad. Sci. USA 1992 89, 4220. [Google Scholar] [CrossRef]
- Teubner, B.; Odermatt, B.; Guldenagel, M.; Sohl, G.; Degen, J.; Bukauskas, F.; Kronengold, J.; Verselis, V.K.; Jung, Y.T.; Kozak, C.A.; et al. Functional expression of the new gap junction gene connexin47 transcribed in mouse brain and spinal cord neurons. J. Neurosci. 2001, 21, 1117–1126. [Google Scholar] [CrossRef]
- Altevogt, B.M.; Kleopa, K.A.; Postma, F.R.; Scherer, S.S.; Paul, D.L. Connexin29 is uniquely distributed within myelinating glial cells of the central and peripheral nervous systems. J. Neurosci. 2002, 22, 6458–6470. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.B.; Ichikawa, H.; Bechberger, J.F.; Valiunas, V.; Ohki, M.; Naus, C.C.; Kunimoto, T.; Tsuda, H.; Miller, W.T.; Goldberg, G.S. Normal cells control the growth of neighboring transformed cells independent of gap junctional communication and SRC activity. Cancer Res. 2004, 64, 1347–1358. [Google Scholar] [CrossRef]
- Nagy, J.I.; Ionescu, A.V.; Lynn, B.D.; Rash, J.E. Connexin29 and connexin32 at oligodendrocyte and astrocyte gap junctions and in myelin of the mouse central nervous system. J. Comp. Neurol. 2003, 464, 356–370. [Google Scholar] [CrossRef]
- Ahn, M.; Lee, J.; Gustafsson, A.; Enriquez, A.; Lancaster, E.; Sul, J.Y.; Haydon, P.G.; Paul, D.L.; Huang, Y.; Abrams, C.K.; et al. Cx29 and Cx32, two connexins expressed by myelinating glia, do not interact and are functionally distinct. J. Neurosci. Res. 2008, 86, 992–1006. [Google Scholar] [CrossRef]
- Sargiannidou, I.; Ahn, M.; Enriquez, A.D.; Peinado, A.; Reynolds, R.; Abrams, C.; Scherer, S.S.; Kleopa, K.A. Human oligodendrocytes express Cx31.3: Function and interactions with Cx32 mutants. Neurobiol. Dis. 2008, 30, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Scherer, S.S.; Deschenes, S.M.; Xu, Y.T.; Grinspan, J.B.; Fischbeck, K.H.; Paul, D.L. Connexin32 is a myelin-related protein in the PNS and CNS. J. Neurosci. 1995, 15, 8281–8294. [Google Scholar] [CrossRef] [PubMed]
- Massa, P.T.; Mugnaini, E. Cell junctions and intramembrane particles of astrocytes and oligodendrocytes: A freeze-fracture study. Neuroscience 1982, 7, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Massa, P.T.; Mugnaini, E. Cell-cell junctional interactions and characteristic plasma membrane features of cultured rat glial cells. Neuroscience 1985, 14, 695–709. [Google Scholar] [CrossRef]
- Wasseff, S.K.; Scherer, S.S. Cx32 and Cx47 mediate oligodendrocyte: Astrocyte and oligodendrocyte:oligodendrocyte gap junction coupling. Neurobiol. dis. 2011, 42, 506–513. [Google Scholar] [CrossRef]
- Maglione, M.; Tress, O.; Haas, B.; Karram, K.; Trotter, J.; Willecke, K.; Kettenmann, H. Oligodendrocytes in mouse corpus callosum are coupled via gap junction channels formed by connexin47 and connexin32. Glia 2010, 58, 1104–1117. [Google Scholar] [CrossRef]
- Abrams, C.K.; Flores-Obando, R.E.; Dungan, G.D.; Cherepanova, E.; Freidin, M.M. Investigating oligodendrocyte connexins: Heteromeric interactions between Cx32 and mutant or wild-type forms of Cx47 do not contribute to or modulate gap junction function. Glia 2021, 69, 1882–1896. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.M. Connexin-specific distribution within gap junctions revealed in living cells. J. Cell. Sci. 2000, 113 Pt 22, 4109–4120. [Google Scholar] [CrossRef] [PubMed]
