1. Introduction
Wound healing can be described as the process of the repair of cutaneous tissue damage or breakdown. It consists of a complex cascade of events that can be summarised into several overlapping stages, each requiring coordination of a variety of signals and cell types [
1,
2,
3,
4,
5,
6]. Wound remodelling involves many cells and signalling pathways and consists of the critical re-epithelialisation of the wound by keratinocytes as they proliferate and migrate in order to close the wound. This is achieved through cell polarisation in the direction of movement according to numerous signals, such as cytokines and growth factors, and the extension of actin-rich membrane structures [
7]. Furthermore, adhesion dynamics, where a coordinated balance in the formation of adhesions at the leading edge of the cell and disassembly at the rear, allows cells to gain traction by connecting the actin cytoskeleton to the surrounding matrix, subsequently enabling the cell to move. An optimal level of adhesion is key in allowing the motility of migrating cells [
6,
8,
9,
10].
In most cases, acute cutaneous wounds, which typically occur suddenly or following surgery, close within a reasonable period. However, underlying pathological complications or other factors that impair the normal healing process can cause chronic or non-healing wounds to form. Many factors, such as vascular compromise, repetitive insult to the tissue or chronic inflammation, can lead to chronic wound formation. This makes aged individuals or patients with diabetes, vascular diseases and auto-immune diseases more vulnerable to developing chronic wounds, the majority of which are classified as leg, pressure or diabetic foot ulcers [
11,
12,
13,
14,
15,
16]. Chronic non-healing wounds present a substantial economic burden to the healthcare system; significant reductions in quality of life for those affected; and often precede serious events, such as limb amputations or even premature deaths [
17,
18]. In addition to the significant costs associated with these wounds, they also have a large impact on the quality of life of patients. Physical symptoms from chronic wounds include leakage, odour and pain, as well as impairments to daily living [
19,
20,
21]. The lack of appropriate investigation and classification of chronic wounds was identified by several studies and healthcare professionals currently often rely on limited, traditional wound care approaches, such as dressings, debridement and compression treatment [
22,
23,
24]. There is a clear need for effective diagnosis and identification of new therapeutic approaches to treat chronic wounds. The complex and diverse underlying genetic and molecular processes involved in the wound healing process and chronic wound development make identifying the cause of a particular chronic wound a difficult task. However, these genetic and protein expression deficiencies in chronic wounds also represent potential diagnostic and therapeutic opportunities. Several studies explored these molecular changes and, as a result, highlighted many novel proteins that could potentially be used to explain the development of chronic wounds, offer prognoses and even influence wound repair [
25,
26,
27]. In one study, nWASP, amongst other molecular markers, was shown to have differential expression in acute, chronic healing and chronic non-healing wound tissues [
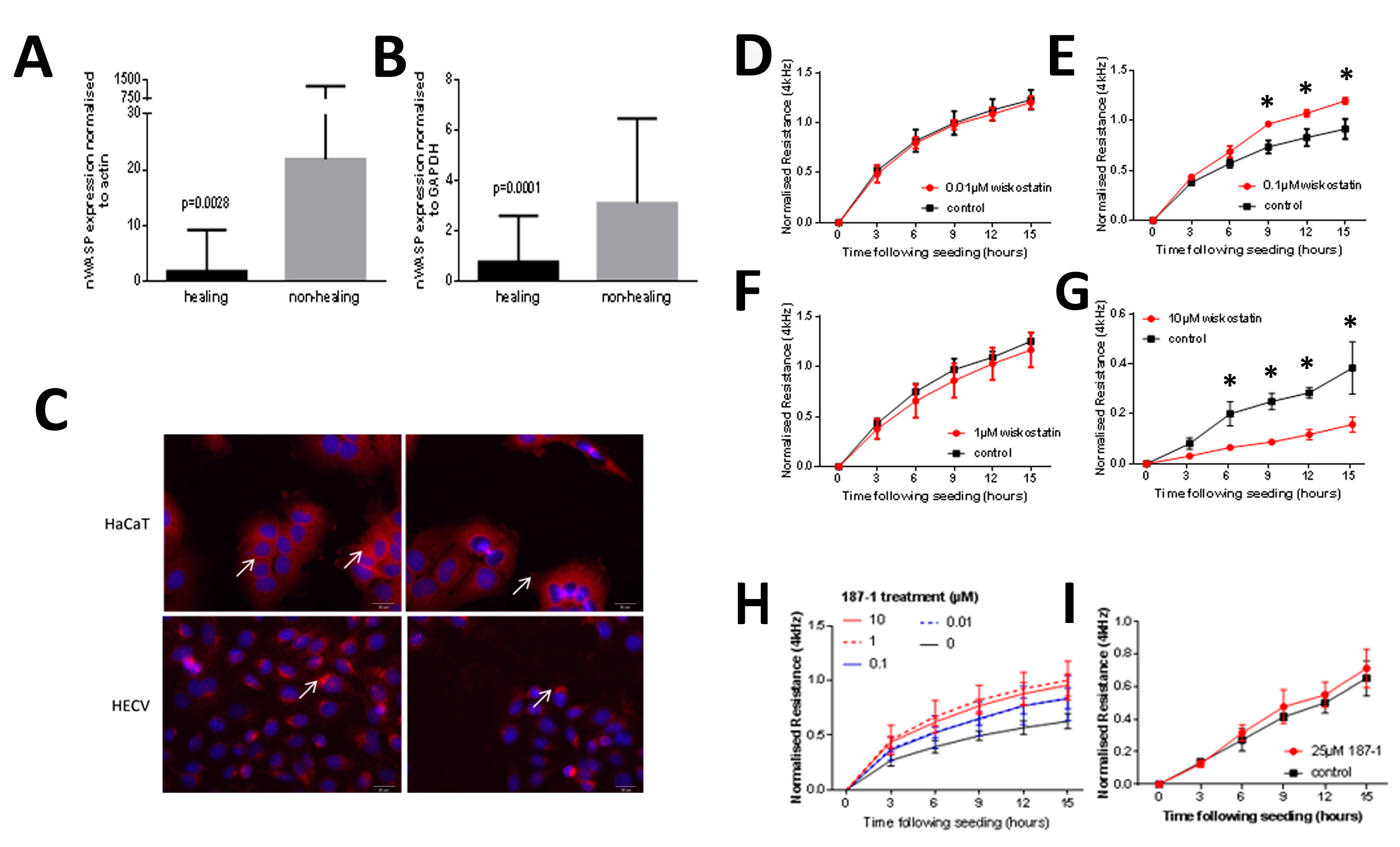
27].
