
Target-Based Small Molecule Drug Discovery for Colorectal Cancer: A Review of Molecular Pathways and In Silico Studies

 , , ,
, , ,  , and
, and 
Abstract
1. Introduction
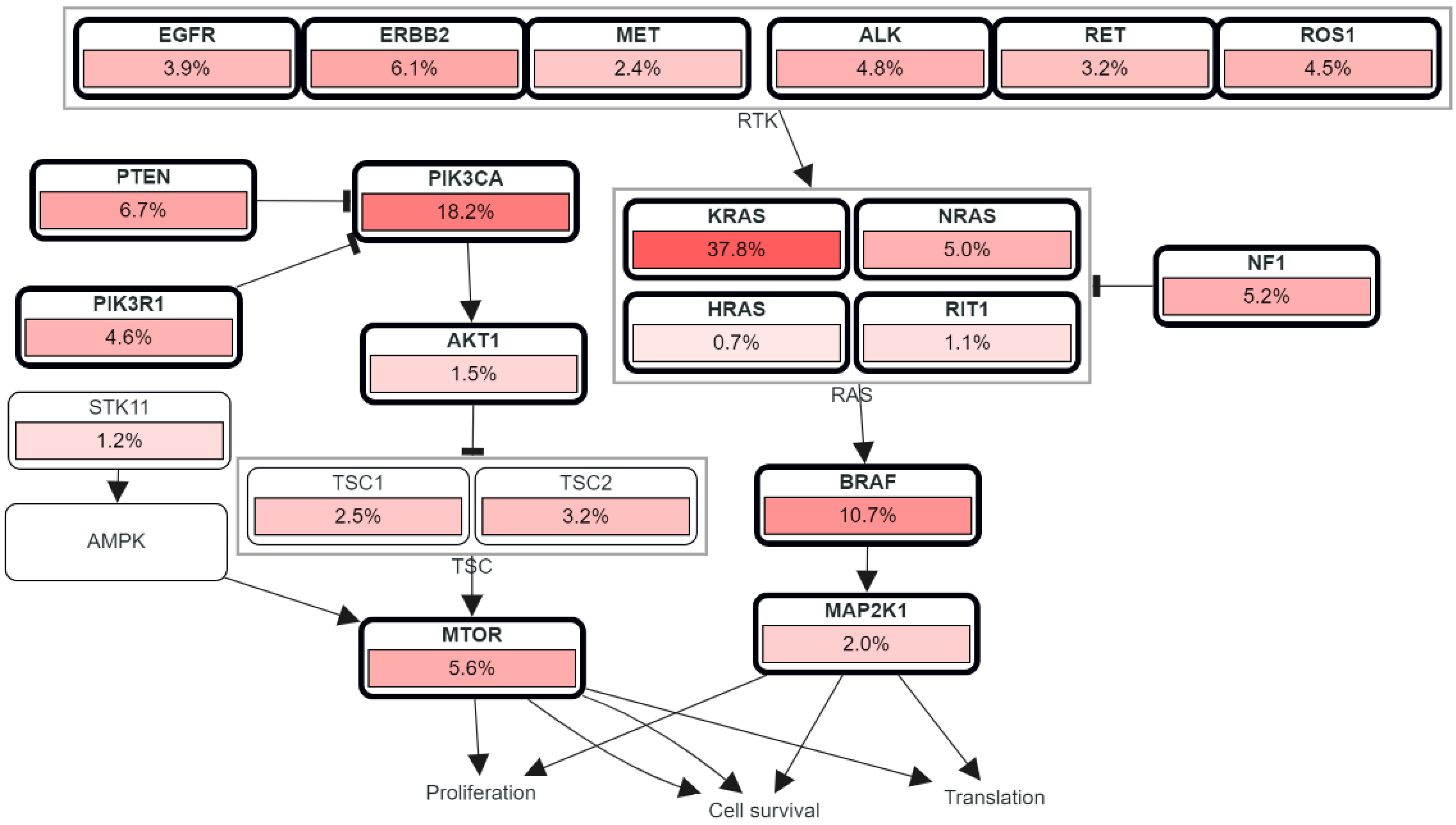
2. Driver Genes in CRC
3. Inactivation of Tumor-Suppressor Genes
3.1. Adenomatous Polyposis Coli (APC)
3.2. TP53 Inactivation Pathway
3.3. TGF-β Tumor Suppressor Pathway
4. Growth Factor Pathways
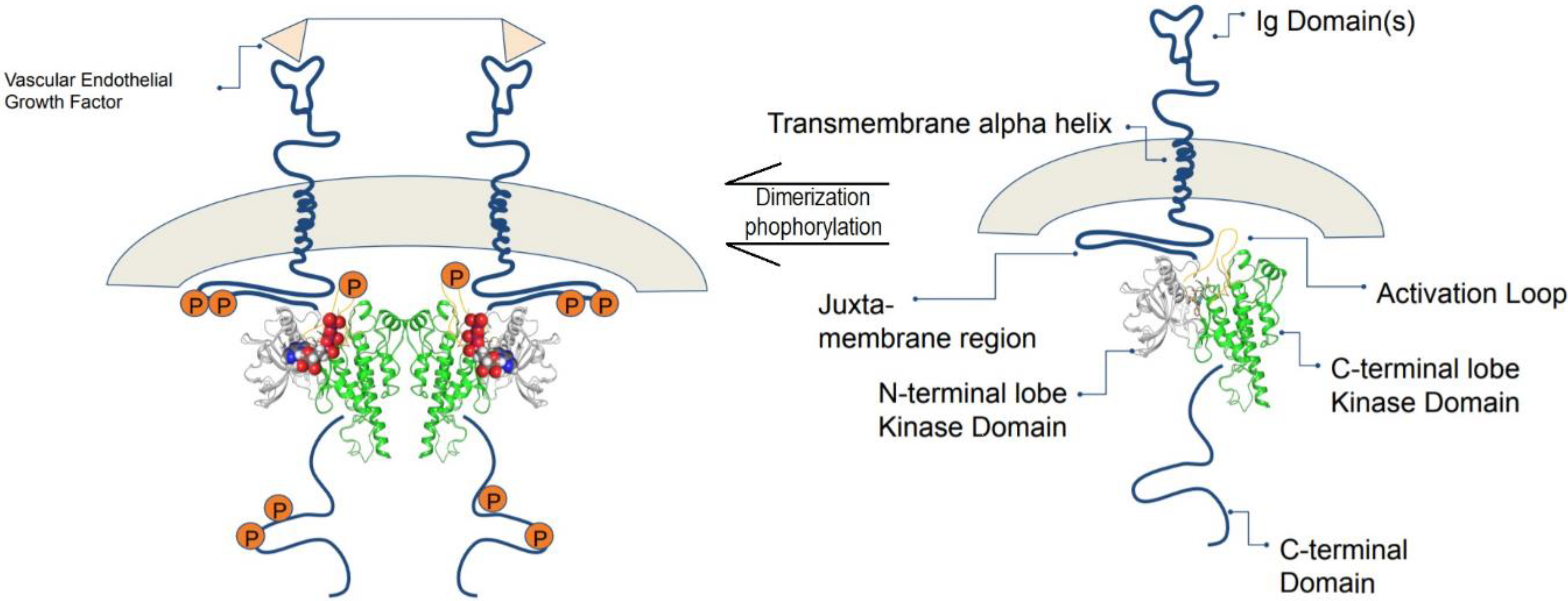
4.1. Vascular Endothelial Growth Factor Receptor-2 (VEGFR-2)
4.2. Epidermal Growth Factor Receptor (EGFR)
4.3. Other Receptor and Protein Kinases in CRC
5. Microsatellite Instability Pathways
5.1. Epigenetic Silencing of Gene Expression
5.2. Base Excision Repair Defects
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Bogaert, J.; Prenen, H. Molecular genetics of colorectal cancer. Ann. Gastroenterol. 2014, 27, 9. [Google Scholar] [PubMed]
- Issa, J.-P. CpG island methylator phenotype in cancer. Nat. Rev. Cancer 2004, 4, 988–993. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Castelli, G.; Pelosi, E. Genetic alterations of metastatic colorectal cancer. Biomedicines 2020, 8, 414. [Google Scholar] [CrossRef]
- Mármol, I.; Sánchez-de-Diego, C.; Pradilla Dieste, A.; Cerrada, E.; Rodriguez Yoldi, M.J. Colorectal carcinoma: A general overview and future perspectives in colorectal cancer. Int. J. Mol. Sci. 2017, 18, 197. [Google Scholar] [CrossRef]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef]
- Willett, C.G.; Chang, D.T.; Czito, B.G.; Meyer, J.; Wo, J. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012.(5). Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 1. [Google Scholar] [CrossRef]
- Lin, M.; Whitmire, S.; Chen, J.; Farrel, A.; Shi, X.; Guo, J.-T. Effects of short indels on protein structure and function in human genomes. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef]
- Garraway, L.A.; Lander, E.S. Lessons from the Cancer Genome. Cell 2013, 153, 17–37. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Dees, N.D.; Zhang, Q.; Kandoth, C.; Wendl, M.C.; Schierding, W.; Koboldt, D.C.; Mooney, T.B.; Callaway, M.B.; Dooling, D.; Mardis, E.R. MuSiC: Identifying mutational significance in cancer genomes. Genome Res. 2012, 22, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Loganathan, L.; Muthusamy, K.; Jayaraj, J.M.; Kajamaideen, A.; Balthasar, J.J. In silico insights on tankyrase protein: A potential target for colorectal cancer. J. Biomol. Struct. Dyn. 2018, 37, 3637–3648. [Google Scholar] [CrossRef]
- Baur, F.; Nietzer, S.L.; Kunz, M.; Saal, F.; Jeromin, J.; Matschos, S.; Linnebacher, M.; Walles, H.; Dandekar, T.; Dandekar, G. Connecting cancer pathways to tumor engines: A stratification tool for colorectal cancer combining human in vitro tissue models with boolean in silico models. Cancers 2019, 12, 28. [Google Scholar] [CrossRef]
- Kather, J.N.; Poleszczuk, J.; Suarez-Carmona, M.; Krisam, J.; Charoentong, P.; Valous, N.A.; Weis, C.-A.; Tavernar, L.; Leiss, F.; Herpel, E. In silico modeling of immunotherapy and stroma-targeting therapies in human colorectal cancer. Cancer Res. 2017, 77, 6442–6452. [Google Scholar] [CrossRef]
- Greenhalgh, K.; Ramiro-Garcia, J.; Heinken, A.; Ullmann, P.; Bintener, T.; Pacheco, M.P.; Baginska, J.; Shah, P.; Frachet, A.; Halder, R.; et al. Integrated In Vitro and In Silico Modeling Delineates the Molecular Effects of a Synbiotic Regimen on Colorectal-Cancer-Derived Cells. Cell Rep. 2019, 27, 1621–1632.e1629. [Google Scholar] [CrossRef]
- Nazempour, N.; Taleqani, M.H.; Taheri, N.; Najafabadi, A.H.H.A.A.; Shokrollahi, A.; Zamani, A.; Fattahi Dolatabadi, N.; Peymani, M.; Mahdevar, M. The role of cell surface proteins gene expression in diagnosis, prognosis, and drug resistance of colorectal cancer: In silico analysis and validation. Exp. Mol. Pathol. 2021, 123, 104688. [Google Scholar] [CrossRef]