- Jordan, K.; Chodock, R.; Hand, A.R.; Laird, D.W. The origin of annular junctions: A mechanism of gap junction internalization. J. Cell Sci. 2001, 114, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Carette, D.; Gilleron, J.; Denizot, J.-P.; Grant, K.; Pointis, G.; Segretain, D. New cellular mechanisms of gap junction degradation and recycling. Biol. Cell 2015, 107, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Lynn, B.D.; Tress, O.; May, D.; Willecke, K.; Nagy, J.I. Ablation of connexin30 in transgenic mice alters expression patterns of connexin26 and connexin32 in glial cells and leptomeninges. Eur. J. Neurosci. 2011, 34, 1783–1793. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Gloor, G.B.; Bai, D. The distribution and functional properties of Pelizaeus-Merzbacher-like disease-linked Cx47 mutations on Cx47/Cx47 homotypic and Cx47/Cx43 heterotypic gap junctions. Biochem. J. 2013, 452, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Orthmann-Murphy, J.L.; Enriquez, A.D.; Abrams, C.K.; Scherer, S.S. Loss-of-function GJA12/Connexin47 mutations cause Pelizaeus-Merzbacher-like disease. Mol. Cell Neurosci. 2007, 34, 629–641. [Google Scholar] [CrossRef]
- Fasciani, I.; Pluta, P.; González-Nieto, D.; Martínez-Montero, P.; Molano, J.; Paíno, C.L.; Millet, O.; Barrio, L.C. Directional coupling of oligodendrocyte connexin-47 and astrocyte connexin-43 gap junctions. Glia 2018, 66, 2340–2352. [Google Scholar] [CrossRef]
- Anzini, P.; Neuberg, D.H.; Schachner, M.; Nelles, E.; Willecke, K.; Zielasek, J.; Toyka, K.V.; Suter, U.; Martini, R. Structural abnormalities and deficient maintenance of peripheral nerve myelin in mice lacking the gap junction protein connexin 32. J. Neurosci. 1997, 17, 4545–4551. [Google Scholar] [CrossRef]
- Scherer, S.S.; Xu, Y.T.; Nelles, E.; Fischbeck, K.; Willecke, K.; Bone, L.J. Connexin32-null mice develop demyelinating peripheral neuropathy. Glia 1998, 24, 8–20. [Google Scholar] [CrossRef]
- Vavlitou, N.; Sargiannidou, I.; Markoullis, K.; Kyriacou, K.; Scherer, S.S.; Kleopa, K.A. Axonal pathology precedes demyelination in a mouse model of X-linked demyelinating/type I Charcot-Marie Tooth neuropathy. J. Neuropathol. Exp. Neurol. 2010, 69, 945–958. [Google Scholar] [CrossRef]
- Sutor, B.; Schmolke, C.; Teubner, B.; Schirmer, C.; Willecke, K. Myelination defects and neuronal hyperexcitability in the neocortex of connexin 32-deficient mice. Cereb. Cortex. 2000, 10, 684–697. [Google Scholar] [CrossRef]
- Sargiannidou, I.; Vavlitou, N.; Aristodemou, S.; Hadjisavvas, A.; Kyriacou, K.; Scherer, S.S.; Kleopa, K.A. Connexin32 mutations cause loss of function in Schwann cells and oligodendrocytes leading to PNS and CNS myelination defects. J. Neurosci. 2009, 29, 4736–4749. [Google Scholar] [CrossRef]
- Markoullis, K.; Sargiannidou, I.; Gardner, C.; Hadjisavvas, A.; Reynolds, R.; Kleopa, K.A. Disruption of oligodendrocyte gap junctions in experimental autoimmune encephalomyelitis. Glia 2012, 60, 1053–1066. [Google Scholar] [CrossRef]
- Papaneophytou, C.P.; Georgiou, E.; Karaiskos, C.; Sargiannidou, I.; Markoullis, K.; Freidin, M.M.; Abrams, C.K.; Kleopa, K.A. Regulatory role of oligodendrocyte gap junctions in inflammatory demyelination. Glia 2018, 66, 2589–2603. [Google Scholar] [CrossRef]
