
Anti-Citrullinated Protein Antibody Reactivity towards Neutrophil-Derived Antigens: Clonal Diversity and Inter-Individual Variation
 , , , , , and
, , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Neutrophil Isolation
2.2. Neutrophil Activation
2.3. Osteoclast Cultures
2.4. ACPA and Control Antibodies
2.5. Fine-Specificity Analysis of the Tested Antibodies
2.6. Flow Cytometry
2.7. Immunofluorescence
2.8. Statistical Analysis
3. Results
3.1. ACPA Binding to NET-Associated Antigens and Nuclear Epitopes in Intact Cells
3.2. ACPA Targets Are Produced in Activated Neutrophils and Show Little Accessibility to Antibodies
3.3. PAD4-Dependent Recognition of Activated Neutrophils by ACPAs
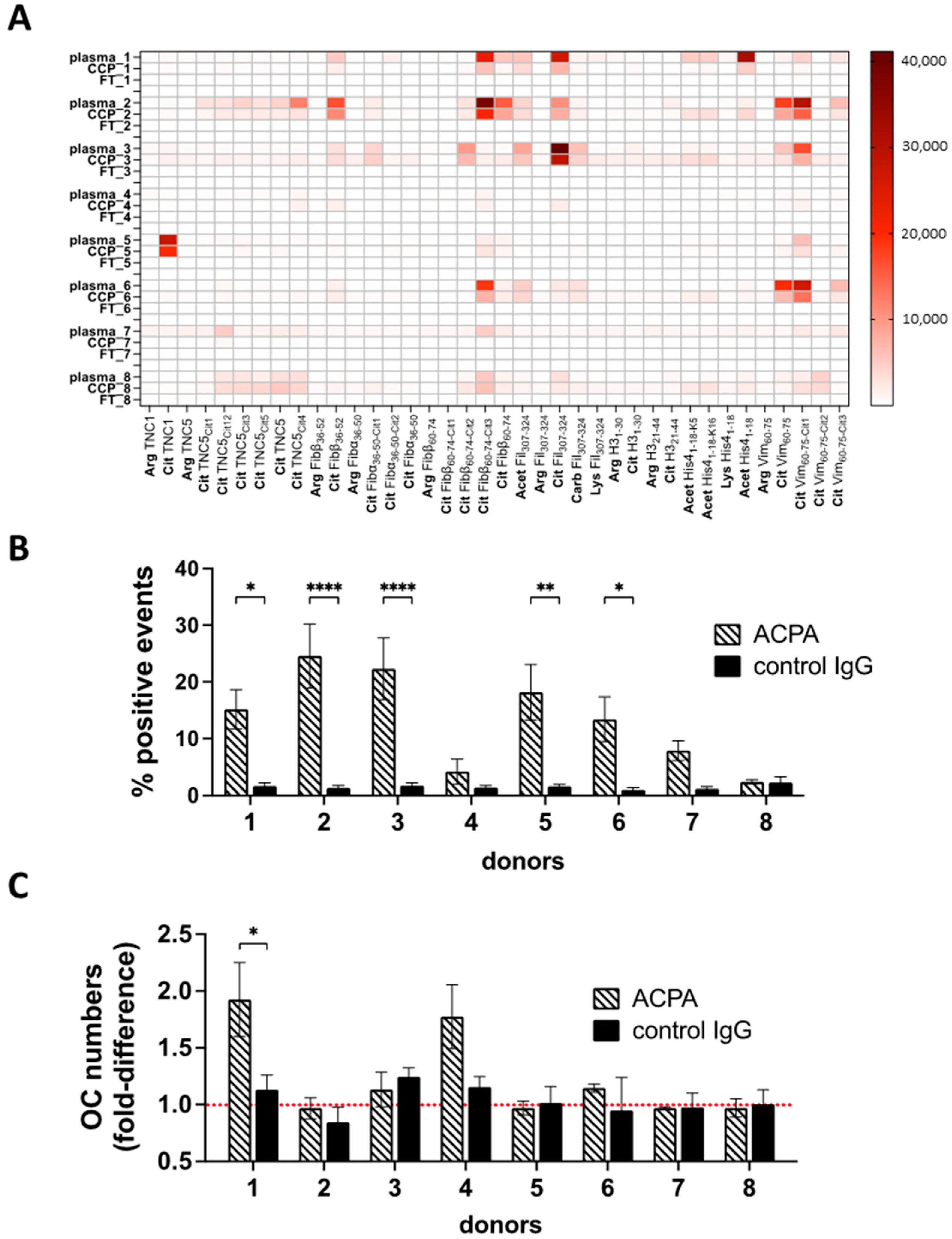
3.4. ACPA-Positive Individuals Differ in ACPA Fine Specificities and Cell-Targeting Antibodies
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. NETs Are a Source of Citrullinated Autoantigens and Stimulate Inflammatory Responses in Rheumatoid Arthritis. Sci. Transl. Med. 2013, 5, 178ra40. [Google Scholar] [CrossRef] [PubMed]
- Catrina, A.I.; Svensson, C.; Malmström, V.; Schett, G.; Klareskog, A.I.C.V.M.L. Mechanisms leading from systemic autoimmunity to joint-specific disease in rheumatoid arthritis. Nat. Rev. Rheumatol. 2017, 13, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Harre, U.; Georgess, D.; Bang, H.; Bozec, A.; Axmann, R.; Ossipova, E.; Jakobsson, P.-J.; Baum, W.; Nimmerjahn, F.; Szarka, E.; et al. Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J. Clin. Investig. 2012, 122, 1791–1802. [Google Scholar] [CrossRef] [PubMed]
- Jurczak, A.; Delay, L.; Barbier, J.; Simon, N.; Krock, E.; Sandor, K.; Agalave, N.M.; Rudjito, R.; Wigerblad, G.; Rogóż, K.; et al. Antibody-induced pain-like behavior and bone erosion: Links to subclinical inflammation, osteoclast activity, and acid-sensing ion channel 3-dependent sensitization. Pain 2021, 163, 1542–1559. [Google Scholar] [CrossRef]
- Krishnamurthy, A.; Circiumaru, A.; Sun, J.; Kisten, Y.; Damberg, P.; Sakuraba, K.; Jarvoll, P.; Zhou, T.; Malmström, V.; Svensson, C.I.; et al. Combination of two monoclonal ACPAs induced tenosynovitis, pain and bone loss in mice in a Peptidyl Arginine Deiminase-4 dependent manner. Arthritis Rheumatol. 2022, 75, 164–170. [Google Scholar] [CrossRef]
- Krishnamurthy, A.; Ytterberg, A.J.; Sun, M.; Sakuraba, K.; Steen, J.; Joshua, V.; Tarasova, N.K.; Malmström, V.; Wähämaa, H.; Réthi, B.; et al. Citrullination Controls Dendritic Cell Transdifferentiation into Osteoclasts. J. Immunol. 2019, 202, 3143–3150. [Google Scholar] [CrossRef]