- Fadaka, A.O.; Klein, A.; Pretorius, A. In silico identification of microRNAs as candidate colorectal cancer biomarkers. Tumor Biol. 2019, 41, 1010428319883721. [Google Scholar] [CrossRef]
- Raskov, H.; Søby, J.H.; Troelsen, J.; Bojesen, R.D.; Gögenur, I. Driver gene mutations and epigenetics in colorectal cancer. Ann. Surg. 2020, 271, 75–85. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. The path to cancer—Three strikes and you’re out. N. Engl. J. Med. 2015, 373, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Sakai, E.; Nakayama, M.; Oshima, H.; Kouyama, Y.; Niida, A.; Fujii, S.; Ochiai, A.; Nakayama, K.I.; Mimori, K.; Suzuki, Y. Combined mutation of Apc, Kras, and Tgfbr2 effectively drives metastasis of intestinal cancer. Cancer Res. 2018, 78, 1334–1346. [Google Scholar] [CrossRef] [PubMed]
- Schell, M.J.; Yang, M.; Teer, J.K.; Lo, F.Y.; Madan, A.; Coppola, D.; Monteiro, A.N.A.; Nebozhyn, M.V.; Yue, B.; Loboda, A.; et al. A multigene mutation classification of 468 colorectal cancers reveals a prognostic role for APC. Nat. Commun. 2016, 7, 11743. [Google Scholar] [CrossRef] [PubMed]
- Haigis, K.M. KRAS Alleles: The Devil Is in the Detail. Trends Cancer 2017, 3, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Joseph, R.; Little, P.; Hayes, D.N.; Lee, M.S. Characterization of the Number and Site of APC Mutations in Sporadic Colorectal Cancer; American Society of Clinical Oncology: Alexandria, VA, USA, 2017. [Google Scholar]
- Fodde, R. The APC gene in colorectal cancer. Eur. J. Cancer 2002, 38, 867–871. [Google Scholar] [CrossRef]
- Malki, A.; ElRuz, R.A.; Gupta, I.; Allouch, A.; Vranic, S.; Al Moustafa, A.-E. Molecular mechanisms of colon cancer progression and metastasis: Recent insights and advancements. Int. J. Mol. Sci. 2021, 22, 130. [Google Scholar] [CrossRef]
- Finch, A.J.; Soucek, L.; Junttila, M.R.; Swigart, L.B.; Evan, G.I. Acute overexpression of Myc in intestinal epithelium recapitulates some but not all the changes elicited by Wnt/β-catenin pathway activation. Mol. Cell. Biol. 2009, 29, 5306–5315. [Google Scholar] [CrossRef][Green Version]
- Utsunomiya, T.; Doki, Y.; Takemoto, H.; Shiozaki, H.; Yano, M.; Sekimoto, M.; Tamura, S.; Yasuda, T.; Fujiwara, Y.; Monden, M. Correlation of beta-catenin and cyclin D1 expression in colon cancers. Oncology 2001, 61, 226–233. [Google Scholar] [CrossRef]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 15, 857–865. [Google Scholar] [CrossRef]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-spondin fusions in colon cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef]
- Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [CrossRef] [PubMed]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e296. [Google Scholar] [CrossRef] [PubMed]
- Brannon, A.R.; Vakiani, E.; Sylvester, B.E.; Scott, S.N.; McDermott, G.; Shah, R.H.; Kania, K.; Viale, A.; Oschwald, D.M.; Vacic, V.; et al. Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome Biol. 2014, 15, 454. [Google Scholar] [CrossRef] [PubMed]
- Mondaca, S.; Walch, H.; Nandakumar, S.; Chatila, W.K.; Schultz, N.; Yaeger, R. Specific Mutations in APC, but Not Alterations in DNA Damage Response, Associate With Outcomes of Patients With Metastatic Colorectal Cancer. Gastroenterology 2020, 159, 1975–1978.e1974. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136.e123. [Google Scholar] [CrossRef]
- Guda, K.; Veigl, M.L.; Varadan, V.; Nosrati, A.; Ravi, L.; Lutterbaugh, J.; Beard, L.; Willson, J.K.; Sedwick, W.D.; Wang, Z.J.; et al. Novel recurrently mutated genes in African American colon cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 1149–1154. [Google Scholar] [CrossRef]
- Vasaikar, S.; Huang, C.; Wang, X.; Petyuk, V.A.; Savage, S.R.; Wen, B.; Dou, Y.; Zhang, Y.; Shi, Z.; Arshad, O.A.; et al. Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 2019, 177, 1035–1049.e1019. [Google Scholar] [CrossRef]
