
Metagenomic Analysis of the Composition of Microbial Consortia Involved in Spruce Degradation over Time in Białowieża Natural Forest

 ,
,  ,
,  ,
,  ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Research Plot in Białowieża Forest
2.2. Sample Collection
2.3. Isolation of DNA and 16S rDNA and ITS1 Sequencing
2.4. Bioinformatic Analysis
2.5. Wood Chemical Analysis
3. Results
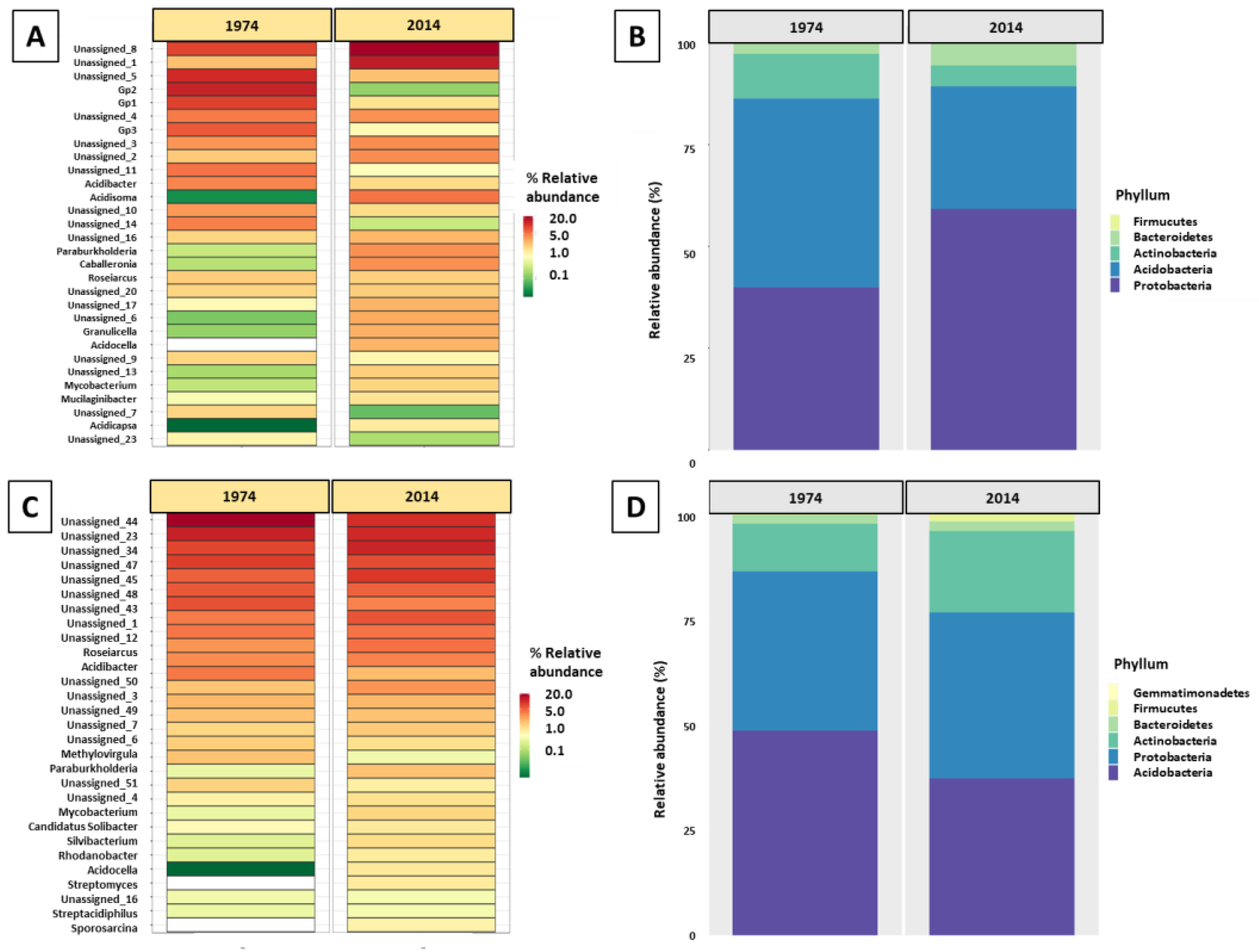
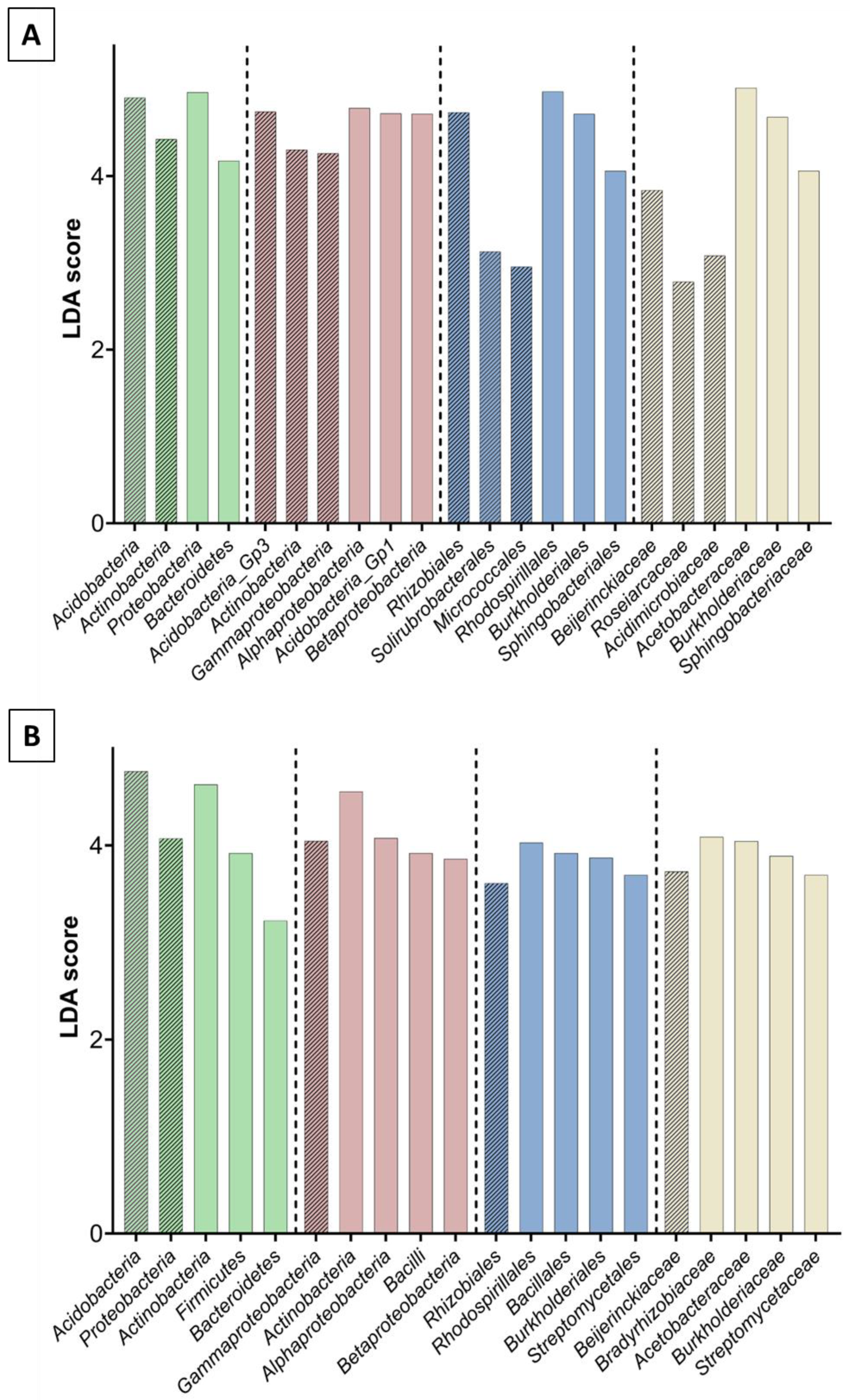
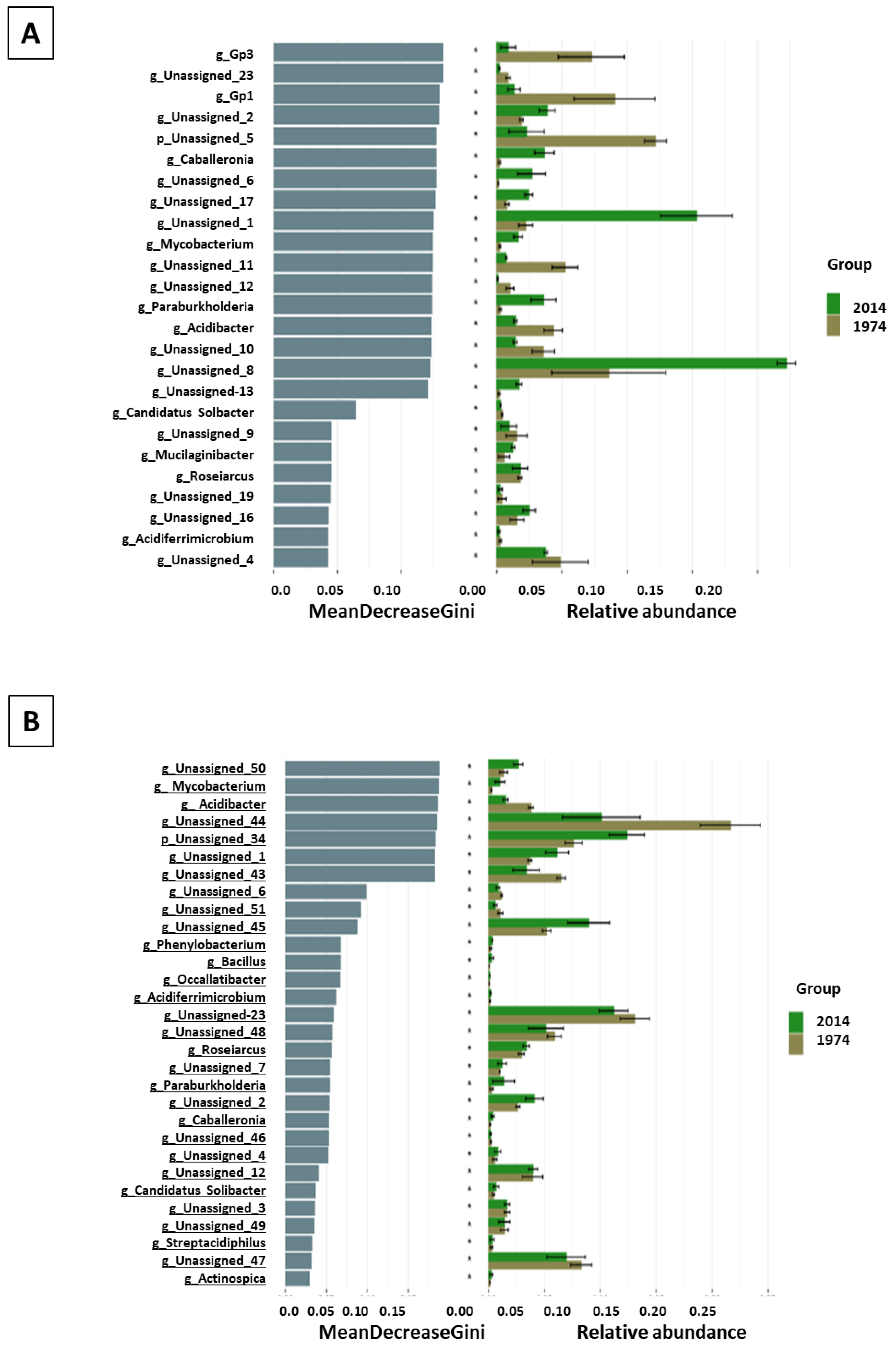
3.1. Metagenomic Analysis
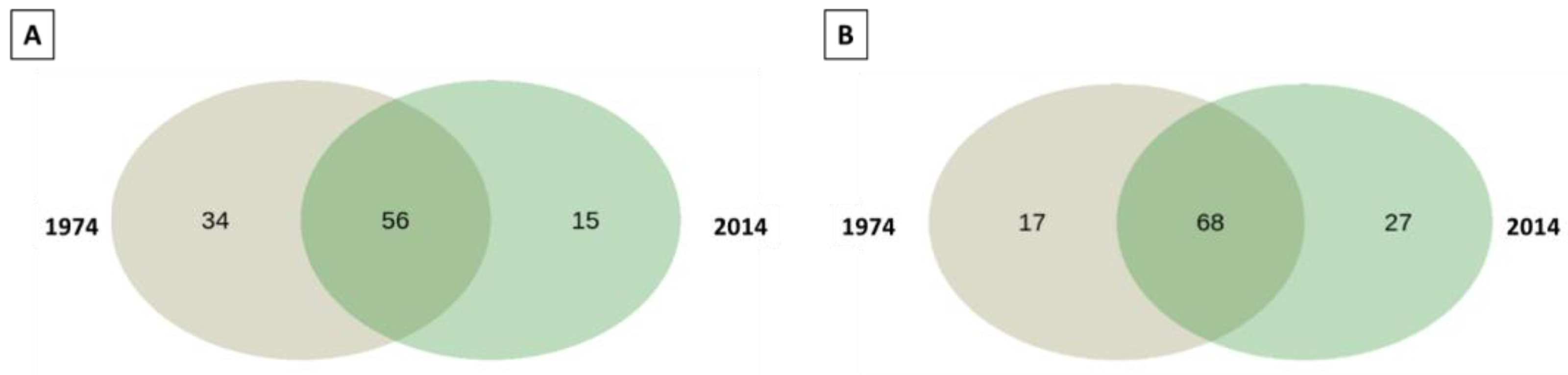
3.2. Bacterial and Fungal Community Diversity and Richness Analysis—16S Profiling of Wood and Soil
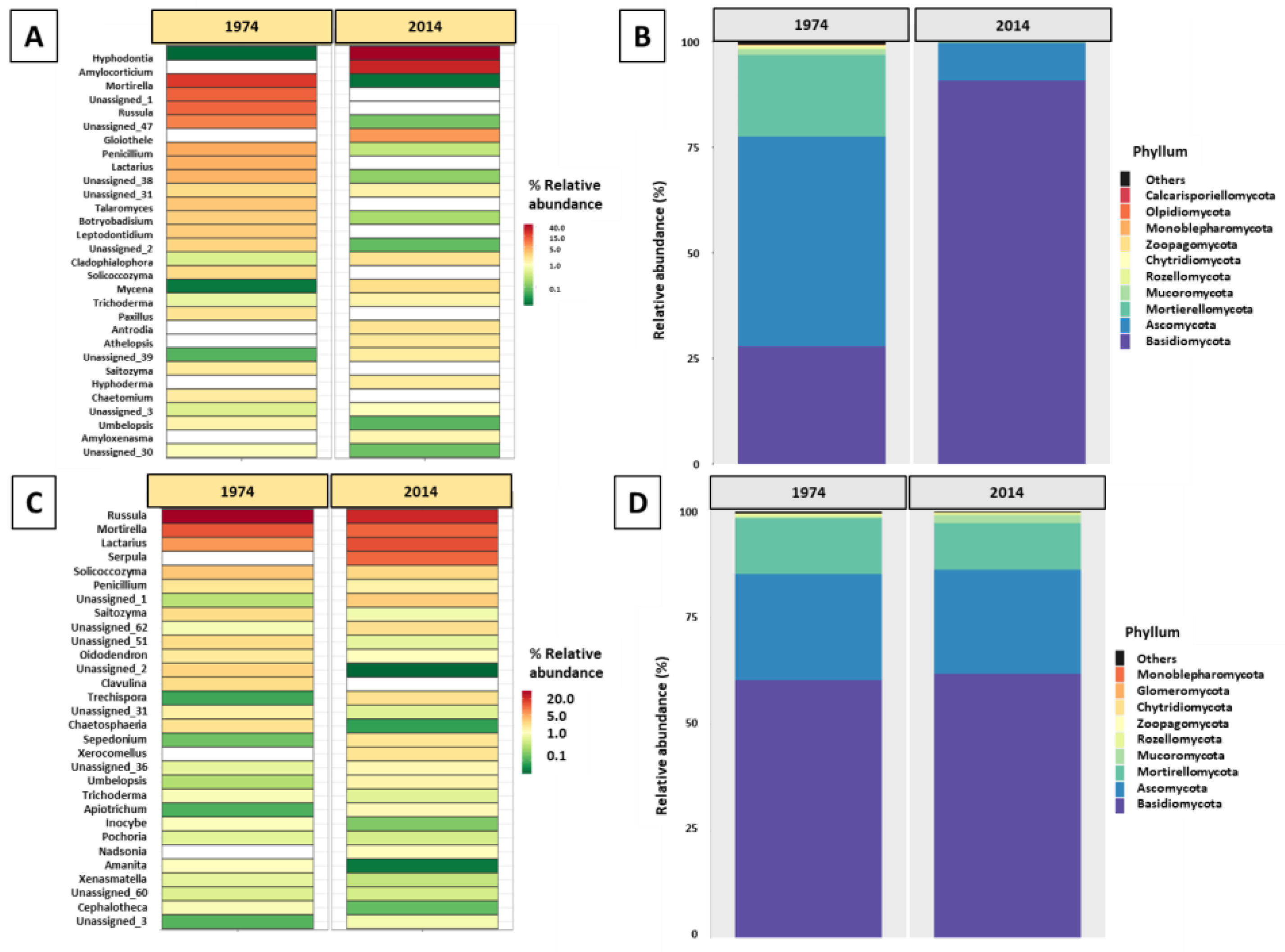
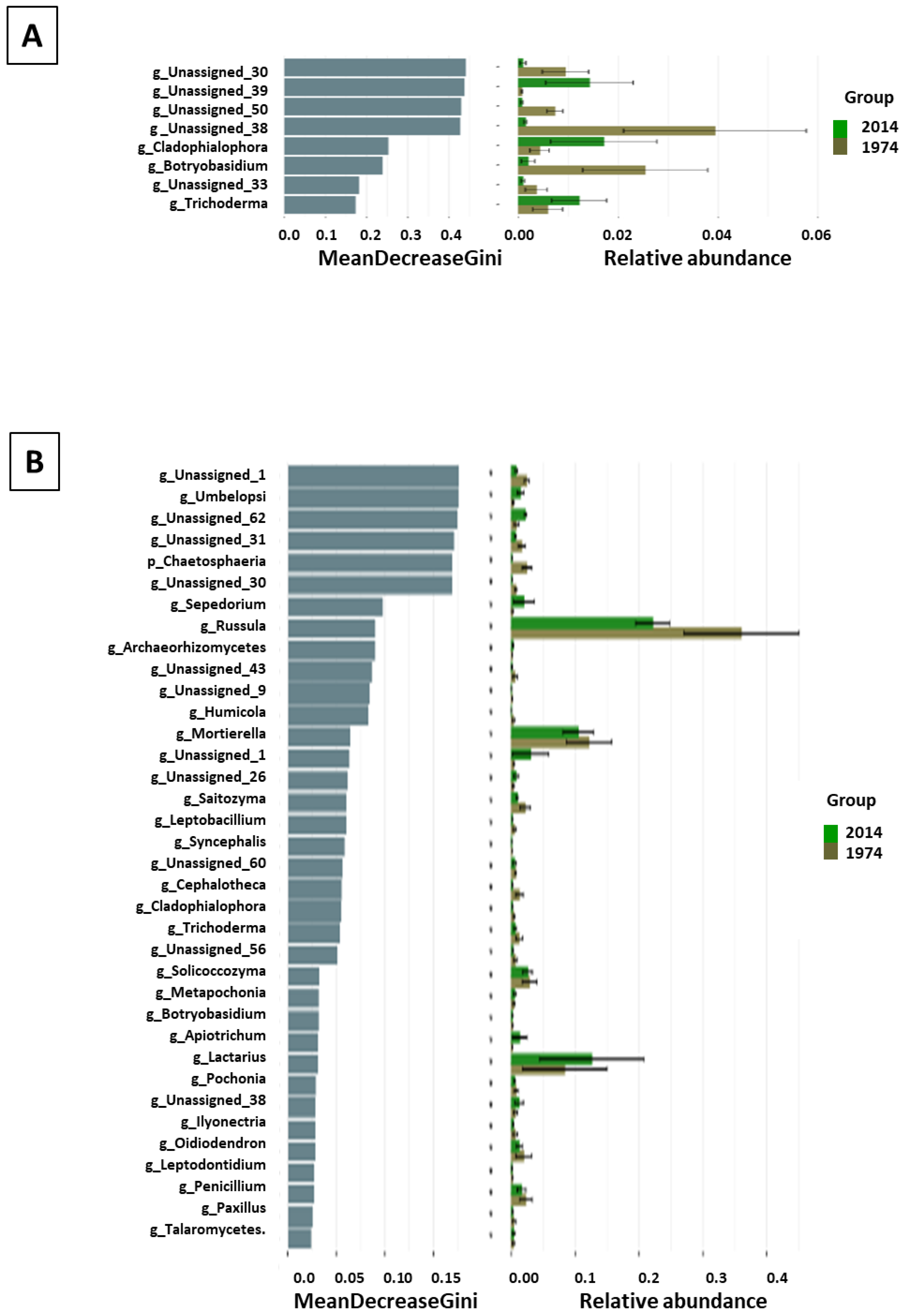
3.3. Fungal Community Composition and Diversity Analysis of Wood and Soil—ITS1 Profiling
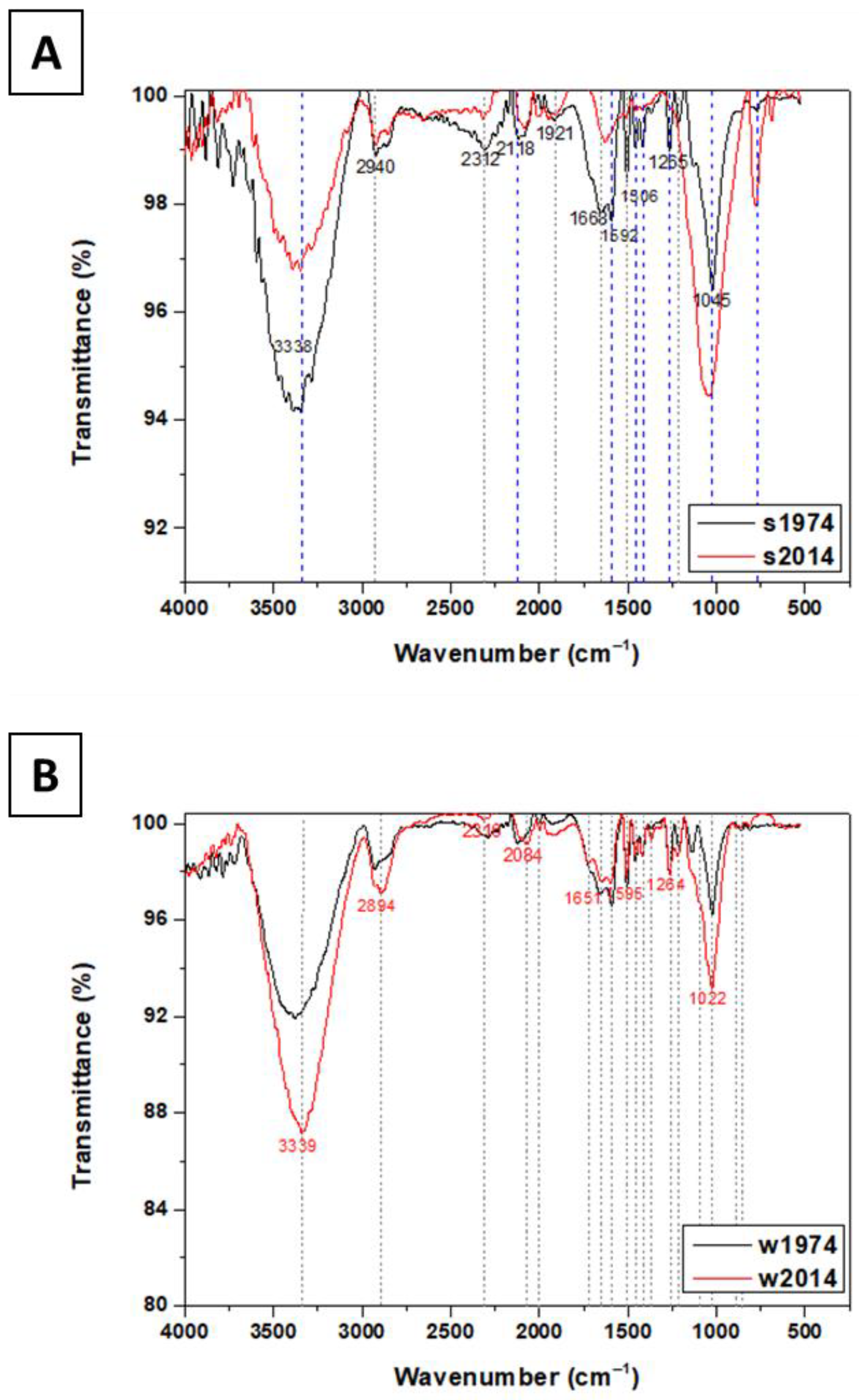
3.4. Chemical Composition of Soil and Wood Samples
3.5. Analysis of Carbon, Hydrogen, and Nitrogen Content (CHN) in Wood and Soil Samples
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sabatini, F.M.; Burrascano, S.; Keeton, W.S.; Levers, C.; Lindner, M.; Pötzschner, F.; Verkerk, P.J.; Bauhus, J.; Buchwald, E.; Chaskovsky, O.; et al. Where are Europe’s last primary forests? Divers. Distrib. 2018, 24, 1426–1439. [Google Scholar] [CrossRef]
- Reichstein, M.; Carvalhais, N. Aspects of Forest Biomass in the Earth System: Its Role and Major Unknowns. Surv. Geophys. 2019, 40, 693–707. [Google Scholar] [CrossRef]
