
Modeling the Competition between Misfolded Aβ Conformers That Produce Distinct Types of Amyloid Pathology in Alzheimer’s Disease


Abstract
:1. Introduction
2. Methods
2.1. Transgenic Animals
2.2. Inoculum Preparation
2.3. Neonatal Intracerebral Seeding
2.4. Tissue Collection
2.5. Histology and Immunochemistry
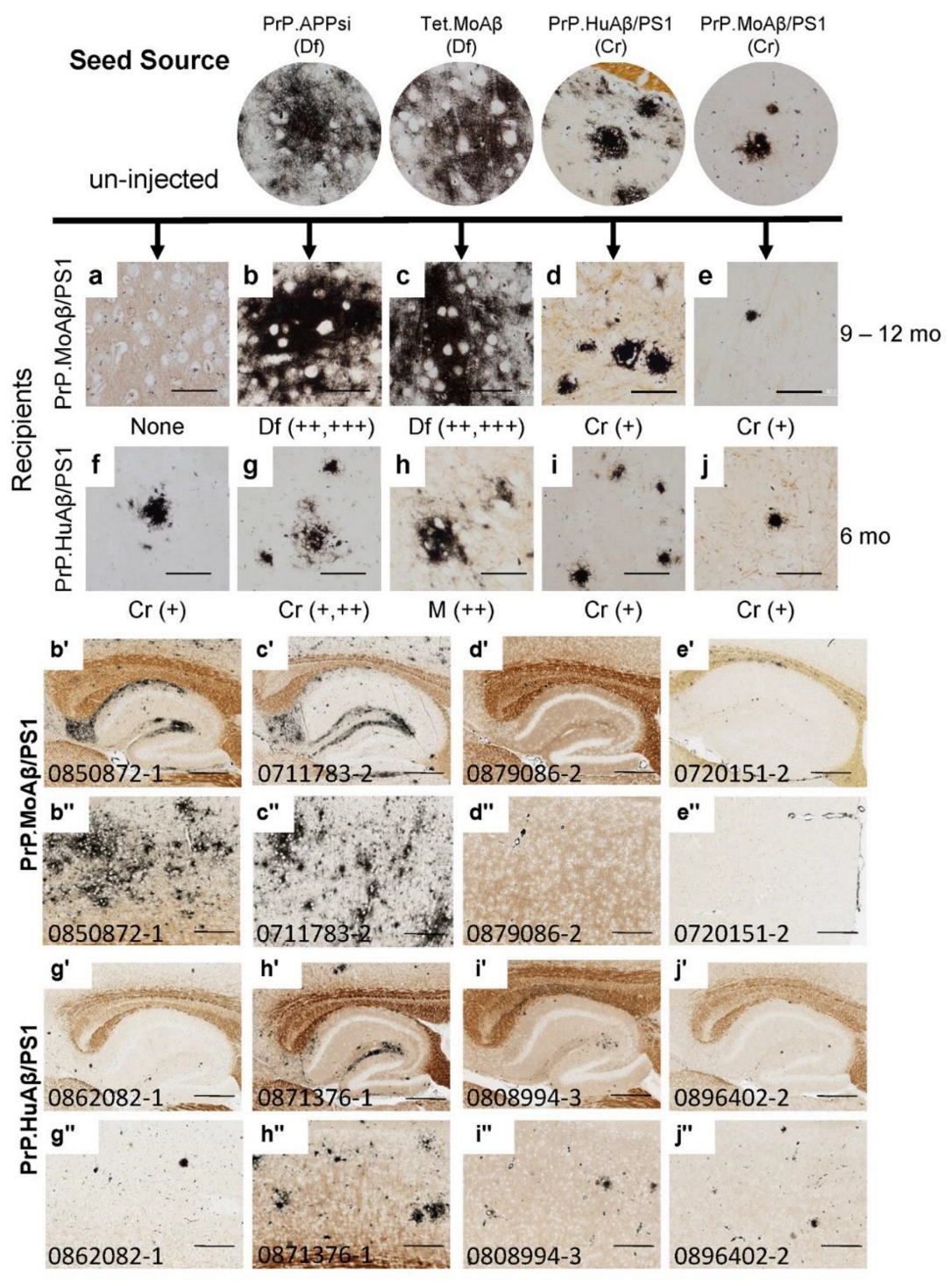
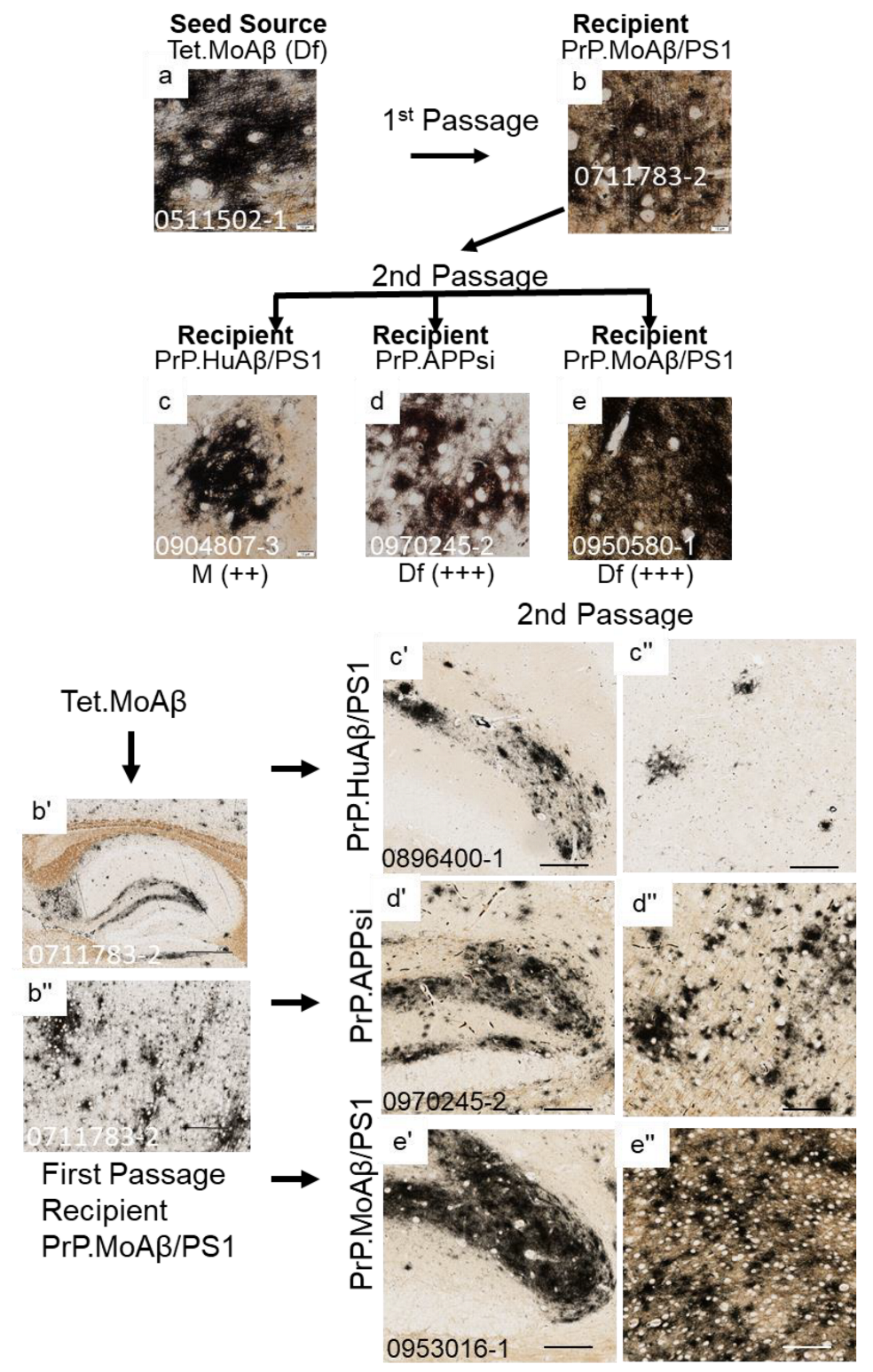
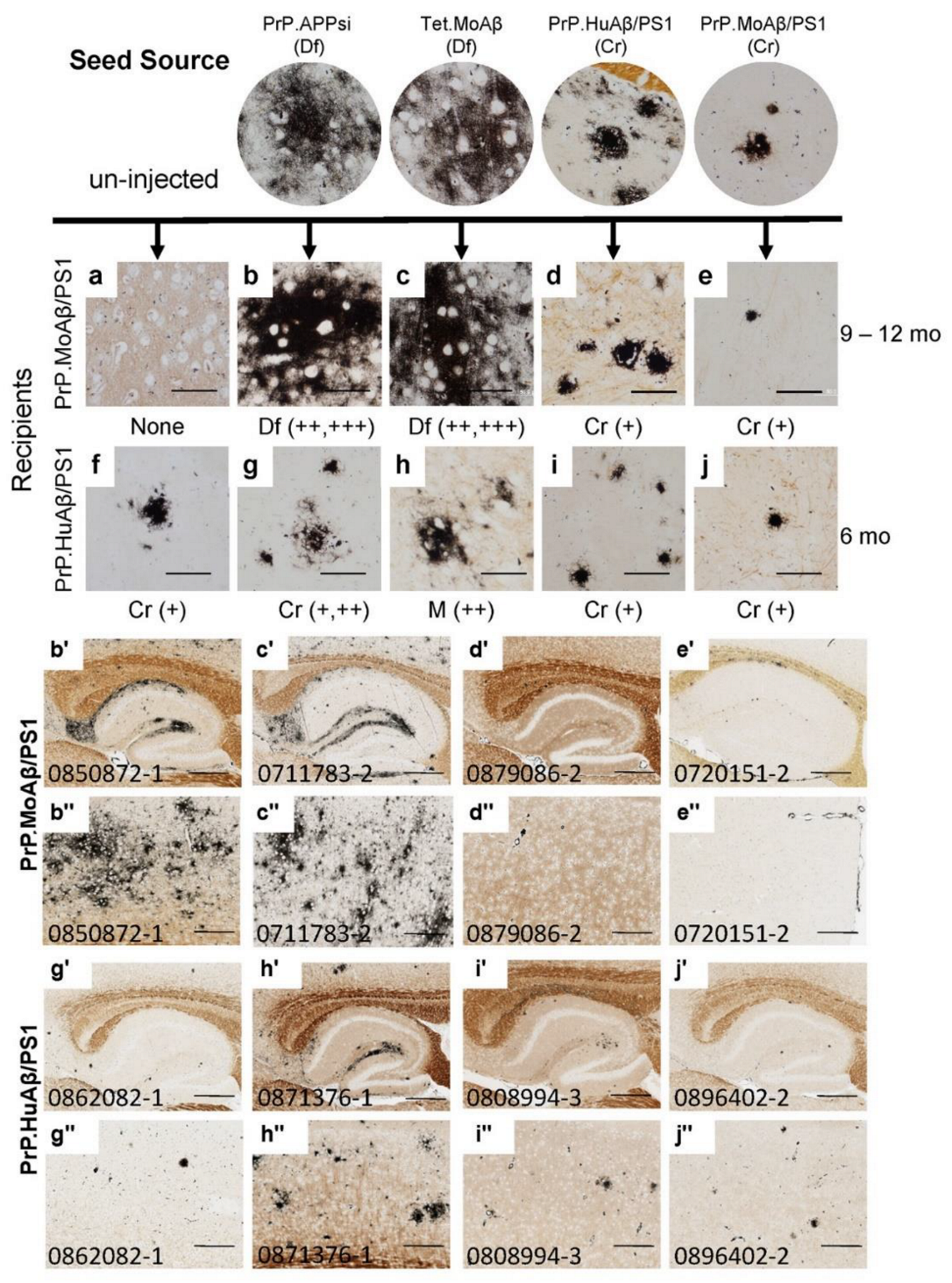
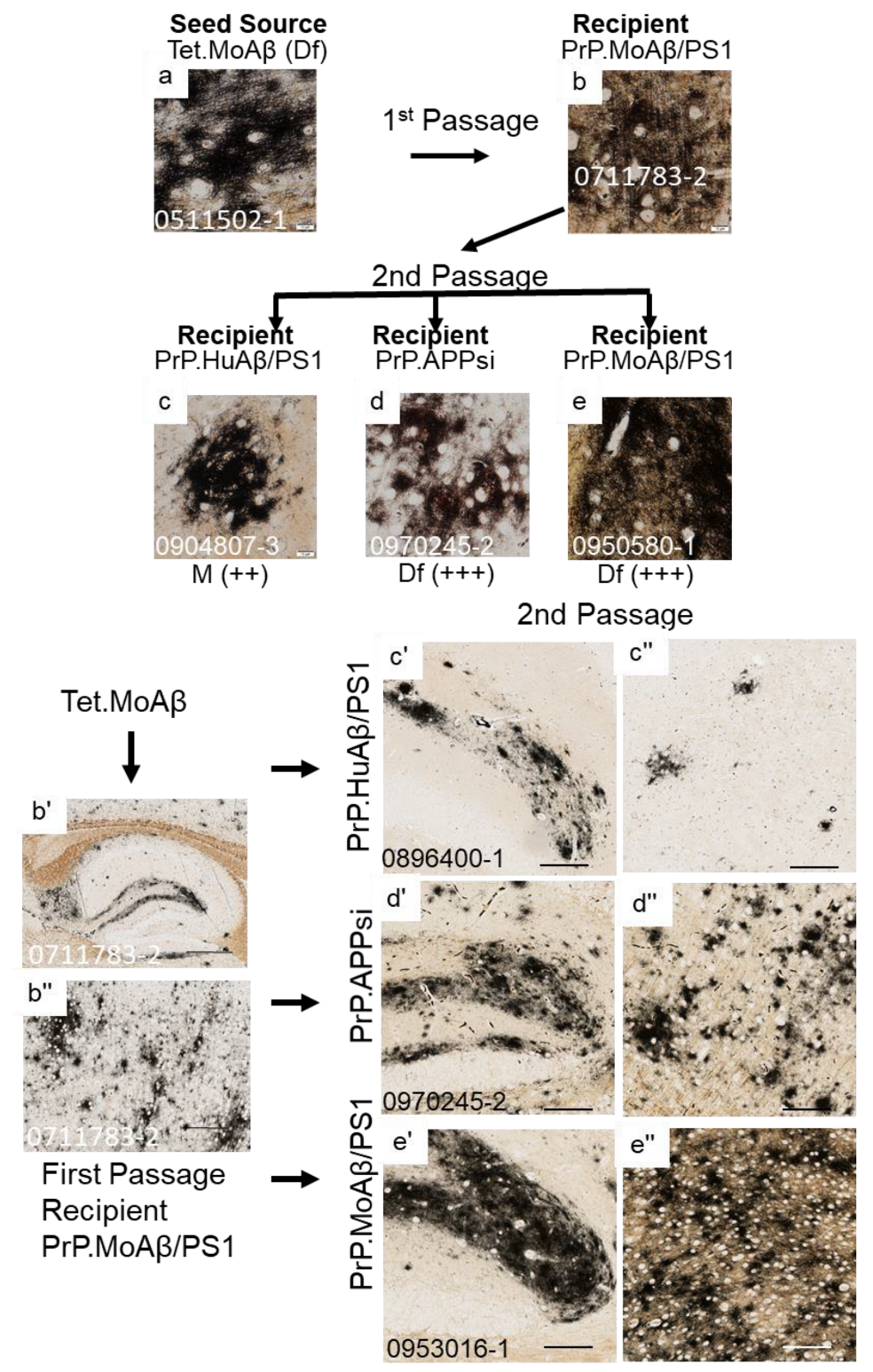
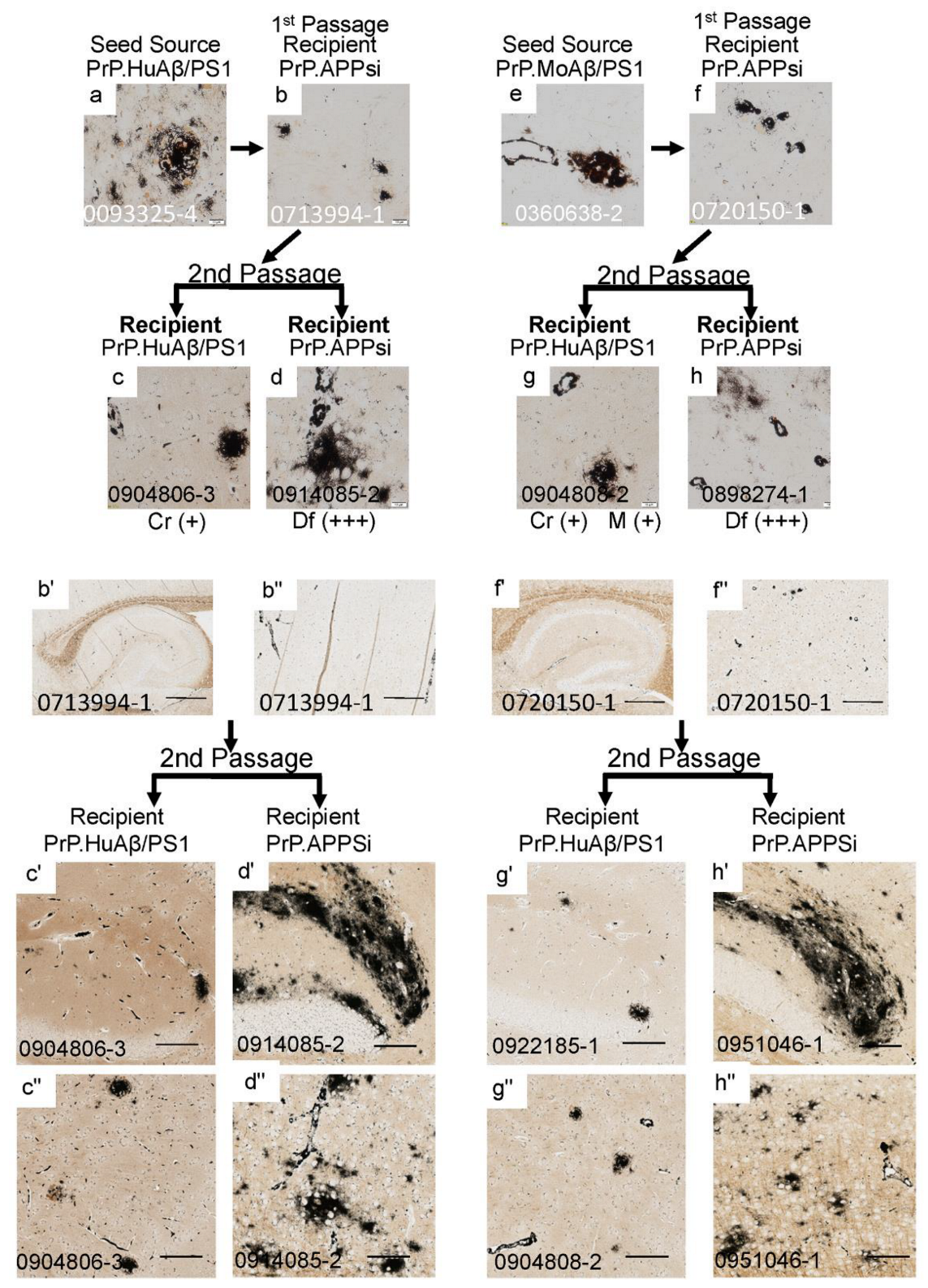
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s. 2021 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Braak, H.; Markesbery, W.R. Neuropathology and cognitive impairment in Alzheimer disease: A complex but coherent relationship. J. Neuropathol. Exp. Neurol. 2009, 68, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Huynh, T.-P.V.; Davis, A.A.; Ulrich, J.D.; Holtzman, D.M. Apolipoprotein E and Alzheimer’s disease: The influence of apolipoprotein E on amyloid-β and other amyloidogenic proteins. J. Lipid Res. 2017, 58, 824–836. [Google Scholar] [CrossRef] [Green Version]
- DeMattos, R.B.; O’dell, M.A.; Parsadanian, M.; Taylor, J.W.; Harmony, J.A.; Bales, K.R.; Paul, S.M.; Aronow, B.J.; Holtzman, D.M. Clusterin promotes amyloid plaque formation and is critical for neuritic toxicity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2002, 99, 10843–10848. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-C.; Zhao, N.; Fu, Y.; Wang, N.; Linares, C.; Tsai, C.-W.; Bu, G. ApoE4 Accelerates Early Seeding of Amyloid Pathology. Neuron 2017, 96, 1024–1032.e3. [Google Scholar] [CrossRef] [Green Version]
- Bales, K.R.; Verina, T.; Cummins, D.J.; Du, Y.; Dodel, R.C.; Saura, J.; Fishman, C.E.; DeLong, C.A.; Piccardo, P.; Petegnief, V.; et al. Apoplipoprotein E is essential for amyloid deposition in the APPV717F transgenic mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 1999, 96, 15233–15238. [Google Scholar] [CrossRef] [Green Version]