- Olympiou, M.; Sargiannidou, I.; Markoullis, K.; Karaiskos, C.; Kagiava, A.; Kyriakoudi, S.; Abrams, C.K.; Kleopa, K.A. Systemic inflammation disrupts oligodendrocyte gap junctions and induces ER stress in a model of CNS manifestations of X-linked Charcot-Marie-Tooth disease. Acta. Neuropathol. Commun. 2016, 4, 95. [Google Scholar] [CrossRef] [PubMed]
- Stavropoulos, F.; Georgiou, E.; Sargiannidou, I.; Kleopa, K.A. Dysregulation of Blood-Brain Barrier and Exacerbated Inflammatory Response in Cx47-Deficient Mice after Induction of EAE. Pharmaceuticals 2021, 14, 621. [Google Scholar] [CrossRef] [PubMed]
- Giepmans, B.N.; Moolenaar, W.H. The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens-1 protein. Curr. Biol. 1998, 8, 931–934. [Google Scholar] [CrossRef]
- Kausalya, P.J.; Reichert, M.; Hunziker, W. Connexin45 directly binds to ZO-1 and localizes to the tight junction region in epithelial MDCK cells. FEBS Lett. 2001, 505, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.A.; Baruch, A.; Shestopalov, V.I.; Giepmans, B.N.; Dunia, I.; Benedetti, E.L.; Kumar, N.M. Lens connexins alpha3Cx46 and alpha8Cx50 interact with zonula occludens protein-1 (ZO-1). Mol. Biol. Cell 2003, 14, 2470–2481. [Google Scholar] [CrossRef]
- Li, X.; Ionescu, A.V.; Lynn, B.D.; Lu, S.; Kamasawa, N.; Morita, M.; Davidson, K.G.; Yasumura, T.; Rash, J.E.; Nagy, J.I. Connexin47, connexin29 and connexin32 co-expression in oligodendrocytes and Cx47 association with zonula occludens-1 (ZO-1) in mouse brain. Neuroscience 2004, 126, 611–630. [Google Scholar] [CrossRef] [PubMed]
- Penes, M.C.; Li, X.; Nagy, J.I. Expression of zonula occludens-1 (ZO-1) and the transcription factor ZO-1-associated nucleic acid-binding protein (ZONAB)-MsY3 in glial cells and colocalization at oligodendrocyte and astrocyte gap junctions in mouse brain. Eur. J. Neurosci. 2005, 22, 404–418. [Google Scholar] [CrossRef] [PubMed]
- Stout, C.; Goodenough, D.A.; Paul, D.L. Connexins: Functions without junctions. Curr. Opin. Cell Biol. 2004, 16, 507–512. [Google Scholar] [CrossRef]
- Li, X.; Penes, M.; Odermatt, B.; Willecke, K.; Nagy, J.I. Ablation of Cx47 in transgenic mice leads to the loss of MUPP1, ZONAB and multiple connexins at oligodendrocyte-astrocyte gap junctions. Eur. J. Neurosci. 2008, 28, 1503–1517. [Google Scholar] [CrossRef] [PubMed]
- Balda, M.S.; Matter, K. The tight junction protein ZO-1 and an interacting transcription factor regulate ErbB-2 expression. Embo. J. 2000, 19, 2024–2033. [Google Scholar] [CrossRef]
- Flores, A.I.; Mallon, B.S.; Matsui, T.; Ogawa, W.; Rosenzweig, A.; Okamoto, T.; Macklin, W.B. Akt-mediated survival of oligodendrocytes induced by neuregulins. J. Neurosci. 2000, 20, 7622–7630. [Google Scholar] [CrossRef]
- Park, S.K.; Miller, R.; Krane, I.; Vartanian, T. The erbB2 gene is required for the development of terminally differentiated spinal cord oligodendrocytes. J. Cell Biol. 2001, 154, 1245–1258. [Google Scholar] [CrossRef]
- Kim, J.Y.; Sun, Q.; Oglesbee, M.; Yoon, S.O. The role of ErbB2 signaling in the onset of terminal differentiation of oligodendrocytes in vivo. J. Neurosci. 2003, 23, 5561–5571. [Google Scholar] [CrossRef]
- Balda, M.S.; Garrett, M.D.; Matter, K. The ZO-1-associated Y-box factor ZONAB regulates epithelial cell proliferation and cell density. J. Cell Biol. 2003, 160, 423–432. [Google Scholar] [CrossRef]