- Wigerblad, G.; Bas, D.B.; Fernades-Cerqueira, C.; Krishnamurthy, A.; Nandakumar, K.S.; Rogoz, K.; Kato1, J.; Sandor, K.; Su, J.; Jimenez-Andrade, J.M.; et al. Autoantibodies to citrullinated proteins induce joint pain independent of inflammation via a chemokine-dependent mechanism. Ann. Rheum. Dis. 2016, 75, 730–738. [Google Scholar] [CrossRef]
- Sahlström, P.; Hansson, M.; Steen, J.; Amara, K.; Titcombe, P.J.; Forsström, B.; Stålesen, R.; Israelsson, L.; Piccoli, L.; Lundberg, K.; et al. Different hierarchies of anti-modified protein autoantibody reactivities in rheumatoid arthritis. Arthritis Rheumatol. 2020, 72, 1643–1657. [Google Scholar] [CrossRef]
- Steen, J.; Forsström, B.; Sahlström, P.; Odowd, V.; Israelsson, L.; Krishnamurthy, A.; Badreh, S.; Mathsson Alm, L.; Compson, J.S.; Ramsköld, D.; et al. Recognition of Amino Acid Motifs, Rather Than Specific Proteins, by Human Plasma Cell-Derived Monoclonal Antibodies to Posttranslationally Modified Proteins in Rheumatoid Arthritis. Arthritis Rheumatol. 2019, 71, 196–209. [Google Scholar] [CrossRef]
- Okamoto, Y.; Devoe, S.; Seto, N.; Minarchick, V.; Wilson, T.; Rothfuss, H.M.; Mohning, M.P.; Arbet, J.; Kroehl, M.; Visser, A.; et al. Association of Sputum Neutrophil Extracellular Trap Subsets With IgA Anti–Citrullinated Protein Antibodies in Subjects at Risk for Rheumatoid Arthritis. Arthritis Rheumatol. 2022, 74, 38–48. [Google Scholar] [CrossRef]
- Bawadekar, M.; Shim, D.; Johnson, C.J.; Warner, T.F.; Rebernick, R.; Damgaard, D.; Nielsen, C.H.; Pruijn, G.J.; Nett, J.E.; Shelef, M.A. Peptidylarginine deiminase 2 is required for tumor necrosis factor alpha-induced citrullination and arthritis, but not neutrophil extracellular trap formation. J. Autoimmun. 2017, 80, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Spengler, J.; Lugonja, B.; Ytterberg, A.J.; Zubarev, R.A.; Creese, A.J.; Pearson, M.J.; Grant, M.M.; Milward, M.; Lundberg, K.; Buckley, C.D.; et al. Release of Active Peptidyl Arginine Deiminases by Neutrophils Can Explain Production of Extracellular Citrullinated Autoantigens in Rheumatoid Arthritis Synovial Fluid. Arthritis Rheumatol. 2015, 67, 3135–3145. [Google Scholar] [CrossRef] [PubMed]
- Pratesi, F.; Dioni, I.; Tommasi, C.; Alcaro, M.C.; Paolini, I.; Barbetti, F.; Boscaro, F.; Panza, F.; Puxeddu, I.; Rovero, P.; et al. Antibodies from patients with rheumatoid arthritis target citrullinated histone 4 contained in neutrophils extracellular traps. Ann. Rheum. Dis. 2014, 73, 1414–1422. [Google Scholar] [CrossRef]
- Corsiero, E.; Bombardieri, M.; Carlotti, E.; Pratesi, F.; Robinson, W.; Migliorini, P.; Pitzalis, C. Single cell cloning and recombinant monoclonal antibodies generation from RA synovial B cells reveal frequent targeting of citrullinated histones of NETs. Ann. Rheum. Dis. 2016, 75, 1866–1875. [Google Scholar] [CrossRef]
