
A Novel and Selective Dopamine Transporter Inhibitor, (S)-MK-26, Promotes Hippocampal Synaptic Plasticity and Restores Effort-Related Motivational Dysfunctions


 , , , , , , , ,
, , , , , , , ,  add
Show full author list
add
Show full author list
Abstract
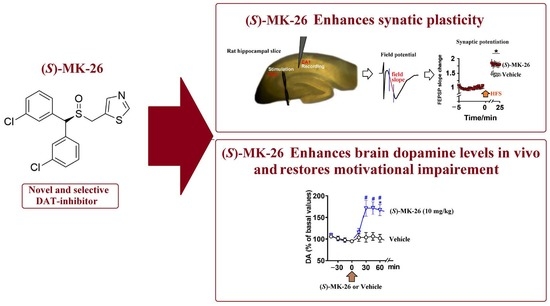
1. Introduction
2. Chemistry
3. Results
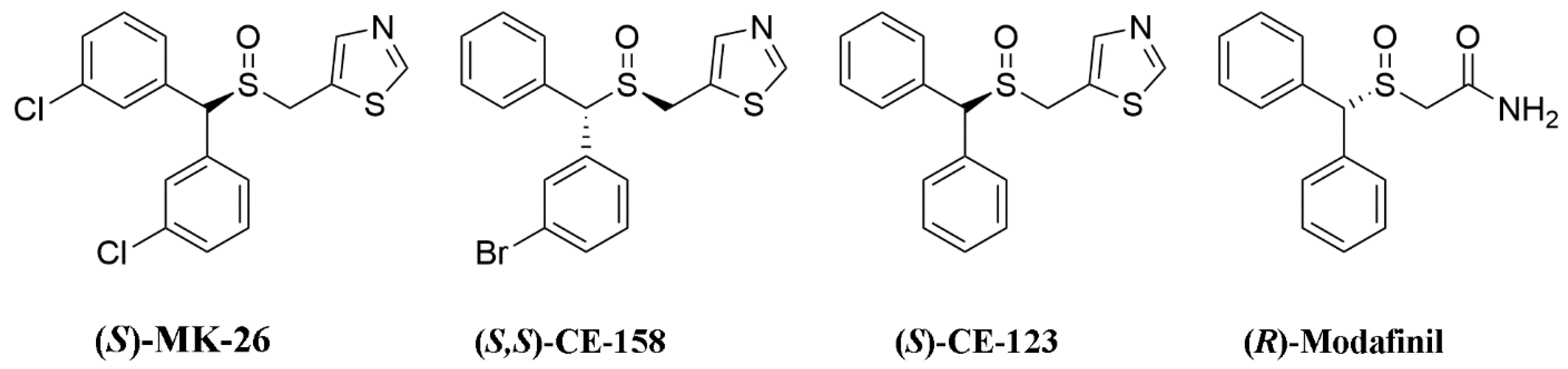
3.1. (S)-MK-26 Is a Potent and Selective DAT Inhibitor
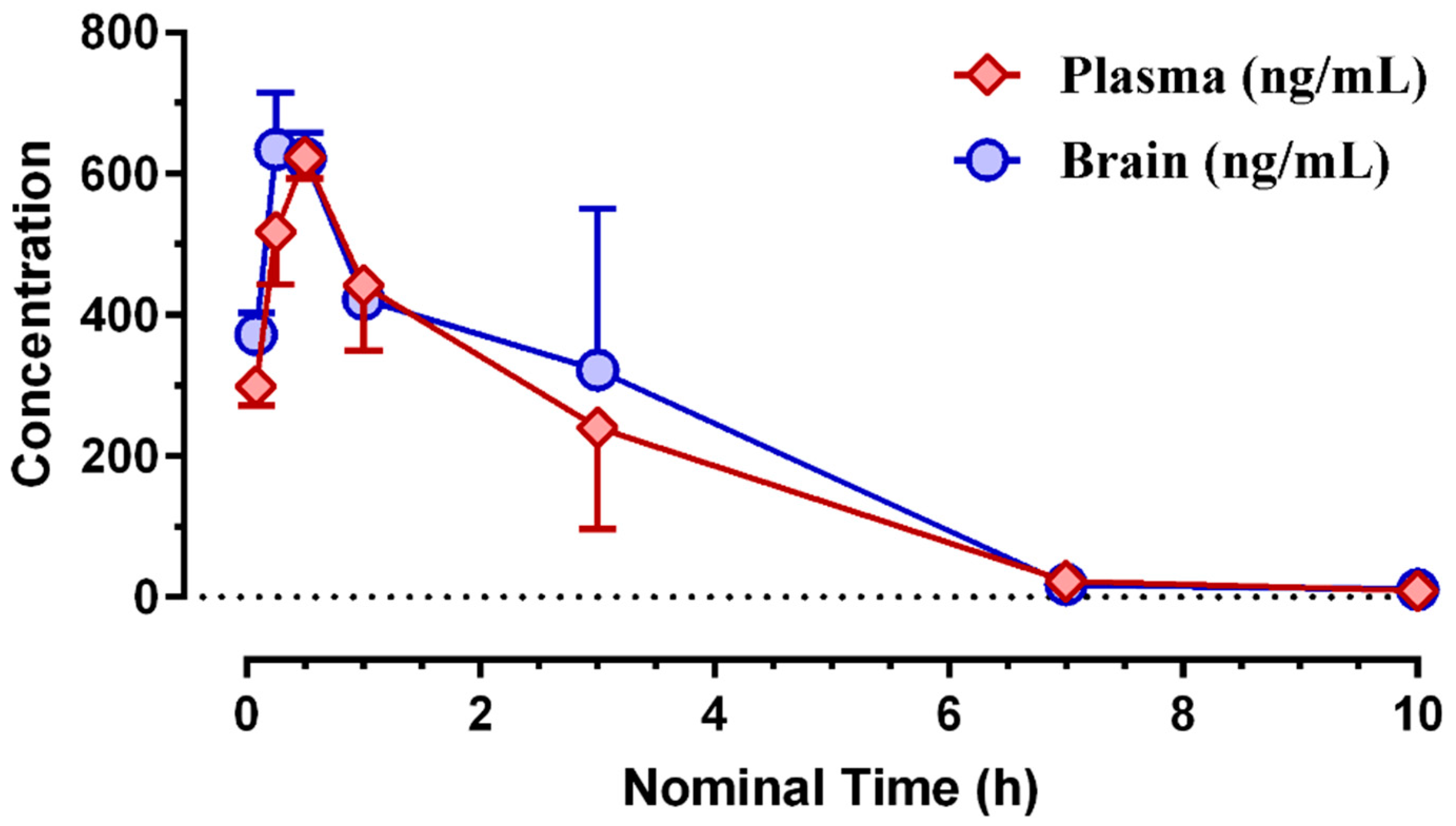
3.2. Pharmacokinetic Profiling of (S)-MK-26
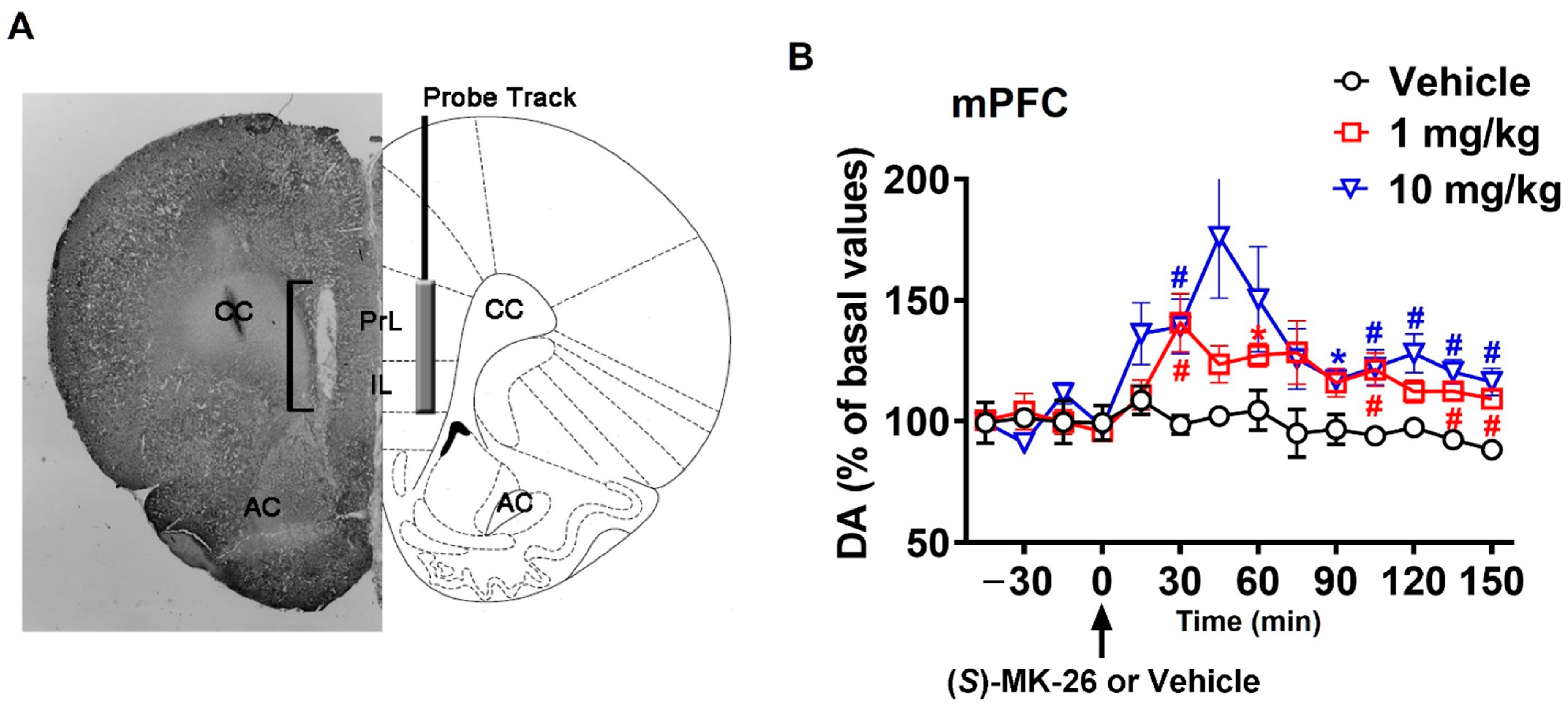
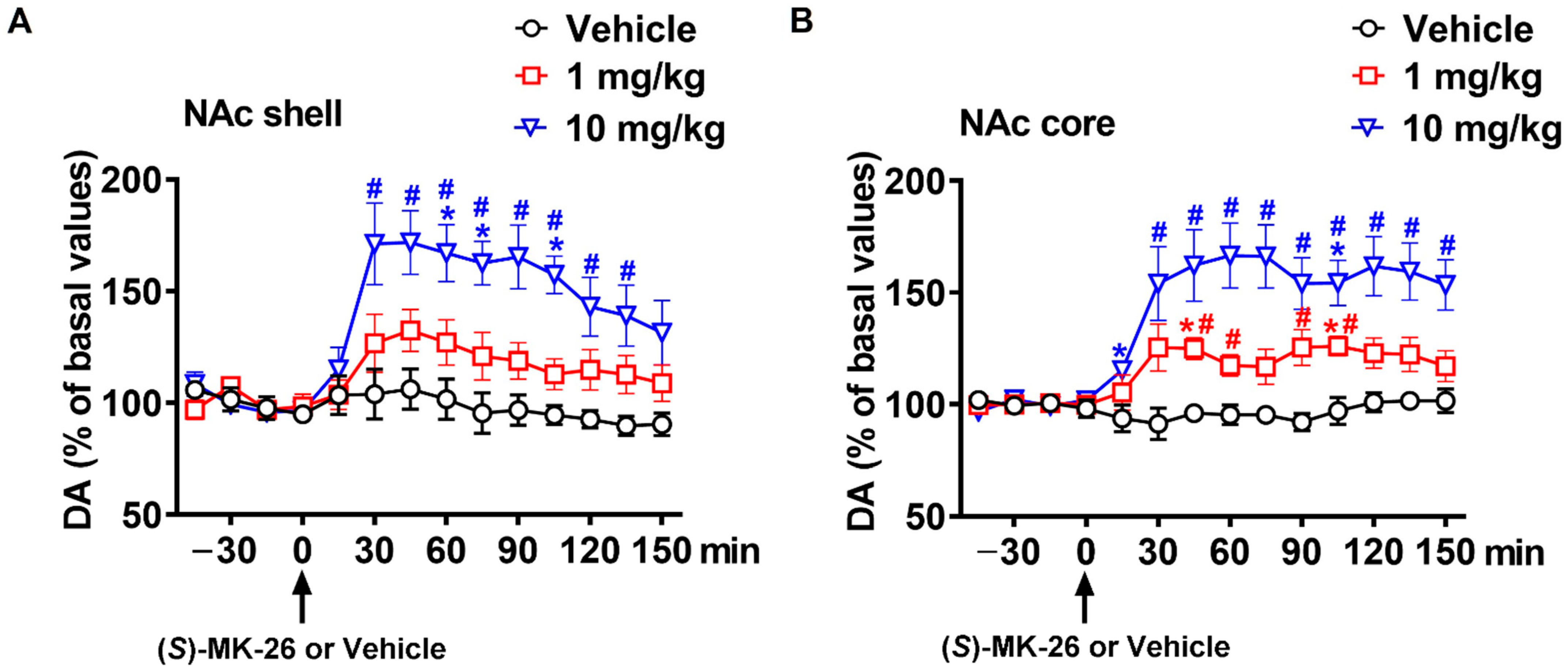
3.3. (S)-MK-26 Increases Extracellular Dopamine Levels In Vivo
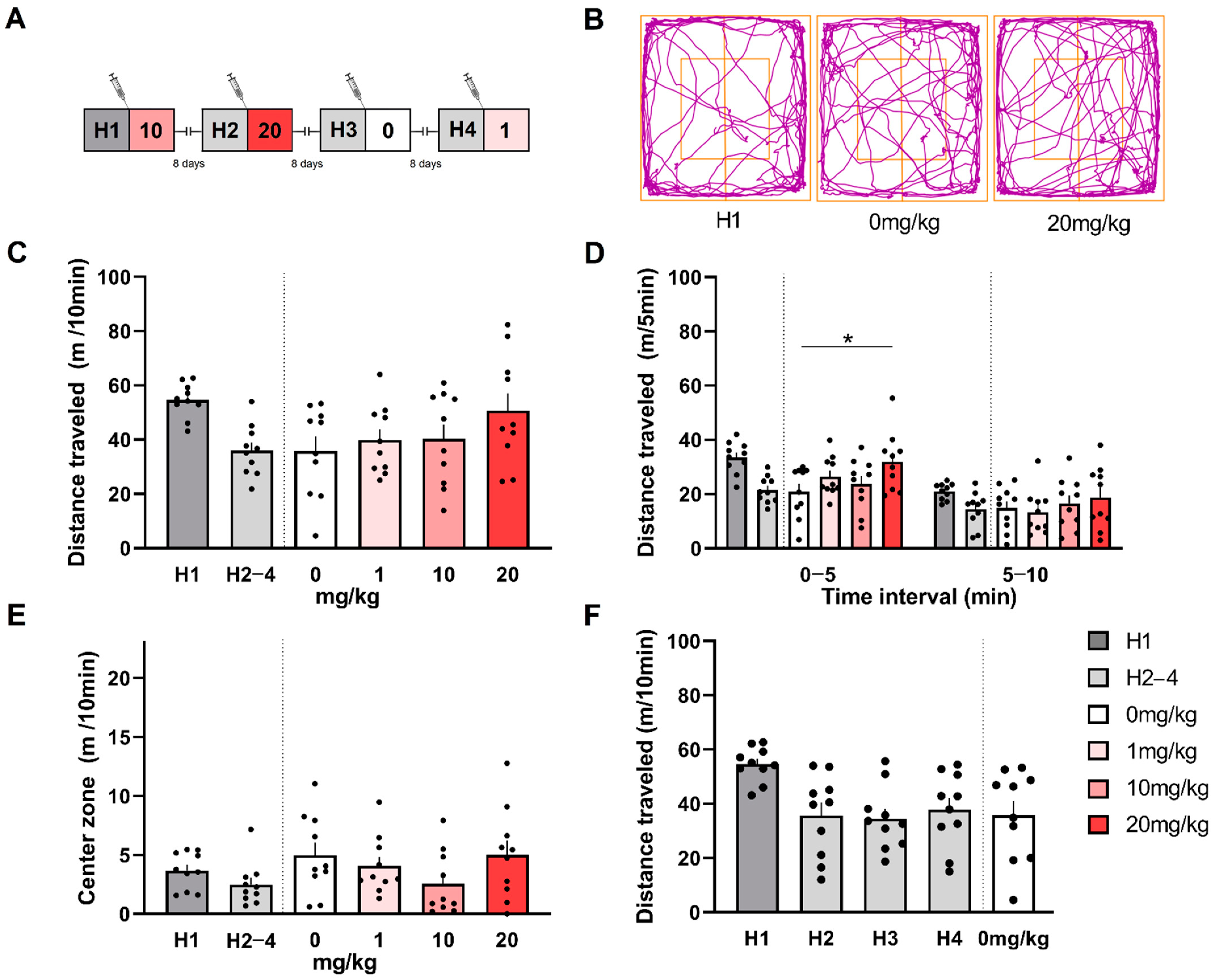
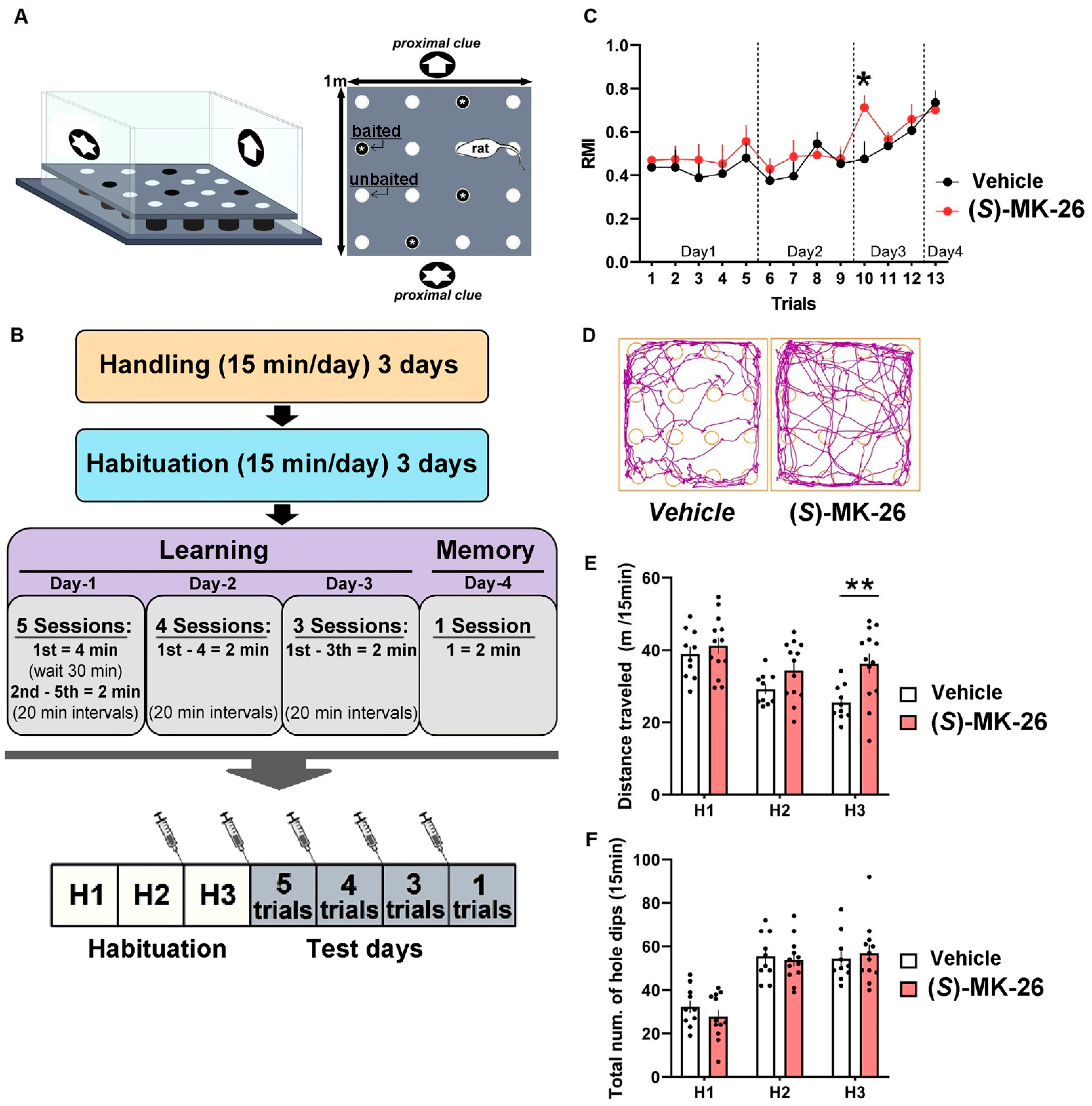
3.4. (S)-MK-26 Has a Transient Effect on Spontaneous Locomotion and Exploration
3.5. (S)-MK-26 Has Lesser Effects on Reference Memory
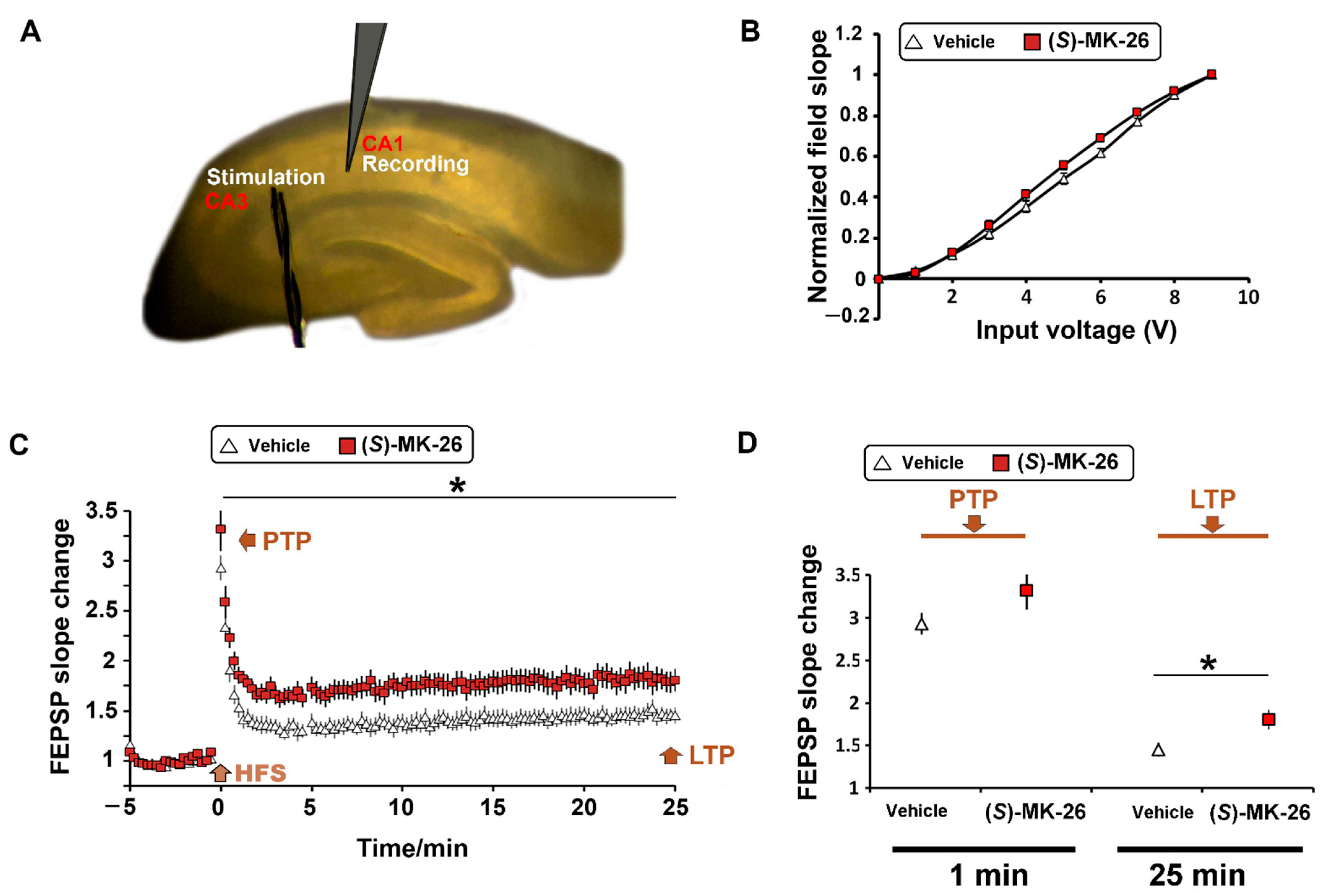
3.6. (S)-MK-26 Enhanced Synaptic Plasticity at CA3-CA1 Synapses of Aged Rats
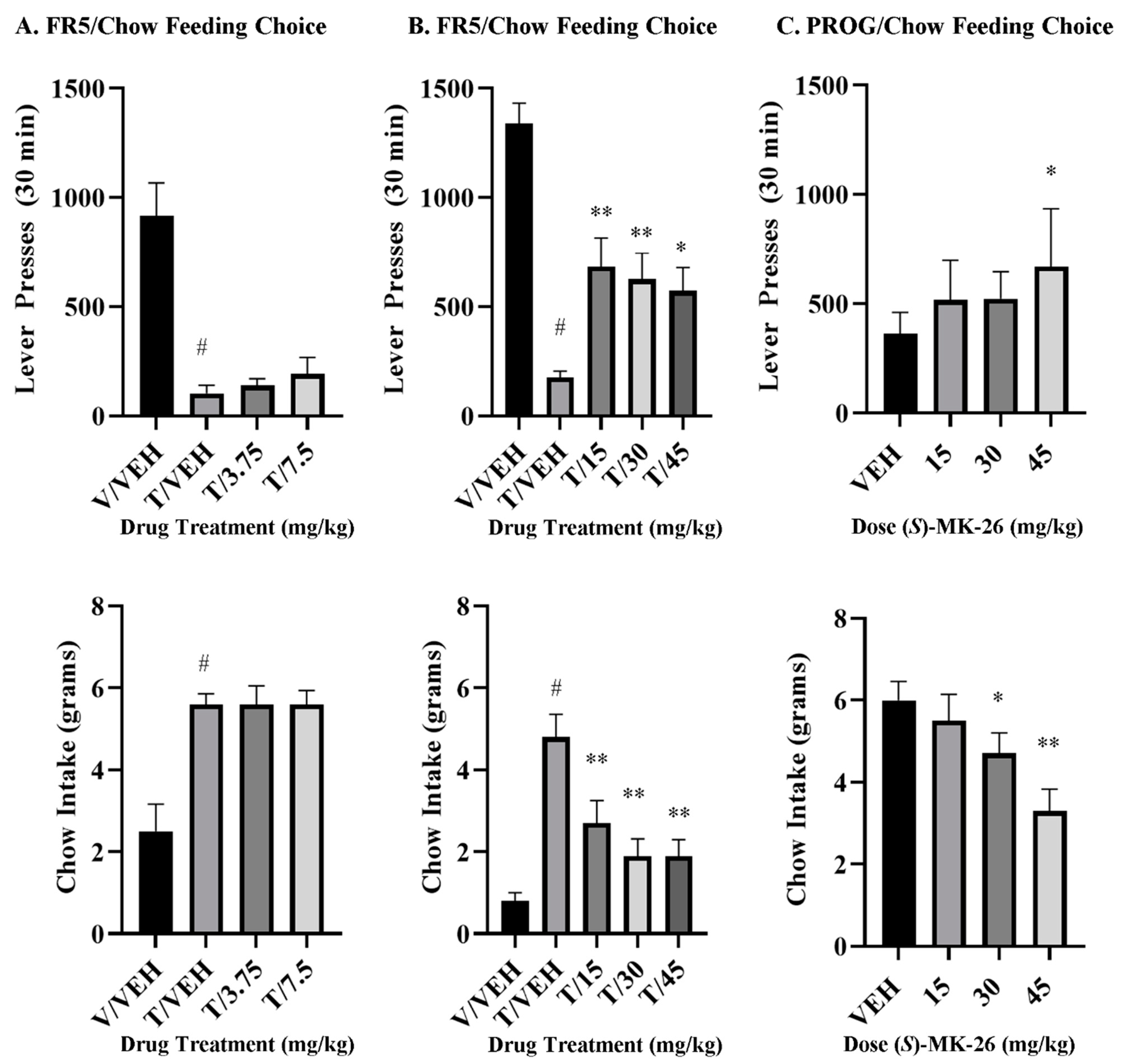
3.7. (S)-MK-26 Restores Motivational Impairments in Effort-Based Choice Tasks
4. Discussion
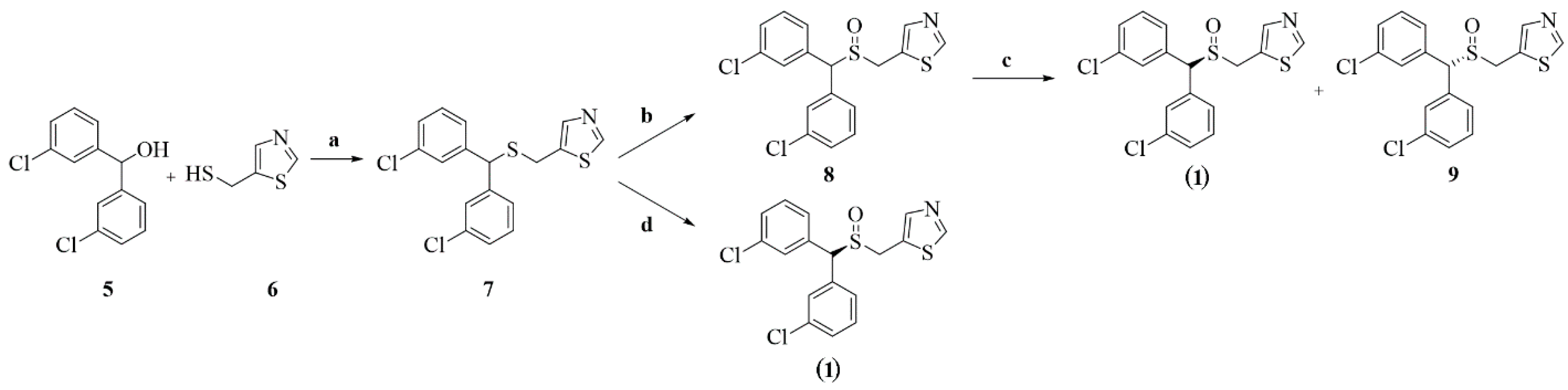
4.1. Design and Synthesis of (S)-MK-26
4.2. Relevance of DAT-Mediated Dopaminergic Regulation in the Young and Aging Brains
4.3. (S)-MK-26 Reverses TBZ-Induced Effort-Related Motivational Dysfunctions and Increases Selection of High-Effort Choices
4.4. (S)-MK-26 and Modafinil
5. Limitations
6. Materials and Methods
6.1. Synthesis
6.2. 5.-(((B(3-chlorophenyl)methyl)thio)methyl)thiazole (7)
6.3. 5.-(((B(3-chlorophenyl)methyl)sulphinyl)methyl)thiazole (8)
6.4. (S)-5-(((B(3-chlorophenyl)methyl)sulphinyl)methyl)thiazole (S)-MK-26 and (R)-5-(((bis(3-chlorophenyl)methyl)sulphinyl)methyl)thiazole (9)
6.5. Determination of Absolute Configuration
6.6. Uptake Inhibition Assays
6.7. hDAT, hNET and hSERT Binding Assays
6.8. Plasma and Brain Compound Levels and Pharmacokinetic Parameters
6.9. In Vivo Microdialysis
6.10. Hippocampal Slice Preparation and Electrophysiology
6.11. Open Field
6.12. Hole-Board Test
6.13. Effort-Based Choice Behavior Procedures
6.14. Statistical Analysis
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DA | Dopamine |
| DAT | Dopamine Transporter |
| hDAT | human dopamine Transporter |
| hNET | human norepinephrine transporter |
| hSERT | human serotonin transporter |
| DIPEA | potassium tert-butoxide |
| BF3·Et2O | boron trifluoride diethyl etherate |
| [3H] MPP+ | methyl-4-phenylpyridinium |
| KHB | Krebs–Henseleit Buffer |
| PDL | Poly-D-Lysine |
| CDCl3 | deuterated chloroform |
| DMSO-d6 | deuterated dimethyl sulfoxide |
| i.p. | intraperitoneal |
| LTP | long-term potentiation |
| PTP | post-tetanic potentiation |
| I/O | input/output |
References
- Salamone, J.D.; Pardo, M.; Yohn, S.E.; Lopez-Cruz, L.; SanMiguel, N.; Correa, M. Mesolimbic Dopamine and the Regulation of Motivated Behavior. Curr. Top. Behav. Neurosci. 2016, 27, 231–257. [Google Scholar] [CrossRef] [PubMed]
- Wise, R.A. Roles for nigrostriatal—Not just mesocorticolimbic—Dopamine in reward and addiction. Trends Neurosci. 2009, 32, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Duan, T.T.; Tan, J.W.; Yuan, Q.; Cao, J.; Zhou, Q.X.; Xu, L. Acute ketamine induces hippocampal synaptic depression and spatial memory impairment through dopamine D1/D5 receptors. Psychopharmacology 2013, 228, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Navakkode, S.; Sajikumar, S.; Korte, M.; Soong, T.W. Dopamine induces LTP differentially in apical and basal dendrites through BDNF and voltage-dependent calcium channels. Learn. Mem. 2012, 19, 294–299. [Google Scholar] [CrossRef]
- Popolo, M.; McCarthy, D.M.; Bhide, P.G. Influence of dopamine on precursor cell proliferation and differentiation in the embryonic mouse telencephalon. Dev. Neurosci. 2004, 26, 229–244. [Google Scholar] [CrossRef]
- Hoops, D.; Flores, C. Making Dopamine Connections in Adolescence. Trends Neurosci. 2017, 40, 709–719. [Google Scholar] [CrossRef]
- Missale, C.; Nash, S.R.; Robinson, S.W.; Jaber, M.; Caron, M.G. Dopamine receptors: From structure to function. Physiol. Rev. 1998, 78, 189–225. [Google Scholar] [CrossRef]
- Sala, A.; Caminiti, S.P.; Presotto, L.; Pilotto, A.; Liguori, C.; Chiaravalloti, A.; Garibotto, V.; Frisoni, G.B.; D’Amelio, M.; Paghera, B.; et al. In vivo human molecular neuroimaging of dopaminergic vulnerability along the Alzheimer’s disease phases. Alzheimer’s Res. Ther. 2021, 13, 187. [Google Scholar] [CrossRef]
- Gibb, W.R.; Mountjoy, C.Q.; Mann, D.M.; Lees, A.J. The substantia nigra and ventral tegmental area in Alzheimer’s disease and Down’s syndrome. J. Neurol. Neurosurg. Psychiatry 1989, 52, 193–200. [Google Scholar] [CrossRef]
- Joyce, J.N.; Smutzer, G.; Whitty, C.J.; Myers, A.; Bannon, M.J. Differential modification of dopamine transporter and tyrosine hydroxylase mRNAs in midbrain of subjects with Parkinson’s, Alzheimer’s with parkinsonism, and Alzheimer’s disease. Mov. Disord. 1997, 12, 885–897. [Google Scholar] [CrossRef]
- Rinne, J.O.; Sako, E.; Paljarvi, L.; Molsa, P.K.; Rinne, U.K. Brain dopamine D-1 receptors in senile dementia. J. Neurol. Sci. 1986, 73, 219–230. [Google Scholar] [CrossRef]
- Storga, D.; Vrecko, K.; Birkmayer, J.G.; Reibnegger, G. Monoaminergic neurotransmitters, their precursors and metabolites in brains of Alzheimer patients. Neurosci. Lett. 1996, 203, 29–32. [Google Scholar] [CrossRef]
- Glaser, T.; Andrejew, R.; Oliveira-Giacomelli, A.; Ribeiro, D.E.; Bonfim Marques, L.; Ye, Q.; Ren, W.J.; Semyanov, A.; Illes, P.; Tang, Y.; et al. Purinergic Receptors in Basal Ganglia Diseases: Shared Molecular Mechanisms between Huntington’s and Parkinson’s Disease. Neurosci. Bull. 2020, 36, 1299–1314. [Google Scholar] [CrossRef] [PubMed]
- Marquie, M.; Locascio, J.J.; Rentz, D.M.; Becker, J.A.; Hedden, T.; Johnson, K.A.; Growdon, J.H.; Gomperts, S.N. Striatal and extrastriatal dopamine transporter levels relate to cognition in Lewy body diseases: An (11)C altropane positron emission tomography study. Alzheimer’s Res. Ther. 2014, 6, 52. [Google Scholar] [CrossRef]
- Rieckmann, A.; Gomperts, S.N.; Johnson, K.A.; Growdon, J.H.; Van Dijk, K.R. Putamen-midbrain functional connectivity is related to striatal dopamine transporter availability in patients with Lewy body diseases. Neuroimage Clin. 2015, 8, 554–559. [Google Scholar] [CrossRef]
- Lees, A.J.; Hardy, J.; Revesz, T. Parkinson’s disease. Lancet 2009, 373, 2055–2066. [Google Scholar] [CrossRef]
