

The Many (Inter)faces of Anti-CRISPRs: Modulation of CRISPR-Cas Structure and Dynamics by Mechanistically Diverse Inhibitors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
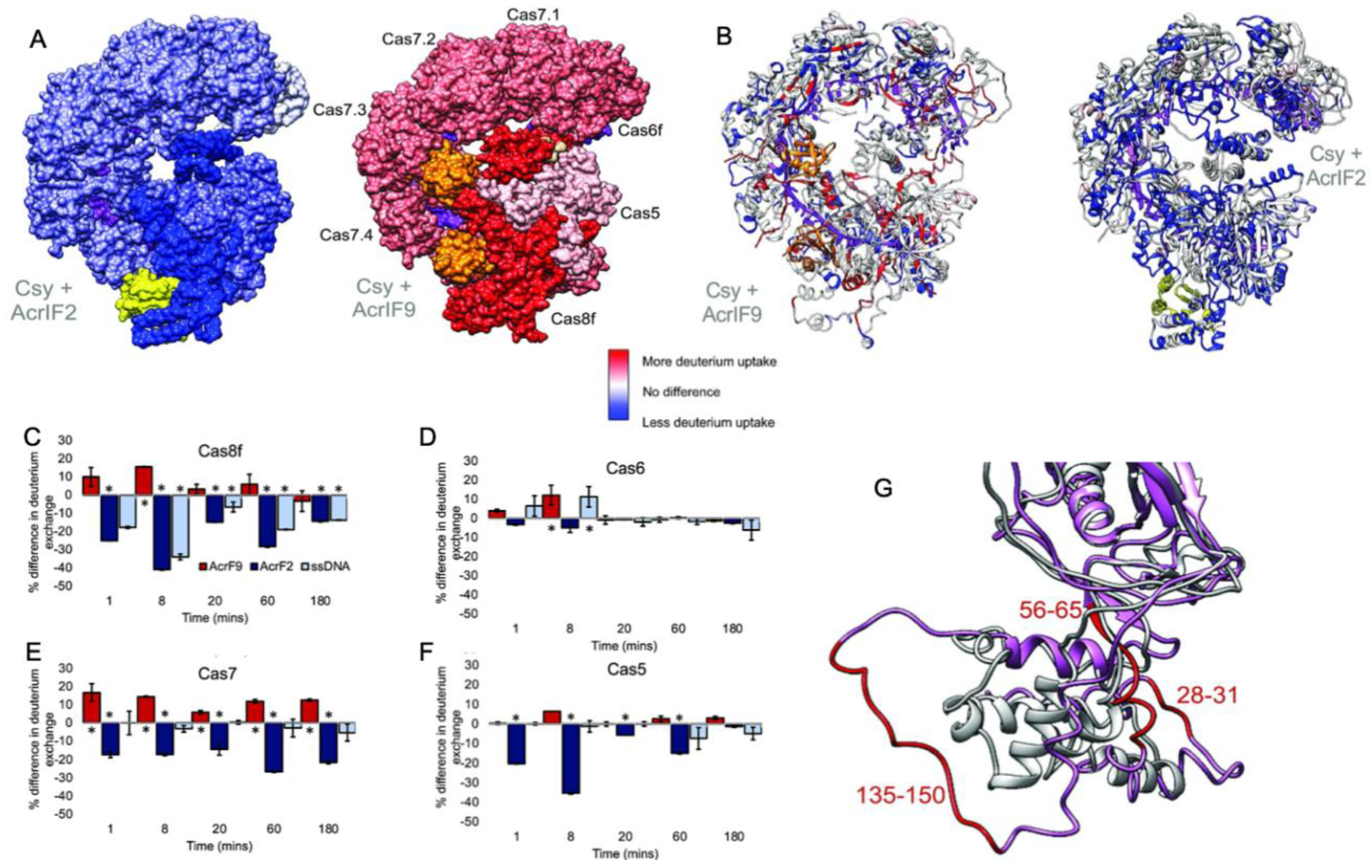
2. General Modes of Cas Inhibition by Acrs
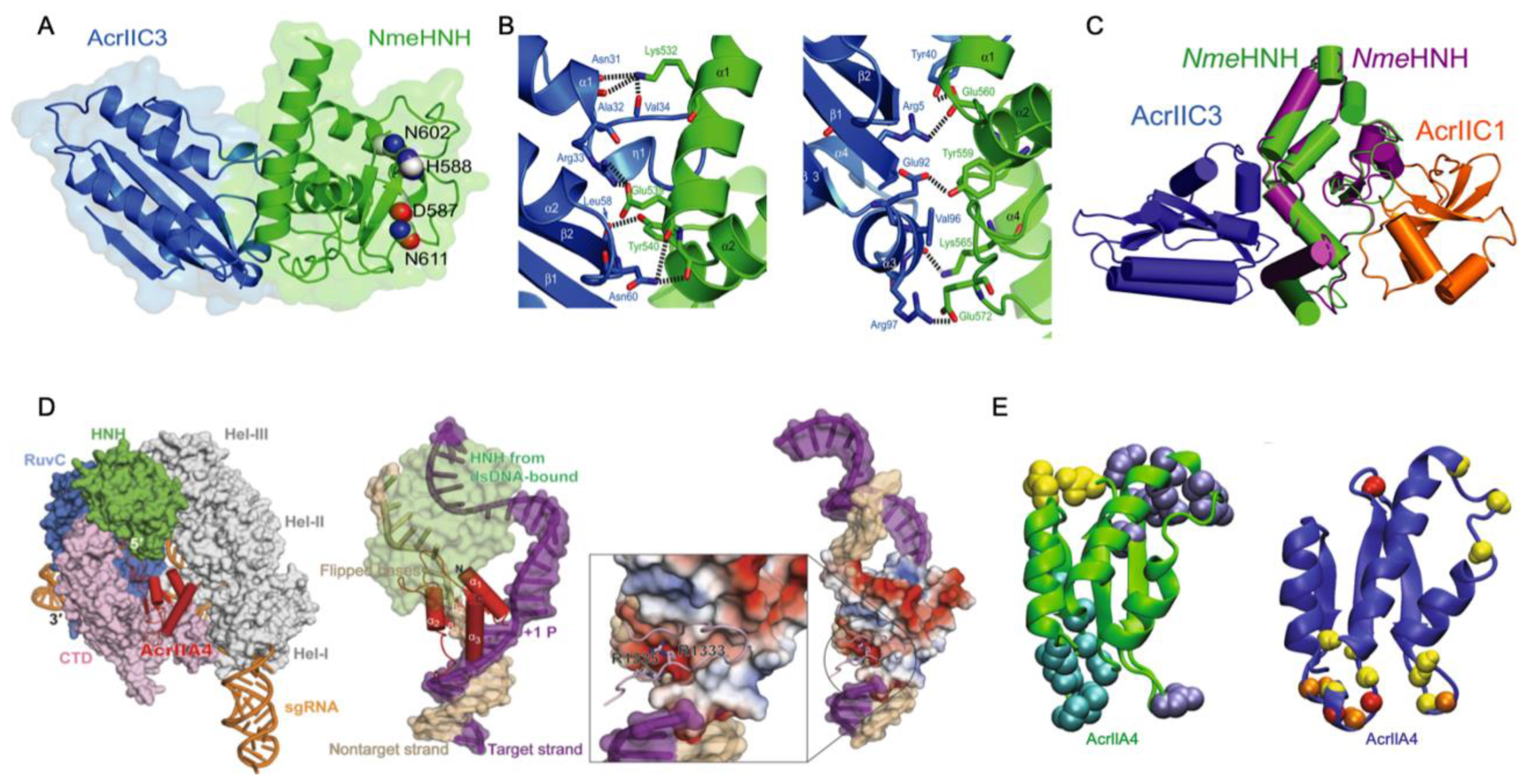
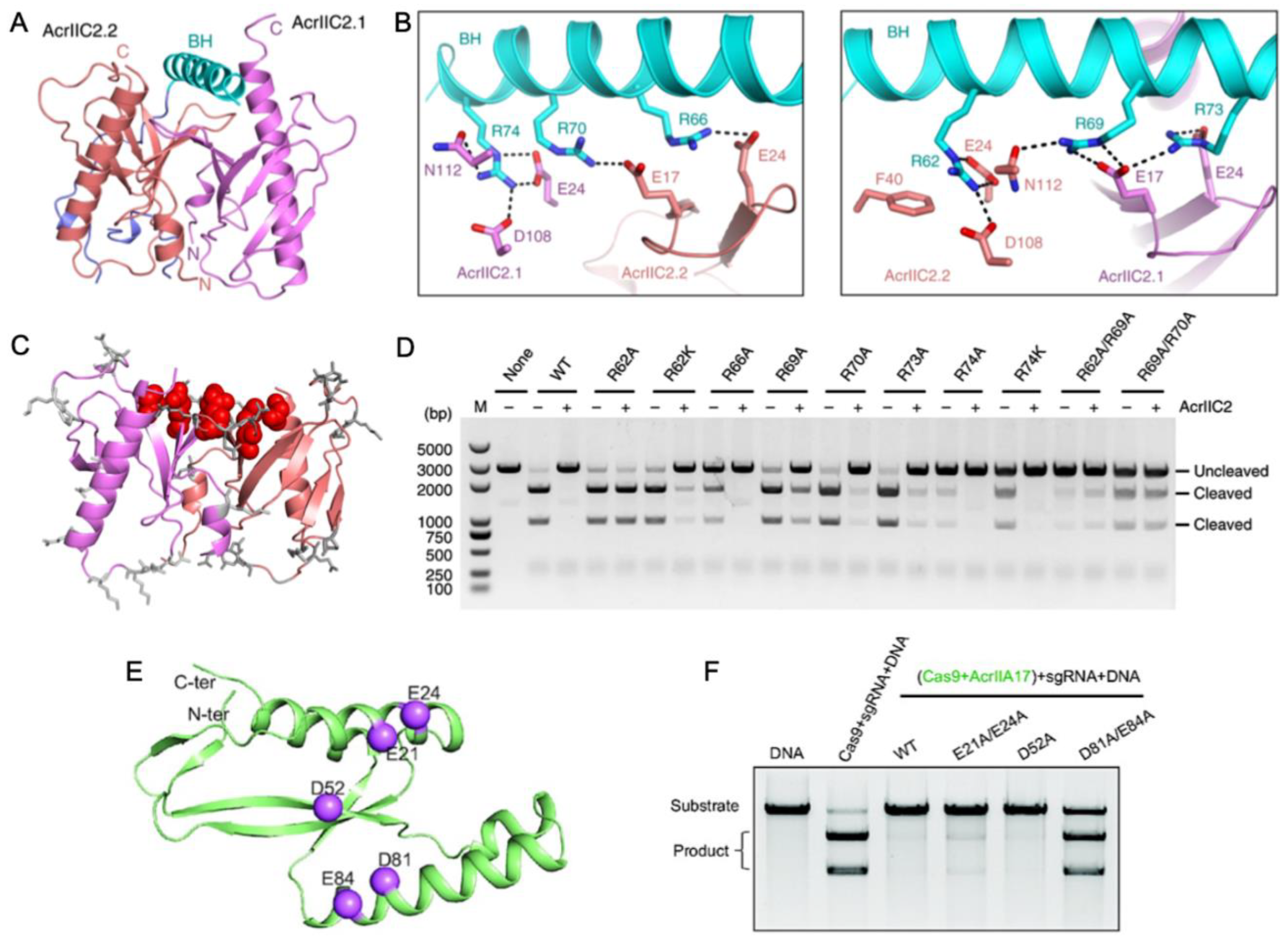
3. Direct Interaction of Acrs with Cas Nucleases (HNH or RuvC)
4. Steric Occlusion of Nucleic Acids by Acrs
5. Interaction with the Nucleic Acid Bridge Helix
6. Acr-Driven Oligomerization of Cas Proteins

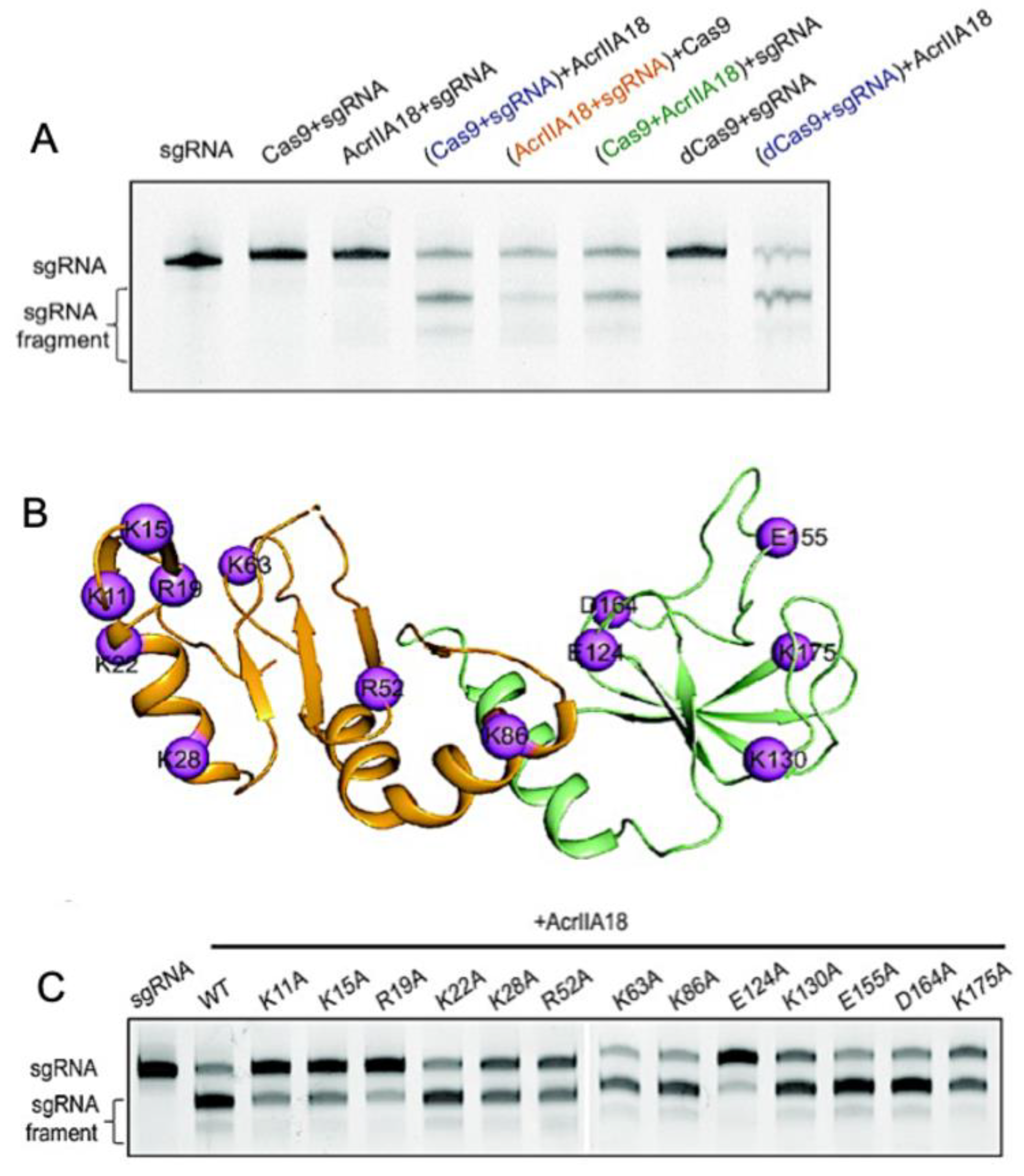
7. Enzymatic Modification of Cas9 Nucleic Acids by Acrs
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kim, J.-S. Unexpected CRISPR on-target effects. Nat. Biotechnol. 2018, 36, 703–704. [Google Scholar] [CrossRef]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. p53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018, 24, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Psatha, N.; Gil, S.; Wang, H.; Papayannopoulou, T.; Lieber, A. HDAd5/35++ Adenovirus Vector Expressing Anti-CRISPR Peptides Decreases CRISPR/Cas9 Toxicity in Human Hematopoietic Stem Cells. Mol. Ther. Methods Clin. Dev. 2018, 9, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Chew, W.L.; Tabebordbar, W.L.C.M.; Cheng, J.; Mali, P.; Wu, E.Y.; Ng, A.; Zhu, K.; Wagers, M.T.J.K.W.C.E.Y.W.K.Z.A.J.; Church, W.L.C.P.M.A.H.M.N.G.M. A multifunctional AAV–CRISPR–Cas9 and its host response. Nat. Methods 2016, 13, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Mou, H.; Li, S.; Li, Y.; Hough, S.; Tran, K.; Li, J.; Yin, H.; Anderson, D.G.; Sontheimer, E.J.; et al. Adenovirus-Mediated Somatic Genome Editing of Pten by CRISPR/Cas9 in Mouse Liver in Spite of Cas9-Specific Immune Responses. Hum. Gene Ther. 2015, 26, 432–442. [Google Scholar] [CrossRef]