nWASP (neural Wiskott–Aldrich syndrome protein) belongs to the WASP/WAVE (WASP family verprolin-homologous protein) family. Mutations in the first WASP family member to be identified, namely, WASP itself, were found to be the cause of WAS, which is a recessive disorder that was initially described as a triad of symptoms, namely, thrombocytopenia, eczema and immunodeficiency [
28,
29,
30]; furthermore, they disrupt the activity of important functional domains, leading to more severe phenotypes [
31,
32]. nWASP was the second protein to be classified as a WASP family member due to the detection of several functional motifs shared with WASP. This 65 kDa protein was named neural WASP due to its shared homology with WASP and abundance in the brain, although it is widely expressed in different tissues throughout the body [
33]. Under resting conditions, nWASP exists in an inactive, auto-inhibited confirmation whereby the main catalytic domain is shielded by the N-terminus regions. The WASP/WAVE family mediates the signals between the Rho GTPase family members, such as Rho, Rac and Cdc42, and the factors that modulate the actin cytoskeleton, in particular the actin-related protein 2/3 (Arp2/3) complex. Cdc42 can bind to the GBD domain, destabilising the folded confirmation of nWASP and exposing its catalytic domain [
34,
35]. Arp2/3 becomes activated when bound to the C-terminus CA region on WASP family proteins, allowing actin polymerisation to be initiated if an actin monomer is bound in conjunction with the V region [
36,
37,
38,
39].
Through its role as an organiser of the actin cytoskeleton, nWASP was also shown to be involved in the formation of membrane protrusions that are important for cell movement and its expression was shown to correlate with certain cancer phenotypes [
34,
40,
41,
42,
43]. Consequently, nWASP has been recognised as a potential therapeutic target in a range of contexts, including wound healing.
The aims of this study were to validate nWASP as a novel therapeutic target for the treatment of chronic wounds by examining expression levels in human wound tissues. The therapeutic potential of targeting nWASP was then explored by examining the effect of inhibiting nWASP using the agents 187-1 and wiskostatin, which act to maintain nWASP in its inactive, auto-inhibited state (small-molecule inhibitors of nWASP, namely, wiskostatin and 187-1, bind to nWASP and allosterically block its activity by stabilising the closed, auto-inhibited conformation of nWASP. Both inhibitors act in the same way to stabilise the autoinhibited state of nWASP, blocking activation and hence actin polymerisation, but wiskostatin was used more extensively in research to examine the effects of nWASP activity. Wiskostatin is a cell-permeable N-alkylated carbazole that interacts with a cleft in the regulatory GBD of nWASP in the solution structure of the complex [
44], as well as on human skin cell behaviour in vitro and in vivo on the closure of wounds in mouse models with impaired healing abilities. We also investigated the potential effects of these treatments on downstream signalling changes in skin cells in order to elucidate mechanistic effects.
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
Human keratinocyte cells (HaCaT cells) from the German Cancer Institute/Cell Service, Germany, human vascular endothelial cells (HECV cells) from Interlab, Italy, and TE 354.T cells (LCG Standards, Bury, UK) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Sigma-Aldrich, Dorset, UK) supplemented with 10% foetal bovine serum (FBS; Sigma-Aldrich, Dorset, UK), 100 units/mL penicillin and 100 µg/mL streptomycin (Sigma-Aldrich, Dorset, UK). The cells were incubated at 37 °C, 5% CO2 and 95% humidity.
2.2. Tissue Collection
Fresh tissues from chronic wounds (
n = 14), acute wounds (
n = 10) and normal skin from healthy volunteers (
n = 10) were collected under the approval of the local ethical committee (ethical approval ID: 04/WSE03/92). Chronic wound tissues were obtained from patients with chronic leg ulcers that had been present for a minimum of 6 months prior to their biopsy with no evidence of healing 6 weeks before the biopsy. Acute wound tissues were obtained from patients with acute surgical wounds following the excision of pilonidal disease and normal tissues were from healthy, unwounded skin. A second cohort of chronic wound tissues was collected from chronic venous leg ulcer patients following ethical approval from a local committee (South East Wales Ethics Committee reference number 09/WSE02/51). Wound size was recorded at the time of the initial biopsy and again after 3 months, during which wounds were treated as per best medical practise (
n-77). Wounds that reduced in size over this period were described as ‘healing/healed’ and those that grew or did not reduce in size were described as ‘non-healing’. All tissues were snap-frozen following collection and stored at −80 °C until processing. Written informed consent was obtained from each patient who agreed for a biopsy to be taken. Wound cohorts were previously described [
45,
46,
47].
2.3. Tissue Processing for the RNA Extraction and Reverse Transcription
Tissues were sectioned on a cryostat (Leica, Microsystems Ltd., Milton Keynes, UK) with either a 7 µm thickness for immune-histochemical analysis or at 20 µm thickness for the extraction of RNA. Approximately 20 sections from the same patient sample biopsy were pooled and homogenised in an ice-cold TRI reagent (Sigma-Aldrich, Poole, UK) using a hand-held homogeniser (Cole Palmer, London, UK). RNA was extracted from the tissues following the manufacturer’s instructions, and the same protocol was also used to extract RNA from cultured cell lines. Samples were standardised and cDNA was subsequently generated (BioRad, Hemel Hempstead, UK).
