

In Vivo Acute Toxicity Studies of Novel Anti-Melanoma Compounds Downregulators of hnRNPH1/H2

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
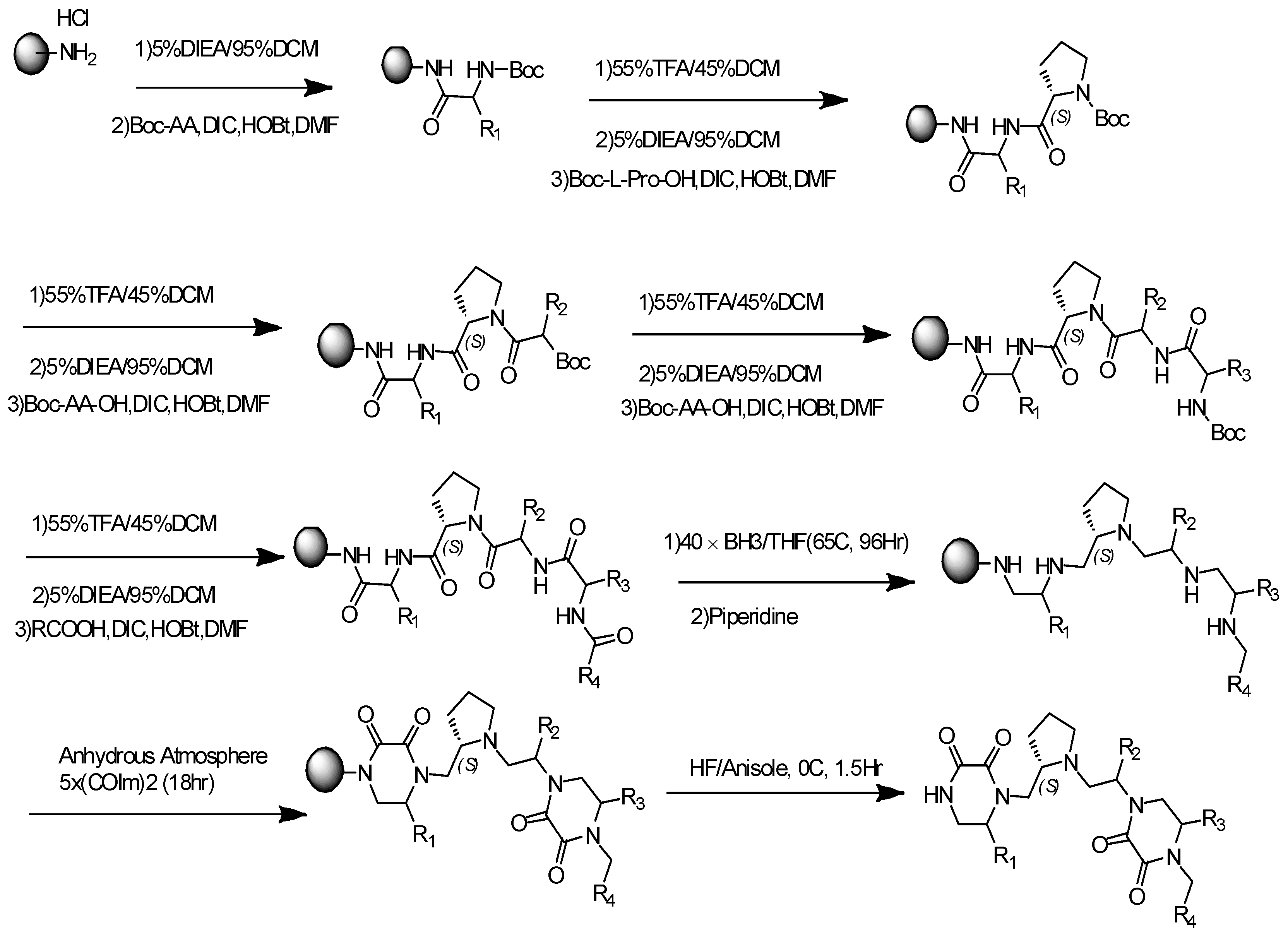
2.1. General Synthesis Procedure for Pyrrolidine-Bis-Diketopiperazine
2.2. Compound Purification and Characterization
2.3. Animal Protocol
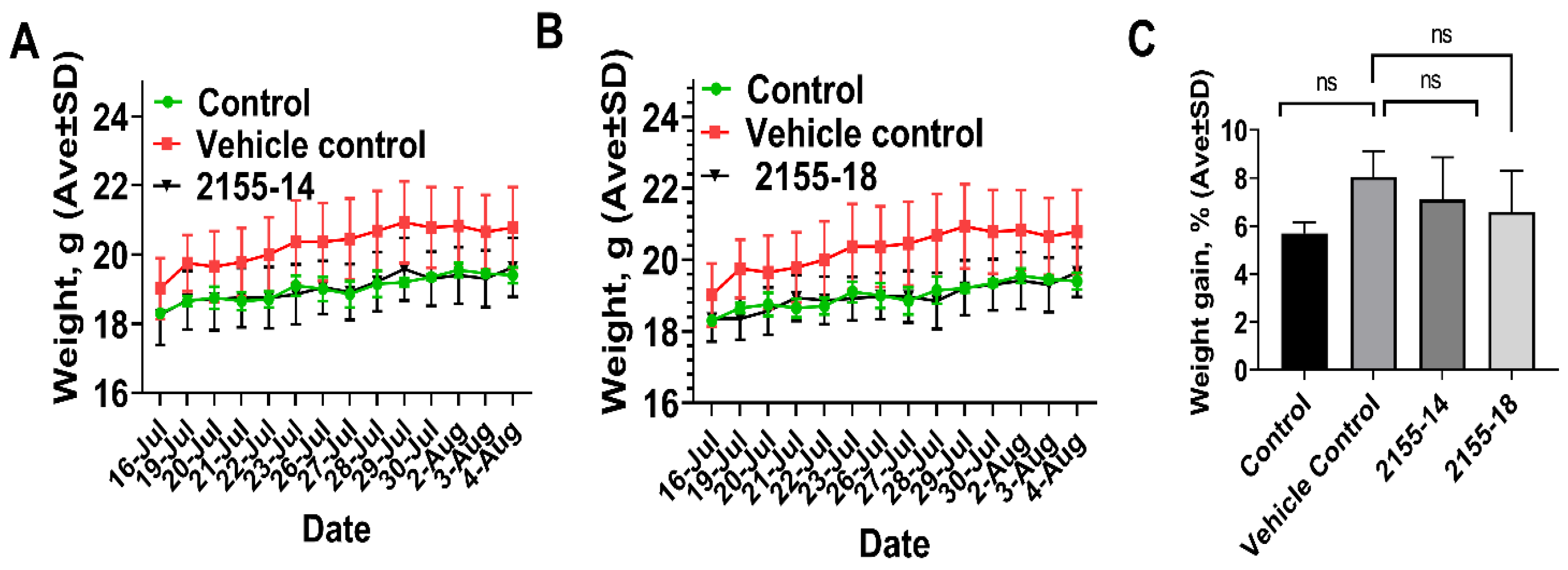
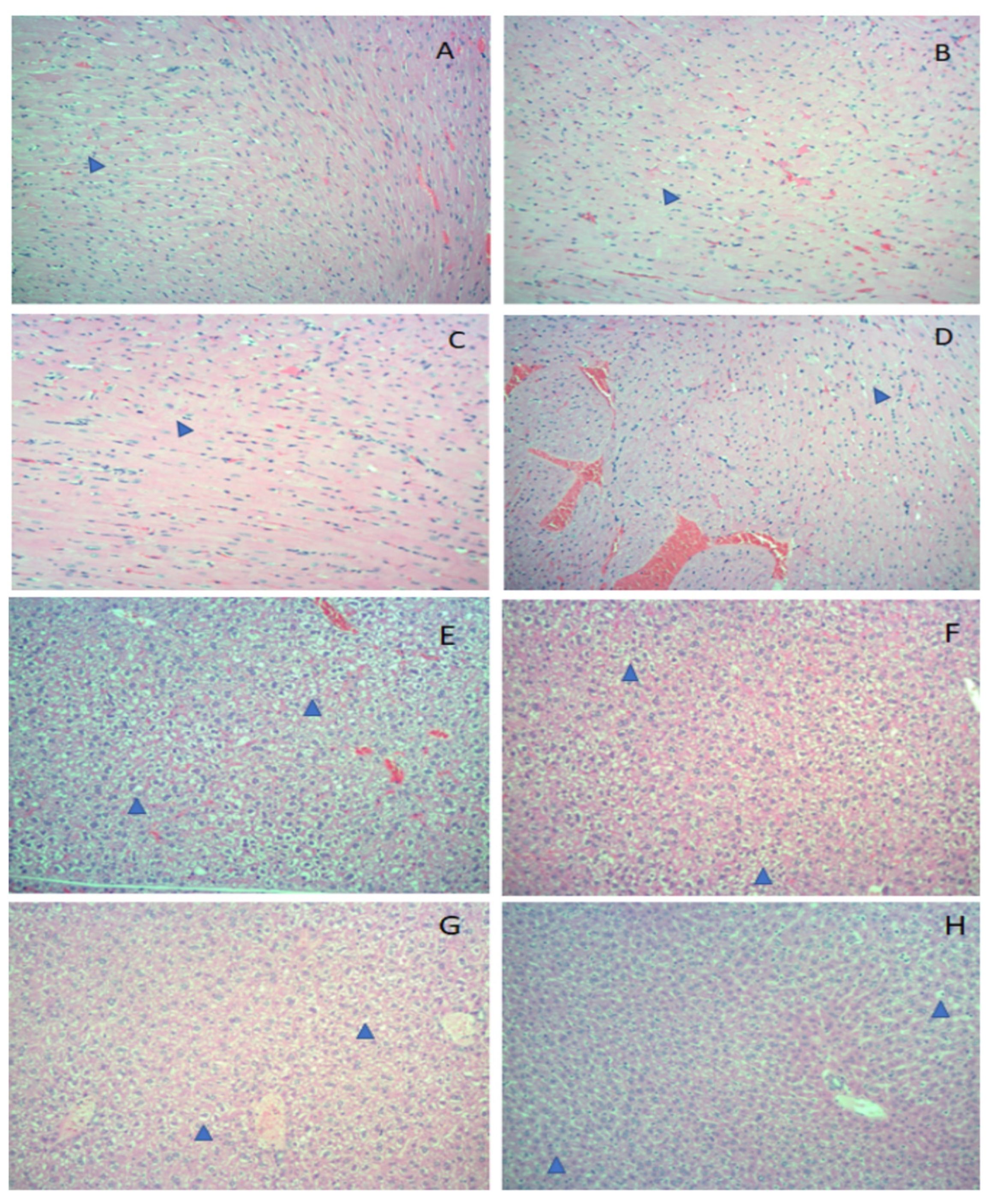
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Welch, H.G.; Kramer, B.S.; Black, W.C. Epidemiologic Signatures in Cancer. Reply. N. Engl. J. Med. 2020, 382, 96–98. [Google Scholar] [CrossRef] [PubMed]
- Welch, H.G.; Kramer, B.S.; Black, W.C. Epidemiologic Signatures in Cancer. N. Engl. J. Med. 2019, 381, 1378–1386. [Google Scholar] [CrossRef] [PubMed]
- Kahlon, N.; Doddi, S.; Yousif, R.; Najib, S.; Sheikh, T.; Abuhelwa, Z.; Burmeister, C.; Hamouda, D.M. Melanoma Treatments and Mortality Rate Trends in the US, 1975 to 2019. JAMA Netw. Open 2022, 5, e2245269. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Fisher, D.E. Treatment of Advanced Melanoma in 2020 and Beyond. J. Investig. Dermatol. 2021, 141, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Long, G.V.; Scolyer, R.A.; Teng, M.W.; Smyth, M.J. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat. Rev. 2017, 52, 71–81. [Google Scholar] [CrossRef]
- Morante, M.; Pandiella, A.; Crespo, P.; Herrero, A. Immune Checkpoint Inhibitors and RAS-ERK Pathway-Targeted Drugs as Combined Therapy for the Treatment of Melanoma. Biomolecules 2022, 12, 1562. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Wei, C.; Xiong, S.; Yu, P.; Zhou, R.; Cheng, L. Spliceosome inhibitor induces human hematopoietic progenitor cell reprogramming toward stemness. Exp. Hematol. Oncol. 2022, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y.; Sagane, K.; Owa, T.; Mimori-Kiyosue, Y.; Shimizu, H.; Uesugi, M.; Ishihama, Y.; Iwata, M.; Mizui, Y. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat. Chem. Biol. 2007, 3, 570–575. [Google Scholar] [CrossRef]