- Tlaskal, V.; Brabcova, V.; Vetrovsky, T.; Lopez-Mondejar, R.; Monteiro, L.M.O.; Saraiva, J.P.; da Rocha, U.N.; Baldrian, P. Metagenomes, metatranscriptomes and microbiomes of naturally decomposing deadwood. Sci. Data 2021, 8, 198. [Google Scholar] [CrossRef]
- Pan, Y.D.; Birdsey, R.A.; Fang, J.Y.; Houghton, R.; Kauppi, P.E.; Kurz, W.A.; Phillips, O.L.; Shvidenko, A.; Lewis, S.L.; Canadell, J.G.; et al. A Large and Persistent Carbon Sink in the World’s Forests. Science 2011, 333, 988–993. [Google Scholar] [CrossRef]
- Morales-Hidalgo, D.; Oswalt, S.N.; Somanathan, E. Status and trends in global primary forest, protected areas, and areas designated for conservation of biodiversity from the Global Forest Resources Assessment 2015. For. Ecol. Manag. 2015, 352, 68–77. [Google Scholar] [CrossRef]
- Luyssaert, S.; Schulze, E.D.; Borner, A.; Knohl, A.; Hessenmoller, D.; Law, B.E.; Ciais, P.; Grace, J. Old-growth forests as global carbon sinks. Nature 2008, 455, 213–215. [Google Scholar] [CrossRef]
- Baldrian, P. Forest microbiome: Diversity, complexity and dynamics. FEMS Microbiol. Rev. 2017, 41, 109–130. [Google Scholar] [CrossRef]
- Martin, A.R.; Domke, G.M.; Doraisami, M.; Thomas, S.C. Carbon fractions in the world’s dead wood. Nat. Commun. 2021, 12, 889. [Google Scholar] [CrossRef]
- Herve, V.; Le Roux, X.; Uroz, S.; Gelhaye, E.; Frey-Klett, P. Diversity and structure of bacterial communities associated with Phanerochaete chrysosporium during wood decay. Environ. Microbiol. 2014, 16, 2238–2252. [Google Scholar] [CrossRef]