- Vinters, H.V.; Miller, B.L.; Pardridge, W.M. Brain amyloid and Alzheimer disease. Ann. Intern. Med. 1988, 109, 41–54. [Google Scholar] [CrossRef]
- Thal, D.R.; Walter, J.; Saido, T.C.; Fandrich, M. Neuropathology and biochemistry of Aβ and its aggregates in Alzheimer’s disease. Acta Neuropathol. 2015, 129, 167–182. [Google Scholar] [CrossRef]
- Dickson, T.C.; Vickers, J.C. The morphological phenotype of β-amyloid plaques and associated neuritic changes in Alzheimer’s disease. Neuroscience 2001, 105, 99–107. [Google Scholar] [CrossRef]
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s Dement. 2012, 8, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol. 2012, 123, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knopman, D.S.; Parisi, J.E.; Salviati, A.; Floriach-Robert, M.; Boeve, B.F.; Ivnik, R.J.; Smith, G.E.; Dickson, D.W.; Johnson, K.A.; Petersen, L.E.; et al. Neuropathology of cognitively normal elderly. J. Neuropathol. Exp. Neurol. 2003, 62, 1087–1095. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Fromholt, S.E.; Chakrabarty, P.; Zhu, F.; Liu, X.; Pace, M.C.; Koh, J.; Golde, T.E.; Levites, Y.; Lewis, J.; et al. Diversity in Aβ deposit morphology and secondary proteome insolubility across models of Alzheimer-type amyloidosis. Acta Neuropathol. Commun. 2020, 8, 43. [Google Scholar] [CrossRef]
- Yang, Y.; Arseni, D.; Zhang, W.; Huang, M.; Lövestam, S.; Schweighauser, M.; Kotecha, A.; Murzin, A.G.; Peak-Chew, S.Y.; Macdonald, J.; et al. Cryo-EM structures of amyloid-β 42 filaments from human brains. Science 2022, 375, 167–172. [Google Scholar] [CrossRef]