- Yokoi, A.; Kotake, Y.; Takahashi, K.; Kadowaki, T.; Matsumoto, Y.; Minoshima, Y.; Sugi, N.H.; Sagane, K.; Hamaguchi, M.; Iwata, M.; et al. Biological validation that SF3b is a target of the antitumor macrolide pladienolide. FEBS J. 2011, 278, 4870–4880. [Google Scholar] [CrossRef]
- Sato, M.; Muguruma, N.; Nakagawa, T.; Okamoto, K.; Kimura, T.; Kitamura, S.; Yano, H.; Sannomiya, K.; Goji, T.; Miyamoto, H.; et al. High antitumor activity of pladienolide B and its derivative in gastric cancer. Cancer Sci. 2014, 105, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Jorge, J.; Petronilho, S.; Alves, R.; Coucelo, M.; Goncalves, A.C.; Nascimento Costa, J.M.; Sarmento-Ribeiro, A.B. Apoptosis induction and cell cycle arrest of pladienolide B in erythroleukemia cell lines. Investig. New Drugs 2020, 38, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Di, C.; Yan, J.; Wang, F.; Qu, T.; Wang, Y.; Chen, Y.; Zhang, X.; Liu, Y.; Yang, H.; et al. Inhibition of SF3b1 by pladienolide B evokes cycle arrest, apoptosis induction and p73 splicing in human cervical carcinoma cells. Artif. Cells Nanomed. Biotechnol. 2019, 47, 1273–1280. [Google Scholar] [CrossRef]
- O’Brien, K.; Matlin, A.J.; Lowell, A.M.; Moore, M.J. The biflavonoid isoginkgetin is a general inhibitor of Pre-mRNA splicing. J. Biol. Chem. 2008, 283, 33147–33154. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.O.; Shin, S.; Lee, H.J.; Chun, H.K.; Chung, A.S. Isoginkgetin inhibits tumor cell invasion by regulating phosphatidylinositol 3-kinase/Akt-dependent matrix metalloproteinase-9 expression. Mol. Cancer Ther. 2006, 5, 2666–2675. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, B.; Xia, Z.M.; Tian, Y.; Zhang, D.; Rui, W.J.; Dong, J.X.; Xiao, F.J. Anticancer Effects of Five Biflavonoids from Ginkgo Biloba L. Male Flowers In Vitro. Molecules 2019, 24, 1496. [Google Scholar] [CrossRef]
- Murphy, A.J.; Li, A.H.; Li, P.; Sun, H. Therapeutic Targeting of Alternative Splicing: A New Frontier in Cancer Treatment. Front. Oncol. 2022, 12, 868664. [Google Scholar] [CrossRef]