- Kim, D.K.J.K.K.W.B.S.-H.Y.J.-S.; Kim, J.; Hur, J.K.; Been, K.W.; Yoon, S.-H.; Kim, J.-S. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat. Biotechnol. 2016, 34, 863–868. [Google Scholar] [CrossRef]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.-S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife 2014, 3, e04766. [Google Scholar] [CrossRef]
- Nihongaki, Y.; Kawano, F.; Nakajima, T.; Sato, M. Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat. Biotechnol. 2015, 33, 755–760. [Google Scholar] [CrossRef]
- Senturk, S.; Shirole, N.H.; Nowak, D.G.; Corbo, V.; Pal, D.; Vaughan, A.; Tuveson, D.A.; Trotman, L.C.; Kinney, J.B.; Sordella, R. Rapid and tunable method to temporally control gene editing based on conditional Cas9 stabilization. Nat. Commun. 2017, 8, 14370. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, T.; Karambelkar, S.; Bondy-Denomy, J.; Wiedenheft, B. Structures and Strategies of Anti-CRISPR-Mediated Immune Suppression. Annu. Rev. Microbiol. 2020, 74, 21–37. [Google Scholar] [CrossRef]
- Bondy-Denomy, J.; Davidson, A.R.; Doudna, J.A.; Fineran, P.C.; Maxwell, K.L.; Moineau, S.; Peng, X.; Sontheimer, E.J.; Wiedenheft, B. A Unified Resource for Tracking Anti-CRISPR Names. CRISPR J. 2018, 1, 304–305. [Google Scholar] [CrossRef]
- Malone, L.M.; Birkholz, N.; Fineran, P.C. Conquering CRISPR: How phages overcome bacterial adaptive immunity. Curr. Opin. Biotechnol. 2020, 68, 30–36. [Google Scholar] [CrossRef]
- Borges, A.L.; Davidson, A.R.; Bondy-Denomy, J. The Discovery, Mechanisms, and Evolutionary Impact of Anti-CRISPRs. Annu. Rev. Virol. 2017, 4, 37–59. [Google Scholar] [CrossRef]
- Hwang, S.; Maxwell, K.L. Meet the Anti-CRISPRs: Widespread Protein Inhibitors of CRISPR-Cas Systems. CRISPR J. 2019, 2, 23–30. [Google Scholar] [CrossRef]