- Jellinger, K.A. Significance of brain lesions in Parkinson disease dementia and Lewy body dementia. Front. Neurol. Neurosci. 2009, 24, 114–125. [Google Scholar] [CrossRef]
- Blagotinsek Cokan, K.; Mavri, M.; Rutland, C.S.; Glisic, S.; Sencanski, M.; Vrecl, M.; Kubale, V. Critical Impact of Different Conserved Endoplasmic Retention Motifs and Dopamine Receptor Interacting Proteins (DRIPs) on Intracellular Localization and Trafficking of the D2 Dopamine Receptor (D2-R) Isoforms. Biomolecules 2020, 10, 1355. [Google Scholar] [CrossRef]
- Dillman, A.A.; Majounie, E.; Ding, J.; Gibbs, J.R.; Hernandez, D.; Arepalli, S.; Traynor, B.J.; Singleton, A.B.; Galter, D.; Cookson, M.R. Transcriptomic profiling of the human brain reveals that altered synaptic gene expression is associated with chronological aging. Sci. Rep. 2017, 7, 16890. [Google Scholar] [CrossRef]
- Karrer, T.M.; Josef, A.K.; Mata, R.; Morris, E.D.; Samanez-Larkin, G.R. Reduced dopamine receptors and transporters but not synthesis capacity in normal aging adults: A meta-analysis. Neurobiol. Aging 2017, 57, 36–46. [Google Scholar] [CrossRef]
- Kaasinen, V.; Rinne, J.O. Functional imaging studies of dopamine system and cognition in normal aging and Parkinson’s disease. Neurosci. Biobehav. Rev. 2002, 26, 785–793. [Google Scholar] [CrossRef]
- Backman, L.; Lindenberger, U.; Li, S.C.; Nyberg, L. Linking cognitive aging to alterations in dopamine neurotransmitter functioning: Recent data and future avenues. Neurosci. Biobehav. Rev. 2010, 34, 670–677. [Google Scholar] [CrossRef]
- Gupta, H.V.; Beach, T.G.; Mehta, S.H.; Shill, H.A.; Driver-Dunckley, E.; Sabbagh, M.N.; Belden, C.M.; Liebsack, C.; Dugger, B.N.; Serrano, G.E.; et al. Clinicopathological Correlation: Dopamine and Amyloid PET Imaging with Neuropathology in Three Subjects Clinically Diagnosed with Alzheimer’s Disease or Dementia with Lewy Bodies. J. Alzheimer’s Dis. 2021, 80, 1603–1612. [Google Scholar] [CrossRef] [PubMed]
- Siderowf, A.; Pontecorvo, M.J.; Shill, H.A.; Mintun, M.A.; Arora, A.; Joshi, A.D.; Lu, M.; Adler, C.H.; Galasko, D.; Liebsack, C.; et al. PET imaging of amyloid with Florbetapir F 18 and PET imaging of dopamine degeneration with 18F-AV-133 (florbenazine) in patients with Alzheimer’s disease and Lewy body disorders. BMC Neurol. 2014, 14, 79. [Google Scholar] [CrossRef] [PubMed]
- Villemagne, V.L.; Okamura, N.; Pejoska, S.; Drago, J.; Mulligan, R.S.; Chetelat, G.; O’Keefe, G.; Jones, G.; Kung, H.F.; Pontecorvo, M.; et al. Differential diagnosis in Alzheimer’s disease and dementia with Lewy bodies via VMAT2 and amyloid imaging. Neuro-Degener. Dis. 2012, 10, 161–165. [Google Scholar] [CrossRef]
- Alexopoulos, G.S. Depression in the elderly. Lancet 2005, 365, 1961–1970. [Google Scholar] [CrossRef]
- Bonaconsa, M.; Colavito, V.; Pifferi, F.; Aujard, F.; Schenker, E.; Dix, S.; Grassi-Zucconi, G.; Bentivoglio, M.; Bertini, G. Cell clocks and neuronal networks: Neuron ticking and synchronization in aging and aging-related neurodegenerative disease. Curr. Alzheimer Res. 2013, 10, 597–608. [Google Scholar] [CrossRef]
- Paulson, H.L.; Igo, I. Genetics of dementia. Semin Neurol. 2011, 31, 449–460. [Google Scholar] [CrossRef]
- Young, B.K.; Camicioli, R.; Ganzini, L. Neuropsychiatric adverse effects of antiparkinsonian drugs. Characteristics, evaluation and treatment. Drugs Aging 1997, 10, 367–383. [Google Scholar] [CrossRef]
- Tye, K.M.; Mirzabekov, J.J.; Warden, M.R.; Ferenczi, E.A.; Tsai, H.C.; Finkelstein, J.; Kim, S.Y.; Adhikari, A.; Thompson, K.R.; Andalman, A.S.; et al. Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature 2013, 493, 537–541. [Google Scholar] [CrossRef]
- Salamone, J.D.; Yohn, S.E.; Lopez-Cruz, L.; San Miguel, N.; Correa, M. Activational and effort-related aspects of motivation: Neural mechanisms and implications for psychopathology. Brain 2016, 139, 1325–1347. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Leri, F.; Rizvi, S.J. Anhedonia as a central factor in depression: Neural mechanisms revealed from preclinical to clinical evidence. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 110, 110289. [Google Scholar] [CrossRef] [PubMed]
- Beracochea, D.; Cagnard, B.; Celerier, A.; le Merrer, J.; Peres, M.; Pierard, C. First evidence of a delay-dependent working memory-enhancing effect of modafinil in mice. Neuroreport 2001, 12, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Karabacak, Y.; Sase, S.; Aher, Y.D.; Sase, A.; Saroja, S.R.; Cicvaric, A.; Hoger, H.; Berger, M.; Bakulev, V.; Sitte, H.H.; et al. The effect of modafinil on the rat dopamine transporter and dopamine receptors D1-D3 paralleling cognitive enhancement in the radial arm maze. Front. Behav. Neurosci. 2015, 9, 215. [Google Scholar] [CrossRef]
- Mereu, M.; Bonci, A.; Newman, A.H.; Tanda, G. The neurobiology of modafinil as an enhancer of cognitive performance and a potential treatment for substance use disorders. Psychopharmacology 2013, 229, 415–434. [Google Scholar] [CrossRef]
- Depue, R.A.; Collins, P.F. Neurobiology of the structure of personality: Dopamine, facilitation of incentive motivation, and extraversion. Behav. Brain Sci. 1999, 22, 491–517; discussion 518–469. [Google Scholar] [CrossRef]
- Fernandez-Espejo, E. How does the nucleus accumbens function? Rev. Neurol. 2000, 30, 845–849. [Google Scholar]
- Goto, Y.; Grace, A.A. Dopaminergic modulation of limbic and cortical drive of nucleus accumbens in goal-directed behavior. Nat. Neurosci. 2005, 8, 805–812. [Google Scholar] [CrossRef]
- Rotolo, R.A.; Dragacevic, V.; Kalaba, P.; Urban, E.; Zehl, M.; Roller, A.; Wackerlig, J.; Langer, T.; Pistis, M.; De Luca, M.A.; et al. The Novel Atypical Dopamine Uptake Inhibitor (S)-CE-123 Partially Reverses the Effort-Related Effects of the Dopamine Depleting Agent Tetrabenazine and Increases Progressive Ratio Responding. Front. Pharmacol. 2019, 10, 682. [Google Scholar] [CrossRef]
- Rotolo, R.A.; Kalaba, P.; Dragacevic, V.; Presby, R.E.; Neri, J.; Robertson, E.; Yang, J.H.; Correa, M.; Bakulev, V.; Volkova, N.N.; et al. Behavioral and dopamine transporter binding properties of the modafinil analog (S, S)-CE-158: Reversal of the motivational effects of tetrabenazine and enhancement of progressive ratio responding. Psychopharmacology 2020, 237, 3459–3470. [Google Scholar] [CrossRef]