- Huszno, J.; Grzybowska, E. TP53 mutations and SNPs as prognostic and predictive factors in patients with breast cancer. Oncol. Lett. 2018, 16, 34–40. [Google Scholar] [CrossRef]
- He, X.; Liao, J.; Liu, F.; Yan, J.; Yan, J.; Shang, H.; Dou, Q.; Chang, Y.; Lin, J.; Song, Y. Functional repair of p53 mutation in colorectal cancer cells using trans-splicing. Oncotarget 2015, 6, 2034. [Google Scholar] [CrossRef]
- Li, X.-L.; Zhou, J.; Chen, Z.-R.; Chng, W.-J. P53 mutations in colorectal cancer-molecular pathogenesis and pharmacological reactivation. World J. Gastroenterol. 2015, 21, 84. [Google Scholar] [CrossRef]
- Shen, J.; Vakifahmetoglu, H.; Stridh, H.; Zhivotovsky, B.; Wiman, K. PRIMA-1 MET induces mitochondrial apoptosis through activation of caspase-2. Oncogene 2008, 27, 6571–6580. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.M.; Phillips, A.C.; Vousden, K.H. Regulation and function of the p53 tumor suppressor protein. Curr. Opin. Cell Biol. 2001, 13, 332–337. [Google Scholar] [CrossRef]
- Taketani, K.; Kawauchi, J.; Tanaka-Okamoto, M.; Ishizaki, H.; Tanaka, Y.; Sakai, T.; Miyoshi, J.; Maehara, Y.; Kitajima, S. Key role of ATF3 in p53-dependent DR5 induction upon DNA damage of human colon cancer cells. Oncogene 2012, 31, 2210–2221. [Google Scholar] [CrossRef] [PubMed]
- Chasov, V.; Mirgayazova, R.; Zmievskaya, E.; Khadiullina, R.; Valiullina, A.; Stephenson Clarke, J.; Rizvanov, A.; Baud, M.G.; Bulatov, E. Key players in the mutant p53 team: Small molecules, gene editing, immunotherapy. Front. Oncol. 2020, 10, 1460. [Google Scholar] [CrossRef] [PubMed]
- Perdrix, A.; Najem, A.; Saussez, S.; Awada, A.; Journe, F.; Ghanem, G.; Krayem, M. PRIMA-1 and PRIMA-1Met (APR-246): From mutant/wild type p53 reactivation to unexpected mechanisms underlying their potent anti-tumor effect in combinatorial therapies. Cancers 2017, 9, 172. [Google Scholar] [CrossRef]
- Bauer, M.R.; Jones, R.N.; Tareque, R.K.; Springett, B.; Dingler, F.A.; Verduci, L.; Patel, K.J.; Fersht, A.R.; Joerger, A.C.; Spencer, J. A structure-guided molecular chaperone approach for restoring the transcriptional activity of the p53 cancer mutant Y220C. Future Med. Chem. 2019, 11, 2491–2504. [Google Scholar] [CrossRef]
- Liu, X.; Wilcken, R.; Joerger, A.C.; Chuckowree, I.S.; Amin, J.; Spencer, J.; Fersht, A.R. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 2013, 41, 6034–6044. [Google Scholar] [CrossRef]
- Principe, D.R.; Doll, J.A.; Bauer, J.; Jung, B.; Munshi, H.G.; Bartholin, L.; Pasche, B.; Lee, C.; Grippo, P.J. TGF-β: Duality of function between tumor prevention and carcinogenesis. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef]
- Yingling, J.M.; Blanchard, K.L.; Sawyer, J.S. Development of TGF-β signalling inhibitors for cancer therapy. Nat. Rev. Drug Discov. 2004, 3, 1011–1022. [Google Scholar] [CrossRef]
- Antony, M.L.; Nair, R.; Sebastian, P.; Karunagaran, D. Changes in expression, and/or mutations in TGF-β receptors (TGF-β RI and TGF-β RII) and Smad 4 in human ovarian tumors. J. Cancer Res. Clin. Oncol. 2010, 136, 351–361. [Google Scholar] [CrossRef]
- Itatani, Y.; Kawada, K.; Sakai, Y. Transforming growth factor-β signaling pathway in colorectal cancer and its tumor microenvironment. Int. J. Mol. Sci. 2019, 20, 5822. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-β-mediated epithelial-mesenchymal transition and cancer metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, J.H.; Enns, R.; Heidelbaugh, J.; Barkun, A.; Adams, M.A.; Dorn, S.D.; Dudley-Brown, S.L.; Flamm, S.L.; Gellad, Z.F.; Gruss, C.B. American Gastroenterological Association Institute guideline on the diagnosis and management of Lynch syndrome. Gastroenterology 2015, 149, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.M.; Davison, J.; Carter, K.T.; O’Leary, R.M.; Trobridge, P.; Knoblaugh, S.E.; Myeroff, L.L.; Markowitz, S.D.; Brett, B.T.; Scheetz, T.E. Transposon mutagenesis identifies candidate genes that cooperate with loss of transforming growth factor-beta signaling in mouse intestinal neoplasms. Int. J. Cancer 2017, 140, 853–863. [Google Scholar] [CrossRef]