- Stanley, S.Y.; Maxwell, K.L. Phage-Encoded Anti-CRISPR Defenses. Annu. Rev. Genet. 2018, 52, 445–464. [Google Scholar] [CrossRef]
- Trasanidou, D.; Gerós, A.S.; Mohanraju, P.; Nieuwenweg, A.C.; Nobrega, F.L.; Staals, R.H.J. Keeping CRISPR in check: Diverse mechanisms of phage-encoded anti-crisprs. FEMS Microbiol. Lett. 2019, 366, fnz098. [Google Scholar] [CrossRef]
- Knott, G.J.; Thornton, B.W.; Lobba, M.J.; Liu, J.-J.; Al-Shayeb, B.; Watters, K.E.; Doudna, J.A. Broad-spectrum enzymatic inhibition of CRISPR-Cas12a. Nat. Struct. Mol. Biol. 2019, 26, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Guan, X.; Li, N.; Zhang, F.; Zhu, Y.; Ren, K.; Yu, L.; Zhou, F.; Han, Z.; Gao, N.; et al. An anti-CRISPR protein disables type V Cas12a by acetylation. Nat. Struct. Mol. Biol. 2019, 26, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Rauch, B.J.; Silvis, M.R.; Hultquist, J.F.; Waters, C.S.; McGregor, M.J.; Krogan, N.J.; Bondy-Denomy, J. Inhibition of CRISPR-Cas9 with Bacteriophage Proteins. Cell 2016, 168, 150–158.e10. [Google Scholar] [CrossRef]
- Pawluk, A.; Amrani, N.; Zhang, Y.; Garcia, B.; Hidalgo-Reyes, Y.; Lee, J.; Edraki, A.; Shah, M.; Sontheimer, E.J.; Maxwell, K.L.; et al. Naturally Occurring Off-Switches for CRISPR-Cas9. Cell 2016, 167, 1829–1838.e9. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Srinivasan, P.; Chavez, M.; Carter, M.A.; Dominguez, A.A.; La Russa, M.; Lau, M.B.; Abbott, T.R.; Xu, X.; Zhao, D.; et al. Anti-CRISPR-mediated control of gene editing and synthetic circuits in eukaryotic cells. Nat. Commun. 2019, 10, 194–211. [Google Scholar] [CrossRef]