- Sourisseau, T.; Georgiadis, A.; Tsapara, A.; Ali, R.R.; Pestell, R.; Matter, K.; Balda, M.S. Regulation of PCNA and cyclin D1 expression and epithelial morphogenesis by the ZO-1-regulated transcription factor ZONAB/DbpA. Mol. Cell Biol. 2006, 26, 2387–2398. [Google Scholar] [CrossRef]
- Neusch, C.; Rozengurt, N.; Jacobs, R.E.; Lester, H.A.; Kofuji, P. Kir4.1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J. Neurosci. 2001, 21, 5429–5438. [Google Scholar] [CrossRef]
- Higashi, K.; Fujita, A.; Inanobe, A.; Tanemoto, M.; Doi, K.; Kubo, T.; Kurachi, Y. An inwardly rectifying K(+) channel, Kir4.1, expressed in astrocytes surrounds synapses and blood vessels in brain. Am. J. Physiol. Cell Physiol. 2001, 281, C922–C931. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Head, V.; Timpe, L.C. Identification of an inward rectifier potassium channel gene expressed in mouse cortical astrocytes. Glia 2001, 33, 57–71. [Google Scholar] [CrossRef]
- Sohl, G.; Eiberger, J.; Jung, Y.T.; Kozak, C.A.; Willecke, K. The mouse gap junction gene connexin29 is highly expressed in sciatic nerve and regulated during brain development. Biol. Chem. 2001, 382, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Eiberger, J.; Kibschull, M.; Strenzke, N.; Schober, A.; Bussow, H.; Wessig, C.; Djahed, S.; Reucher, H.; Koch, D.A.; Lautermann, J.; et al. Expression pattern and functional characterization of connexin29 in transgenic mice. Glia 2006, 53, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Zhang, Y.; Chang, Q.; Ahmad, S.; Dahlke, I.; Yi, H.; Chen, P.; Paul, D.L.; Lin, X. Connexin29 is highly expressed in cochlear Schwann cells, and it is required for the normal development and function of the auditory nerve of mice. J. Neurosci. 2006, 26, 1991–1999. [Google Scholar] [CrossRef] [PubMed]
- Adadey, S.M.; Esoh, K.K.; Quaye, O.; Amedofu, G.K.; Awandare, G.A.; Wonkam, A. GJB4 and GJC3 variants in non-syndromic hearing impairment in Ghana. Exp. Biol. Med. (Maywood) 2020, 245, 1355–1367. [Google Scholar] [CrossRef]
- Wang, W.H.; Yang, J.J.; Lin, Y.C.; Yang, J.T.; Chan, C.H.; Li, S.Y. Identification of novel variants in the Cx29 gene of nonsyndromic hearing loss patients using buccal cells and restriction fragment length polymorphism method. Audiol. Neurootol. 2010, 15, 81–87. [Google Scholar] [CrossRef]
- Yang, J.J.; Huang, S.H.; Chou, K.H.; Liao, P.J.; Su, C.C.; Li, S.Y. Identification of mutations in members of the connexin gene family as a cause of nonsyndromic deafness in Taiwan. Audiol. Neurootol. 2007, 12, 198–208. [Google Scholar] [CrossRef]
- Wong, S.H.; Wang, W.H.; Chen, P.H.; Li, S.Y.; Yang, J.J. Functional analysis of a nonsyndromic hearing loss-associated mutation in the transmembrane II domain of the GJC3 gene. Int. J. Med. Sci. 2017, 14, 246–256. [Google Scholar] [CrossRef]
- Lee, H.J.; Jeong, H.; Hyun, J.; Ryu, B.; Park, K.; Lim, H.H.; Yoo, J.; Woo, J.S. Cryo-EM structure of human Cx31.3/GJC3 connexin hemichannel. Sci. Adv. 2020, 6, eaba4996. [Google Scholar] [CrossRef] [PubMed]
- Su, C.C.; Li, S.Y.; Yen, Y.C.; Nian, J.H.; Liang, W.G.; Yang, J.J. Mechanism of two novel human GJC3 missense mutations in causing non-syndromic hearing loss. Cell Biochem. Biophys. 2013, 66, 277–286. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic. Acids. Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed]
