
HOXA Amplification Defines a Genetically Distinct Subset of Angiosarcomas


Abstract
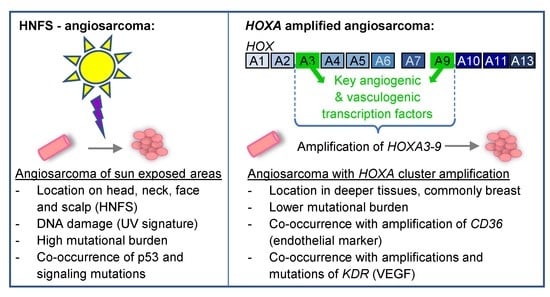
1. Introduction
Methodology
2. Results


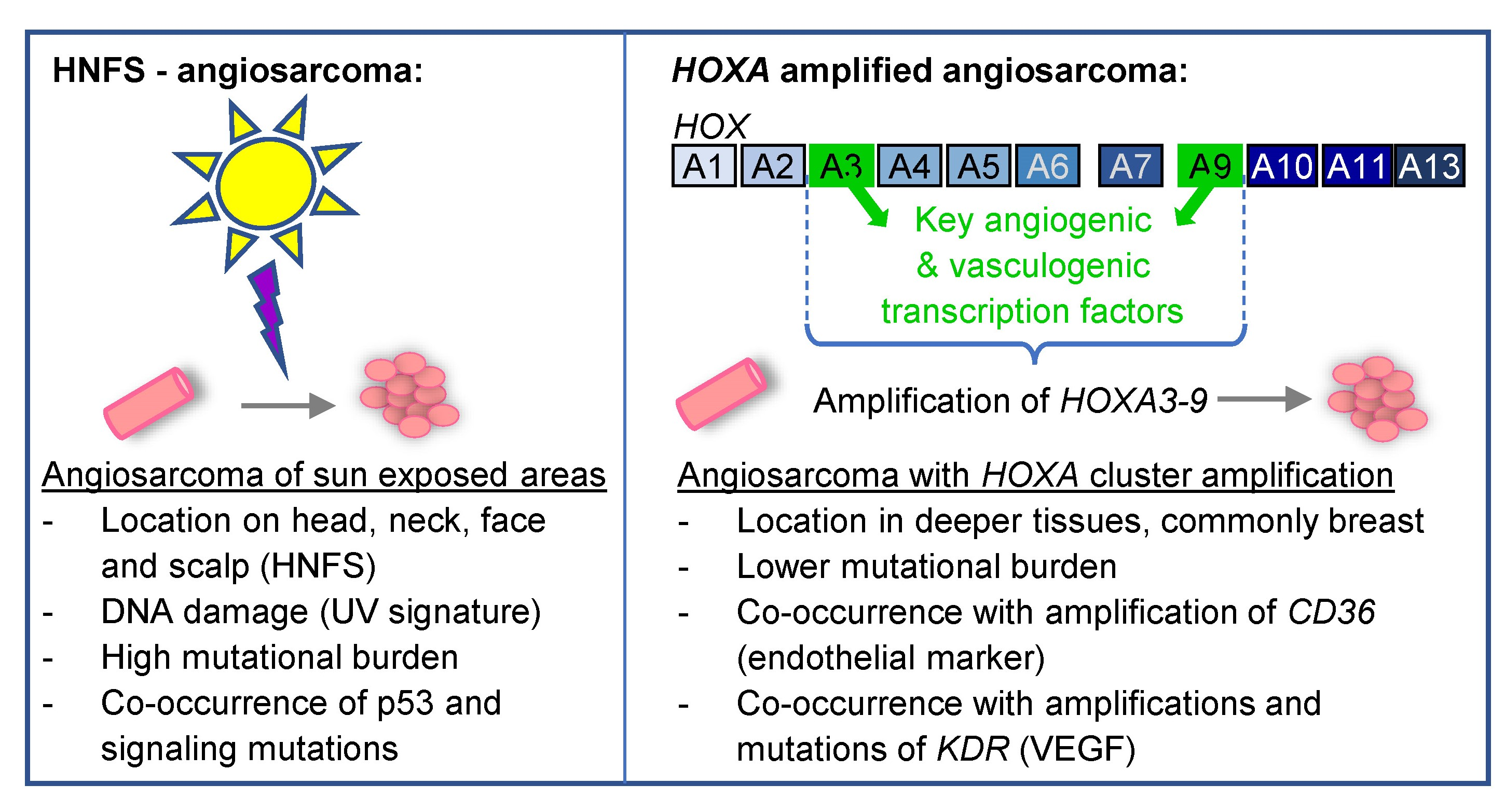{kind=link}
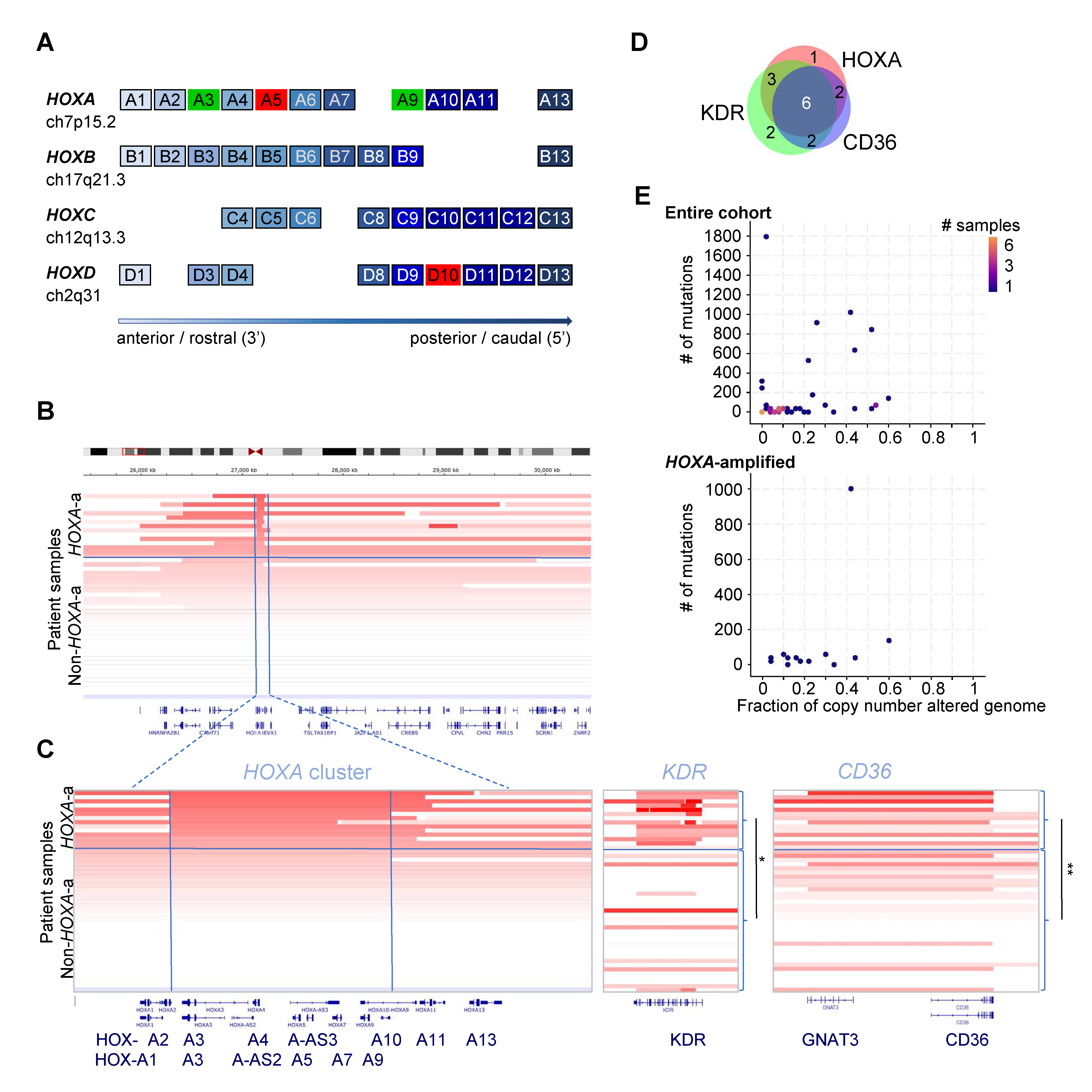{kind=link}
| Patient Characteristics (36 Patients Total) | HOXA-Amplified (12) # (%) | Non-HOXA-Amp (24) # (%) | Significance p-Value FE (MV) |
|---|---|---|---|
| Gender | |||
| Male | 2 (16.7) | 8 (33.3) | ns |
| Female | 10 (83.3) | 16 (66.7) | ns |
| Location | |||
| HNFS | 2 (16.7) | 8 (33.3) | ns |
| Breast | 8 (66.7) | 11 (45.8) | ns |
| intrathoracic/abd | 2 (16.7) | 4 (16.7) | ns |
| LIMB | 0 (0) | 1 (4.2) | ns |
| Prior Radiation | |||
| Yes | 2 (16.7) | 5 (20.8) | ns |
| Age | |||
| Peak 1: 26–40 Years | 5 (41.6) | 12 (50) | ns |
| 41–50 Years | 0 (0) | 2 (8.3) | ns |
| Peak 2: 51–70 Years | 6 (50) | 8 (33.3) | ns |
| >70 Years | 1 (8.3) | 2 (8.3) | ns |
| Pathology | |||
| Vasoformative | 10 (83.3) | 24 (100) | ns |
| Epithelioid | 7 (58.3) | 8 (33.3) | ns |
| Spindle Cell | 6 (50) | 17 (70.8) | ns |
| Mutation Burden | |||
| >200 Mutations | 1 (8.3) | 7 (29.2) | ns |
| Amplifications | |||
| CD36 | 8 (66.7) | 2 (8.3) | 5.59 × 10−4 (1.62 × 10−2) |
| KDR | 9 (75) | 4 (16.7) | 1.07 × 10−3 (3.11 × 10−2) |
| PHF1 | 9(75) | 10 (41.7) | ns |
| Mutations | |||
| TP53 | 4 (33.3) | 7 (29.2) | ns |
| KDR | 3 (25) | 5 (20.8) | ns |
| PIK3CA | 2 (16.7) | 4 (16.7) | ns |
| NRAS | 0 | 3 (12.5) | ns |
| HRAS | 1 (8.3) | 1 (4.1) | ns |
| BRAF | 0 | 2 (8.3) | ns |
| RAF1 | 0 | 1 (4.1) | ns |
| NF1 | 2 (16.7) | 2 (8.3) | ns |
| MAP3K13 | 0 | 1 (4.1) | ns |
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HNFS | head, neck, face and scalp |
| HOX | homeobox |
References