- Bubeck, F.; Hoffmann, M.D.; Harteveld, Z.; Aschenbrenner, S.; Bietz, A.; Waldhauer, M.C.; Börner, K.; Fakhiri, J.; Schmelas, C.; Dietz, L.; et al. Engineered anti-CRISPR proteins for optogenetic control of CRISPR–Cas9. Nat. Methods 2018, 15, 924–927. [Google Scholar] [CrossRef]
- Harrington, L.B.; Doxzen, K.W.; Ma, E.; Liu, J.-J.; Knott, G.J.; Edraki, A.; Garcia, B.; Amrani, N.; Chen, J.S.; Cofsky, J.C.; et al. A Broad-Spectrum Inhibitor of CRISPR-Cas9. Cell 2017, 170, 1224–1233.e15. [Google Scholar] [CrossRef] [PubMed]
- Marino, N.D.; Zhang, J.Y.; Borges, A.L.; Sousa, A.A.; Leon, L.M.; Rauch, B.J.; Walton, R.T.; Berry, J.D.; Joung, J.K.; Kleinstiver, B.P.; et al. Discovery of widespread type I and type V CRISPR-Cas inhibitors. Science 2018, 362, 240–242. [Google Scholar] [CrossRef]
- Hynes, A.P.; Rousseau, G.M.; Agudelo, D.; Goulet, A.; Amigues, B.; Loehr, J.; Romero, D.A.; Fremaux, C.; Horvath, P.; Doyon, Y.; et al. Widespread anti-CRISPR proteins in virulent bacteriophages inhibit a range of Cas9 proteins. Nat. Commun. 2018, 9, 2919. [Google Scholar] [CrossRef]
- Watters, K.E.; Fellmann, C.; Bai, H.B.; Ren, S.M.; Doudna, J.A. Systematic discovery of natural CRISPR-Cas12a inhibitors. Science 2018, 362, 236–239. [Google Scholar] [CrossRef]
- Kim, I.; Jeong, M.; Ka, D.; Han, M.; Kim, N.-K.; Bae, E.; Suh, J.-Y. Solution structure and dynamics of anti-CRISPR AcrIIA4, the Cas9 inhibitor. Sci. Rep. 2018, 8, 3883. [Google Scholar] [CrossRef] [PubMed]
- Patterson, A.; White, A.; Waymire, E.; Fleck, S.; Golden, S.; A Wilkinson, R.; Wiedenheft, B.; Bothner, B. Anti-CRISPR proteins function through thermodynamic tuning and allosteric regulation of CRISPR RNA-guided surveillance complex. Nucleic Acids Res. 2022, 50, 11243–11254. [Google Scholar] [CrossRef] [PubMed]