- Konig, M.F.; Andrade, F. A Critical Reappraisal of Neutrophil Extracellular Traps and NETosis Mimics Based on Differential Requirements for Protein Citrullination. Front. Immunol. 2016, 7, 461. [Google Scholar] [CrossRef]
- Holmes, C.L.; Shim, D.; Kernien, J.; Johnson, C.J.; Nett, J.E.; Shelef, M.A. Insight into Neutrophil Extracellular Traps through Systematic Evaluation of Citrullination and Peptidylarginine Deiminases. J. Immunol. Res. 2019, 2019, 2160192. [Google Scholar] [CrossRef]
- Chirivi, R.G.S.; van Rosmalen, J.W.G.; van der Linden, M.; Euler, M.; Schmets, G.; Bogatkevich, G.; Kambas, K.; Hahn, J.; Braster, Q.; Soehnlein, O.; et al. Therapeutic ACPA inhibits NET formation: A potential therapy for neutrophil-mediated inflammatory diseases. Cell Mol. Immunol. 2021, 18, 1528–1544. [Google Scholar] [CrossRef]
- Hensvold, A.K.; Kisten, Y.; Circiumaru, A.; Hansson, M.; Sun, M.; Fei, G.; Vivar, N.; Af Klint, E.; Rezaei, H.; Klareskor, L.; et al. Development of Ultrasound Detectable Arthritis Among ACPA Positive Subjects with Musculoskeletal Symptoms: The Risk RA Prospective Study. Arthritis Rheumatol. 2019, 71 (Suppl. S10), 310. [Google Scholar]
- Gupta, S.; Chan, D.W.; Zaal, K.J.; Kaplan, M.J. A High-Throughput Real-Time Imaging Technique To Quantify NETosis and Distinguish Mechanisms of Cell Death in Human Neutrophils. J. Immunol. 2018, 200, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Mócsai, A.; Ligeti, E.A.; Lowell, C.; Berton, G. Adhesion-dependent degranulation of neutrophils requires the Src family kinases Fgr and Hck. J. Immunol. 1999, 162, 1120–1126. [Google Scholar] [CrossRef] [PubMed]
- Mócsai, A.; Zhang, H.; Jakus, Z.; Kitaura, J.; Kawakami, T.; Lowell, C.A. G-protein–coupled receptor signaling in Syk-deficient neutrophils and mast cells. Blood 2003, 101, 4155–4163. [Google Scholar] [CrossRef] [PubMed]
- Raijmakers, R.; Vogelzangs, J.; Raats, J.; Panzenbeck, M.; Corby, M.; Jiang, H.; Thibodeau, M.; Haynes, N.; Van Venrooij, W.J.; Pruijn, G.J.; et al. Experimental autoimmune encephalomyelitis induction in peptidylarginine deiminase 2 knockout mice. J. Comp. Neurol. 2006, 498, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Lin, C.; Deng, H.; Strnad, J.; Bernabei, L.; Vogl, D.T.; Burke, J.J.; Nefedova, Y. A Novel Peptidylarginine Deiminase 4 (PAD4) Inhibitor BMS-P5 Blocks Formation of Neutrophil Extracellular Traps and Delays Progression of Multiple Myeloma. Mol. Cancer Ther. 2020, 19, 1530–1538. [Google Scholar] [CrossRef] [PubMed]