- Kane, M.D.; Lipinski, W.J.; Callahan, M.J.; Bian, F.; Durham, R.A.; Schwarz, R.D.; Roher, A.E.; Walker, L.C. Evidence for seeding of β-amyloid by intracerebral infusion of Alzheimer brain extracts in β-amyloid precursor protein-transgenic mice. J. Neurosci. 2000, 20, 3606–3611. [Google Scholar] [CrossRef] [Green Version]
- Walker, L.C. Prion-like mechanisms in Alzheimer disease. Handb. Clin. Neurol. 2018, 153, 303–319. [Google Scholar] [CrossRef]
- Watts, J.C.; Prusiner, S.B. β-Amyloid Prions and the Pathobiology of Alzheimer’s Disease. Cold Spring Harb. Perspect. Med. 2018, 8, a023507. [Google Scholar] [CrossRef] [Green Version]
- Lau, H.H.C.; Ingelsson, M.; Watts, J.C. The existence of Aβ strains and their potential for driving phenotypic heterogeneity in Alzheimer’s disease. Acta Neuropathol. 2021, 142, 17–39. [Google Scholar] [CrossRef]
- Ulm, B.S.; Borchelt, D.R.; Moore, B.D. Remodeling Alzheimer-amyloidosis models by seeding. Mol. Neurodegener. 2021, 16, 8. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.C.; Schelle, J.; Jucker, M. The Prion-like Properties of Amyloid-β Assemblies: Implications for Alzheimer’s Disease. Cold Spring Harb. Perspect. Med. 2016, 6, a024398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jankowsky, J.L.; Slunt, H.H.; Ratovitski, T.; Jenkins, N.A.; Copeland, N.G.; Borchelt, D.R. Co-expression of multiple transgenes in mouse CNS: A comparison of strategies. Biomol. Eng. 2001, 17, 157–165. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Fadale, D.J.; Anderson, J.; Xu, G.M.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Lee, M.K.; Younkin, L.H.; Wagner, S.L.; et al. Mutant presenilins specifically elevate the levels of the 42 residue β-amyloid peptide in vivo: Evidence for augmentation of a 42-specific γ secretase. Hum. Mol. Genet. 2004, 13, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Ran, Y.; Fromholt, S.E.; Fu, C.; Yachnis, A.T.; Golde, T.E.; Borchelt, D.R. Murine Aβ over-production produces diffuse and compact Alzheimer-type amyloid deposits. Acta Neuropathol. Commun. 2015, 3, 72. [Google Scholar] [CrossRef] [Green Version]