2.4. Tissue Processing for the Ex Vivo Model
The
ex vivo effects of the nWASP inhibitor 187-1 in chronic human wound tissues were examined using a method previously established [
48]. Briefly, fresh biopsies from human chronic wounds were immediately placed in a buffer that mimics physiological fluid and contains a mixture of antibiotics. The tissues were finely minced using a sterile scalpel to sizes less than 1 mm in diameter. After extensive washing in the buffer, the living tissues were immediately embedded in an extracellular matrix gel. The gels and the topping solution included the treatments (nWASP inhibitor). The tissues were photographed daily. The degree of expansion from the implanted tissues was calculated using the imager, as previously reported [
48].
2.5. Quantitative, Real-Time Polymerase Chain Reaction (qPCR) and Conventional PCR
Analysis of gene transcript expression was carried out using qPCR with cDNA produced from a human wound and skin tissues and conventional PCR using cDNA produced from RNA extracted from cell lines. This study adopted Ampliflor quantitation technology in which one set of gene-specific primers (designed using Beacon Design software, PREMIER Biosoft, Palo Alto, CA, USA) and Uniprimer probes (Intergen Inc., New York, NY, USA) were used. The reaction was carried out using the ICycleIQ (BioRad, UK). The real-time qPCR conditions were 95 °C for 15 min, followed by 60 cycles of 95 °C for 20 s, 55 °C for 30 s and 72 °C for 20 s. In all the assays, GAPDH and actin were amplified and used as housekeeping controls, and an internal standard was also employed for quantitation purposes. The nWASP primers were as follows: BNDF (F8: TTCATACTTTGGTTGCATGA, R8: TTCAGTTGGCCTTTTGATAC), GAPDH (F8: GGCTGCTTTTAACTCTGGTA, R8: GACTGTGGTCATGAGTCCTT), nWASP (F8: AGTCCCTCTTCACTTTCCTC, R8: GCTTTTCCCTTCTTCTTTTC), TrkB (F2a: CCCACTCACATGAACAATGG, R2a: TCAGTGACGTCTGTGGAAGG), Actin (F8: GGACCTGACTGACTACCTCA, zR8: ACTGAACCTGACCGTACAAGCTTCTCCTTAATGTCACG), BDNF (F8: TTCATACTTTGGTTGCATGA, Rz8: ACTGAACCTGACCGTACACTCTTGAACCTGCCTTGG), GAPDH (F: CTAGTACGTCGTGGAGTC, zR: ACTGAACCTGACCGTACACAGAGATGATGATGACCCTTTTG), PDPL (F: GAATCATCGTTGTGGTTATG, zR: ACTGAACCTGACCGTACACTTTCATTTGCCTATCACAT) and nWASPKD (F8: AGTCCCTCTTCACTTTCCTC, zR8a: ACTGAACCTGACCGTACAAGATCTCTGTGGATTGTCCT).
2.6. Reagents and Treatments
nWASP inhibitors, wiskostatin (Merck Pharmaceuticals, Watford, UK) and 187-1 (TOCRIS, Bristol, UK) were used. For the in vitro studies, wiskostatin was dissolved in 30% dimethyl sulfoxide (DMSO, Sigma-Aldrich, Dorset, UK) in normal cell culture medium to a stock concentration of 300 µM, whereas 187-1 was diluted in BSS. For the in vivo studies, nWASP inhibitor compounds, 187-1 and wiskostatin were formulated for systemic and topical application. For systemic application, 187-1 was dissolved and diluted in BSS to the required concentration. Wiskostatin was first dissolved in DMSO (Sigma-Aldrich, Gillingham, UK) at a concentration of 5 mg/mL. The DMSO solution was then gradually diluted in BSS in order to avoid precipitation. The solutions were prepared so that each 100 µL contained the correct amount of compounds and was stored at −20 °C until used. The primary antibodies used were as follows: actin (sc-16515, Insight Biotechnology, Middlesex, UK), Erk1/2 (v114A, Promega, Southampton, UK), GAPDH (sc32233, Insight Biotechnology, Middlesex, UK), PLCγ1 (sc81, Insight Biotechnology, Middlesex, UK), p-PLCγ1 (sc22141, Insight Biotechnology, Middlesex, UK), nWASP (NBP1-82512, Novus Biologicals, Abingdon, UK), TrkB Y816 (ABN1381, EMD-Millipore, Watford, UK), TrkB pan (07-225, EMD-Millipore, Watford, UK) and TrkB pan (sc-136990, Insight Biotechnology, Middlesex, UK). The secondary antibodies were as follows: goat anti-mouse IgG (A4416, Sigma-Aldrich, Dorset, UK), goat anti-rabbit IgG (A6154, Sigma-Aldrich, Dorset, UK), rabbit anti-goat IgG (A5420, Sigma-Aldrich, Dorset, UK), DAPI (D1306, Life Technologies, Warrington, UK), anti-rabbit AlexaFluor 488 (A21206, Life Technologies, Warrington, UK), anti-mouse AlexaFluor 488 (A21202, Life Technologies, Warrington, UK), anti-rabbit AlexaFluor 594 (A21207, Life Technologies, Warrington, UK), anti-mouse AlexaFluor 594 (A21203, Life Technologies, Oxford, UK) and anti-goat AlexaFluor 594 (A11058, Life Technologies, Warrington, UK).