- ElHady, A.K.; Abdel-Halim, M.; Abadi, A.H.; Engel, M. Development of Selective Clk1 and -4 Inhibitors for Cellular Depletion of Cancer-Relevant Proteins. J. Med. Chem. 2017, 60, 5377–5391. [Google Scholar] [CrossRef]
- Iwai, K.; Yaguchi, M.; Nishimura, K.; Yamamoto, Y.; Tamura, T.; Nakata, D.; Dairiki, R.; Kawakita, Y.; Mizojiri, R.; Ito, Y.; et al. Anti-tumor efficacy of a novel CLK inhibitor via targeting RNA splicing and MYC-dependent vulnerability. EMBO Mol. Med. 2018, 10, e8289. [Google Scholar] [CrossRef] [PubMed]
- Riggs, J.R.; Nagy, M.; Elsner, J.; Erdman, P.; Cashion, D.; Robinson, D.; Harris, R.; Huang, D.; Tehrani, L.; Deyanat-Yazdi, G.; et al. The Discovery of a Dual TTK Protein Kinase/CDC2-Like Kinase (CLK2) Inhibitor for the Treatment of Triple Negative Breast Cancer Initiated from a Phenotypic Screen. J. Med. Chem. 2017, 60, 8989–9002. [Google Scholar] [CrossRef]
- Zhu, D.; Xu, S.; Deyanat-Yazdi, G.; Peng, S.X.; Barnes, L.A.; Narla, R.K.; Tran, T.; Mikolon, D.; Ning, Y.; Shi, T.; et al. Synthetic Lethal Strategy Identifies a Potent and Selective TTK and CLK1/2 Inhibitor for Treatment of Triple-Negative Breast Cancer with a Compromised G(1)-S Checkpoint. Mol. Cancer Ther. 2018, 17, 1727–1738. [Google Scholar] [CrossRef]
- Fukuhara, T.; Hosoya, T.; Shimizu, S.; Sumi, K.; Oshiro, T.; Yoshinaka, Y.; Suzuki, M.; Yamamoto, N.; Herzenberg, L.A.; Herzenberg, L.A.; et al. Utilization of host SR protein kinases and RNA-splicing machinery during viral replication. Proc. Natl. Acad. Sci. USA 2006, 103, 11329–11333. [Google Scholar] [CrossRef]
- Gammons, M.V.; Fedorov, O.; Ivison, D.; Du, C.; Clark, T.; Hopkins, C.; Hagiwara, M.; Dick, A.D.; Cox, R.; Harper, S.J.; et al. Topical antiangiogenic SRPK1 inhibitors reduce choroidal neovascularization in rodent models of exudative AMD. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6052–6062. [Google Scholar] [CrossRef]