- Thavalingam, A.; Cheng, Z.; Garcia, B.; Huang, X.; Shah, M.; Sun, W.; Wang, M.; Harrington, L.; Hwang, S.; Hidalgo-Reyes, Y.; et al. Inhibition of CRISPR-Cas9 ribonucleoprotein complex assembly by anti-CRISPR AcrIIC2. Nat. Commun. 2019, 10, 2806. [Google Scholar] [CrossRef] [PubMed]
- An, S.Y.; Ka, D.; Kim, I.; Kim, E.-H.; Kim, N.-K.; Bae, E.; Suh, J.-Y. Intrinsic disorder is essential for Cas9 inhibition of anti-CRISPR AcrIIA5. Nucleic Acids Res. 2020, 48, 7584–7594. [Google Scholar] [CrossRef] [PubMed]
- Fuchsbauer, O.; Swuec, P.; Zimberger, C.; Amigues, B.; Levesque, S.; Agudelo, D.; Duringer, A.; Chaves-Sanjuan, A.; Spinelli, S.; Rousseau, G.M.; et al. Cas9 Allosteric Inhibition by the Anti-CRISPR Protein AcrIIA6. Mol. Cell 2019, 76, 922–937.e927. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, S.J.; Yoon, H.; Kim, N.; Lee, B.; Suh, J. Anti-CRISPR AcrIIC3 discriminates between Cas9 orthologs via targeting the variable surface of the HNH nuclease domain. FEBS J. 2019, 286, 4661–4674. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Z.; Daczkowski, C.M.; Gabel, C.; Mesecar, A.D.; Chang, L. Structural Basis for the Inhibition of CRISPR-Cas12a by Anti-CRISPR Proteins. Cell Host Microbe 2019, 25, 815–826.e814. [Google Scholar] [CrossRef]
- Pawluk, A.; Shah, M.; Mejdani, M.; Calmettes, C.; Moraes, T.F.; Davidson, A.R.; Maxwell, K.L. Disabling a Type I-E CRISPR-Cas Nuclease with a Bacteriophage-Encoded Anti-CRISPR Protein. Mbio 2017, 8, e01751-17. [Google Scholar] [CrossRef]
- Wang, X.; Yao, D.; Xu, J.-G.; Li, A.-R.; Xu, J.; Fu, P.; Zhou, Y.; Zhu, Y. Structural basis of Cas3 inhibition by the bacteriophage protein AcrF3. Nat. Struct. Mol. Biol. 2016, 23, 868–870. [Google Scholar] [CrossRef]
- Shin, J.; Jiang, F.; Liu, J.-J.; Bray, N.L.; Rauch, B.J.; Baik, S.H.; Nogales, E.; Bondy-Denomy, J.; Corn, J.E.; Doudna, J.A. Disabling Cas9 by an anti-CRISPR DNA mimic. Sci. Adv. 2017, 3, e1701620. [Google Scholar] [CrossRef]