2.7. Functional Assays
2.7.1. Electric Cell–Substrate Impedance Sensing (ECIS)
ECIS instruments (Applied Biophysics Inc., New York, NJ, USA) were used to electrically monitor the coverage of gold electrodes by cells by measuring the resistance and impedance at a frequency of 4 kHz unless otherwise specified. Cells were seeded in a culture medium containing treatments. Cells were then constantly monitored following seeding and electrical wounds were applied where described. Wound settings were a time of 30 s, current of 2400 μA and frequency of 60,000 Hz. Increased resistance and impedance illustrate a greater number of cells present on the electrode. This can therefore be used to assess attachment and motility.
2.7.2. In Vitro Cell Viability Test
Briefly, cells were seeded in a 96-well plate in varying treatment concentrations/controls and incubated for 92 h. After a further 4 h incubation in 1:10 MTT (Thiazolyl Blue Tetrazolium Bromide, Sigma-Aldrich, Gillingham, UK) solution (5 mg/mL in phosphate buffered solution (PBS)), acidified isopropanol (Sigma-Aldrich, Dorset, UK) was applied and the optical density (OD) was determined using an absorbance reader (Biotek ELx800, Swindon, UK) at 540 nm and used to calculate relative cell viability at each treatment concentration [
49].
2.7.3. In Vitro Growth Assay
Cells were seeded into a 96-well plate with appropriate treatments and fixed after 1/2/4 day incubation periods using 4% formalin (Sigma-Aldrich, Dorset, UK). Plates were stained with 1% crystal violet (Sigma-Aldrich, Gillingham, UK), which was then extracted from cells using 10% acetic acid (Sigma-Aldrich, Dorset, UK) in distilled water (v/v). Absorbance was determined at a 540 nm wavelength on an absorbance plate reader (Biotek ELx800, Swindon, UK).
2.7.4. In Vitro Carrier Bead Assay
A total of 100 μL of Cytodex-2 carrier beads (Sigma-Aldrich Ltd., Gillingham, UK) were added to 10 mL of 1 × 104 cells/mL and gently mixed. The cells were then left at 37 °C for 2 h. Following a wash with medium, cells were allowed to settle until aliquoted into a 96-well plate and treated with inhibitors. After incubation overnight, the beads were washed off in PBS and the cells that had migrated onto the culture vessel floor were counted after visualisation with crystal violet.
2.7.5. In Vitro Scratch Assay
Cells were seeded in appropriate treatments into each well on a 24-well plate. Upon reaching confluence, the monolayer was scratched to create a linear wound. The plate was placed in an EVOS® FL Auto Imaging System (Life Technologies, Oxford, UK), which maintained the plate in normal culture conditions throughout the experiment. Images were captured of the wound every 30 min for up to 24 h.
2.7.6. In Vitro Adhesion Assay
Wells were pre-coated with Matrigel basement membrane matrix (BD Biosciences, Oxford, UK) at 50 μg/mL in a normal culture medium. Cells were then seeded into each well onto the Matrigel membrane in treatments and incubated for 25 min. Adherent cells were fixed, stained and quantified as above.
2.7.7. Tubule Formation Assay
A Matrigel matrix (10 mg/mL) was gently pipetted into the bottom of each well of a 96-well plate. The plate was placed at 37 °C for 40 min whilst the Matrigel polymerised. Then, 4 × 104 cells (and treatments/control reagents where appropriate) were plated on top of the Matrigel in each well in triplicate and incubated at 37 °C, 5% CO2 and 95% humidity. Images of each well were captured at X5 magnification and intervals ranging from 30 to 1170 min using a Leica DMi1 microscope equipped with an MC120 HD camera and Leica Application Suite version 3.0.0 software (Leica Microsystems, Milton Keynes, UK). Analysis of the images was carried out using ImageJ, where the lengths of cell structures that form part of a tubule structure were measured to track the progress of tubule formation.
2.8. In Vivo Tolerance Test
The main tolerance tests were conducted using CD-1 athymic mice (Charles River Laboratories, Oxford, UK), owing to their slow and steady rate of growth and the ease of observing changes in the skin (hairless) and other possible side effects. Briefly, CD-1 mice that were 4–6 weeks old and 20 g in weight were housed in filter-topped cages. 187-1 (MW 1784, dissolved in BSS buffer) and wiskostatin (MW 426, dissolved in DMSO and diluted in BSS) were injected via the intraperitoneal route on a daily basis. Dosages administered were 1 and 10 µM in 100 µL volumes for each compound, equivalent to 1.8 g/kg/day and 17.8 g/kg/day for 187-1 and 0.43 g/kg/day and 4.3 g/kg/day for wiskostatin. CD-1 mice were observed daily and weighed twice weekly based on the previous in vitro experiments. An additional tolerance and efficacy test was carried out using the diabetic db/db strain obtained from Harlan Laboratories (Cambridge, UK), which exhibited impaired wound healing abilities. These mice were 4–6 weeks old and when their body weight reached 20 g they were used for tolerance and efficacy tests.
2.9. In Vivo Efficacy Test and Wound Healing
An ear punch method previously described [
50] was used to create wounds in the mice that were 1 mm in diameter. Treatment was given systemically or topically. For topical application, two carrier gels that are currently used in wound care were used. The inhibitors were diluted from the master stock in the gel at a concentration of 1 mg/g followed by low-speed homogenisation using a hand-held homogeniser. For use, small amounts (150 µL) of the gel were rubbed into the wound area. Both treatments were given every other day. For systemic application, 187-1 was applied at 0.5 and 5 µM (equivalent to 0.89 g/kg/day and 8.9 g/kg/day) and wiskostatin was applied at 1 and 10 µM (equivalent to 0.43 g/kg/day and 4.3 g/kg/day). Images were obtained weekly. The sizes of the wounds were determined using image analysis software. Data are presented in two ways: as the area of the wounds in pixels, where two sample Student t-tests were used for statistical analysis, or as the change in the size of the wound from the starting point calculated using the following formula: (area at a given point—area at the starting point)/(area of the starting point) × 100, in which case, the Bonferroni model was used for data analysis.