- Weidema, M.E.; Versleijen-Jonkers, Y.M.H.; Flucke, U.E.; Desar, I.M.E.; van der Graaf, W.T.A. Targeting angiosarcomas of the soft tissues: A challenging effort in a heterogeneous and rare disease. Crit. Rev. Oncol. Hematol. 2019, 138, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Weidema, M.E.; Flucke, U.E.; van der Graaf, W.T.A.; Ho, V.K.Y.; Hillebrandt-Roeffen, M.H.S.; Dutch Nationwide Network and Registry of Histo- and Cytopathology (PALGA)-Group; Versleijen-Jonkers, Y.M.H.; Husson, O.; Desar, I.M.E. Prognostic Factors in a Large Nationwide Cohort of Histologically Confirmed Primary and Secondary Angiosarcomas. Cancers 2019, 11, 1780. [Google Scholar] [CrossRef] [PubMed]
- Painter, C.A.; Jain, E.; Tomson, B.N.; Dunphy, M.; Stoddard, R.E.; Thomas, B.S.; Damon, A.L.; Shah, S.; Kim, D.; Gomez Tejeda Zanudo, J.; et al. The Angiosarcoma Project: Enabling genomic and clinical discoveries in a rare cancer through patient-partnered research. Nat. Med. 2020, 26, 181–187. [Google Scholar] [CrossRef]
- Behjati, S.; Tarpey, P.S.; Sheldon, H.; Martincorena, I.; Van Loo, P.; Gundem, G.; Wedge, D.C.; Ramakrishna, M.; Cooke, S.L.; Pillay, N.; et al. Recurrent PTPRB and PLCG1 mutations in angiosarcoma. Nat. Genet. 2014, 46, 376–379. [Google Scholar] [CrossRef]
- Murali, R.; Chandramohan, R.; Moller, I.; Scholz, S.L.; Berger, M.; Huberman, K.; Viale, A.; Pirun, M.; Socci, N.D.; Bouvier, N.; et al. Targeted massively parallel sequencing of angiosarcomas reveals frequent activation of the mitogen activated protein kinase pathway. Oncotarget 2015, 6, 36041–36052. [Google Scholar] [CrossRef]
- Italiano, A.; Chen, C.L.; Thomas, R.; Breen, M.; Bonnet, F.; Sevenet, N.; Longy, M.; Maki, R.G.; Coindre, J.M.; Antonescu, C.R. Alterations of the p53 and PIK3CA/AKT/mTOR pathways in angiosarcomas: A pattern distinct from other sarcomas with complex genomics. Cancer 2012, 118, 5878–5887. [Google Scholar] [CrossRef]
- Andersen, N.J.; Boguslawski, E.B.; Kuk, C.Y.; Chambers, C.M.; Duesbery, N.S. Combined inhibition of MEK and mTOR has a synergic effect on angiosarcoma tumorgrafts. Int. J. Oncol. 2015, 47, 71–80. [Google Scholar] [CrossRef]