- Lubec, J.; Kalaba, P.; Hussein, A.M.; Feyissa, D.D.; Kotob, M.H.; Mahmmoud, R.R.; Wieder, O.; Garon, A.; Sagheddu, C.; Ilic, M.; et al. Reinstatement of synaptic plasticity in the aging brain through specific dopamine transporter inhibition. Mol. Psychiatry 2021, 26, 7076–7090. [Google Scholar] [CrossRef] [PubMed]
- Scoriels, L.; Jones, P.B.; Sahakian, B.J. Modafinil effects on cognition and emotion in schizophrenia and its neurochemical modulation in the brain. Neuropharmacology 2013, 64, 168–184. [Google Scholar] [CrossRef] [PubMed]
- Sagheddu, C.; Pintori, N.; Kalaba, P.; Dragacevic, V.; Piras, G.; Lubec, J.; Simola, N.; De Luca, M.A.; Lubec, G.; Pistis, M. Neurophysiological and Neurochemical Effects of the Putative Cognitive Enhancer (S)-CE-123 on Mesocorticolimbic Dopamine System. Biomolecules 2020, 10, 779. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ge, S.; Li, N.; Chen, L.; Zhang, S.; Wang, J.; Wu, H.; Wang, X.; Wang, X. NMDA and dopamine D1 receptors within NAc-shell regulate IEG proteins expression in reward circuit during cocaine memory reconsolidation. Neuroscience 2016, 315, 45–69. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Watson, C. Paxino’s and Watson’s The Rat Brain in Stereotaxic Coordinates, 7th ed.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 1. [Google Scholar]
- Kuc, K.A.; Gregersen, B.M.; Gannon, K.S.; Dodart, J.C. Holeboard discrimination learning in mice. Genes Brain Behav. 2006, 5, 355–363. [Google Scholar] [CrossRef]
- Post, A.M.; Wultsch, T.; Popp, S.; Painsipp, E.; Wetzstein, H.; Kittel-Schneider, S.; Sontag, T.A.; Lesch, K.P.; Reif, A. The COGITAT holeboard system as a valuable tool to assess learning, memory and activity in mice. Behav. Brain Res. 2011, 220, 152–158. [Google Scholar] [CrossRef]
- van der Staay, F.J.; van Nies, J.; Raaijmakers, W. The effects of aging in rats on working and reference memory performance in a spatial holeboard discrimination task. Behav. Neural. Biol. 1990, 53, 356–370. [Google Scholar] [CrossRef]
- Milner, B.; Klein, D. Loss of recent memory after bilateral hippocampal lesions: Memory and memories-looking back and looking forward. J. Neurol. Neurosurg. Psychiatry 2016, 87, 230. [Google Scholar] [CrossRef]
- Swant, J.; Chirwa, S.; Stanwood, G.; Khoshbouei, H. Methamphetamine reduces LTP and increases baseline synaptic transmission in the CA1 region of mouse hippocampus. PLoS ONE 2010, 5, e11382. [Google Scholar] [CrossRef]
- Huang, Y.Y.; Kandel, E.R. D1/D5 receptor agonists induce a protein synthesis-dependent late potentiation in the CA1 region of the hippocampus. Proc. Natl. Acad. Sci. USA 1995, 92, 2446–2450. [Google Scholar] [CrossRef]
- Bach, M.E.; Barad, M.; Son, H.; Zhuo, M.; Lu, Y.F.; Shih, R.; Mansuy, I.; Hawkins, R.D.; Kandel, E.R. Age-related defects in spatial memory are correlated with defects in the late phase of hippocampal long-term potentiation in vitro and are attenuated by drugs that enhance the cAMP signaling pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 5280–5285. [Google Scholar] [CrossRef] [PubMed]
- Rossato, J.I.; Bevilaqua, L.R.; Izquierdo, I.; Medina, J.H.; Cammarota, M. Dopamine controls persistence of long-term memory storage. Science 2009, 325, 1017–1020. [Google Scholar] [CrossRef]
- Cicvaric, A.; Bulat, T.; Bormann, D.; Yang, J.; Auer, B.; Milenkovic, I.; Cabatic, M.; Milicevic, R.; Monje, F.J. Sustained consumption of cocoa-based dark chocolate enhances seizure-like events in the mouse hippocampus. Food Funct. 2018, 9, 1532–1544. [Google Scholar] [CrossRef] [PubMed]
- Cicvaric, A.; Yang, J.; Bulat, T.; Zambon, A.; Dominguez-Rodriguez, M.; Kuhn, R.; Sadowicz, M.G.; Siwert, A.; Egea, J.; Pollak, D.D.; et al. Enhanced synaptic plasticity and spatial memory in female but not male FLRT2-haplodeficient mice. Sci. Rep. 2018, 8, 3703. [Google Scholar] [CrossRef] [PubMed]
- Cicvaric, A.; Yang, J.; Krieger, S.; Khan, D.; Kim, E.J.; Dominguez-Rodriguez, M.; Cabatic, M.; Molz, B.; Acevedo Aguilar, J.P.; Milicevic, R.; et al. The brain-tumor related protein podoplanin regulates synaptic plasticity and hippocampus-dependent learning and memory. Ann. Med. 2016, 1–17. [Google Scholar] [CrossRef]
- Bormann, D.; Stojanovic, T.; Cicvaric, A.; Schuld, G.J.; Cabatic, M.; Ankersmit, H.J.; Monje, F.J. miRNA-132/212 Gene-Deletion Aggravates the Effect of Oxygen-Glucose Deprivation on Synaptic Functions in the Female Mouse Hippocampus. Cells 2021, 10, 1709. [Google Scholar] [CrossRef]
- Malenka, R.C.; Nicoll, R.A. Long-term potentiation—A decade of progress? Science 1999, 285, 1870–1874. [Google Scholar] [CrossRef]
- Gruart, A.; Munoz, M.D.; Delgado-Garcia, J.M. Involvement of the CA3-CA1 synapse in the acquisition of associative learning in behaving mice. J. Neurosci. 2006, 26, 1077–1087. [Google Scholar] [CrossRef]
- Wang, C.C.; Weyrer, C.; Paturu, M.; Fioravante, D.; Regehr, W.G. Calcium-Dependent Protein Kinase C Is Not Required for Post-Tetanic Potentiation at the Hippocampal CA3 to CA1 Synapse. J. Neurosci. 2016, 36, 6393–6402. [Google Scholar] [CrossRef]
- Saroja, S.R.; Aher, Y.D.; Kalaba, P.; Aher, N.Y.; Zehl, M.; Korz, V.; Subramaniyan, S.; Miklosi, A.G.; Zanon, L.; Neuhaus, W.; et al. A novel heterocyclic compound targeting the dopamine transporter improves performance in the radial arm maze and modulates dopamine receptors D1-D3. Behav. Brain Res. 2016, 312, 127–137. [Google Scholar] [CrossRef]
- Hussein, A.M.; Aher, Y.D.; Kalaba, P.; Aher, N.Y.; Dragacevic, V.; Radoman, B.; Ilic, M.; Leban, J.; Beryozkina, T.; Ahmed, A.; et al. A novel heterocyclic compound improves working memory in the radial arm maze and modulates the dopamine receptor D1R in frontal cortex of the Sprague-Dawley rat. Behav. Brain Res. 2017, 332, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Kristofova, M.; Aher, Y.D.; Ilic, M.; Radoman, B.; Kalaba, P.; Dragacevic, V.; Aher, N.Y.; Leban, J.; Korz, V.; Zanon, L.; et al. A daily single dose of a novel modafinil analogue CE-123 improves memory acquisition and memory retrieval. Behav. Brain Res. 2018, 343, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Camats-Perna, J.; Kalaba, P.; Ebner, K.; Sartori, S.B.; Vuyyuru, H.; Aher, N.Y.; Dragacevic, V.; Singewald, N.; Engelmann, M.; Lubec, G. Differential Effects of Novel Dopamine Reuptake Inhibitors on Interference With Long-Term Social Memory in Mice. Front. Behav. Neurosci. 2019, 13, 63. [Google Scholar] [CrossRef] [PubMed]
- Kalaba, P.; Aher, N.Y.; Ilic, M.; Dragacevic, V.; Wieder, M.; Miklosi, A.G.; Zehl, M.; Wackerlig, J.; Roller, A.; Beryozkina, T.; et al. Heterocyclic Analogues of Modafinil as Novel, Atypical Dopamine Transporter Inhibitors. J. Med. Chem. 2017, 60, 9330–9348. [Google Scholar] [CrossRef] [PubMed]