2.10. Microarray Analysis
This study used the KinexusTM KAM880 protein array service provided by Kinexus Bioinformatics Ltd. (Vancouver, BC, Canada). Signal quantification was performed with ImaGene 8.0 by Kinexus Bioinformatics Ltd., which has predetermined settings for spot segmentation and background correction. The background-corrected raw intensity data were then globally normalised by summing the intensities of all the net signal median values for a sample to obtain the globally normalised signal intensities for each protein. The percentage change of the treated samples from the control was calculated based on the globally normalised intensity for each protein using the following calculation: %CFC = (globally normalised treated − globally normalised control)/globally normalised control) × 100. The percentage error range was also calculated to examine how tightly the globally normalised net signal intensity varied for duplicate spots of the same protein in the sample. The z-scores were also calculated by subtracting the overall average intensity of all replicate spots from the raw intensity for each spot and then dividing it by the standard deviation of all the measured intensities within each sample. The z-ratio was calculated by dividing the differences between the observed z-scores by the standard deviation of all the differences for that comparison. Several factors were used to determine the most important changes in protein expression and phosphorylation including %CFC, error ranges, value for the globally normalised intensity of one of the samples of >1500 and significance based on z-ratios of <−1.64 or >1.64.
2.11. Immunofluorescence
Cells were cultured in Millicell EZ 8-well chamber slides (Merck Millipore, Watford, UK). To fix the cells, the culture medium was removed and the cells were washed with PBS and then fixed in 100% ice-cold ethanol. To proceed with immunofluorescence staining, the cells were washed 3 times in PBS and then permeabilised using 0.1% Triton X-100 (Sigma-Aldrich, Dorset, UK). Blocking buffer (5–10% donkey serum (D9663, Sigma-Aldrich, Dorset, UK)) in PBS was added to each well. The slide was left for 3 h at room temperature in a blocking buffer. The chamber slide was then incubated with primary antibodies for an hour on the bench or at 4 °C overnight. Secondary antibodies were (1:500) with the addition of DAPI (1:100), and each primary antibody was incubated with the corresponding secondary for a further hour. Following three washes in PBS, the slides were mounted in FluorSave™ (Calbiochem, Nottingham, UK) and allowed to dry before being visualised using an Olympus BX51 microscope with a Hamamatsu Orca ER digital camera at ×40. Images were analysed using ImageJ.
2.12. Immunohistochemistry (IHC)
Immunohistological analysis using an avidin–biotin peroxidase technique was performed on human tissue samples collected from cohort 2. The frozen sections were fixed in dried acetone (10162180, Fisher Scientific, Loughborough, UK), air-dried and washed in PBS. Sections were then incubated in 0.1% BSA/10% horse serum in PBS (referred to as blocking solution) in a humidified box at room temperature, followed by primary antibody solutions (diluted in blocking buffer to a final concentration of 2 µg/mL). Following washing with PBS, biotinylated horse anti-mouse/rabbit IgG secondary antibody (Vector Laboratories, Oxford, UK) was applied for 30 min, followed by ABC reagent for 30 min, both of which were provided in the VECTASTAIN® Elite ABC Kit (Vector Laboratories, Peterborough, UK). 3,3′-Diaminobenzidine (DAB)(Sigma-Aldrich, Gillingham, UK) substrate (5 mg/mL) was used to develop the final reaction and the sections were counterstained with Gill’s haematoxylin (Vector Laboratories, Oxford, UK). Following dehydration and clearing in xylene, sections were mounted in Distyrene Plasticizer Xylene (DPX, Merck Pharmaceuticals, Gillingham, UK). Staining was visualised using a Leica DM1000LED microscope with an MC120 HD camera and Leica Application Suite (version 3.0.0) software (Leica Microsystems, Milton Keynes, UK). The localisation and intensity of staining were judged blindly by two people independently. Positive staining was seen as a brown/black deposit, whilst negatively stained cells could be clearly distinguished using a blue nucleated stain.
2.13. Protein Extraction, SDS-PAGE and Western Blotting
A lysis buffer was used to extract protein from cells, which was then used for SDS-PAGE. Proteins were transferred onto Immobilon® PVDF membranes (Merck Millipore, Watford, UK), which were blocked and probed with primary antibodies and then incubated with the corresponding peroxidase-conjugated secondary antibodies. Proteins were visualised using an EZ-ECL Kit (Biological Industries, Beit Haemk, Israel).
2.14. siRNA Silencing of nWASP
nWASP siRNA (sc36006) was obtained from Insight Biotechnology (UK) and non-targeting siRNA (NT) was obtained from Dharmacon (D001810; Layfayette, CO, USA). Cells were transfected with nWASP siRNA (sc360006; Insight Biotechnology, UK) at varying concentrations or NT control siRNA at the same concentration, as described. siRNA, which was diluted according to the desired end concentration in SFM, was combined with equal volumes of Lipofectamine 3000 reagent (ThermoFisher Scientific, Boston, MA, USA). This siRNA/Lipofectamine 3000 mix in SFM (with no antibiotics) was allowed to stand at room temperature for 30–40 min. Antibiotic-free DMEM supplemented with 5% FBS was then added to the siRNA/Lipofectamine 3000 solution to achieve the desired end concentration of reagents and make up the required volume for application to cells; then, the final solution was gently applied to each well. Cells were incubated under normal culture conditions and a normal culture medium was used 24 h after transfection for any further culturing or assays unless stated otherwise.
2.15. Statistical Analysis
Statistical analysis was conducted using Minitab, SPSS, GraphPad 6.0 Prism (PRISM, Boston, MA, USA) and an online chi-square service tool. Transcript levels from qPCR experiments are reported as median ± SEM and the Mann–Whitney test was used to analyse qPCR data. Representative data are presented. A p-value < 0.05 was considered statistically significant.