- Tlaskal, V.; Baldrian, P. Deadwood-Inhabiting Bacteria Show Adaptations to Changing Carbon and Nitrogen Availability during Decomposition. Front. Microbiol. 2021, 12, 685303. [Google Scholar] [CrossRef]
- Odriozola, I.; Abrego, N.; Tlaskal, V.; Zrustova, P.; Morais, D.; Vetrovsky, T.; Ovaskainen, O.; Baldrian, P. Fungal Communities Are Important Determinants of Bacterial Community Composition in Deadwood. mSystems 2021, 6, e01017-20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.B.; Yang, M.X.; Tu, R. Unexpectedly high bacterial diversity in decaying wood of a conifer as revealed by a molecular method. Int. Biodeterior. Biodegrad. 2008, 62, 471–474. [Google Scholar] [CrossRef]
- Lopez-Mondejar, R.; Tlaskal, V.; Vetrovsky, T.; Stursova, M.; Toscan, R.; da Rocha, U.N.; Baldrian, P. Metagenomics and stable isotope probing reveal the complementary contribution of fungal and bacterial communities in the recycling of dead biomass in forest soil. Soil Biol. Biochem. 2020, 148, 107875. [Google Scholar] [CrossRef]
- Daniel, G. Chapter 8—Fungal Degradation of Wood Cell Walls. In Secondary Xylem Biology; Kim, Y.S., Funada, R., Singh, A.P., Eds.; Academic Press: Boston, MA, USA, 2016; pp. 131–167. [Google Scholar]