- Adadey, S.M.; Wonkam-Tingang, E.; Twumasi Aboagye, E.; Nayo-Gyan, D.W.; Boatemaa Ansong, M.; Quaye, O.; Awandare, G.A.; Wonkam, A. Connexin Genes Variants Associated with Non-Syndromic Hearing Impairment: A Systematic Review of the Global Burden. Life 2020, 10, 258. [Google Scholar] [CrossRef] [PubMed]
- Reaume, A.G.; De Sousa, P.A.; Kulkarni, S.; Langille, B.L.; Zhu, D.; Davies, T.C.; Juneja, S.C.; Kidder, G.M.; Rossant, J. Cardiac malformation in neonatal mice lacking connexin43. Science 1995, 267, 1831–1834. [Google Scholar] [CrossRef]
- Frisch, C.; Theis, M.; De Souza Silva, M.A.; Dere, E.; Sohl, G.; Teubner, B.; Namestkova, K.; Willecke, K.; Huston, J.P. Mice with astrocyte-directed inactivation of connexin43 exhibit increased exploratory behaviour, impaired motor capacities, and changes in brain acetylcholine levels. Eur. J. Neurosci. 2003, 18, 2313–2318. [Google Scholar] [CrossRef]
- Theis, M.; Jauch, R.; Zhuo, L.; Speidel, D.; Wallraff, A.; Doring, B.; Frisch, C.; Sohl, G.; Teubner, B.; Euwens, C.; et al. Accelerated hippocampal spreading depression and enhanced locomotory activity in mice with astrocyte-directed inactivation of connexin43. J. Neurosci. 2003, 23, 766–776. [Google Scholar] [CrossRef]
- Dere, E.; De Souza-Silva, M.A.; Frisch, C.; Teubner, B.; Sohl, G.; Willecke, K.; Huston, J.P. Connexin30-deficient mice show increased emotionality and decreased rearing activity in the open-field along with neurochemical changes. Eur. J. Neurosci. 2003, 18, 629–638. [Google Scholar] [CrossRef]
- Lutz, S.E.; Zhao, Y.; Gulinello, M.; Lee, S.C.; Raine, C.S.; Brosnan, C.F. Deletion of astrocyte connexins 43 and 30 leads to a dysmyelinating phenotype and hippocampal CA1 vacuolation. J. Neurosci. 2009, 29, 7743–7752. [Google Scholar] [CrossRef]
- Rouach, N.; Koulakoff, A.; Abudara, V.; Willecke, K.; Giaume, C. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science 2008, 322, 1551–1555. [Google Scholar] [CrossRef]
- Magnotti, L.M.; Goodenough, D.A.; Paul, D.L. Deletion of oligodendrocyte Cx32 and astrocyte Cx43 causes white matter vacuolation, astrocyte loss and early mortality. Glia 2011, 59, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- May, D.; Tress, O.; Seifert, G.; Willecke, K. Connexin47 protein phosphorylation and stability in oligodendrocytes depend on expression of Connexin43 protein in astrocytes. J. Neurosci. 2013, 33, 7985–7996. [Google Scholar] [CrossRef] [PubMed]
- Tress, O.; Maglione, M.; May, D.; Pivneva, T.; Richter, N.; Seyfarth, J.; Binder, S.; Zlomuzica, A.; Seifert, G.; Theis, M.; et al. Panglial gap junctional communication is essential for maintenance of myelin in the CNS. J. Neurosci. 2012, 32, 7499–7518. [Google Scholar] [CrossRef] [PubMed]
- Garbern, J.Y. Pelizaeus-Merzbacher disease: Genetic and cellular pathogenesis. Cell Mol. Life Sci. 2007, 64, 50–65. [Google Scholar] [CrossRef]
- Ferrell, R.E.; Baty, C.J.; Kimak, M.A.; Karlsson, J.M.; Lawrence, E.C.; Franke-Snyder, M.; Meriney, S.D.; Feingold, E.; Finegold, D.N. GJC2 missense mutations cause human lymphedema. Am. J. Hum. Genet. 2010, 86, 943–948. [Google Scholar] [CrossRef]
- Michelini, S.; Vettori, A.; Maltese, P.E.; Cardone, M.; Bruson, A.; Fiorentino, A.; Cappellino, F.; Sainato, V.; Guerri, G.; Marceddu, G.; et al. Genetic Screening in a Large Cohort of Italian Patients Affected by Primary Lymphedema Using a Next Generation Sequencing (NGS) Approach. Lymphology 2016, 49, 57–72. [Google Scholar]