4. Discussion
This study identified nWASP as a molecule that is important in human wound healing and recognises nWASP as a new molecular target to encourage healing in human chronic wounds. Non-healing chronic wounds showed higher expression of nWASP than healing chronic wounds. This quantitative transcript analysis suggests that a balance of nWASP activity in human wounds may be key for healthy wound healing behaviour with over-expression above a certain level indicative of impaired wound healing. Hence, this study proposes that targeting nWASP in a clinical setting may provide a means to encourage healing behaviour in chronic wounds.
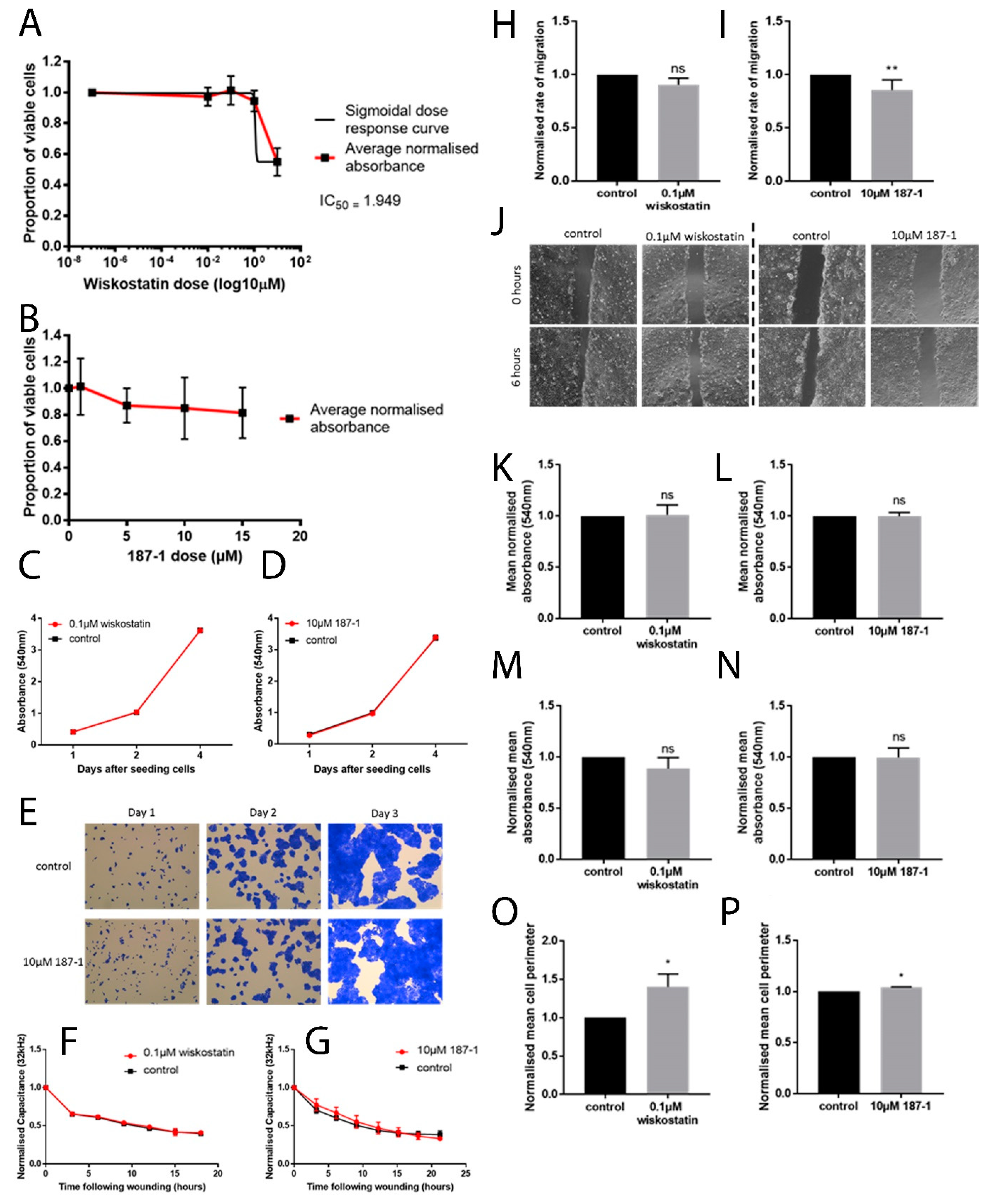
In vitro work carried out in this study supported the idea that altering nWASP activity through inhibitor treatment and at a transcript expression level can affect human keratinocyte and endothelial behaviour. The reduction of nWASP activity through nWASP inhibition and transcript expression knockdown affected the cell spreading and attachment properties of cells. Decreased nWASP activity from wild-type keratinocyte levels through transcript knockdown or inhibition appeared to increase the adherent properties of cells; furthermore, the knockdown of nWASP in endothelial cells caused the opposite effect in that attachment and spreading appeared to be impaired. This suggests that nWASP may have a role in the attachment properties of cells to a surface. This could explain how a balance of nWASP expression is important for correct wound healing behaviour, as the optimal level of adhesion and attachment of cells were shown to be a key factor in their migratory properties [
9,
10]. The implication that nWASP may be involved in affecting these properties of cells may explain how this molecule is involved in wound healing in humans. The molecular mechanisms involved in nWASP activity affecting the attachment properties of keratinocytes in this context are currently unknown but the diverse roles of nWASP and the effects of the actin polymerisation that can result from its activation offer many potential explanations for how nWASP may influence cell attachment and motility.