- Gammons, M.V.; Lucas, R.; Dean, R.; Coupland, S.E.; Oltean, S.; Bates, D.O. Targeting SRPK1 to control VEGF-mediated tumour angiogenesis in metastatic melanoma. Br. J. Cancer 2014, 111, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Mavrou, A.; Brakspear, K.; Hamdollah-Zadeh, M.; Damodaran, G.; Babaei-Jadidi, R.; Oxley, J.; Gillatt, D.A.; Ladomery, M.R.; Harper, S.J.; Bates, D.O.; et al. Serine-arginine protein kinase 1 (SRPK1) inhibition as a potential novel targeted therapeutic strategy in prostate cancer. Oncogene 2015, 34, 4311–4319. [Google Scholar] [CrossRef] [PubMed]
- Palrasu, M.; Knapinska, A.M.; Diez, J.; Smith, L.; LaVoi, T.; Giulianotti, M.; Houghten, R.A.; Fields, G.B.; Minond, D. A Novel Probe for Spliceosomal Proteins that Induces Autophagy and Death of Melanoma Cells Reveals New Targets for Melanoma Drug Discovery. Cell Physiol. Biochem. 2019, 53, 656–686. [Google Scholar] [CrossRef] [PubMed]
- Uren, P.J.; Bahrami-Samani, E.; de Araujo, P.R.; Vogel, C.; Qiao, M.; Burns, S.C.; Smith, A.D.; Penalva, L.O. High-throughput analyses of hnRNP H1 dissects its multi-functional aspect. RNA Biol. 2016, 13, 400–411. [Google Scholar] [CrossRef]
- Herviou, P.; Le Bras, M.; Dumas, L.; Hieblot, C.; Gilhodes, J.; Cioci, G.; Hugnot, J.P.; Ameadan, A.; Guillonneau, F.; Dassi, E.; et al. hnRNP H/F drive RNA G-quadruplex-mediated translation linked to genomic instability and therapy resistance in glioblastoma. Nat. Commun. 2020, 11, 2661. [Google Scholar] [CrossRef]
- Tyson-Capper, A.; Gautrey, H. Regulation of Mcl-1 alternative splicing by hnRNP F, H1 and K in breast cancer cells. RNA Biol. 2018, 15, 1448–1457. [Google Scholar] [CrossRef]
- Braun, S.; Enculescu, M.; Setty, S.T.; Cortes-Lopez, M.; de Almeida, B.P.; Sutandy, F.X.R.; Schulz, L.; Busch, A.; Seiler, M.; Ebersberger, S.; et al. Decoding a cancer-relevant splicing decision in the RON proto-oncogene using high-throughput mutagenesis. Nat. Commun. 2018, 9, 3315. [Google Scholar] [CrossRef]