- Chadwick, M.L.; Lane, A.; Thomas, D.; Smith, A.R.; White, A.R.; Davidson, D.; Feng, Y.; Boscolo, E.; Zheng, Y.; Adams, D.M.; et al. Combined mTOR and MEK inhibition is an effective therapy in a novel mouse model for angiosarcoma. Oncotarget 2018, 9, 24750–24765. [Google Scholar] [CrossRef]
- Rothweiler, S.; Dill, M.T.; Terracciano, L.; Makowska, Z.; Quagliata, L.; Hlushchuk, R.; Djonov, V.; Heim, M.H.; Semela, D. Generation of a murine hepatic angiosarcoma cell line and reproducible mouse tumor model. Lab. Investig. 2015, 95, 351–362. [Google Scholar] [CrossRef]
- Guo, T.; Zhang, L.; Chang, N.E.; Singer, S.; Maki, R.G.; Antonescu, C.R. Consistent MYC and FLT4 gene amplification in radiation-induced angiosarcoma but not in other radiation-associated atypical vascular lesions. Genes Chromosomes Cancer 2011, 50, 25–33. [Google Scholar] [CrossRef]
- Kunze, K.; Spieker, T.; Gamerdinger, U.; Nau, K.; Berger, J.; Dreyer, T.; Sindermann, J.R.; Hoffmeier, A.; Gattenlohner, S.; Brauninger, A. A recurrent activating PLCG1 mutation in cardiac angiosarcomas increases apoptosis resistance and invasiveness of endothelial cells. Cancer Res. 2014, 74, 6173–6183. [Google Scholar] [CrossRef] [PubMed]
- Shon, W.; Sukov, W.R.; Jenkins, S.M.; Folpe, A.L. MYC amplification and overexpression in primary cutaneous angiosarcoma: A fluorescence in-situ hybridization and immunohistochemical study. Mod. Pathol. 2014, 27, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Kachgal, S.; Mace, K.A.; Boudreau, N.J. The dual roles of homeobox genes in vascularization and wound healing. Cell Adhes. Migr. 2012, 6, 457–470. [Google Scholar] [CrossRef]
- Terracciano, D.; Terreri, S.; de Nigris, F.; Costa, V.; Calin, G.A.; Cimmino, A. The role of a new class of long noncoding RNAs transcribed from ultraconserved regions in cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 449–455. [Google Scholar] [CrossRef]
- Bejerano, G.; Pheasant, M.; Makunin, I.; Stephen, S.; Kent, W.J.; Mattick, J.S.; Haussler, D. Ultraconserved elements in the human genome. Science 2004, 304, 1321–1325. [Google Scholar] [CrossRef]