Using ex vivo and in vivo models, this study also demonstrated how nWASP inhibition can encourage wound healing behaviour in tissues with impaired healing abilities and thus provides a new management model for chronic wounds. Using mouse models to study the effect of nWASP inhibitor treatment on wound healing has not only supported previous findings that nWASP has potential as a target in wound healing but highlights a very simple but possibly life-changing therapy option that has potential in human cases. The fact that commercially available nWASP inhibitors can be applied so simply using carrier gels that are already used to treat chronic wounds in the clinic but with such a dramatic effect on encouraging healing in wounds in mice that naturally exhibit impaired wound healing abilities is very promising.
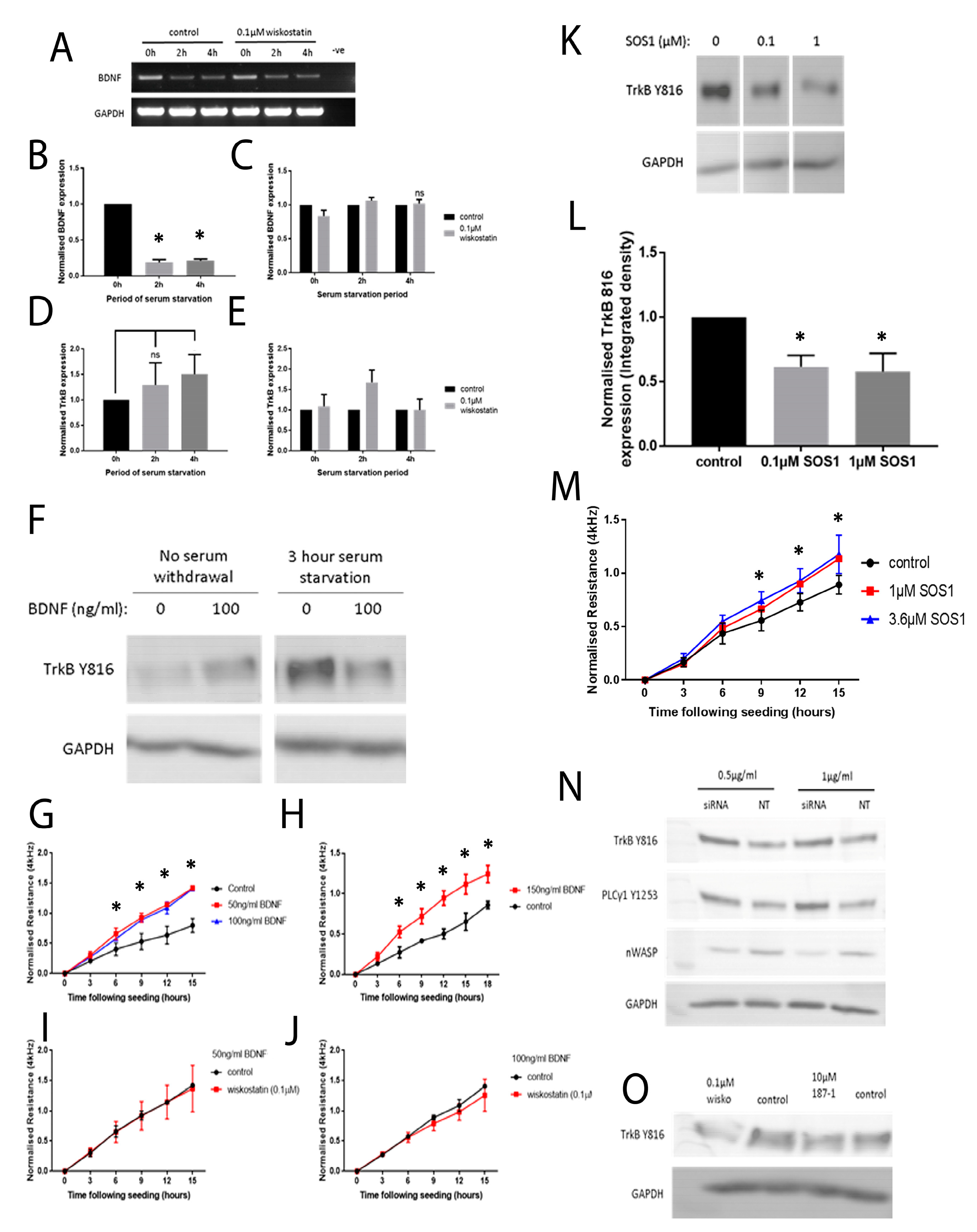
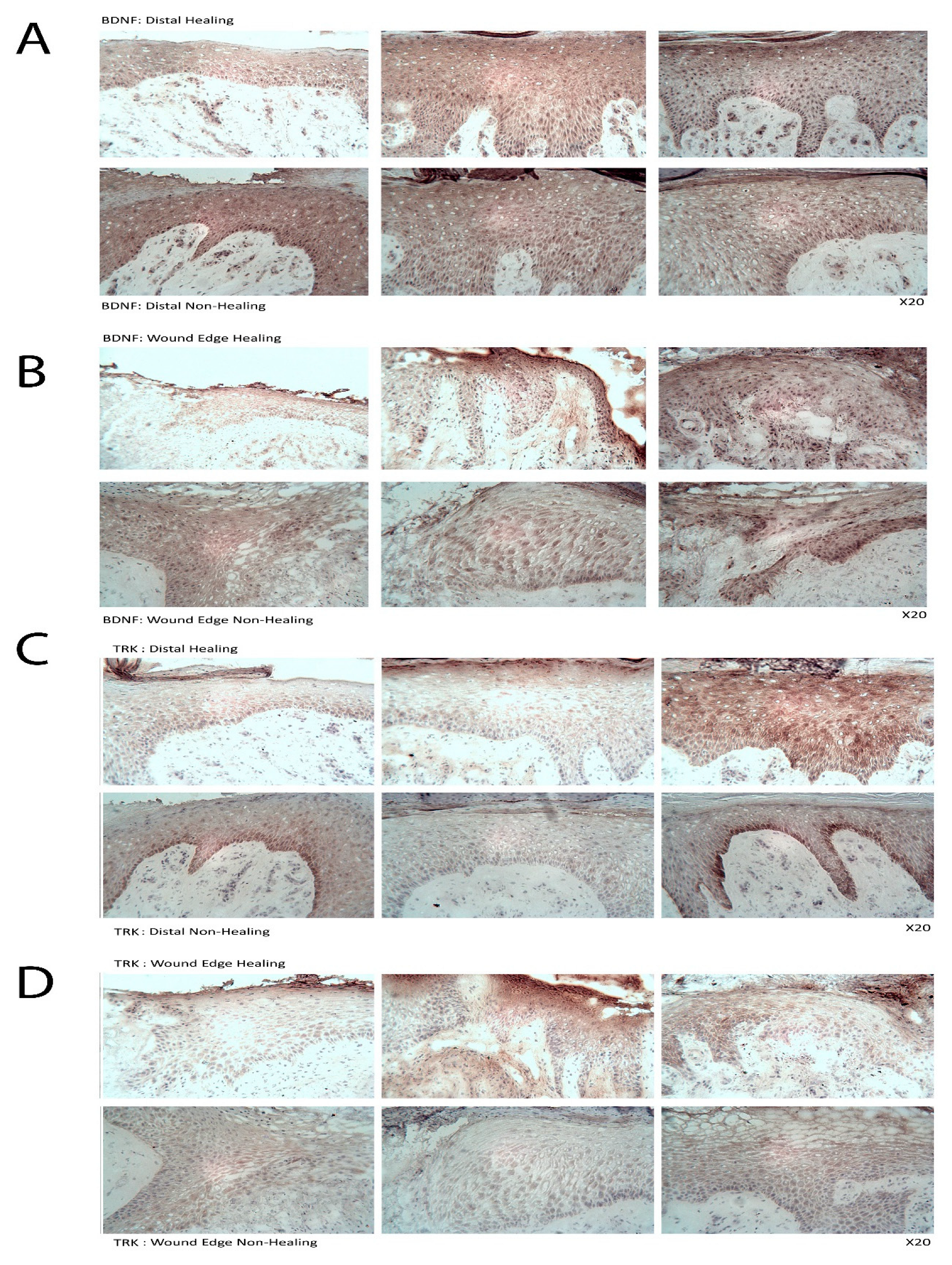
To begin to understand how nWASP inhibitors affect cell behaviour and may act in the context of a chronic wound, an investigation into the effect of altering nWASP activity on the signalling mechanisms in cell models that represent the skin and wound environment was carried out. Initially, a protein array highlighted changes in the expression and phosphorylation of hundreds of kinases and other common signalling proteins following treatment with wiskostatin in HaCaT cells. Of the most significantly altered proteins, several proteins belonging to very common signalling pathways were highlighted, including members of the Ras–Raf–Mek–Erk, Akt–mTOR and Jak–STAT pathways. Numerous receptors and membrane-bound proteins were also identified as being significantly altered by wiskostatin treatment, including VEGFR2 and -3, EGFR and the progesterone receptor, which have been shown to be upstream of these common signalling cascades. TrkB signalling was found to be significantly altered by nWASP inhibitor treatments in HaCaT cells and, as such, the effect of nWASP activity on TrkB signalling became the focus. The findings that were reported here appeared to demonstrate that the TrkB activity in HaCaT cells was extremely sensitive to confluency, serum starvation, BDNF application and nWASP inhibition. Furthermore, the TrkB activity is found to be increased in basal keratinocytes in chronic wound tissues. How TrkB, BDNF and nWASP activity may be linked in the context of chronic wounds requires significant further investigation, especially considering the sensitivity to