- Voorneveld, P.W.; Kodach, L.L.; Jacobs, R.J.; Liv, N.; Zonnevylle, A.C.; Hoogenboom, J.P.; Biemond, I.; Verspaget, H.W.; Hommes, D.W.; De Rooij, K. Loss of SMAD4 alters BMP signaling to promote colorectal cancer cell metastasis via activation of Rho and ROCK. Gastroenterology 2014, 147, 196–208.e113. [Google Scholar] [CrossRef]
- Nicklas, D.; Saiz, L. In silico identification of potential therapeutic targets in the TGF-β signal transduction pathway. Mol. BioSystems 2014, 10, 537–548. [Google Scholar] [CrossRef]
- Wang, J.; Tucker-Kellogg, L.; Ng, I.C.; Jia, R.; Thiagarajan, P.; White, J.K.; Yu, H. The self-limiting dynamics of TGF-β signaling in silico and in vitro, with negative feedback through PPM1A upregulation. PLoS Comput. Biol. 2014, 10, e1003573. [Google Scholar] [CrossRef]
- Jadav, S.S.; Macalino, S.J.Y.; Alluri, R. Structure-based discovery of small molecule APC-Asef interaction inhibitors: In silico approaches and molecular dynamics simulations. J. Mol. Modeling 2020, 26, 1–11. [Google Scholar] [CrossRef]
- Li, B.; Liang, J.; Lu, F.; Zeng, G.; Zhang, J.; Ma, Y.; Liu, P.; Wang, Q.; Zhou, Q.; Chen, L. Discovery of novel inhibitor for Wnt/β-catenin pathway by tankyrase 1/2 structure-based virtual screening. Molecules 2020, 25, 1680. [Google Scholar] [CrossRef]
- Zhang, W.; Lu, W.; Ananthan, S.; Suto, M.J.; Li, Y. Discovery of novel frizzled-7 inhibitors by targeting the receptor’s transmembrane domain. Oncotarget 2017, 8, 91459. [Google Scholar] [CrossRef]
- Lee, H.-M.; Chan, D.S.-H.; Yang, F.; Lam, H.-Y.; Yan, S.-C.; Che, C.-M.; Ma, D.-L.; Leung, C.-H. Identification of natural product Fonsecin B as a stabilizing ligand of c-myc G-quadruplex DNA by high-throughput virtual screening. Chem. Commun. 2010, 46, 4680–4682. [Google Scholar] [CrossRef] [PubMed]
- Carabet, L.A.; Lallous, N.; Leblanc, E.; Ban, F.; Morin, H.; Lawn, S.; Ghaidi, F.; Lee, J.; Mills, I.G.; Gleave, M.E.; et al. Computer-aided drug discovery of Myc-Max inhibitors as potential therapeutics for prostate cancer. Eur. J. Med. Chem. 2018, 160, 108–119. [Google Scholar] [CrossRef]
- Mokgautsi, N.; Wang, Y.-C.; Lawal, B.; Khedkar, H.; Sumitra, M.R.; Wu, A.T.; Huang, H.-S. Network pharmacological analysis through a bioinformatics approach of novel NSC765600 and NSC765691 compounds as potential inhibitors of CCND1/CDK4/PLK1/CD44 in cancer types. Cancers 2021, 13, 2523. [Google Scholar] [CrossRef] [PubMed]
- Leung, W.-H.; Shih, J.-W.; Chen, J.-S.; Mokgautsi, N.; Wei, P.-L.; Huang, Y.-J. Preclinical Identification of Sulfasalazine’s Therapeutic Potential for Suppressing Colorectal Cancer Stemness and Metastasis through Targeting KRAS/MMP7/CD44 Signaling. Biomedicines 2022, 10, 377. [Google Scholar] [CrossRef] [PubMed]
- Chaurasia, P.; Mezei, M.; Zhou, M.-M.; Ossowski, L. Computer aided identification of small molecules disrupting uPAR/α5β1-integrin interaction: A new paradigm for metastasis prevention. PLoS ONE 2009, 4, e4617. [Google Scholar] [CrossRef]
- Dolezal, R.; Melikova, M.; Mesicek, J.; Kuca, K. Rational discovery of GSK3-beta modulators aided by protein pocket prediction and high-throughput molecular docking. In Proceedings of the International Conference on Computational Collective Intelligence, Wrocław, Poland, 5–7 October 2009; pp. 429–439. [Google Scholar]
- Nagaraj, A.; Wang, Q.; Joseph, P.; Zheng, C.; Chen, Y.; Kovalenko, O.; Singh, S.; Armstrong, A.; Resnick, K.; Zanotti, K. Using a novel computational drug-repositioning approach (DrugPredict) to rapidly identify potent drug candidates for cancer treatment. Oncogene 2018, 37, 403–414. [Google Scholar] [CrossRef]
- Tian, W.; Han, X.; Yan, M.; Xu, Y.; Duggineni, S.; Lin, N.; Luo, G.; Li, Y.M.; Han, X.; Huang, Z. Structure-based discovery of a novel inhibitor targeting the β-catenin/Tcf4 interaction. Biochemistry 2012, 51, 724–731. [Google Scholar] [CrossRef]