- Moore, B.D.; Levites, Y.; Xu, G.; Hampton, H.; Adamo, M.F.; Croft, C.L.; Futch, H.S.; Moran, C.; Fromholt, S.; Janus, C.; et al. Soluble brain homogenates from diverse human and mouse sources preferentially seed diffuse Aβ plaque pathology when injected into newborn mouse hosts. Free Neuropathol. 2022, 3, 9. [Google Scholar] [CrossRef]
- McGowan, E.; Pickford, F.; Kim, J.; Onstead, L.; Eriksen, J.; Yu, C.; Skipper, L.; Murphy, M.P.; Beard, J.; Das, P.; et al. Aβ42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron 2005, 47, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, G.; Adams, M.E.; Jaunmuktane, Z.; Alistair Lammie, G.; Turner, B.; Wani, M.; Sawhney, I.M.S.; Houlden, H.; Mead, S.; Brandner, S.; et al. Early onset cerebral amyloid angiopathy following childhood exposure to cadaveric dura. Ann. Neurol. 2019, 85, 284–290. [Google Scholar] [CrossRef]
- Cali, I.; Cohen, M.L.; Haik, S.; Parchi, P.; Giaccone, G.; Collins, S.J.; Kofskey, D.; Wang, H.; McLean, C.A.; Brandel, J.-P.; et al. Iatrogenic Creutzfeldt-Jakob disease with Amyloid-β pathology: An international study. Acta Neuropathol. Commun. 2018, 6, 5. [Google Scholar] [CrossRef] [Green Version]
- Caroppo, P.; Marucci, G.; Maccagnano, E.; Gobbo, C.L.; Bizzozero, I.; Tiraboschi, P.; Redaelli, V.; Catania, M.; Di Fede, G.; Caputi, L.; et al. Cerebral amyloid angiopathy in a 51-year-old patient with embolization by dura mater extract and surgery for nasopharyngeal angiofibroma at age 17. Amyloid 2021, 28, 142–143. [Google Scholar] [CrossRef]