- Finegold, D.N.; Baty, C.J.; Knickelbein, K.Z.; Perschke, S.; Noon, S.E.; Campbell, D.; Karlsson, J.M.; Huang, D.; Kimak, M.A.; Lawrence, E.C.; et al. Connexin 47 mutations increase risk for secondary lymphedema following breast cancer treatment. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 2382–2390. [Google Scholar] [CrossRef]
- Ehrlich, A.; Molica, F.; Hautefort, A.; Kwak, B.R. Lymphatic Connexins and Pannexins in Health and Disease. Int. J. Mol. Sci. 2021, 22, 5734. [Google Scholar] [CrossRef]
- Henneke, M.; Combes, P.; Diekmann, S.; Bertini, E.; Brockmann, K.; Burlina, A.P.; Kaiser, J.; Ohlenbusch, A.; Plecko, B.; Rodriguez, D.; et al. GJA12 mutations are a rare cause of Pelizaeus-Merzbacher-like disease. Neurology 2008, 70, 748–754. [Google Scholar] [CrossRef]
- Meyer, E.; Kurian, M.A.; Morgan, N.V.; McNeill, A.; Pasha, S.; Tee, L.; Younis, R.; Norman, A.; van der Knaap, M.S.; Wassmer, E.; et al. Promoter mutation is a common variant in GJC2-associated Pelizaeus-Merzbacher-like disease. Mol. Genet. Metab. 2011, 104, 637–643. [Google Scholar] [CrossRef]
- Feinstein, M.; Markus, B.; Noyman, I.; Shalev, H.; Flusser, H.; Shelef, I.; Liani-Leibson, K.; Shorer, Z.; Cohen, I.; Khateeb, S.; et al. Pelizaeus-Merzbacher-like disease caused by AIMP1/p43 homozygous mutation. Am. J. Hum. Genet. 2010, 87, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Biancheri, R.; Rossi, A.; Zara, F.; Filocamo, M. AIMP1/p43 mutation and PMLD. Am. J. Hum. Genet. 2011, 88, 391. [Google Scholar] [CrossRef] [PubMed]
- Boespflug-Tanguy, O.; Aubourg, P.; Dorboz, I.; Bégou, M.; Giraud, G.; Sarret, C.; Vaurs-Barrière, C. Neurodegenerative disorder related to AIMP1/p43 mutation is not a PMLD. Am. J. Hum. Genet. 2011, 88, 392–393; author reply 393–395. [Google Scholar] [CrossRef] [PubMed]
- Llaci, L.; Ramsey, K.; Belnap, N.; Claasen, A.M.; Balak, C.D.; Szelinger, S.; Jepsen, W.M.; Siniard, A.L.; Richholt, R.; Izat, T.; et al. Compound heterozygous mutations in SNAP29 is associated with Pelizaeus-Merzbacher-like disorder (PMLD). Hum. Genet. 2019, 138, 1409–1417. [Google Scholar] [CrossRef]
- Fu, H.; Wang, Q.; Liu, H. Novel Mutations in NPC1 are Associated with Pelizaeus-Merzbacher-Like Disease: A Case Report. Int. J. Gen. Med. 2021, 14, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Nafisinia, M.; Sobreira, N.; Riley, L.; Gold, W.; Uhlenberg, B.; Weiß, C.; Boehm, C.; Prelog, K.; Ouvrier, R.; Christodoulou, J. Mutations in RARS cause a hypomyelination disorder akin to Pelizaeus-Merzbacher disease. Eur. J. Hum. Genet. 2017, 25, 1134–1141. [Google Scholar] [CrossRef]
- Salviati, L.; Trevisson, E.; Baldoin, M.C.; Toldo, I.; Sartori, S.; Calderone, M.; Tenconi, R.; Laverda, A. A novel deletion in the GJA12 gene causes Pelizaeus-Merzbacher-like disease. Neurogenetics 2007, 8, 57–60. [Google Scholar] [CrossRef]
- Wolf, N.I.; Cundall, M.; Rutland, P.; Rosser, E.; Surtees, R.; Benton, S.; Chong, W.K.; Malcolm, S.; Ebinger, F.; Bitner-Glindzicz, M.; et al. Frameshift mutation in GJA12 leading to nystagmus, spastic ataxia and CNS dys-/demyelination. Neurogenetics 2007, 8, 39–44. [Google Scholar] [CrossRef]