- Jiang, F.; Zhou, K.; Ma, L.; Gressel, S.; Doudna, J.A. Structural biology. A Cas9–guide RNA complex preorganized for target DNA recognition. Science 2015, 348, 1477–1481. [Google Scholar] [CrossRef]
- Anders, C.; Niewoehner, O.; Duerst, A.; Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 2014, 513, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Taylor, D.W.; Chen, J.S.; Kornfeld, J.E.; Zhou, K.; Thompson, A.J.; Nogales, E.; Doudna, J.A. Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science 2016, 351, 867–871. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Guo, M.; Wang, S.; Zhu, Y.; Wang, S.; Xiong, Z.; Yang, J.; Xu, Z.; Huang, Z. Structural basis of CRISPR–SpyCas9 inhibition by an anti-CRISPR protein. Nature 2017, 546, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Patel, D.J. Inhibition Mechanism of an Anti-CRISPR Suppressor AcrIIA4 Targeting SpyCas9. Mol. Cell 2017, 67, 117–127.e115. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.W.; Bartesaghi, A.; Yang, H.; Falconieri, V.; Rao, P.; Merk, A.; Eng, E.T.; Raczkowski, A.M.; Fox, T.; Earl, L.A.; et al. Cryo-EM Structures Reveal Mechanism and Inhibition of DNA Targeting by a CRISPR-Cas Surveillance Complex. Cell 2017, 171, 414–426.e12. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Liu, J.-J.; Osuna, B.A.; Xu, M.; Berry, J.D.; Rauch, B.J.; Nogales, E.; Bondy-Denomy, J.; Doudna, J.A. Temperature-Responsive Competitive Inhibition of CRISPR-Cas9. Mol. Cell 2018, 73, 601–610.e605. [Google Scholar] [CrossRef]
- Liu, L.; Yin, M.; Wang, M.; Wang, Y. Phage AcrIIA2 DNA Mimicry: Structural Basis of the CRISPR and Anti-CRISPR Arms Race. Mol. Cell 2018, 73, 611–620. [Google Scholar] [CrossRef]