- Frontzek, K.; Lutz, M.I.; Aguzzi, A.; Kovacs, G.G.; Budka, H. Amyloid-β pathology and cerebral amyloid angiopathy are frequent in iatrogenic Creutzfeldt-Jakob disease after dural grafting. Swiss Med. Wkly. 2016, 146, w14287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamaguchi, T.; Taniguchi, Y.; Sakai, K.; Kitamoto, T.; Takao, M.; Murayama, S.; Iwasaki, Y.; Yoshida, M.; Shimizu, H.; Kakita, A.; et al. Significant association of cadaveric dura mater grafting with subpial Aβ deposition and meningeal amyloid angiopathy. Acta Neuropathol. 2016, 132, 313–315. [Google Scholar] [CrossRef] [PubMed]
- Hervé, D.; Porché, M.; Cabrejo, L.; Guidoux, C.; Tournier-Lasserve, E.; Nicolas, G.; Adle-Biassette, H.; Plu, I.; Chabriat, H.; Duyckaerts, C. Fatal Aβ cerebral amyloid angiopathy 4 decades after a dural graft at the age of 2 years. Acta Neuropathol. 2018, 135, 801–803. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, Y.; Imamura, K.; Iwai, K.; Kobayashi, Y.; Akagi, A.; Mimuro, M.; Miyahara, H.; Kitamoto, T.; Yoshida, M. Autopsied case of non-plaque-type dura mater graft-associated Creutzfeldt-Jakob disease presenting with extensive amyloid-β deposition. Neuropathology 2018, 38, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Lutz, M.I.; Ricken, G.; Ströbel, T.; Höftberger, R.; Preusser, M.; Regelsberger, G.; Hönigschnabl, S.; Reiner, A.; Fischer, P.; et al. Dura mater is a potential source of Aβ seeds. Acta Neuropathol. 2016, 131, 911–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raposo, N.; Planton, M.; Siegfried, A.; Calviere, L.; Payoux, P.; Albucher, J.-F.; Viguier, A.; Delisle, M.-B.; Uro-Coste, E.; Chollet, F.; et al. Amyloid-β transmission through cardiac surgery using cadaveric dura mater patch. J. Neurol. Neurosurg. Psychiatry 2020, 91, 440–441. [Google Scholar] [CrossRef]
- Jaunmuktane, Z.; Banerjee, G.; Paine, S.; Parry-Jones, A.; Rudge, P.; Grieve, J.; Toma, A.K.; Farmer, S.F.; Mead, S.; Houlden, H.; et al. Alzheimer’s disease neuropathological change three decades after iatrogenic amyloid-β transmission. Acta Neuropathol. 2021, 142, 211–215. [Google Scholar] [CrossRef]