- Folman, L.B.; Gunnewiek, P.J.A.K.; Boddy, L.; de Boer, W. Impact of white-rot fungi on numbers and community composition of bacteria colonizing beech wood from forest soil. FEMS Microbiol. Ecol. 2008, 63, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Urbanova, M.; Snajdr, J.; Baldrian, P. Composition of fungal and bacterial communities in forest litter and soil is largely determined by dominant trees. Soil Biol. Biochem. 2015, 84, 53–64. [Google Scholar] [CrossRef]
- Purahong, W.; Wubet, T.; Lentendu, G.; Schloter, M.; Pecyna, M.J.; Kapturska, D.; Hofrichter, M.; Kruger, D.; Buscot, F. Life in leaf litter: Novel insights into community dynamics of bacteria and fungi during litter decomposition. Mol. Ecol. 2016, 25, 4059–4074. [Google Scholar] [CrossRef] [PubMed]
- Purahong, W.; Wubet, T.; Lentendu, G.; Hoppe, B.; Jariyavidyanont, K.; Arnstadt, T.; Baber, K.; Otto, P.; Kellner, H.; Hofrichter, M.; et al. Determinants of Deadwood-Inhabiting Fungal Communities in Temperate Forests: Molecular Evidence from a Large Scale Deadwood Decomposition Experiment. Front. Microbiol. 2018, 9, 2120. [Google Scholar] [CrossRef]
- Faliński, J.B. Uprooted Trees, Their Distribution and Influence in the Primeval Forest Biotope. Vegetatio 1978, 38, 175–183. [Google Scholar] [CrossRef]
- Jaroszewicz, B.; Cholewińska, O.; Gutowski, J.M.; Zimny, M.; Samojlik, T.; Latałowa, M. Białowieża Forest—A Relic of the High Naturalness of European Forests. Forests 2019, 10, 849. [Google Scholar] [CrossRef]