- Enayatkhani, M.; Salimi, M.; Azadmanesh, K.; Teimoori-Toolabi, L. In-silico identification of new inhibitors for Low-density lipoprotein receptor-related protein6 (LRP6). J. Biomol. Struct. Dyn. 2020, 40, 1–11. [Google Scholar] [CrossRef]
- Li, X.; Zhang, X.-X.; Lin, Y.-X.; Xu, X.-M.; Li, L.; Yang, J.-B. Virtual Screening Based on Ensemble Docking Targeting Wild-Type p53 for Anticancer Drug Discovery. Chem. Biodivers. 2019, 16, e1900170. [Google Scholar] [CrossRef]
- Park, I.-S.; Seo, H.R.; Kim, K.; Lee, H.; Shum, D.; Choi, I.; Kim, J. Identification of inhibitors of Bcl-2 family protein-protein interaction by combining the BRET screening platform with virtual screening. Biochem. Biophys. Res. Commun. 2020, 527, 709–715. [Google Scholar] [CrossRef]
- Atatreh, N.; Ghattas, M.A.; Bardaweel, S.K.; Al Rawashdeh, S.; Al Sorkhy, M. Identification of new inhibitors of Mdm2–p53 interaction via pharmacophore and structure-based virtual screening. Drug Des. Dev. Ther. 2018, 12, 3741. [Google Scholar] [CrossRef] [PubMed]
- Mostafavi, S.M.; Bagherzadeh, K.; Amanlou, M. A new attempt to introduce efficient inhibitors for Caspas-9 according to structure-based Pharmacophore Screening strategy and Molecular Dynamics Simulations. Medbiotech J. 2017, 1, 1–8. [Google Scholar]
- Lakshmi, P.J.; Kumar, B.S.; Nayana, R.S.; Mohan, M.S.; Bolligarla, R.; Das, S.K.; Bhanu, M.U.; Kondapi, A.K.; Ravikumar, M. Design, synthesis, and discovery of novel non-peptide inhibitor of Caspase-3 using ligand based and structure based virtual screening approach. Bioorganic Med. Chem. 2009, 17, 6040–6047. [Google Scholar] [CrossRef]
- Tahir, R.A.; Sehgal, S.A.; Khattak, N.A.; Khan Khattak, J.Z.; Mir, A. Tumor necrosis factor receptor superfamily 10B (TNFRSF10B): An insight from structure modeling to virtual screening for designing drug against head and neck cancer. Theor. Biol. Med. Model. 2013, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sessions, R.B.; Prime, S.S.; Shoemark, D.K.; Allen, S.J.; Hong, W.; Narayanan, S.; Paterson, I.C. Identification of novel small molecule TGF-β antagonists using structure-based drug design. J. Comput. -Aided Mol. Des. 2013, 27, 365–372. [Google Scholar] [CrossRef]
- Singh, J.; Chuaqui, C.E.; Boriack-Sjodin, P.A.; Lee, W.-C.; Pontz, T.; Corbley, M.J.; Cheung, H.K.; Arduini, R.M.; Mead, J.N.; Newman, M.N.; et al. Successful shape-Based virtual screening: The discovery of a potent inhibitor of the type I TGFβ receptor kinase (TβRI). Bioorganic Med. Chem. Lett. 2003, 13, 4355–4359. [Google Scholar] [CrossRef]
- Huang, S.; Mei, H.; Lu, L.; Qiu, M.; Liang, X.; Xu, L.; Kuang, Z.; Heng, Y.; Pan, X. De Novo Molecular Design of Caspase-6 Inhibitors by a GRU-Based Recurrent Neural Network Combined with a Transfer Learning Approach. Pharmaceuticals 2021, 14, 1249. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Sussman, J.L.; Lin, D.; Jiang, J.; Manning, N.O.; Prilusky, J.; Ritter, O.; Abola, E.E. Protein Data Bank (PDB): Database of three-dimensional structural information of biological macromolecules. Acta Crystallogr. Sect. D. 1998, 54, 1078–1084. [Google Scholar] [CrossRef]
- Nagy, J.A.; Dvorak, A.M.; Dvorak, H.F. VEGF-A and the induction of pathological angiogenesis. Annu. Rev. Pathol. Mech. Dis. 2007, 2, 251–275. [Google Scholar] [CrossRef]
- Ferrara, N. Vascular endothelial growth factor as a target for anticancer therapy. Oncol. 2004, 9, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Hari, S.B.; Merritt, E.A.; Maly, D.J. Sequence Determinants of a Specific Inactive Protein Kinase Conformation. Chem. Biol. 2013, 20, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2022-1. Maestro; Schrödinger LLC: New York, NY, USA. Available online: https://www.schrodinger.com/learn/training/schrodinger-online-learning (accessed on 10 November 2021).