- Duyckaerts, C.; Sazdovitch, V.; Ando, K.; Seilhean, D.; Privat, N.; Yilmaz, Z.; Peckeu, L.; Amar, E.; Comoy, E.; Maceski, A.; et al. Neuropathology of iatrogenic Creutzfeldt-Jakob disease and immunoassay of French cadaver-sourced growth hormone batches suggest possible transmission of tauopathy and long incubation periods for the transmission of Abeta pathology. Acta Neuropathol. 2018, 135, 201–212. [Google Scholar] [CrossRef]
- Jaunmuktane, Z.; Mead, S.; Ellis, M.; Wadsworth, J.D.F.; Nicoll, A.J.; Kenny, J.; Launchbury, F.; Linehan, J.; Richard-Loendt, A.; Walker, A.S.; et al. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature 2015, 525, 247–250. [Google Scholar] [CrossRef]
- Ritchie, D.L.; Adlard, P.; Peden, A.H.; Lowrie, S.; Le Grice, M.; Burns, K.; Jackson, R.J.; Yull, H.; Keogh, M.J.; Wei, W.; et al. Amyloid-β accumulation in the CNS in human growth hormone recipients in the UK. Acta Neuropathol. 2017, 134, 221–240. [Google Scholar] [CrossRef] [Green Version]
- Lauwers, E.; Lalli, G.; Brandner, S.; Collinge, J.; Compernolle, V.; Duyckaerts, C.; Edgren, G.; Haïk, S.; Hardy, J.; Helmy, A.; et al. Potential human transmission of amyloid β pathology: Surveillance and risks. Lancet Neurol. 2020, 19, 872–878. [Google Scholar] [CrossRef]
- Lee, J.-H.; Yang, D.-S.; Goulbourne, C.N.; Im, E.; Stavrides, P.; Pensalfini, A.; Chan, H.; Bouchet-Marquis, C.; Bleiwas, C.; Berg, M.J.; et al. Faulty autolysosome acidification in Alzheimer’s disease mouse models induces autophagic build-up of Aβ in neurons, yielding senile plaques. Nat. Neurosci. 2022, 25, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Takami, M.; Nagashima, Y.; Sano, Y.; Ishihara, S.; Morishima-Kawashima, M.; Funamoto, S.; Ihara, Y. γ-Secretase: Successive tripeptide and tetrapeptide release from the transmembrane domain of β-carboxyl terminal fragment. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 13042–13052. [Google Scholar] [CrossRef] [PubMed]