- Collins, E.M.; Thompson, A. HOX genes in normal, engineered and malignant hematopoiesis. Int. J. Dev. Biol. 2018, 62, 847–856. [Google Scholar] [CrossRef]
- Chisaka, O.; Capecchi, M.R. Regionally restricted developmental defects resulting from targeted disruption of the mouse homeobox gene hox-1.5. Nature 1991, 350, 473–479. [Google Scholar] [CrossRef]
- Bahrami, S.B.; Veiseh, M.; Dunn, A.A.; Boudreau, N.J. Temporal changes in Hox gene expression accompany endothelial cell differentiation of embryonic stem cells. Cell Adhes. Migr. 2011, 5, 133–141. [Google Scholar] [CrossRef]
- Iacovino, M.; Chong, D.; Szatmari, I.; Hartweck, L.; Rux, D.; Caprioli, A.; Cleaver, O.; Kyba, M. HoxA3 is an apical regulator of haemogenic endothelium. Nat. Cell Biol. 2011, 13, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Rossig, L.; Urbich, C.; Bruhl, T.; Dernbach, E.; Heeschen, C.; Chavakis, E.; Sasaki, K.; Aicher, D.; Diehl, F.; Seeger, F.; et al. Histone deacetylase activity is essential for the expression of HoxA9 and for endothelial commitment of progenitor cells. J. Exp. Med. 2005, 201, 1825–1835. [Google Scholar] [CrossRef]
- Crompton, M.R.; Bartlett, T.J.; MacGregor, A.D.; Manfioletti, G.; Buratti, E.; Giancotti, V.; Goodwin, G.H. Identification of a novel vertebrate homeobox gene expressed in haematopoietic cells. Nucleic Acids Res. 1992, 20, 5661–5667. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mace, K.A.; Hansen, S.L.; Myers, C.; Young, D.M.; Boudreau, N. HOXA3 induces cell migration in endothelial and epithelial cells promoting angiogenesis and wound repair. J. Cell Sci. 2005, 118, 2567–2577. [Google Scholar] [CrossRef]
- Mace, K.A.; Restivo, T.E.; Rinn, J.L.; Paquet, A.C.; Chang, H.Y.; Young, D.M.; Boudreau, N.J. HOXA3 modulates injury-induced mobilization and recruitment of bone marrow-derived cells. Stem Cells 2009, 27, 1654–1665. [Google Scholar] [CrossRef]