- Liu, Y.; Gray, N.S. Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2006, 2, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Rathi, E.; Kumar, A.; Kini, S.G. Molecular dynamics guided insight, binding free energy calculations and pharmacophore-based virtual screening for the identification of potential VEGFR2 inhibitors. J. Recept. Signal Transduct. 2019, 39, 415–433. [Google Scholar] [CrossRef]
- Treiber, D.K.; Shah, N.P. Ins and outs of kinase DFG motifs. Chem. Biol. 2013, 20, 745–746. [Google Scholar] [CrossRef]
- Mol, C.D.; Fabbro, D.; Hosfield, D.J. Structural insights into the conformational selectivity of STI-571 and related kinase inhibitors. Curr. Opin. Drug Discov. Dev. 2004, 7, 639–648. [Google Scholar]
- Traxler, P.; Furet, P. Strategies toward the design of novel and selective protein tyrosine kinase inhibitors. Pharmacol. Ther. 1999, 82, 195–206. [Google Scholar] [CrossRef]
- Sharma, S.; Johnson, D.; Abouammoh, M.; Hollands, S.; Brissette, A. Rate of serious adverse effects in a series of bevacizumab and ranibizumab injections. Can. J. Ophthalmol. 2012, 47, 275–279. [Google Scholar] [CrossRef]
- Li, X.-S.; Wu, X.; Zhao, P.-J.; Huang, L.-H.; Song, Y.; Gong, K.; Shen, C.Y.W.; Song, G.; Zhao, Z.; Zhang, Z. Efficacy and safety of sunitinib in the treatment of metastatic renal cell carcinoma. Chin. Med. J. 2011, 124, 2920–2924. [Google Scholar]
- Sharma, N.; Sharma, M.; Rahman, Q.I.; Akhtar, S.; Muddassir, M. Quantitative structure activity relationship and molecular simulations for the exploration of natural potent VEGFR-2 inhibitors: An in silico anti-angiogenic study. J. Biomol. Struct. Dyn. 2021, 39, 2806–2823. [Google Scholar] [CrossRef]
- Parveen, S. In silico drug repurposing of fda-approved artemisinins as potent chemotherapeutics targeting BCL-2, CDK-6 & VEGFR-2: Density functional exploration and molecular docking study. Biointerface Res. Appl. Chem. 2021, 11, 9604–9618. [Google Scholar]
- Varma, D.A.; Singh, M.; Wakode, S.; Dinesh, N.; Vinaik, S.; Asthana, S.; Tiwari, M. Structure-based pharmacophore mapping and virtual screening of natural products to identify polypharmacological inhibitor against c-MET/EGFR/VEGFR-2. J. Biomol. Struct. Dyn. 2022, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Ban, H.S.; Kawada, J.; Hirokawa, T.; Nakamura, H. Discovery of indenopyrazoles as EGFR and VEGFR-2 tyrosine kinase inhibitors by in silico high-throughput screening. Bioorganic Med. Chem. Lett. 2008, 18, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, S.; Jiao, Y.; Liu, H.; Yuan, H.; Lu, S.; Ran, T.; Yao, S.; Ke, Z.; Xu, J. An integrated virtual screening approach for VEGFR-2 inhibitors. J. Chem. Inf. Modeling 2013, 53, 3163–3177. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Patidar, K.; Ali, M.A.; Patil, P.; Goud, H.; Hussain, T.; Nayarisseri, A.; Singh, S.K. Structure-based virtual screening for the identification of high affinity compounds as potent VEGFR2 inhibitors for the treatment of renal cell carcinoma. Curr. Top. Med. Chem. 2018, 18, 2174–2185. [Google Scholar] [CrossRef]
- Li, J.; Zhou, N.; Luo, K.; Zhang, W.; Li, X.; Wu, C.; Bao, J. In silico discovery of potential VEGFR-2 inhibitors from natural derivatives for anti-angiogenesis therapy. Int. J. Mol. Sci. 2014, 15, 15994–16011. [Google Scholar] [CrossRef]
- Harris, P.A.; Cheung, M.; Hunter, R.N.; Brown, M.L.; Veal, J.M.; Nolte, R.T.; Wang, L.; Liu, W.; Crosby, R.M.; Johnson, J.H. Discovery and evaluation of 2-anilino-5-aryloxazoles as a novel class of VEGFR2 kinase inhibitors. J. Med. Chem. 2005, 48, 1610–1619. [Google Scholar] [CrossRef]
- Lee, K.; Jeong, K.-W.; Lee, Y.; Song, J.Y.; Kim, M.S.; Lee, G.S.; Kim, Y. Pharmacophore modeling and virtual screening studies for new VEGFR-2 kinase inhibitors. Eur. J. Med. Chem. 2010, 45, 5420–5427. [Google Scholar] [CrossRef]
- Kankanala, J.; Latham, A.; Johnson, A.; Homer-Vanniasinkam, S.; Fishwick, C.; Ponnambalam, S. A combinatorial in silico and cellular approach to identify a new class of compounds that target VEGFR2 receptor tyrosine kinase activity and angiogenesis. Br. J. Pharmacol. 2012, 166, 737–748. [Google Scholar] [CrossRef]
- Goldstein, N.S.; Armin, M. Epidermal growth factor receptor immunohistochemical reactivity in patients with American Joint Committee on Cancer Stage IV colon adenocarcinoma: Implications for a standardized scoring system. Cancer 2001, 92, 1331–1346. [Google Scholar] [CrossRef]