- Checko, E.; Jaroszewicz, B.; Olejniczak, K.; Kwiatkowska-Falinska, A.J. The importance of coarse woody debris for vascular plants in temperate mixed deciduous forests. Can. J. For. Res. 2015, 45, 1154–1163. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Cui, Y.M.; Li, X.Z.; Yao, M.J. microeco: An R package for data mining in microbial community ecology. FEMS Microbiol. Ecol. 2021, 97, fiaa255. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Song, Z.W.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Margenot, A.J.; Calderon, F.J.; Parikh, S.J. Limitations and Potential of Spectral Subtractions in Fourier-Transform Infrared Spectroscopy of Soil Samples. Soil Sci. Soc. Am. J. 2016, 80, 10–26. [Google Scholar] [CrossRef]
- Younis, S.M.Z.; Iqbal, J. Estimation of soil moisture using multispectral and FTIR techniques. Egypt. J. Remote Sens. Space Sci. 2015, 18, 151–161. [Google Scholar] [CrossRef]
- Prewitt, L.; Kang, Y.; Kakumanu, M.L.; Williams, M. Fungal and bacterial community succession differs for three wood types during decay in a forest soil. Microb. Ecol. 2014, 68, 212–221. [Google Scholar] [CrossRef]
- Kuramae, E.E.; Leite, M.F.A.; Suleiman, A.K.A.; Gough, C.M.; Castillo, B.T.; Faller, L.; Franklin, R.B.; Syring, J. Wood Decay Characteristics and Interspecific Interactions Control Bacterial Community Succession in Populus grandidentata (Bigtooth Aspen). Front. Microbiol. 2019, 10, 979. [Google Scholar] [CrossRef]
- Stokland, J.N.; Siitonen, J.; Jonsson, B.G. Biodiversity in Dead Wood. In Biodiversity in Dead Wood; Cambridge University Press: Cambridge, UK, 2012. [Google Scholar]