- Bugiani, M.; Al Shahwan, S.; Lamantea, E.; Bizzi, A.; Bakhsh, E.; Moroni, I.; Balestrini, M.R.; Uziel, G.; Zeviani, M. GJA12 mutations in children with recessive hypomyelinating leukoencephalopathy. Neurology 2006, 67, 273–279. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H.; Wang, Y.; Chen, T.; Wu, X.; Jiang, Y. Two novel gap junction protein alpha 12 gene mutations in two Chinese patients with Pelizaeus-Merzbacher-like disease. Brain Dev. 2010, 32, 236–243. [Google Scholar] [CrossRef]
- Biancheri, R.; Rosano, C.; Denegri, L.; Lamantea, E.; Pinto, F.; Lanza, F.; Severino, M.; Filocamo, M. Expanded spectrum of Pelizaeus-Merzbacher-like disease: Literature revision and description of a novel GJC2 mutation in an unusually severe form. Eur. J. Hum. Genet. 2013, 21, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Shimojima, K.; Tanaka, R.; Shimada, S.; Sangu, N.; Nakayama, J.; Iwasaki, N.; Yamamoto, T. A novel homozygous mutation of GJC2 derived from maternal uniparental disomy in a female patient with Pelizaeus-Merzbacher-like disease. J. Neurol. Sci. 2013. [Google Scholar] [CrossRef] [PubMed]
- Kammoun Jellouli, N.; Salem, I.H.; Ellouz, E.; Louhichi, N.; Tlili, A.; Kammoun, F.; Triki, C.; Fakhfakh, F. Molecular confirmation of founder mutation c.-167A>G in Tunisian patients with PMLD disease. Gene 2013, 513, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Ji, T.; Li, D.; Wu, Y.; Xiao, J.; Ji, H.; Wu, X.; Wang, J.; Jiang, Y. Identification of GJC2 gene mutations in chinese patients with Pelizaeus-Merzbacher-like disease. Minerva Pediatr. 2016. [Google Scholar] [CrossRef]
- Ji, H.; Li, D.; Wu, Y.; Zhang, Q.; Gu, Q.; Xie, H.; Ji, T.; Wang, H.; Zhao, L.; Zhao, H.; et al. Hypomyelinating disorders in China: The clinical and genetic heterogeneity in 119 patients. PLoS ONE 2018, 13, e0188869. [Google Scholar] [CrossRef]
- Henneke, M.; Gegner, S.; Hahn, A.; Plecko-Startinig, B.; Weschke, B.; Gartner, J.; Brockmann, K. Clinical neurophysiology in GJA12-related hypomyelination vs Pelizaeus-Merzbacher disease. Neurology 2010, 74, 1785–1789. [Google Scholar] [CrossRef]
- Bilir, B.; Yapici, Z.; Yalcinkaya, C.; Baris, I.; Carvalho, C.M.; Bartnik, M.; Ozes, B.; Eraksoy, M.; Lupski, J.R.; Battaloglu, E. High frequency of GJA12/GJC2 mutations in Turkish patients with Pelizaeus-Merzbacher disease. Clin. Genet. 2013, 83, 66–72. [Google Scholar] [CrossRef]
- Strauss, K.A.; Puffenberger, E.G. Genetics, medicine, and the Plain people. Annu. Rev. Genomics. Hum. Genet. 2009, 10, 513–536. [Google Scholar] [CrossRef]
- Owczarek-Lipska, M.; Mulahasanovic, L.; Obermaier, C.D.; Hortnagel, K.; Neubauer, B.A.; Korenke, G.C.; Biskup, S.; Neidhardt, J. Novel mutations in the GJC2 gene associated with Pelizaeus-Merzbacher-like disease. Mol. Biol. Rep. 2019, 46, 4507–4516. [Google Scholar] [CrossRef]
- Diekmann, S.; Henneke, M.; Burckhardt, B.C.; Gartner, J. Pelizaeus-Merzbacher-like disease is caused not only by a loss of connexin47 function but also by a hemichannel dysfunction. Eur. J. Hum. Genet. 2010, 18, 985–992. [Google Scholar] [CrossRef]
- Karimzadeh, P.; Ahmadabadi, F.; Aryani, O.; Houshmand, M.; Khatami, A. New mutation of pelizaeus–Merzbacher-like disease; A report from Iran. Iran. J. Radiol. Q. J. Publ. Iran. Radiol. Soc. 2014, 11, e6913. [Google Scholar] [CrossRef]
- Javadikooshesh, S.; Zaimkohan, H.; Pourghorban, P.; Bahramim, F.; Ebadi, N. Pelizaeus-Merzbacher-Like Disease 1 Caused by a Novel Mutation in GJC2 Gene: A Case Report. Iran. J. Med. Sci. 2021, 46, 493–497. [Google Scholar] [CrossRef]