- Spano, J.P.; Fagard, R.; Soria, J.C.; Rixe, O.; Khayat, D.; Milano, G. Epidermal growth factor receptor signaling in colorectal cancer: Preclinical data and therapeutic perspectives. Ann. Oncol. 2005, 16, 189–194. [Google Scholar] [CrossRef]
- Cohen, R.B. Epidermal growth factor receptor as a therapeutic target in colorectal cancer. Clin. Colorectal Cancer 2003, 2, 246–251. [Google Scholar] [CrossRef]
- Messa, C.; Russo, F.; Gabriella Caruso, M.; Di Leo, A. EGF, TGF-a, and EGF-R in human colorectal adenocarcinoma. Acta Oncol. 1998, 37, 285–289. [Google Scholar] [CrossRef]
- Markman, B.; Javier Ramos, F.; Capdevila, J.; Tabernero, J. EGFR and KRAS in colorectal cancer. In Advances in Clinical Chemistry; Academic Press: New York, NY, USA, 2010; Volume 51, p. 72. [Google Scholar]
- Snyder, L.C.; Astsaturov, I.; Weiner, L.M. Overview of monoclonal antibodies and small molecules targeting the epidermal growth factor receptor pathway in colorectal cancer. Clin. Colorectal Cancer 2005, 5, S71–S80. [Google Scholar] [CrossRef]
- Ferguson, K.M.; Berger, M.B.; Mendrola, J.M.; Cho, H.-S.; Leahy, D.J.; Lemmon, M.A. EGF Activates Its Receptor by Removing Interactions that Autoinhibit Ectodomain Dimerization. Mol. Cell 2003, 11, 507–517. [Google Scholar] [CrossRef]
- Yarom, N.; Jonker, D.J. The role of the epidermal growth factor receptor in the mechanism and treatment of colorectal cancer. Discov. Med. 2011, 11, 95–105. [Google Scholar]
- Berg, M.; Soreide, K. EGFR and downstream genetic alterations in KRAS/BRAF and PI3K/AKT pathways in colorectal cancer—Implications for targeted therapy. Discov. Med. 2012, 14, 207–214. [Google Scholar]
- Patel, H.M.; Ahmad, I.; Pawara, R.; Shaikh, M.; Surana, S. In silico search of triple mutant T790M/C797S allosteric inhibitors to conquer acquired resistance problem in non-small cell lung cancer (NSCLC): A combined approach of structure-based virtual screening and molecular dynamics simulation. J. Biomol. Struct. Dyn. 2021, 39, 1491–1505. [Google Scholar] [CrossRef]
- Karnik, K.S.; Sarkate, A.P.; Lokwani, D.K.; Narula, I.S.; Burra, P.V.; Wakte, P.S. Development of triple mutant T790M/C797S allosteric EGFR inhibitors: A computational approach. J. Biomol. Struct. Dyn. 2021, 39, 5376–5398. [Google Scholar] [CrossRef]
- Patel, H.; Pawara, R.; Surana, S. In-silico evidences for binding of Glucokinase activators to EGFR C797S to overcome EGFR resistance obstacle with mutant-selective allosteric inhibition. Comput. Biol. Chem. 2018, 74, 167–189. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Et. Biophys. Acta (BBA)-Mol. Cell Res. 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.; Vikis, H.G.; Guan, K.-L. Mechanisms of regulating the Raf kinase family. Cell. Signal. 2003, 15, 463–469. [Google Scholar] [CrossRef]
- Mebratu, Y.; Tesfaigzi, Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle 2009, 8, 1168–1175. [Google Scholar] [CrossRef]
- Arcaro, A.; Guerreiro, A.S. The phosphoinositide 3-kinase pathway in human cancer: Genetic alterations and therapeutic implications. Curr. Genom. 2007, 8, 271–306. [Google Scholar] [CrossRef] [PubMed]
- Wojtalla, A.; Salm, F.; Christiansen, D.G.; Cremona, T.; Cwiek, P.; Shalaby, T.; Gross, N.; Grotzer, M.A.; Arcaro, A. Novel agents targeting the IGF-1R/PI3K pathway impair cell proliferation and survival in subsets of medulloblastoma and neuroblastoma. PLoS ONE 2012, 7, e47109. [Google Scholar] [CrossRef] [PubMed]
- Peters, G.; Gongoll, S.; Langner, C.; Mengel, M.; Piso, P.; Klempnauer, J.; Rüschoff, J.; Kreipe, H.; von Wasielewski, R. IGF-1R, IGF-1 and IGF-2 expression as potential prognostic and predictive markers in colorectal-cancer. Virchows Arch. 2003, 443, 139–145. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. Fibroblast growth factors. Genome Biol. 2001, 2, 1–12. [Google Scholar] [CrossRef]
- Katoh, M.; Nakagama, H. FGF receptors: Cancer biology and therapeutics. Med. Res. Rev. 2014, 34, 280–300. [Google Scholar] [CrossRef]
- Chen, Y.; Li, X.; Eswarakumar, V.P.; Seger, R.; Lonai, P. Fibroblast growth factor (FGF) signaling through PI 3-kinase and Akt/PKB is required for embryoid body differentiation. Oncogene 2000, 19, 3750–3756. [Google Scholar] [CrossRef]