- Mahdipour, E.; Charnock, J.C.; Mace, K.A. Hoxa3 promotes the differentiation of hematopoietic progenitor cells into proangiogenic Gr-1+CD11b+ myeloid cells. Blood 2011, 117, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Bruhl, T.; Urbich, C.; Aicher, D.; Acker-Palmer, A.; Zeiher, A.M.; Dimmeler, S. Homeobox A9 transcriptionally regulates the EphB4 receptor to modulate endothelial cell migration and tube formation. Circ. Res. 2004, 94, 743–751. [Google Scholar] [CrossRef]
- Miano, J.M.; Firulli, A.B.; Olson, E.N.; Hara, P.; Giachelli, C.M.; Schwartz, S.M. Restricted expression of homeobox genes distinguishes fetal from adult human smooth muscle cells. Proc. Natl. Acad. Sci. USA 1996, 93, 900–905. [Google Scholar] [CrossRef]
- Rhoads, K.; Arderiu, G.; Charboneau, A.; Hansen, S.L.; Hoffman, W.; Boudreau, N. A role for Hox A5 in regulating angiogenesis and vascular patterning. Lymphat. Res. Biol. 2005, 3, 240–252. [Google Scholar] [CrossRef]
- Arderiu, G.; Cuevas, I.; Chen, A.; Carrio, M.; East, L.; Boudreau, N.J. HoxA5 stabilizes adherens junctions via increased Akt1. Cell Adhes. Migr. 2007, 1, 185–195. [Google Scholar] [CrossRef][Green Version]
- Chen, A.; Cuevas, I.; Kenny, P.A.; Miyake, H.; Mace, K.; Ghajar, C.; Boudreau, A.; Bissell, M.J.; Boudreau, N. Endothelial cell migration and vascular endothelial growth factor expression are the result of loss of breast tissue polarity. Cancer Res. 2009, 69, 6721–6729. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.W.; Song, E.; Feng, Z.; Sinha, A.; Chen, C.W.; Deshpande, A.J.; Cusan, M.; Farnoud, N.; Mupo, A.; Grove, C.; et al. Targeting Chromatin Regulators Inhibits Leukemogenic Gene Expression in NPM1 Mutant Leukemia. Cancer Discov. 2016, 6, 1166–1181. [Google Scholar] [CrossRef] [PubMed]