- Chowdhury, S.; Carter, J.; Rollins, M.F.; Golden, S.M.; Jackson, R.N.; Hoffmann, C.; Nosaka, L.; Bondy-Denomy, J.; Maxwell, K.L.; Davidson, A.R.; et al. Structure Reveals Mechanisms of Viral Suppressors that Intercept a CRISPR RNA-Guided Surveillance Complex. Cell 2017, 169, 47–57.e11. [Google Scholar] [CrossRef]
- Jinek, M.; Jiang, F.; Taylor, D.W.; Sternberg, S.H.; Kaya, E.; Ma, E.; Anders, C.; Hauer, M.; Zhou, K.; Lin, S.; et al. Structures of Cas9 Endonucleases Reveal RNA-Mediated Conformational Activation. Science 2014, 343, 1247997. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, X.; Ma, Y.; He, J.; Liu, X.; Yu, G.; Yin, H.; Zhang, H. Inhibition mechanisms of CRISPR-Cas9 by AcrIIA17 and AcrIIA18. Nucleic Acids Res. 2021, 50, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, R.A.; Martin, C.; Nemudryi, A.; Wiedenheft, B. CRISPR RNA-guided autonomous delivery of Cas9. Nat. Struct. Mol. Biol. 2019, 26, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Palermo, G.; Miao, Y.; Walker, R.C.; Jinek, M.; McCammon, J.A. Striking Plasticity of CRISPR-Cas9 and Key Role of Non-target DNA, as Revealed by Molecular Simulations. ACS Central Sci. 2016, 2, 756–763. [Google Scholar] [CrossRef]
- Mekler, V.; Minakhin, L.; Severinov, K. Mechanism of duplex DNA destabilization by RNA-guided Cas9 nuclease during target interrogation. Proc. Natl. Acad. Sci. USA 2017, 114, 5443–5448. [Google Scholar] [CrossRef]
- Szczelkun, M.D.; Tikhomirova, M.S.; Sinkunas, T.; Gasiunas, G.; Karvelis, T.; Pschera, P.; Siksnys, V.; Seidel, R. Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proc. Natl. Acad. Sci. USA 2014, 111, 9798–9803. [Google Scholar] [CrossRef]
- Knott, G.J.; Cress, B.F.; Liu, J.-J.; Thornton, B.W.; Lew, R.J.; Al-Shayeb, B.; Rosenberg, D.J.; Hammel, M.; Adler, B.A.; Lobba, M.J.; et al. Structural basis for AcrVA4 inhibition of specific CRISPR-Cas12a. Elife 2019, 8. [Google Scholar] [CrossRef]