- Sakuraba, K.; Krishnamurthy, A.; Sun, J.; Zheng, X.; Xu, C.; Peng, B.; Engström, M.; Jakobsson, P.-J.; Wermeling, F.; Catrina, S.; et al. Autoantibodies targeting malondialdehyde-modifications in rheumatoid arthritis regulate osteoclasts via inducing glycolysis and lipid biosynthesis. J. Autoimmun. 2022, 133, 102903. [Google Scholar] [CrossRef]
- Lloyd, K.A.; Steen, J.; Amara, K.; Titcombe, P.J.; Israelsson, L.; Lundström, S.L.; Zhou, D.; Zubarev, R.; Reed, E.; Piccoli, L.; et al. Variable domain N-linked glycosylation and negative surface charge are key features of monoclonal ACPA: Implications for B-cell selection. Eur. J. Immunol. 2018, 48, 1030–1045. [Google Scholar] [CrossRef]
- Titcombe, P.J.; Wigerblade, G.; Sippl, N.; Zhang, N.; Shmagel, A.K.; Sahlström, P.; Zhang, Y.; Barsness, L.O.; Ghodke, Y.; Baharpoor, A.; et al. Pathogenic Citrulline-Multispecific B Cell Receptor Clades in Rheumatoid Arthritis. Arthritis Rheumatol. 2018, 70, 1933–1945. [Google Scholar] [CrossRef]
- Amara, K.; Israelsson, L.; Stålesen, R.; Sahlström, P.; Steen, J.; Malmström, V.; Grönwall, C. A Refined Protocol for Identifying Citrulline-specific Monoclonal Antibodies from Single Human B Cells from Rheumatoid Arthritis Patient Material. Bio-Protocol 2019, 9, e3347. [Google Scholar] [CrossRef]
- Ossipova, E.; Cerqueira, C.F.; Reed, E.; Kharlamova, N.; Israelsson, L.; Holmdahl, R.; Nandakumar, K.S.; Engström, M.; Harre, U.; Schett, G.; et al. Affinity purified anti-citrullinated protein/peptide antibodies target antigens expressed in the rheumatoid joint. Thromb. Haemost. 2014, 16, R167. [Google Scholar] [CrossRef]
- Sun, M.; Rethi, B.; Krishnamurthy, A.; Joshua, V.; Circiumaru, A.; Hensvold, A.H.; Ossipova, E.; Grönwall, C.; Liu, Y.; Engstrom, M.; et al. Anticitrullinated protein antibodies facilitate migration of synovial tissue-derived fibroblasts. Ann. Rheum. Dis. 2019, 78, 1621–1631. [Google Scholar] [CrossRef]
- Krishnamurthy, A.; Joshua, V.; Hensvold, A.H.; Jin, T.; Sun, M.; Vivar, N.; Ytterberg, A.J.; Engström, M.; Fernandes-Cerqueira, C.; Amara, K.; et al. Identification of a novel chemokine-dependent molecular mechanism underlying rheumatoid arthritis-associated autoantibody-mediated bone loss. Ann. Rheum. Dis. 2016, 75, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Wigerblad, G.; Kaplan, M.J. Neutrophil extracellular traps in systemic autoimmune and autoinflammatory diseases. Nat. Rev. Immunol. 2022, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Raposo, B.; Afonso, M.; Israelsson, L.; Wähämaa, H.; Stålesen, R.; Wermeling, F.; Hensvold, A.H.; Grönwall, C.; Rethi, B.; Klareskog, L.; et al. Divergent and dominant anti-inflammatory effects of patient-derived anticitrullinated protein antibodies (ACPA) in arthritis development. Ann. Rheum. Dis. 2023; Published online first. [Google Scholar] [CrossRef]