- Kalaba, P.; Ilic, M.; Aher, N.Y.; Dragacevic, V.; Wieder, M.; Zehl, M.; Wackerlig, J.; Beyl, S.; Sartori, S.B.; Ebner, K.; et al. Structure-Activity Relationships of Novel Thiazole-Based Modafinil Analogues Acting at Monoamine Transporters. J. Med. Chem. 2020, 63, 391–417. [Google Scholar] [CrossRef] [PubMed]
- Iversen, L.L. Role of transmitter uptake mechanisms in synaptic neurotransmission. Br. J. Pharmacol. 1971, 41, 571–591. [Google Scholar] [CrossRef]
- Giros, B.; Caron, M.G. Molecular characterization of the dopamine transporter. Trends Pharmacol. Sci. 1993, 14, 43–49. [Google Scholar] [CrossRef]
- Giros, B.; el Mestikawy, S.; Godinot, N.; Zheng, K.; Han, H.; Yang-Feng, T.; Caron, M.G. Cloning, pharmacological characterization, and chromosome assignment of the human dopamine transporter. Mol. Pharmacol. 1992, 42, 383–390. [Google Scholar]
- Jones, S.R.; Gainetdinov, R.R.; Jaber, M.; Giros, B.; Wightman, R.M.; Caron, M.G. Profound neuronal plasticity in response to inactivation of the dopamine transporter. Proc. Natl. Acad. Sci. USA 1998, 95, 4029–4034. [Google Scholar] [CrossRef]
- Kurian, M.A.; Zhen, J.; Cheng, S.Y.; Li, Y.; Mordekar, S.R.; Jardine, P.; Morgan, N.V.; Meyer, E.; Tee, L.; Pasha, S.; et al. Homozygous loss-of-function mutations in the gene encoding the dopamine transporter are associated with infantile parkinsonism-dystonia. J. Clin. Investig. 2009, 119, 1595–1603. [Google Scholar] [CrossRef]
- Reith, M.E.A.; Kortagere, S.; Wiers, C.E.; Sun, H.; Kurian, M.A.; Galli, A.; Volkow, N.D.; Lin, Z. The dopamine transporter gene SLC6A3: Multidisease risks. Mol. Psychiatry 2022, 27, 1031–1046. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, Y.; Pektas, E.; Tokatli, A.; Haliloglu, G. Hereditary Dopamine Transporter Deficiency Syndrome: Challenges in Diagnosis and Treatment. Neuropediatrics 2017, 48, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Maier, W.; Minges, J.; Eckstein, N.; Brodski, C.; Albus, M.; Lerer, B.; Hallmayer, J.; Fimmers, R.; Ackenheil, M.; Ebstein, R.E.; et al. Genetic relationship between dopamine transporter gene and schizophrenia: Linkage and association. Schizophr. Res. 1996, 20, 175–180. [Google Scholar] [CrossRef]
- Surasi, D.S.; Peller, P.J.; Szabo, Z.; Mercier, G.; Subramaniam, R.M. Dopamine Transporter SPECT Imaging in Parkinson Disease and Dementia. PET Clin. 2013, 8, 459–467. [Google Scholar] [CrossRef]
- Sweatt, J.D. Chapter 11—Aging-Related Memory Disorders: Alzheimer’s Disease. In Mechanisms of Memory; Academic Press: San Diego, CA, USA, 2003; pp. 337–366. [Google Scholar]
- Ennis, G.E.; Hess, T.M.; Smith, B.T. The impact of age and motivation on cognitive effort: Implications for cognitive engagement in older adulthood. Psychol. Aging 2013, 28, 495–504. [Google Scholar] [CrossRef]
- Prull, M.W.; Gabrieli, J.D.E.; Bunge, S.A. Age-related changes in memory: A cognitive neuroscience perspective. In The Handbook of Aging and Cognition, 2nd ed.; Craik, F.I.M., Salthouse, T.A., Eds.; Lawrence Erlbaum Associates Publishers: Mahwah, NJ, USA, 2000; pp. 91–153. [Google Scholar]
- Wimmer, M.E.; Hernandez, P.J.; Blackwell, J.; Abel, T. Aging impairs hippocampus-dependent long-term memory for object location in mice. Neurobiol. Aging 2012, 33, 2220–2224. [Google Scholar] [CrossRef]
- Niello, M.; Gradisch, R.; Loland, C.J.; Stockner, T.; Sitte, H.H. Allosteric Modulation of Neurotransmitter Transporters as a Therapeutic Strategy. Trends Pharmacol. Sci. 2020, 41, 446–463. [Google Scholar] [CrossRef]
- Schapira, A.H.; Bezard, E.; Brotchie, J.; Calon, F.; Collingridge, G.L.; Ferger, B.; Hengerer, B.; Hirsch, E.; Jenner, P.; Le Novere, N.; et al. Novel pharmacological targets for the treatment of Parkinson’s disease. Nat. Rev. Drug Discov. 2006, 5, 845–854. [Google Scholar] [CrossRef]
- Nutt, J.G.; Woodward, W.R.; Hammerstad, J.P.; Carter, J.H.; Anderson, J.L. The “on-off” phenomenon in Parkinson’s disease. Relation to levodopa absorption and transport. N. Engl. J. Med. 1984, 310, 483–488. [Google Scholar] [CrossRef]
- Hauser, R.A.; LeWitt, P.A.; Comella, C.L. On demand therapy for Parkinson’s disease patients: Opportunities and choices. Postgrad. Med. 2021, 133, 721–727. [Google Scholar] [CrossRef]
- Jenner, P. Dopamine agonists, receptor selectivity and dyskinesia induction in Parkinson’s disease. Curr. Opin. Neurol. 2003, 16 (Suppl. 1), S3–S7. [Google Scholar] [CrossRef] [PubMed]
- Cyron, D. Mental Side Effects of Deep Brain Stimulation (DBS) for Movement Disorders: The Futility of Denial. Front. Integr. Neurosci. 2016, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Politis, M.; Wu, K.; Loane, C.; Quinn, N.P.; Brooks, D.J.; Rehncrona, S.; Bjorklund, A.; Lindvall, O.; Piccini, P. Serotonergic neurons mediate dyskinesia side effects in Parkinson’s patients with neural transplants. Sci. Transl. Med. 2010, 2, 38ra46. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, A.S.; Groiss, S.J.; Elben, S.; Sudmeyer, M.; Schnitzler, A.; Wojtecki, L. The treatment of Parkinson’s disease with deep brain stimulation: Current issues. Neural. Regen. Res. 2015, 10, 1018–1022. [Google Scholar] [CrossRef]
- Freed, C.R.; Greene, P.E.; Breeze, R.E.; Tsai, W.Y.; DuMouchel, W.; Kao, R.; Dillon, S.; Winfield, H.; Culver, S.; Trojanowski, J.Q.; et al. Transplantation of embryonic dopamine neurons for severe Parkinson’s disease. N. Engl. J. Med. 2001, 344, 710–719. [Google Scholar] [CrossRef]
- Milner, B. The medial temporal-lobe amnesic syndrome. Psychiatr. Clin. N. Am. 2005, 28, 599–611. [Google Scholar] [CrossRef]
- Lee, J.Q.; Sutherland, R.J.; McDonald, R.J. Hippocampal damage causes retrograde but not anterograde memory loss for context fear discrimination in rats. Hippocampus 2017, 27, 951–958. [Google Scholar] [CrossRef]
- Lehmann, H.; Lacanilao, S.; Sutherland, R.J. Complete or partial hippocampal damage produces equivalent retrograde amnesia for remote contextual fear memories. Eur. J. Neurosci. 2007, 25, 1278–1286. [Google Scholar] [CrossRef]
- Sutherland, R.J.; Weisend, M.P.; Mumby, D.; Astur, R.S.; Hanlon, F.M.; Koerner, A.; Thomas, M.J.; Wu, Y.; Moses, S.N.; Cole, C.; et al. Retrograde amnesia after hippocampal damage: Recent vs. remote memories in two tasks. Hippocampus 2001, 11, 27–42. [Google Scholar] [CrossRef]
- Erickson, C.A.; Barnes, C.A. The neurobiology of memory changes in normal aging. Exp. Gerontol. 2003, 38, 61–69. [Google Scholar] [CrossRef]