- Gautrey, H.; Jackson, C.; Dittrich, A.L.; Browell, D.; Lennard, T.; Tyson-Capper, A. SRSF3 and hnRNP H1 regulate a splicing hotspot of HER2 in breast cancer cells. RNA Biol. 2015, 12, 1139–1151. [Google Scholar] [CrossRef]
- Lefave, C.V.; Squatrito, M.; Vorlova, S.; Rocco, G.L.; Brennan, C.W.; Holland, E.C.; Pan, Y.X.; Cartegni, L. Splicing factor hnRNPH drives an oncogenic splicing switch in gliomas. EMBO J. 2011, 30, 4084–4097. [Google Scholar] [CrossRef]
- Stark, M.; Bram, E.E.; Akerman, M.; Mandel-Gutfreund, Y.; Assaraf, Y.G. Heterogeneous nuclear ribonucleoprotein H1/H2-dependent unsplicing of thymidine phosphorylase results in anticancer drug resistance. J. Biol. Chem. 2011, 286, 3741–3754. [Google Scholar] [CrossRef]
- Rauch, J.; O’Neill, E.; Mack, B.; Matthias, C.; Munz, M.; Kolch, W.; Gires, O. Heterogeneous nuclear ribonucleoprotein H blocks MST2-mediated apoptosis in cancer cells by regulating A-Raf transcription. Cancer Res. 2010, 70, 1679–1688. [Google Scholar] [CrossRef]
- Takahashi, K.; Fujiya, M.; Konishi, H.; Murakami, Y.; Iwama, T.; Sasaki, T.; Kunogi, T.; Sakatani, A.; Ando, K.; Ueno, N.; et al. Heterogenous Nuclear Ribonucleoprotein H1 Promotes Colorectal Cancer Progression through the Stabilization of mRNA of Sphingosine-1-Phosphate Lyase 1. Int. J. Mol. Sci. 2020, 21, 4514. [Google Scholar] [CrossRef]
- Eskens, F.A.; Ramos, F.J.; Burger, H.; O’Brien, J.P.; Piera, A.; de Jonge, M.J.; Mizui, Y.; Wiemer, E.A.; Carreras, M.J.; Baselga, J.; et al. Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin. Cancer Res. 2013, 19, 6296–6304. [Google Scholar] [CrossRef]
- Hong, D.S.; Kurzrock, R.; Naing, A.; Wheler, J.J.; Falchook, G.S.; Schiffman, J.S.; Faulkner, N.; Pilat, M.J.; O’Brien, J.; LoRusso, P. A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Investig. New Drugs 2014, 32, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Houghten, R.A. General method for the rapid solid-phase synthesis of large numbers of peptides: Specificity of antigen-antibody interaction at the level of individual amino acids. Proc. Natl. Acad. Sci. USA 1985, 82, 5131–5135. [Google Scholar] [CrossRef]