- Tlaskal, V.; Brabcova, V.; Vetrovsky, T.; Jomura, M.; Lopez-Mondejar, R.; Oliveira Monteiro, L.M.; Saraiva, J.P.; Human, Z.R.; Cajthaml, T.; Nunes da Rocha, U.; et al. Complementary Roles of Wood-Inhabiting Fungi and Bacteria Facilitate Deadwood Decomposition. mSystems 2021, 6, e01078-20. [Google Scholar] [CrossRef] [PubMed]
- Bebber, S.W.D.; Darrah, P.; Fricker, M.; Tlalka, M.; Boddy, L. The role of wood decay fungi in the carbon and nitrogen dynamics of the forest floor. In Fungi in Biogeochemical Cycles; Gadd, G.M., Ed.; Cambridge University Press: Cambridge, UK, 2006. [Google Scholar]
- Kielak, A.M.; Scheublin, T.R.; Mendes, L.W.; van Veen, J.A.; Kuramae, E.E. Bacterial Community Succession in Pine-Wood Decomposition. Front. Microbiol. 2016, 7, 231. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, H.; Han, H.; Zhang, B.; Zhang, C.; He, J.; Li, S.; Cao, H. Microbial Community Succession Associated with Poplar Wood Discoloration. Plants 2022, 11, 2420. [Google Scholar] [CrossRef] [PubMed]
- Herzog, C.; Hartmann, M.; Frey, B.; Stierli, B.; Rumpel, C.; Buchmann, N.; Brunner, I. Microbial succession on decomposing root litter in a drought-prone Scots pine forest. ISME J. 2019, 13, 2346–2362. [Google Scholar] [CrossRef]
- Pettifor, B.J.; McDonald, J.E. Chapter 9—Bacterial biota of forest trees. In Forest Microbiology; Asiegbu, F.O., Kovalchuk, A., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 161–173. [Google Scholar]