- Levin, E.D.; Torry, D. Acute and chronic nicotine effects on working memory in aged rats. Psychopharmacology 1996, 123, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Terry, A.V., Jr.; Kutiyanawalla, A.; Pillai, A. Age-dependent alterations in nerve growth factor (NGF)-related proteins, sortilin, and learning and memory in rats. Physiol. Behav. 2011, 102, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Okubo, Y.; Olsson, H.; Ito, H.; Lofti, M.; Suhara, T.; Halldin, C.; Farde, L. PET mapping of extrastriatal D2-like dopamine receptors in the human brain using an anatomic standardization technique and [11C]FLB 457. NeuroImage 1999, 10, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Cortes, R.; Camps, M.; Gueye, B.; Probst, A.; Palacios, J.M. Dopamine receptors in human brain: Autoradiographic distribution of D1 and D2 sites in Parkinson syndrome of different etiology. Brain Res. 1989, 483, 30–38. [Google Scholar] [CrossRef]
- Calabresi, P.; Castrioto, A.; Di Filippo, M.; Picconi, B. New experimental and clinical links between the hippocampus and the dopaminergic system in Parkinson’s disease. Lancet Neurol. 2013, 12, 811–821. [Google Scholar] [CrossRef]
- Allen, T.A.; Fortin, N.J. The evolution of episodic memory. Proc. Natl. Acad. Sci. USA 2013, 110 (Suppl. 2), 10379–10386. [Google Scholar] [CrossRef]
- Clark, R.E.; Squire, L.R. Similarity in form and function of the hippocampus in rodents, monkeys, and humans. Proc. Natl. Acad. Sci. USA 2013, 110 (Suppl. 2), 10365–10370. [Google Scholar] [CrossRef]
- Dupont, S. The anatomy of episodic memory: Evolution of concepts. Morphologie 2003, 87, 5–9. [Google Scholar]
- Reiter, S.; Liaw, H.P.; Yamawaki, T.M.; Naumann, R.K.; Laurent, G. On the Value of Reptilian Brains to Map the Evolution of the Hippocampal Formation. Brain Behav. Evol. 2017, 90, 41–52. [Google Scholar] [CrossRef]
- Rosenzweig, E.S.; Barnes, C.A. Impact of aging on hippocampal function: Plasticity, network dynamics, and cognition. Prog. Neurobiol. 2003, 69, 143–179. [Google Scholar] [CrossRef]
- Sikora, E.; Bielak-Zmijewska, A.; Dudkowska, M.; Krzystyniak, A.; Mosieniak, G.; Wesierska, M.; Wlodarczyk, J. Cellular Senescence in Brain Aging. Front. Aging Neurosci. 2021, 13, 646924. [Google Scholar] [CrossRef] [PubMed]
- Penner, M.R.; Roth, T.L.; Barnes, C.A.; Sweatt, J.D. An epigenetic hypothesis of aging-related cognitive dysfunction. Front. Aging Neurosci. 2010, 2, 9. [Google Scholar] [CrossRef] [PubMed]
- Arias-Cavieres, A.; Adasme, T.; Sanchez, G.; Munoz, P.; Hidalgo, C. Aging Impairs Hippocampal- Dependent Recognition Memory and LTP and Prevents the Associated RyR Up-regulation. Front. Aging Neurosci. 2017, 9, 111. [Google Scholar] [CrossRef] [PubMed]
- Barnes, C.A. Aging and the physiology of spatial memory. Neurobiol. Aging 1988, 9, 563–568. [Google Scholar] [CrossRef]
- Swant, J.; Wagner, J.J. Dopamine transporter blockade increases LTP in the CA1 region of the rat hippocampus via activation of the D3 dopamine receptor. Learn. Mem. 2006, 13, 161–167. [Google Scholar] [CrossRef]
- Whitlock, J.R.; Heynen, A.J.; Shuler, M.G.; Bear, M.F. Learning induces long-term potentiation in the hippocampus. Science 2006, 313, 1093–1097. [Google Scholar] [CrossRef]
- Kahn, I.; Shohamy, D. Intrinsic connectivity between the hippocampus, nucleus accumbens, and ventral tegmental area in humans. Hippocampus 2013, 23, 187–192. [Google Scholar] [CrossRef]
- Lisman, J.; Grace, A.A.; Duzel, E. A neoHebbian framework for episodic memory; role of dopamine-dependent late LTP. Trends Neurosci. 2011, 34, 536–547. [Google Scholar] [CrossRef]
- Winblad, B.; Hardy, J.; Backman, L.; Nilsson, L.G. Memory function and brain biochemistry in normal aging and in senile dementia. Ann. N. Y. Acad. Sci. 1985, 444, 255–268. [Google Scholar] [CrossRef]
- Zang, X.; Cheng, Z.Y.; Sun, Y.; Hua, N.; Zhu, L.H.; He, L. The ameliorative effects and underlying mechanisms of dopamine D1-like receptor agonist SKF38393 on Abeta1-42-induced cognitive impairment. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 81, 250–261. [Google Scholar] [CrossRef]
- Adcock, R.A.; Thangavel, A.; Whitfield-Gabrieli, S.; Knutson, B.; Gabrieli, J.D. Reward-motivated learning: Mesolimbic activation precedes memory formation. Neuron 2006, 50, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Shohamy, D.; Adcock, R.A. Dopamine and adaptive memory. Trends Cogn. Sci. 2010, 14, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Shohamy, D.; Wagner, A.D. Integrating memories in the human brain: Hippocampal-midbrain encoding of overlapping events. Neuron 2008, 60, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Floresco, S.B.; Todd, C.L.; Grace, A.A. Glutamatergic afferents from the hippocampus to the nucleus accumbens regulate activity of ventral tegmental area dopamine neurons. J. Neurosci. 2001, 21, 4915–4922. [Google Scholar] [CrossRef] [PubMed]
- Luo, A.H.; Tahsili-Fahadan, P.; Wise, R.A.; Lupica, C.R.; Aston-Jones, G. Linking context with reward: A functional circuit from hippocampal CA3 to ventral tegmental area. Science 2011, 333, 353–357. [Google Scholar] [CrossRef]
- Valenti, O.; Lodge, D.J.; Grace, A.A. Aversive stimuli alter ventral tegmental area dopamine neuron activity via a common action in the ventral hippocampus. J. Neurosci. 2011, 31, 4280–4289. [Google Scholar] [CrossRef]
- Gasbarri, A.; Packard, M.G.; Campana, E.; Pacitti, C. Anterograde and retrograde tracing of projections from the ventral tegmental area to the hippocampal formation in the rat. Brain Res. Bull. 1994, 33, 445–452. [Google Scholar] [CrossRef]
- Samson, Y.; Wu, J.J.; Friedman, A.H.; Davis, J.N. Catecholaminergic innervation of the hippocampus in the cynomolgus monkey. J. Comp. Neurol. 1990, 298, 250–263. [Google Scholar] [CrossRef]
- Loy, R.; Koziell, D.A.; Lindsey, J.D.; Moore, R.Y. Noradrenergic innervation of the adult rat hippocampal formation. J. Comp. Neurol. 1980, 189, 699–710. [Google Scholar] [CrossRef]
- Edelmann, E.; Lessmann, V. Dopaminergic innervation and modulation of hippocampal networks. Cell Tissue Res. 2018, 373, 711–727. [Google Scholar] [CrossRef]
- Brouwer, N.; Van Dijken, H.; Ruiters, M.H.; Van Willigen, J.D.; Ter Horst, G.J. Localization of dopamine D2 receptor mRNA with non-radioactive in situ hybridization histochemistry. Neurosci. Lett. 1992, 142, 223–227. [Google Scholar] [CrossRef]