- Osaka, H.; Hamanoue, H.; Yamamoto, R.; Nezu, A.; Sasaki, M.; Saitsu, H.; Kurosawa, K.; Shimbo, H.; Matsumoto, N.; Inoue, K. Disrupted SOX10 regulation of GJC2 transcription causes Pelizaeus-Merzbacher-like disease. Ann. Neurol. 2010, 68, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Combes, P.; Kammoun, N.; Monnier, A.; Gonthier-Guéret, C.; Giraud, G.; Bertini, E.; Chahnez, T.; Fakhfakh, F.; Boespflug-Tanguy, O.; Vaurs-Barrière, C. Relevance of GJC2 promoter mutation in Pelizaeus-Merzbacher-like disease. Ann. Neurol. 2012, 71, 146–148. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, L.; Inoue, K.; Helman, G.; Mora, S.; Maski, K.; Soul, J.S.; Bloom, M.; Evans, S.H.; Goto, Y.; Caldovic, L.; et al. GJC2 promoter mutations causing Pelizaeus-Merzbacher-like disease. Mol. Genet. Metab. 2014, 111, 393–398. [Google Scholar] [CrossRef]
- Al-Yahyaee, S.A.; Al-Kindi, M.; Jonghe, P.D.; Al-Asmi, A.; Al-Futaisi, A.; Vriendt, E.D.; Deconinck, T.; Chand, P. Pelizaeus-Merzbacher-Like Disease in a Family With Variable Phenotype and a Novel Splicing GJC2 Mutation. J. Child. Neurol. 2013, 28, 1467–1473. [Google Scholar] [CrossRef]
- Osório, M.J.; Goldman, S.A. Neurogenetics of Pelizaeus-Merzbacher disease. Handb. Clin. Neurol. 2018, 148, 701–722. [Google Scholar] [CrossRef]
- Bonkowsky, J.L.; Nelson, C.; Kingston, J.L.; Filloux, F.M.; Mundorff, M.B.; Srivastava, R. The burden of inherited leukodystrophies in children. Neurology 2010, 75, 718–725. [Google Scholar] [CrossRef]
- Singh, R.; Debopam, S. Pelizaeus-Merzbacher Disease. In StatPearls [Internet]. Available online: https://www.ncbi.nlm.nih.gov/books/NBK560522/ (accessed on 4 July 2022).
- Kuipers, D.J.S.; Tufekcioglu, Z.; Bilgiç, B.; Olgiati, S.; Dremmen, M.H.G.; van IJcken, W.F.J.; Breedveld, G.J.; Mancini, G.M.S.; Hanagasi, H.A.; Emre, M.; et al. Late-onset phenotype associated with a homozygous GJC2 missense mutation in a Turkish family. Park. Relat. Disord. 2019, 66, 228–231. [Google Scholar] [CrossRef]
- Flores-Obando, R.E.; Freidin, M.M.; Hernández, A.I.; Abrams, C.K. Activation of the unfolded protein response by Connexin47 mutations associated with Pelizaeus-Merzbacher-like disease. Mol. Cell. Neurosci. 2022, 120, 103716. [Google Scholar] [CrossRef]
- Chen, N.; Wang, J.; Jiang, Y.; Wu, Y.; Hao, H.; Ji, T. Different Mutations of Gap Junction Connexin 47 Lead to Discrepant Activation of Unfolded Protein Response Pathway in Pelizaeus-Merzbacher-Like Disease. Neuropediatrics 2017, 48, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Koval, M.; Molina, S.A.; Burt, J.M. Mix and match: Investigating heteromeric and heterotypic gap junction channels in model systems and native tissues. FEBS. Lett. 2014, 588, 1193–1204. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abrams, C.K. Mechanisms of Diseases Associated with Mutation in GJC2/Connexin 47. Biomolecules 2023, 13, 712. https://doi.org/10.3390/biom13040712
Abrams CK. Mechanisms of Diseases Associated with Mutation in GJC2/Connexin 47. Biomolecules. 2023; 13(4):712. https://doi.org/10.3390/biom13040712
Chicago/Turabian StyleAbrams, Charles K. 2023. "Mechanisms of Diseases Associated with Mutation in GJC2/Connexin 47" Biomolecules 13, no. 4: 712. https://doi.org/10.3390/biom13040712
APA StyleAbrams, C. K. (2023). Mechanisms of Diseases Associated with Mutation in GJC2/Connexin 47. Biomolecules, 13(4), 712. https://doi.org/10.3390/biom13040712