- Pinilla, C.; Edwards, B.S.; Appel, J.R.; Yates-Gibbins, T.; Giulianotti, M.A.; Medina-Franco, J.L.; Young, S.M.; Santos, R.G.; Sklar, L.A.; Houghten, R.A. Selective agonists and antagonists of formylpeptide receptors: Duplex flow cytometry and mixture-based positional scanning libraries. Mol. Pharmacol. 2013, 84, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Onwuha-Ekpete, L.; Tack, L.; Knapinska, A.; Smith, L.; Kaushik, G.; Lavoi, T.; Giulianotti, M.; Houghten, R.A.; Fields, G.B.; Minond, D. Novel pyrrolidine diketopiperazines selectively inhibit melanoma cells via induction of late-onset apoptosis. J. Med. Chem. 2014, 57, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lushington, G.H.; Huan, J. Characterizing the diversity and biological relevance of the MLPCN assay manifold and screening set. J. Chem. Inf. Model. 2011, 51, 1205–1215. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; McDonald, P.R.; Sittampalam, S.; Chaguturu, R. Open access high throughput drug discovery in the public domain: A Mount Everest in the making. Curr. Pharm. Biotechnol. 2010, 11, 764–778. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Parameter | Control | Vehicle | 2155-14 | 2155-18 | Reference Range | Units |
|---|---|---|---|---|---|---|
| WBC | 11.4 ± 0.42 | 6.8 ± 2.5 | 5.1 ± 1.6 | 4.2 ± 2.7 | 3.48–14.03 | K/µL |
| RBC | 9.0 ± 1.1 | 9.6 ± 1.9 | 10.6 ± 2.1 | 7.9 ± 1.4 | 6.93–12.24 | M/µL |
| Hgb | 15.5 ± 0.5 | 15 ± 1.7 | 14.1 ± 1.6 | * 11.9 ± 1.6 | 12.6–20.5 | g/dL |
| HCT | 53.8 ± 4.6 | 56.8 ± 12.2 | 61.1± | 46.9 ± 6.4 | 42.1–68.3 | % |
| MCV | 59.5 ± 0.7 | 59 ± 1.4 | 61 ± 2.2 | 59.3 ± 2.1 | 50.7–64.4 | fL |
| MCH | 17 ± 1.4 | 15.6 ± 2.3 | 14 ± 2.3 | 14.7 ± 0.57 | 13.2–17.6 | pg |
| MCHC | 28.5 ± 2.1 | 27 ± 4.2 | 23 ± 5.1 | 25.3 ± 0.58 | 23.3–32.7 | g/dL |
| Segmented Neutrophils | 18.5 ± 3.5 | 23.3 ± 7.2 | 18.8 ± 8.0 | 24.7 ± 10.9 | 9.86–39.11 | % |
| Banded Neutrophils | 0 | 0 | 0 | 0 | 0–1 | % |
| Lymphocytes | 73.5 ± 6.4 | 67.3 ± 8.4 | 78.3 ± 7.5 | 70.7 ± 11.3 | 48.81–83.19 | % |