- Bernt, K.M.; Zhu, N.; Sinha, A.U.; Vempati, S.; Faber, J.; Krivtsov, A.V.; Feng, Z.; Punt, N.; Daigle, A.; Bullinger, L.; et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 2011, 20, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Majer, C.R.; Sneeringer, C.J.; Song, J.; Johnston, L.D.; Scott, M.P.; Smith, J.J.; Xiao, Y.; et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 2011, 20, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; Garcia-Manero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Savona, M.R.; Jongen-Lavrenic, M.; Altman, J.K.; Thomson, B.; Blakemore, S.J.; et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 2018, 131, 2661–2669. [Google Scholar] [CrossRef]
- Svoboda, L.K.; Bailey, N.; Van Noord, R.A.; Krook, M.A.; Harris, A.; Cramer, C.; Jasman, B.; Patel, R.M.; Thomas, D.; Borkin, D.; et al. Tumorigenicity of Ewing sarcoma is critically dependent on the trithorax proteins MLL1 and menin. Oncotarget 2017, 8, 458–471. [Google Scholar] [CrossRef]
- Kempinska, K.; Malik, B.; Borkin, D.; Klossowski, S.; Shukla, S.; Miao, H.; Wang, J.; Cierpicki, T.; Grembecka, J. Pharmacologic Inhibition of the Menin-MLL Interaction Leads to Transcriptional Repression of PEG10 and Blocks Hepatocellular Carcinoma. Mol. Cancer Ther. 2018, 17, 26–38. [Google Scholar] [CrossRef]
- The Angiosarcoma Project—Count Me In (Nature Medicine, 2020). Available online: https://www.cbioportal.org/study/summary?id=angs_project_painter_2018 (accessed on 4 July 2022).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, H.M.; Bernt, K.M. HOXA Amplification Defines a Genetically Distinct Subset of Angiosarcomas. Biomolecules 2022, 12, 1124. https://doi.org/10.3390/biom12081124
Xie HM, Bernt KM. HOXA Amplification Defines a Genetically Distinct Subset of Angiosarcomas. Biomolecules. 2022; 12(8):1124. https://doi.org/10.3390/biom12081124
Chicago/Turabian StyleXie, Hongbo M., and Kathrin M. Bernt. 2022. "HOXA Amplification Defines a Genetically Distinct Subset of Angiosarcomas" Biomolecules 12, no. 8: 1124. https://doi.org/10.3390/biom12081124
APA StyleXie, H. M., & Bernt, K. M. (2022). HOXA Amplification Defines a Genetically Distinct Subset of Angiosarcomas. Biomolecules, 12(8), 1124. https://doi.org/10.3390/biom12081124