- Arber, C.; Toombs, J.; Lovejoy, C.; Ryan, N.S.; Paterson, R.W.; Willumsen, N.; Gkanatsiou, E.; Portelius, E.; Blennow, K.; Heslegrave, A.; et al. Familial Alzheimer’s disease patient-derived neurons reveal distinct mutation-specific effects on amyloid beta. Mol. Psychiatry 2020, 25, 2919–2931. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Designation | Aβ Species | Transgenes | Onset Aβ Deposition (mo) | Neuropath Features of Aged Animals | Role in Study | Reference |
|---|---|---|---|---|---|---|
| PrP.HuAβ/PS1 | Human | APPswe, PS1dE9 | ~6 | Cored > > diffuse, Thio+ with CAA | Host/ Donor | [23,24] |
| PrP.MoAβ/PS1 | Mouse | APPswe, PS1dE9 | ~14 | Cored, Thio-S+ with CAA | Host/ Donor | [25] |
| PrP.APPsi | Human | APPswe/ind, GFP | ~12 | Diffuse > > Cored, Weakly Thio-S+ | Host/ Donor | [25] |
| Tet.MoAβ | Mouse | APPswe, tTA, GFP | ~13 | Diffuse, mainly Thio-S neg | Donor | [25] |
| First Passage to PrP.MoAβ/PS1(Cr) Mice Harvested at 9–12 Months of Age | |||
|---|---|---|---|
| Number of Mice Injected | Seed Source | Recipient Pathology Type at Harvest | Pathology Burden Score a |
| 5 | None | None | - |
| 3 | PrP.APPsi (Df) | Df | ++ to +++ |
| 4 | Tet.MoAβ (Df) | Df | ++ to +++ |
| 5 | PrP.HuAβ/PS1 (Cr) | Cr | + * |
| 6 | PrP.MoAβ/PS1 (Cr) | Cr | + ** |
| First Passage to PrP.HuAβ/PS1(Cr) Mice Harvested at 6 Months of Age | |||
| 3 | None | Cr | + |
| 4 | PrP.APPsi (Df) | Cr | + to ++ |
| 5 | Tet.MoAβ (Df) | M | ++ |
| 9 | PrP.HuAβ/PS1 (Cr) | Cr | + |
| 7 | PrP.MoAβ/PS1 (Cr) | Cr | + |
| Number of Mice Injected | Passage History (Host Strain) | Last Recipient Pathology Type at Harvest | Pathology Burden Score a |