| Monocytes | 4.5 ± 0.7 | 5.83 ± 2.22 | 4.25 ± 4.27 | 6.7 ± 2.3 | 3.29–12.48 | % |
| Eosinohils | 3.0 ± 1.4 | 2.7 ± 1.2 | 0.5 ± 1.2 | 2 ± 1 | 0.11–4.91 | % |
| Basophils | 0.5 ± 0.7 | 1 ± 0 | 0 | 0 | 0–1.84 | % |
| Platelets | 686 ± 279 | 535 ± 88 | 542 ± 95 | 741 ± 305 | 420–1698 | K/µL |
| Parameter | Control | Vehicle | 2155-14 | 2155-18 | Reference Range | Units |
|---|---|---|---|---|---|---|
| Total protein | 8.4 ± 3.5 | 8.1 ± 0.6 | 7.1 ± 0.6 | 6.7 ± 0.48 | 4.6–8.9 | g/dL |
| Creatinine | 0.5 ± 0 | 0.5 ± 0 | * 0.44 ± 0.19 | * 0.46 ± 0.11 | 0.2–0.4 | mg/dL |
| BUN | 18.5 ± 2.1 | 17 ± 1.1 | 18.3 ± 2.5 | 16.7 ± 1.6 | 7.0–26 | mg/dL |
| Glucose | 170 ± 39.7 | 199 ± 49.7 | 181 ± 25.7 | 212 ± 50.5 | 129–329 | mg/dL |
| Calcium | 10.5 ± 0.25 | 9.4 ± 0.2 | 9.1 ± 0.85 | 8.7 ± 0.75 | 9.4–12.5 | mg/dL |
| Phosphorus | 11.5 ± 0.2 | 13.2 ± 0.8 | 13.6 ± 0.4 | * 15.3 ± 0.25 | 8.2–14.7 | mg/dL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velayutham, S.; Seal, T.; Danthurthy, S.; Zaias, J.; Smalley, K.S.M.; Minond, D. In Vivo Acute Toxicity Studies of Novel Anti-Melanoma Compounds Downregulators of hnRNPH1/H2. Biomolecules 2023, 13, 349. https://doi.org/10.3390/biom13020349
Velayutham S, Seal T, Danthurthy S, Zaias J, Smalley KSM, Minond D. In Vivo Acute Toxicity Studies of Novel Anti-Melanoma Compounds Downregulators of hnRNPH1/H2. Biomolecules. 2023; 13(2):349. https://doi.org/10.3390/biom13020349
Chicago/Turabian StyleVelayutham, Sadeeshkumar, Trisha Seal, Samaya Danthurthy, Julia Zaias, Keiran S. M. Smalley, and Dmitriy Minond. 2023. "In Vivo Acute Toxicity Studies of Novel Anti-Melanoma Compounds Downregulators of hnRNPH1/H2" Biomolecules 13, no. 2: 349. https://doi.org/10.3390/biom13020349
APA StyleVelayutham, S., Seal, T., Danthurthy, S., Zaias, J., Smalley, K. S. M., & Minond, D. (2023). In Vivo Acute Toxicity Studies of Novel Anti-Melanoma Compounds Downregulators of hnRNPH1/H2. Biomolecules, 13(2), 349. https://doi.org/10.3390/biom13020349