- Gasbarri, A.; Sulli, A.; Packard, M.G. The dopaminergic mesencephalic projections to the hippocampal formation in the rat. Prog. Neuropsychopharmacol. Biol. Psychiatry 1997, 21, 1–22. [Google Scholar] [CrossRef]
- Legault, M.; Rompre, P.P.; Wise, R.A. Chemical stimulation of the ventral hippocampus elevates nucleus accumbens dopamine by activating dopaminergic neurons of the ventral tegmental area. J. Neurosci. 2000, 20, 1635–1642. [Google Scholar] [CrossRef] [PubMed]
- Scatton, B.; Simon, H.; Le Moal, M.; Bischoff, S. Origin of dopaminergic innervation of the rat hippocampal formation. Neurosci. Lett. 1980, 18, 125–131. [Google Scholar] [CrossRef]
- Frey, U.; Matthies, H.; Reymann, K.G.; Matthies, H. The effect of dopaminergic D1 receptor blockade during tetanization on the expression of long-term potentiation in the rat CA1 region in vitro. Neurosci. Lett. 1991, 129, 111–114. [Google Scholar] [CrossRef]
- Frey, U.; Schroeder, H.; Matthies, H. Dopaminergic antagonists prevent long-term maintenance of posttetanic LTP in the CA1 region of rat hippocampal slices. Brain Res. 1990, 522, 69–75. [Google Scholar] [CrossRef]
- Otmakhova, N.A.; Lisman, J.E. D1/D5 dopamine receptor activation increases the magnitude of early long-term potentiation at CA1 hippocampal synapses. J. Neurosci. 1996, 16, 7478–7486. [Google Scholar] [CrossRef]
- Swanson-Park, J.L.; Coussens, C.M.; Mason-Parker, S.E.; Raymond, C.R.; Hargreaves, E.L.; Dragunow, M.; Cohen, A.S.; Abraham, W.C. A double dissociation within the hippocampus of dopamine D1/D5 receptor and beta-adrenergic receptor contributions to the persistence of long-term potentiation. Neuroscience 1999, 92, 485–497. [Google Scholar] [CrossRef]
- Legault, M.; Wise, R.A. Novelty-evoked elevations of nucleus accumbens dopamine: Dependence on impulse flow from the ventral subiculum and glutamatergic neurotransmission in the ventral tegmental area. Eur. J. Neurosci. 2001, 13, 819–828. [Google Scholar] [CrossRef]
- Lisman, J.E.; Grace, A.A. The hippocampal-VTA loop: Controlling the entry of information into long-term memory. Neuron 2005, 46, 703–713. [Google Scholar] [CrossRef]
- Mennicken, F.; Savasta, M.; Peretti-Renucci, R.; Feuerstein, C. Autoradiographic localization of dopamine uptake sites in the rat brain with 3H-GBR 12935. J. Neural. Transm. Gen. Sect. 1992, 87, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Przedborski, S. Parkinson’s disease: Animal models and dopaminergic cell vulnerability. Front. Neuroanat. 2014, 8, 155. [Google Scholar] [CrossRef]
- Nunes, E.J.; Randall, P.A.; Hart, E.E.; Freeland, C.; Yohn, S.E.; Baqi, Y.; Muller, C.E.; Lopez-Cruz, L.; Correa, M.; Salamone, J.D. Effort-related motivational effects of the VMAT-2 inhibitor tetrabenazine: Implications for animal models of the motivational symptoms of depression. J. Neurosci. 2013, 33, 19120–19130. [Google Scholar] [CrossRef] [PubMed]
- Randall, P.A.; Lee, C.A.; Nunes, E.J.; Yohn, S.E.; Nowak, V.; Khan, B.; Shah, P.; Pandit, S.; Vemuri, V.K.; Makriyannis, A.; et al. The VMAT-2 inhibitor tetrabenazine affects effort-related decision making in a progressive ratio/chow feeding choice task: Reversal with antidepressant drugs. PLoS ONE 2014, 9, e99320. [Google Scholar] [CrossRef] [PubMed]
- Randall, P.A.; Lee, C.A.; Podurgiel, S.J.; Hart, E.; Yohn, S.E.; Jones, M.; Rowland, M.; Lopez-Cruz, L.; Correa, M.; Salamone, J.D. Bupropion increases selection of high effort activity in rats tested on a progressive ratio/chow feeding choice procedure: Implications for treatment of effort-related motivational symptoms. Int. J. Neuropsychopharmacol. 2015, 18, pyu017. [Google Scholar] [CrossRef]
- Ballon, J.S.; Feifel, D. A systematic review of modafinil: Potential clinical uses and mechanisms of action. J. Clin. Psychiatry 2006, 67, 554–566. [Google Scholar] [CrossRef]
- Gerrard, P.; Malcolm, R. Mechanisms of modafinil: A review of current research. Neuropsychiatr. Dis. Treat. 2007, 3, 349–364. [Google Scholar]
- Madras, B.K.; Xie, Z.; Lin, Z.; Jassen, A.; Panas, H.; Lynch, L.; Johnson, R.; Livni, E.; Spencer, T.J.; Bonab, A.A.; et al. Modafinil occupies dopamine and norepinephrine transporters in vivo and modulates the transporters and trace amine activity in vitro. J. Pharmacol. Exp. Ther. 2006, 319, 561–569. [Google Scholar] [CrossRef]
- Kim, W.; Tateno, A.; Arakawa, R.; Sakayori, T.; Ikeda, Y.; Suzuki, H.; Okubo, Y. In vivo activity of modafinil on dopamine transporter measured with positron emission tomography and [(1)(8)F]FE-PE2I. Int. J. Neuropsychopharmacol. 2014, 17, 697–703. [Google Scholar] [CrossRef]
- Jasinski, D.R.; Kovacevic-Ristanovic, R. Evaluation of the abuse liability of modafinil and other drugs for excessive daytime sleepiness associated with narcolepsy. Clin. Neuropharmacol. 2000, 23, 149–156. [Google Scholar] [CrossRef]
- Myrick, H.; Malcolm, R.; Taylor, B.; LaRowe, S. Modafinil: Preclinical, clinical, and post-marketing surveillance—A review of abuse liability issues. Ann. Clin. Psychiatry 2004, 16, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Kruszewski, S.P.; Klotz, S.G. Modafinil: Mischaracterization. J. Clin. Psychiatry 2007, 68, 970–971, author reply 971–972. [Google Scholar] [CrossRef] [PubMed]
- Hersey, M.; Bacon, A.K.; Bailey, L.G.; Coggiano, M.A.; Newman, A.H.; Leggio, L.; Tanda, G. Psychostimulant Use Disorder, an Unmet Therapeutic Goal: Can Modafinil Narrow the Gap? Front. Neurosci. 2021, 15, 656475. [Google Scholar] [CrossRef]
- Anderson, A.L.; Li, S.H.; Biswas, K.; McSherry, F.; Holmes, T.; Iturriaga, E.; Kahn, R.; Chiang, N.; Beresford, T.; Campbell, J.; et al. Modafinil for the treatment of methamphetamine dependence. Drug Alcohol. Depend. 2012, 120, 135–141. [Google Scholar] [CrossRef]
- Heinzerling, K.G.; Swanson, A.N.; Kim, S.; Cederblom, L.; Moe, A.; Ling, W.; Shoptaw, S. Randomized, double-blind, placebo-controlled trial of modafinil for the treatment of methamphetamine dependence. Drug Alcohol. Depend. 2010, 109, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Wang, G.J.; Telang, F.; Fowler, J.S.; Logan, J.; Childress, A.R.; Jayne, M.; Ma, Y.; Wong, C. Cocaine cues and dopamine in dorsal striatum: Mechanism of craving in cocaine addiction. J. Neurosci. 2006, 26, 6583–6588. [Google Scholar] [CrossRef]
- Keighron, J.D.; Quarterman, J.C.; Cao, J.; DeMarco, E.M.; Coggiano, M.A.; Gleaves, A.; Slack, R.D.; Zanettini, C.; Newman, A.H.; Tanda, G. Effects of (R)-Modafinil and Modafinil Analogues on Dopamine Dynamics Assessed by Voltammetry and Microdialysis in the Mouse Nucleus Accumbens Shell. ACS Chem. Neurosci. 2019, 10, 2012–2021. [Google Scholar] [CrossRef]