|---|---|---|---|
| 6 | Tet.MoAβ to PrP.APPsi to PrPHuAβ/PS1 (Df to Df to Cr) | M | ++ |
| 3 | Tet.MoAβ to PrP.APPsi to PrP.APPsi (Df to Df to Df) | DF | +++ |
| 5 | Tet.MoAβ to PrP.APPsi to PrP.MoAβ/PS1 (Df to Df to Cr) | Df | +++ |
| 5 | Tet.MoAβ to PrP.MoAβ/PS1 to PrP.HuAβ/PS1 (Df to Cr to Cr) | M | ++ |
| 6 | Tet.MoAβ to PrP.MoAβ/PS1 to PrPAPPsi (Df to Cr to Df) | Df | +++ |
| 3 | Tet.MoAβ to PrP.MoAβ/PS1 to PrP.MoAβ/PS1 (Df to Cr to Cr) | Df | +++ |
| 6 | PrP.HuAβ/PS1 to PrP.APPsi to PrP.HuAβ/PS1 (Cr to Df to Cr) | Cr | + |
| 5 | PrP.HuAβ/PS1 to PrP.APPsi to PrP.APPsi (Cr to Df to Df) | Df | +++ |
| 6 | PrP.MoAβ/PS1 to PrP.APPsi to PrP.HuAβ/PS1 (Cr to Df to Cr) | Cr (4), M (2) | + |
| 3 | PrP.MoAβ/PS1 to PrP.APPsi to PrP.APPsi (Cr to Df to Df) | Df | +++ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, G.; Fromholt, S.; Borchelt, D.R. Modeling the Competition between Misfolded Aβ Conformers That Produce Distinct Types of Amyloid Pathology in Alzheimer’s Disease. Biomolecules 2022, 12, 886. https://doi.org/10.3390/biom12070886
Xu G, Fromholt S, Borchelt DR. Modeling the Competition between Misfolded Aβ Conformers That Produce Distinct Types of Amyloid Pathology in Alzheimer’s Disease. Biomolecules. 2022; 12(7):886. https://doi.org/10.3390/biom12070886
Chicago/Turabian StyleXu, Guilian, Susan Fromholt, and David R. Borchelt. 2022. "Modeling the Competition between Misfolded Aβ Conformers That Produce Distinct Types of Amyloid Pathology in Alzheimer’s Disease" Biomolecules 12, no. 7: 886. https://doi.org/10.3390/biom12070886
APA StyleXu, G., Fromholt, S., & Borchelt, D. R. (2022). Modeling the Competition between Misfolded Aβ Conformers That Produce Distinct Types of Amyloid Pathology in Alzheimer’s Disease. Biomolecules, 12(7), 886. https://doi.org/10.3390/biom12070886