- de Bont, C.M.; Stokman, M.E.M.; Faas, P.; Thurlings, R.M.; Boelens, W.C.; Wright, H.L.; Pruijn, G.J. Autoantibodies to neutrophil extracellular traps represent a potential serological biomarker in rheumatoid arthritis. J. Autoimmun. 2020, 113, 102484. [Google Scholar] [CrossRef]
- Iobagiu, C.; Magyar, A.; Nogueira, L.; Cornillet, M.; Sebbag, M.; Arnaud, J.; Hudecz, F.; Serre, G. The antigen specificity of the rheumatoid arthritis-associated ACPA directed to citrullinated fibrin is very closely restricted. J. Autoimmun. 2011, 37, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Sebbag, M.; Moinard, N.; Auger, I.; Clavel, C.; Arnaud, J.; Nogueira, L.; Roudier, J.; Serre, G. Epitopes of human fibrin recognized by the rheumatoid arthritis-specific autoantibodies to citrullinated proteins. Eur. J. Immunol. 2006, 36, 2250–2263. [Google Scholar] [CrossRef]
- Verpoort, K.N.; Cheung, K.; Ioan-Facsinay, A.; Mil, A.H.M.V.D.H.-V.; De Vries-Bouwstra, J.K.; Allaart, C.F.; Drijfhout, J.W.; De Vries, R.R.P.; Breedveld, F.C.; Huizinga, T.W.J.; et al. Fine specificity of the anti-citrullinated protein antibody response is influenced by the shared epitope alleles. Arthritis Rheum. 2007, 56, 3949–3952. [Google Scholar] [CrossRef]
- Schellekens, C.A.; Visser, H.; Jong, B.A.; Hoogen, F.H.; Hazes, J.M.; Breedveld, F.C.; Venrooij, W.J. The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum. 2000, 43, 155–163. [Google Scholar] [CrossRef]
- Janssen, K.M.J.; Smit, M.J.; Withaar, C.; Brouwer, E.; Winkelhoff, A.J.; Vissink, A.; Westra, J. Autoantibodies against citrullinated histone H3 in rheumatoid arthritis and periodontitis patients. J. Clin. Periodontol. 2017, 44, 577–584. [Google Scholar] [CrossRef]
- Schwenzer, A.; Jiang, X.; Mikuls, T.R.; Payne, J.; Sayles, H.R.; Quirke, A.-M.; Kessler, B.; Fischer, R.; Venables, P.J.; Lundberg, K.; et al. Identification of an immunodominant peptide from citrullinated tenascin-C as a major target for autoantibodies in rheumatoid arthritis. Ann. Rheum. Dis. 2015, 75, 1876–1883. [Google Scholar] [CrossRef]
- Lloyd, K.A.; Wigerblad, G.; Sahlström, P.; Garimella, M.G.; Chemin, K.; Steen, J.; Titcombe, P.J.; Marklein, B.; Zhou, D.; Stålesen, R.; et al. Differential ACPA Binding to Nuclear Antigens Reveals a PAD-Independent Pathway and a Distinct Subset of Acetylation Cross-Reactive Autoantibodies in Rheumatoid Arthritis. Front. Immunol. 2019, 9, 3033. [Google Scholar] [CrossRef]