factors such as confluency and serum availability of the TrkB pathway. The novel relationship between nWASP and TrkB and the downstream PLCγ1 signalling pathway that is affected by changes in signalling may have implications in terms of cell differentiation. As such, further investigation into the factors that affect this signalling and how this may translate into the wound environment needs to be explored.
It is clear that nWASP and TrkB signalling are linked in HaCaT cells and possibly in the chronic wound environment. How nWASP activity alters TrkB signalling and why this pathway is sensitive to factors such as confluency and serum starvation are yet to be answered. The established role of nWASP in endocytosis and receptor trafficking could represent one such potential link. nWASP and TrkB were shown to share numerous interaction partners that may facilitate this activity, for instance, Grb2, pacsin and Nck. A link between nWASP and TrkB through the common binding partner Grb2 has begun to be explored through the use of SOS1. This is an inhibitor of Grb2 activity that acts via blocking the SH3 binding domain of the guanine nucleotide exchange factor SOS, which is the ligand for the adaptor protein Grb2. This blocks the SOS/Grb2 interaction and prevents Ras activation via receptor tyrosine kinases, such as TrkB. SOS1 was found to act in the same way as nWASP inhibition in that TrkB Y816 phosphorylation was significantly reduced at low confluency and after 4 h serum starvation in HaCaT cells. Similarly, Grb2 inhibition was found to increase the resistance of HaCaT cells using ECIS, similar to nWASP inhibitors. This is only the first step in exploring the link between TrkB and nWASP, but this work identified an avenue for further study.
In summary, this study demonstrated that nWASP activity in human wounds can be indicative of its ability to heal effectively and, as a result, using nWASP inhibitors can influence the healing behaviour of wounds. Functional assays indicated that the mechanism through which nWASP may influence wound healing behaviour is potentially via affecting the attachment properties of cells and that this can be mediated through the BDNf/TrkB pathway. This study recognised nWASP as an important therapeutic target in human chronic wounds and proposes the use of nWASP inhibitors as a simple and effective means to encourage wound healing in difficult wounds.


 and
and 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}