- Zhu, Y.; Gao, A.; Zhan, Q.; Wang, Y.; Feng, H.; Liu, S.; Gao, G.; Serganov, A.; Gao, P. Diverse Mechanisms of CRISPR-Cas9 Inhibition by Type IIC Anti-CRISPR Proteins. Mol. Cell 2019, 74, 296–309.e297. [Google Scholar] [CrossRef]
- Sun, W.; Yang, J.; Cheng, Z.; Amrani, N.; Liu, C.; Wang, K.; Ibraheim, R.; Edraki, A.; Huang, X.; Wang, M.; et al. Structures of Neisseria meningitidis Cas9 Complexes in Catalytically Poised and Anti-CRISPR-Inhibited States. Mol. Cell 2019, 76, 938–952.e935. [Google Scholar] [CrossRef]
- Chen, J.S.; Dagdas, Y.S.; Kleinstiver, B.P.; Welch, M.M.; Sousa, A.A.; Harrington, L.B.; Sternberg, S.H.; Joung, J.K.; Yildiz, A.; Doudna, J.A. Enhanced proofreading governs CRISPR–Cas9 targeting accuracy. Nature 2017, 550, 407–410. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belato, H.B.; Lisi, G.P. The Many (Inter)faces of Anti-CRISPRs: Modulation of CRISPR-Cas Structure and Dynamics by Mechanistically Diverse Inhibitors. Biomolecules 2023, 13, 264. https://doi.org/10.3390/biom13020264
Belato HB, Lisi GP. The Many (Inter)faces of Anti-CRISPRs: Modulation of CRISPR-Cas Structure and Dynamics by Mechanistically Diverse Inhibitors. Biomolecules. 2023; 13(2):264. https://doi.org/10.3390/biom13020264
Chicago/Turabian StyleBelato, Helen B., and George P. Lisi. 2023. "The Many (Inter)faces of Anti-CRISPRs: Modulation of CRISPR-Cas Structure and Dynamics by Mechanistically Diverse Inhibitors" Biomolecules 13, no. 2: 264. https://doi.org/10.3390/biom13020264
APA StyleBelato, H. B., & Lisi, G. P. (2023). The Many (Inter)faces of Anti-CRISPRs: Modulation of CRISPR-Cas Structure and Dynamics by Mechanistically Diverse Inhibitors. Biomolecules, 13(2), 264. https://doi.org/10.3390/biom13020264