- Tian, J.H.; Pourcher, A.M.; Bouchez, T.; Gelhaye, E.; Peu, P. Occurrence of lignin degradation genotypes and phenotypes among prokaryotes. Appl. Microbiol. Biotechnol. 2014, 98, 9527–9544. [Google Scholar] [CrossRef]
- Skrede, I. Chapter One—Diversity and distribution of ligninolytic fungi. In Advances in Botanical Research; Morel-Rouhier, M., Sormani, R., Eds.; Academic Press: Cambridge, MA, USA, 2021; Volume 99, pp. 1–36. [Google Scholar]
- Swift, M.J. The ecology of wood decomposition. Sci. Prog. 1977, 64, 175–199. [Google Scholar]
- Kwaśna, H.; Mazur, A.; Łabędzki, A.; Kuźmiński, R.; Łakomy, P. Communities of fungi in decomposed wood of oak and pine. For. Res. Pap./Leśne Pr. Badaw. 2016, 77, 261–275. [Google Scholar] [CrossRef]
- Colom, X.; Carrillo, F.; Nogues, F.; Garriga, P. Structural analysis of photodegraded wood by means of FTIR spectroscopy. Polym. Degrad. Stab. 2003, 80, 543–549. [Google Scholar] [CrossRef]
- Shi, J.T.; Xing, D.; Li, J. FTIR Studies of the Changes in Wood Chemistry from Wood Forming Tissue under Inclined Treatment. Energy Procedia 2012, 16 Pt B, 758–762. [Google Scholar] [CrossRef]


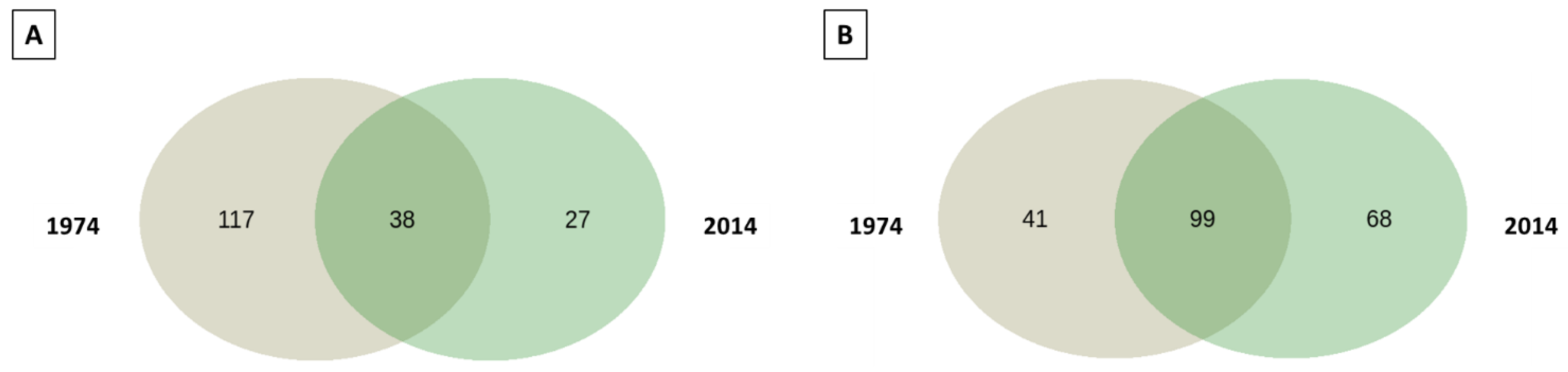


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1974 | 2014 | |||
|---|---|---|---|---|
| Wood | Soil | Wood | Soil | |
| N [%] | 0.83 ± 0.24 | 0.94 ± 0.11 | 0.23 ± 0.10 | 0.75 ± 0.11 |
| C [%] | 51.77 ± 2.79 | 37.95 ± 14.75 | 49.60 ± 1.77 | 13.13 ± 2.76 |