- Figueiredo, C.P.; Bang, H.; Cobra, J.F.; Englbrecht, M.; Hueber, A.; Haschka, J.; Manger, B.; Kleyer, A.; Reiser, M.; Finzel, S.; et al. Antimodified protein antibody response pattern influences the risk for disease relapse in patients with rheumatoid arthritis tapering disease modifying antirheumatic drugs. Ann. Rheum. Dis. 2016, 76, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Hansson, M.; Mathsson, L.; Schlederer, T.; Israelsson, L.; Matsson, P.; Nogueira, L.; Jakobsson, P.-J.; Lundberg, K.; Malmström, V.; Serre, G.; et al. Validation of a multiplex chip-based assay for the detection of autoantibodies against citrullinated peptides. Thromb. Haemost. 2012, 14, R201. [Google Scholar] [CrossRef] [PubMed]
- Rönnelid, J.; Hansson, M.; Mathsson-Alm, L.; Cornillet, M.; Reed, E.; Jakobsson, P.-J.; Alfredsson, L.; Holmdahl, R.; Skriner, K.; Serre, G.; et al. Anticitrullinated protein/peptide antibody multiplexing defines an extended group of ACPA-positive rheumatoid arthritis patients with distinct genetic and environmental determinants. Ann. Rheum. Dis. 2017, 77, 203–211. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cîrciumaru, A.; Afonso, M.G.; Wähämaa, H.; Krishnamurthy, A.; Hansson, M.; Mathsson-Alm, L.; Keszei, M.; Stålesen, R.; Ottosson, L.; de Vries, C.; et al. Anti-Citrullinated Protein Antibody Reactivity towards Neutrophil-Derived Antigens: Clonal Diversity and Inter-Individual Variation. Biomolecules 2023, 13, 630. https://doi.org/10.3390/biom13040630
Cîrciumaru A, Afonso MG, Wähämaa H, Krishnamurthy A, Hansson M, Mathsson-Alm L, Keszei M, Stålesen R, Ottosson L, de Vries C, et al. Anti-Citrullinated Protein Antibody Reactivity towards Neutrophil-Derived Antigens: Clonal Diversity and Inter-Individual Variation. Biomolecules. 2023; 13(4):630. https://doi.org/10.3390/biom13040630
Chicago/Turabian StyleCîrciumaru, Alexandra, Marcelo Gomes Afonso, Heidi Wähämaa, Akilan Krishnamurthy, Monika Hansson, Linda Mathsson-Alm, Márton Keszei, Ragnhild Stålesen, Lars Ottosson, Charlotte de Vries, and et al. 2023. "Anti-Citrullinated Protein Antibody Reactivity towards Neutrophil-Derived Antigens: Clonal Diversity and Inter-Individual Variation" Biomolecules 13, no. 4: 630. https://doi.org/10.3390/biom13040630
APA StyleCîrciumaru, A., Afonso, M. G., Wähämaa, H., Krishnamurthy, A., Hansson, M., Mathsson-Alm, L., Keszei, M., Stålesen, R., Ottosson, L., de Vries, C., Shelef, M. A., Malmström, V., Klareskog, L., Catrina, A. I., Grönwall, C., Hensvold, A., & Réthi, B. (2023). Anti-Citrullinated Protein Antibody Reactivity towards Neutrophil-Derived Antigens: Clonal Diversity and Inter-Individual Variation. Biomolecules, 13(4), 630. https://doi.org/10.3390/biom13040630