- Minzenberg, M.J.; Carter, C.S. Modafinil: A review of neurochemical actions and effects on cognition. Neuropsychopharmacology 2008, 33, 1477–1502. [Google Scholar] [CrossRef]
- Billiard, M.; Broughton, R. Modafinil: Its discovery, the early European and North American experience in the treatment of narcolepsy and idiopathic hypersomnia, and its subsequent use in other medical conditions. Sleep Med. 2018, 49, 69–72. [Google Scholar] [CrossRef]
- Linton, S.R.; Murphy, M.; Schroder, H.S.; Breiger, M.; Iturra-Mena, A.M.; Kangas, B.D.; Bergman, J.; Carlezon, W.A., Jr.; Risbrough, V.B.; Barnes, S.A.; et al. Effects of modafinil on electroencephalographic microstates in healthy adults. Psychopharmacology 2022. [Google Scholar] [CrossRef]
- Yepez, J.E.; Juarez, J. Modafinil acquires reinforcing effects when combined with citalopram. Pharmacol. Biochem. Behav. 2022, 217, 173407. [Google Scholar] [CrossRef] [PubMed]
- Damkier, P.; Broe, A. First-Trimester Pregnancy Exposure to Modafinil and Risk of Congenital Malformations. JAMA 2020, 323, 374–376. [Google Scholar] [CrossRef] [PubMed]
- Cesta, C.E.; Engeland, A.; Karlsson, P.; Kieler, H.; Reutfors, J.; Furu, K. Incidence of Malformations After Early Pregnancy Exposure to Modafinil in Sweden and Norway. JAMA 2020, 324, 895–897. [Google Scholar] [CrossRef] [PubMed]
- Schifano, F.; Catalani, V.; Sharif, S.; Napoletano, F.; Corkery, J.M.; Arillotta, D.; Fergus, S.; Vento, A.; Guirguis, A. Benefits and Harms of ‘Smart Drugs’ (Nootropics) in Healthy Individuals. Drugs 2022, 82, 633–647. [Google Scholar] [CrossRef]
- Miller, D.R.; Guenther, D.T.; Maurer, A.P.; Hansen, C.A.; Zalesky, A.; Khoshbouei, H. Dopamine Transporter Is a Master Regulator of Dopaminergic Neural Network Connectivity. J. Neurosci. 2021, 41, 5453–5470. [Google Scholar] [CrossRef]
- Chaudhry, S.; Bernardes, M.; Harris, P.E.; Maffei, A. Gastrointestinal dopamine as an anti-incretin and its possible role in bypass surgery as therapy for type 2 diabetes with associated obesity. Minerva Endocrinol. 2016, 41, 43–56. [Google Scholar]
- Maffei, A.; Segal, A.M.; Alvarez-Perez, J.C.; Garcia-Ocana, A.; Harris, P.E. Anti-incretin, Anti-proliferative Action of Dopamine on beta-Cells. Mol. Endocrinol. 2015, 29, 542–557. [Google Scholar] [CrossRef]
- Ustione, A.; Piston, D.W.; Harris, P.E. Minireview: Dopaminergic regulation of insulin secretion from the pancreatic islet. Mol. Endocrinol. 2013, 27, 1198–1207. [Google Scholar] [CrossRef]
- Rubi, B.; Maechler, P. Minireview: New roles for peripheral dopamine on metabolic control and tumor growth: Let’s seek the balance. Endocrinology 2010, 151, 5570–5581. [Google Scholar] [CrossRef]
- Jackson, S.J.; Andrews, N.; Ball, D.; Bellantuono, I.; Gray, J.; Hachoumi, L.; Holmes, A.; Latcham, J.; Petrie, A.; Potter, P.; et al. Does age matter? The impact of rodent age on study outcomes. Lab. Anim. 2017, 51, 160–169. [Google Scholar] [CrossRef]
- Quinn, R. Comparing rat’s to human’s age: How old is my rat in people years? Nutrition 2005, 21, 775–777. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, P. The Laboratory Rat: Relating Its Age With Human’s. Int. J. Prev. Med. 2013, 4, 624–630. [Google Scholar] [PubMed]
- Andreollo, N.A.; Santos, E.F.; Araujo, M.R.; Lopes, L.R. Rat’s age versus human’s age: What is the relationship? Arq. Bras. Cir. Dig. 2012, 25, 49–51. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Sengupta, P. Men and mice: Relating their ages. Life Sci. 2016, 152, 244–248. [Google Scholar] [CrossRef]
- Pollak, D.D.; John, J.; Bubna-Littitz, H.; Schneider, A.; Hoeger, H.; Lubec, G. Components of the protein quality control system are expressed in a strain-dependent manner in the mouse hippocampus. Neurochem. Int. 2006, 49, 500–507. [Google Scholar] [CrossRef]
- Pollak, D.D.; John, J.; Scharl, T.; Leisch, F.; Schneider, A.; Hoeger, H.; Lubec, G. Strain-dependent regulation of neurotransmission and actin-remodelling proteins in the mouse hippocampus. Genes Brain Behav. 2006, 5, 200–204. [Google Scholar] [CrossRef]
- Pollak, D.D.; John, J.; Schneider, A.; Hoeger, H.; Lubec, G. Strain-dependent expression of signaling proteins in the mouse hippocampus. Neuroscience 2006, 138, 149–158. [Google Scholar] [CrossRef]
- Pollak, D.D.; Scharl, T.; Leisch, F.; Herkner, K.; Villar, S.R.; Hoeger, H.; Lubec, G. Strain-dependent regulation of plasticity-related proteins in the mouse hippocampus. Behav. Brain Res. 2005, 165, 240–246. [Google Scholar] [CrossRef]
- Balazsfi, D.; Farkas, L.; Csikota, P.; Fodor, A.; Zsebok, S.; Haller, J.; Zelena, D. Sex-dependent role of vesicular glutamate transporter 3 in stress-regulation and related anxiety phenotype during the early postnatal period. Stress 2016, 19, 434–438. [Google Scholar] [CrossRef]
- Caruso, M.J.; Crowley, N.A.; Reiss, D.E.; Caulfield, J.I.; Luscher, B.; Cavigelli, S.A.; Kamens, H.M. Adolescent Social Stress Increases Anxiety-like Behavior and Alters Synaptic Transmission, Without Influencing Nicotine Responses, in a Sex-Dependent Manner. Neuroscience 2018, 373, 182–198. [Google Scholar] [CrossRef]
- Chmielarz, P.; Kreiner, G.; Nalepa, I. Selective ablation of glucocorticoid receptors in the noradrenergic system affects evening corticosterone levels in a sex-dependent manner. Pharmacol. Rep. 2015, 67, 1201–1203. [Google Scholar] [CrossRef] [PubMed]
- Grech, A.M.; Ratnayake, U.; Hannan, A.J.; van den Buuse, M.; Hill, R.A. Sex-Dependent Effects of Environmental Enrichment on Spatial Memory and Brain-Derived Neurotrophic Factor (BDNF) Signaling in a Developmental “Two-Hit” Mouse Model Combining BDNF Haploinsufficiency and Chronic Glucocorticoid Stimulation. Front. Behav. Neurosci. 2018, 12, 227. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.B.; Chartoff, E. Sex differences in neural mechanisms mediating reward and addiction. Neuropsychopharmacology 2019, 44, 166–183. [Google Scholar] [CrossRef]
- Gillies, G.E.; Pienaar, I.S.; Vohra, S.; Qamhawi, Z. Sex differences in Parkinson’s disease. Front. Neuroendocrinol. 2014, 35, 370–384. [Google Scholar] [CrossRef]
- Gillies, G.E.; Virdee, K.; McArthur, S.; Dalley, J.W. Sex-dependent diversity in ventral tegmental dopaminergic neurons and developmental programing: A molecular, cellular and behavioral analysis. Neuroscience 2014, 282, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Williams, O.O.F.; Coppolino, M.; George, S.R.; Perreault, M.L. Sex Differences in Dopamine Receptors and Relevance to Neuropsychiatric Disorders. Brain Sci. 2021, 11, 1199. [Google Scholar] [CrossRef] [PubMed]