| H [%] | 6.29 ± 0.16 | 4.77 ± 1.77 | 6.61 ± 0.16 | 1.90 ± 0.34 |
| O [%] | 41.12 ± 2.75 | 56.33 ± 16.60 | 43.55 ± 1.60 | 84.22 ± 3.20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janusz, G.; Mazur, A.; Pawlik, A.; Kołodyńska, D.; Jaroszewicz, B.; Marzec-Grządziel, A.; Koper, P. Metagenomic Analysis of the Composition of Microbial Consortia Involved in Spruce Degradation over Time in Białowieża Natural Forest. Biomolecules 2023, 13, 1466. https://doi.org/10.3390/biom13101466
Janusz G, Mazur A, Pawlik A, Kołodyńska D, Jaroszewicz B, Marzec-Grządziel A, Koper P. Metagenomic Analysis of the Composition of Microbial Consortia Involved in Spruce Degradation over Time in Białowieża Natural Forest. Biomolecules. 2023; 13(10):1466. https://doi.org/10.3390/biom13101466
Chicago/Turabian StyleJanusz, Grzegorz, Andrzej Mazur, Anna Pawlik, Dorota Kołodyńska, Bogdan Jaroszewicz, Anna Marzec-Grządziel, and Piotr Koper. 2023. "Metagenomic Analysis of the Composition of Microbial Consortia Involved in Spruce Degradation over Time in Białowieża Natural Forest" Biomolecules 13, no. 10: 1466. https://doi.org/10.3390/biom13101466
APA StyleJanusz, G., Mazur, A., Pawlik, A., Kołodyńska, D., Jaroszewicz, B., Marzec-Grządziel, A., & Koper, P. (2023). Metagenomic Analysis of the Composition of Microbial Consortia Involved in Spruce Degradation over Time in Białowieża Natural Forest. Biomolecules, 13(10), 1466. https://doi.org/10.3390/biom13101466