- Nikiforuk, A.; Kalaba, P.; Ilic, M.; Korz, V.; Dragacevic, V.; Wackerlig, J.; Langer, T.; Hoger, H.; Golebiowska, J.; Popik, P.; et al. A Novel Dopamine Transporter Inhibitor CE-123 Improves Cognitive Flexibility and Maintains Impulsivity in Healthy Male Rats. Front. Behav. Neurosci. 2017, 11, 222. [Google Scholar] [CrossRef] [PubMed]
- Niello, M.; Cintulova, D.; Raithmayr, P.; Holy, M.; Jantsch, K.; Colas, C.; Ecker, G.F.; Sitte, H.H.; Mihovilovic, M.D. Effects of Hydroxylated Mephedrone Metabolites on Monoamine Transporter Activity in vitro. Front. Pharmacol. 2021, 12, 654061. [Google Scholar] [CrossRef]
- Maier, J.; Rauter, L.; Rudin, D.; Niello, M.; Holy, M.; Schmid, D.; Wilson, J.; Blough, B.E.; Gannon, B.M.; Murnane, K.S.; et al. alpha-PPP and its derivatives are selective partial releasers at the human norepinephrine transporter: A pharmacological characterization of interactions between pyrrolidinopropiophenones and high and low affinity monoamine transporters. Neuropharmacology 2021, 190, 108570. [Google Scholar] [CrossRef]
- Sanna, F.; Bratzu, J.; Piludu, M.A.; Corda, M.G.; Melis, M.R.; Giorgi, O.; Argiolas, A. Dopamine, Noradrenaline and Differences in Sexual Behavior between Roman High and Low Avoidance Male Rats: A Microdialysis Study in the Medial Prefrontal Cortex. Front. Behav. Neurosci. 2017, 11, 108. [Google Scholar] [CrossRef]
- Sanna, F.; Piludu, M.A.; Corda, M.G.; Melis, M.R.; Giorgi, O.; Argiolas, A. Involvement of dopamine in the differences in sexual behaviour between Roman high and low avoidance rats: An intracerebral microdialysis study. Behav. Brain Res. 2015, 281, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Stojanovic, T.; Benes, H.; Awad, A.; Bormann, D.; Monje, F.J. Nicotine abolishes memory-related synaptic strengthening and promotes synaptic depression in the neurogenic dentate gyrus of miR-132/212 knockout mice. Addict. Biol. 2021, 26, e12905. [Google Scholar] [CrossRef] [PubMed]
- Maggio, N.; Segal, M. Striking variations in corticosteroid modulation of long-term potentiation along the septotemporal axis of the hippocampus. J. Neurosci. 2007, 27, 5757–5765. [Google Scholar] [CrossRef]
- Maggio, N.; Segal, M. Unique regulation of long term potentiation in the rat ventral hippocampus. Hippocampus 2007, 17, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Titulaer, J.; Bjorkholm, C.; Feltmann, K.; Malmlof, T.; Mishra, D.; Bengtsson Gonzales, C.; Schilstrom, B.; Konradsson-Geuken, A. The Importance of Ventral Hippocampal Dopamine and Norepinephrine in Recognition Memory. Front. Behav. Neurosci. 2021, 15, 667244. [Google Scholar] [CrossRef]
- Nguyen, P.V.; Abel, T.; Kandel, E.R. Requirement of a critical period of transcription for induction of a late phase of LTP. Science 1994, 265, 1104–1107. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.V.; Kandel, E.R. Brief theta-burst stimulation induces a transcription-dependent late phase of LTP requiring cAMP in area CA1 of the mouse hippocampus. Learn. Mem. 1997, 4, 230–243. [Google Scholar] [CrossRef]
- Vorhees, C.V.; Williams, M.T. Assessing spatial learning and memory in rodents. ILAR J. 2014, 55, 310–332. [Google Scholar] [CrossRef]
- Shanmugasundaram, B.; Aher, Y.D.; Aradska, J.; Ilic, M.; Daba Feyissa, D.; Kalaba, P.; Aher, N.Y.; Dragacevic, V.; Saber Marouf, B.; Langer, T.; et al. R-Modafinil exerts weak effects on spatial memory acquisition and dentate gyrus synaptic plasticity. PLoS ONE 2017, 12, e0179675. [Google Scholar] [CrossRef]
- Lubec, J.; Smidak, R.; Malikovic, J.; Feyissa, D.D.; Korz, V.; Hoger, H.; Lubec, G. Dentate Gyrus Peroxiredoxin 6 Levels Discriminate Aged Unimpaired From Impaired Rats in a Spatial Memory Task. Front. Aging Neurosci. 2019, 11, 198. [Google Scholar] [CrossRef]
- Keppel, G. Design and Analysis: A Researcher’s Handbook, 3rd ed.; Prentice Hall: Englewood Cliffs, NJ, USA, 1991; Volume xiii, 594p. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Matrix | Tmax (h) | Cmax (ng/mL) | Cmax (nM) | Tlast (h) | Clast (nM) | AUClast (h*ng/mL) | AUClast (h*nM) |
|---|---|---|---|---|---|---|---|
| brain | 0.25 | 634 | 1665 | 10.0 | 19.2 | 1959 | 5143 |
| plasma | 0.50 | 624 | 1637 | 10.0 | 15.7 | 1713 | 4497 |
| DA Concentrations (as Determined from AUCs) | ||||
|---|---|---|---|---|
| Treatment | Vehicle (Kolliphor 30%) 1.5 mL/kg | (S)-MK-26 1 mg/kg | (S)-MK-26 10 mg/kg | |
| Brain Area | ||||
| NAc shell | 664 ± 525 | 2972 ± 1094 | 7730 ± 1389 *# | |
| NAc core | 276 ± 104 | 3056 ± 644 | 7861 ± 1697 *# | |
| mPFC | 530 ± 129 | 3159 ± 223 | 4983 ± 547 * | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kouhnavardi, S.; Ecevitoglu, A.; Dragačević, V.; Sanna, F.; Arias-Sandoval, E.; Kalaba, P.; Kirchhofer, M.; Lubec, J.; Niello, M.; Holy, M.; et al. A Novel and Selective Dopamine Transporter Inhibitor, (S)-MK-26, Promotes Hippocampal Synaptic Plasticity and Restores Effort-Related Motivational Dysfunctions. Biomolecules 2022, 12, 881. https://doi.org/10.3390/biom12070881
Kouhnavardi S, Ecevitoglu A, Dragačević V, Sanna F, Arias-Sandoval E, Kalaba P, Kirchhofer M, Lubec J, Niello M, Holy M, et al. A Novel and Selective Dopamine Transporter Inhibitor, (S)-MK-26, Promotes Hippocampal Synaptic Plasticity and Restores Effort-Related Motivational Dysfunctions. Biomolecules. 2022; 12(7):881. https://doi.org/10.3390/biom12070881
Chicago/Turabian StyleKouhnavardi, Shima, Alev Ecevitoglu, Vladimir Dragačević, Fabrizio Sanna, Edgar Arias-Sandoval, Predrag Kalaba, Michael Kirchhofer, Jana Lubec, Marco Niello, Marion Holy, and et al. 2022. "A Novel and Selective Dopamine Transporter Inhibitor, (S)-MK-26, Promotes Hippocampal Synaptic Plasticity and Restores Effort-Related Motivational Dysfunctions" Biomolecules 12, no. 7: 881. https://doi.org/10.3390/biom12070881
APA StyleKouhnavardi, S., Ecevitoglu, A., Dragačević, V., Sanna, F., Arias-Sandoval, E., Kalaba, P., Kirchhofer, M., Lubec, J., Niello, M., Holy, M., Zehl, M., Pillwein, M., Wackerlig, J., Murau, R., Mohrmann, A., Beard, K. R., Sitte, H. H., Urban, E., Sagheddu, C., ... Monje, F. J. (2022). A Novel and Selective Dopamine Transporter Inhibitor, (S)-MK-26, Promotes Hippocampal Synaptic Plasticity and Restores Effort-Related Motivational Dysfunctions. Biomolecules, 12(7), 881. https://doi.org/10.3390/biom12070881