- Chae, Y.K.; Ranganath, K.; Hammerman, P.S.; Vaklavas, C.; Mohindra, N.; Kalyan, A.; Matsangou, M.; Costa, R.; Carneiro, B.; Villaflor, V.M. Inhibition of the fibroblast growth factor receptor (FGFR) pathway: The current landscape and barriers to clinical application. Oncotarget 2017, 8, 16052. [Google Scholar] [CrossRef] [PubMed]
- Mahajanakatti, A.B.; Murthy, G.; Sharma, N.; Skariyachan, S. Exploring inhibitory potential of Curcumin against various cancer targets by in silico virtual screening. Interdiscip. Sci. 2014, 6, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Sathishkumar, N.; Karpagam, V.; Sathiyamoorthy, S.; Woo, M.J.; Kim, Y.-J.; Yang, D.-C. Computer-aided identification of EGFR tyrosine kinase inhibitors using ginsenosides from Panax ginseng. Comput. Biol. Med. 2013, 43, 786–797. [Google Scholar] [CrossRef] [PubMed]
- Rasyid, H.; Purwono, B.; Pranowo, H.D. Design of New Quinazoline Derivative as EGFR (Epidermal Growth Factor Receptor) Inhibitor through Molecular Docking and Dynamics Simulation. Indones. J. Chem. 2021, 21, 201–211. [Google Scholar] [CrossRef]
- Gómez-Ganau, S.; Castillo, J.; Cervantes, A.; de Julián-Ortiz, J.V.; Gozalbes, R. Computational Evaluation and In Vitro Validation of New Epidermal Growth Factor Receptor Inhibitors. Curr. Top. Med. Chem. 2020, 20, 1628–1639. [Google Scholar] [CrossRef]
- Sharda, S.; Khandelwal, R.; Adhikary, R.; Sharma, D.; Majhi, M.; Hussain, T.; Nayarisseri, A.; Singh, S.K. A Computer-Aided Drug Designing for Pharmacological Inhibition of Mutant ALK for the Treatment of Non-small Cell Lung Cancer. Curr. Top. Med. Chem. 2019, 19, 1129–1144. [Google Scholar] [CrossRef] [PubMed]
- Arulanandam, C.D.; Prathiviraj, R.; Kaveriyappan, G.R. Repurposing of an Antifungal Drug against Gastrointestinal Stromal Tumors. Biorxiv 2021. [Google Scholar] [CrossRef]
- Rocca, R.; Moraca, F.; Costa, G.; Talarico, C.; Ortuso, F.; Da Ros, S.; Nicoletto, G.; Sissi, C.; Alcaro, S.; Artese, A. In silico identification of piperidinyl-amine derivatives as novel dual binders of oncogene c-myc/c-Kit G-quadruplexes. ACS Med. Chem. Lett. 2018, 9, 848–853. [Google Scholar] [CrossRef]
- Zhu, J.; Li, K.; Xu, L.; Cai, Y.; Chen, Y.; Zhao, X.; Li, H.; Huang, G.; Jin, J. Discovery of novel selective PI3Kγ inhibitors through combining machine learning-based virtual screening with multiple protein structures and bio-evaluation. J. Adv. Res. 2022, 36, 1–13. [Google Scholar] [CrossRef]
- Liu, X.; Ma, X.H.; Tan, C.; Jiang, Y.; Go, M.; Low, B.C.; Chen, Y.Z. Virtual screening of Abl inhibitors from large compound libraries by support vector machines. J. Chem. Inf. Modeling 2009, 49, 2101–2110. [Google Scholar] [CrossRef]
- Singh, V.K.; Chang, H.-H.; Kuo, C.-C.; Shiao, H.-Y.; Hsieh, H.-P.; Coumar, M.S. Drug repurposing for chronic myeloid leukemia: In silico and in vitro investigation of DrugBank database for allosteric Bcr-Abl inhibitors. J. Biomol. Struct. Dyn. 2017, 35, 1833–1848. [Google Scholar] [CrossRef]
- Kumar, H.; Raj, U.; Gupta, S.; Varadwaj, P.K. In-silico identification of inhibitors against mutated BCR-ABL protein of chronic myeloid leukemia: A virtual screening and molecular dynamics simulation study. J. Biomol. Struct. Dyn. 2016, 34, 2171–2183. [Google Scholar] [CrossRef]
- Corradi, V.; Mancini, M.; Manetti, F.; Petta, S.; Santucci, M.A.; Botta, M. Identification of the first non-peptidic small molecule inhibitor of the c-Abl/14-3-3 protein–protein interactions able to drive sensitive and Imatinib-resistant leukemia cells to apoptosis. Bioorganic Med. Chem. Lett. 2010, 20, 6133–6137. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Xie, P.; Marmorstein, R. Identification of BRAF inhibitors through in silico screening. J. Med. Chem. 2008, 51, 6121–6127. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Umar, A.B.; Uzairu, A.; Shallangwa, G.A.; Uba, S. In silico evaluation of some 4-(quinolin-2-yl) pyrimidin-2-amine derivatives as potent V600E-BRAF inhibitors with pharmacokinetics ADMET and drug-likeness predictions. Future J. Pharm. Sci. 2020, 6, 1–10. [Google Scholar] [CrossRef]
- Kulkarni, A.M.; Kumar, V.; Parate, S.; Lee, G.; Yoon, S.; Lee, K.W. Identification of New KRAS G12D Inhibitors through Computer-Aided Drug Discovery Methods. Int. J. Mol. Sci. 2022, 23, 1309. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhuang, C.; Lu, J.; Jiang, Y.; Sheng, C. Discovery of novel KRAS-PDEδ inhibitors by fragment-based drug design. J. Med. Chem. 2018, 61, 2604–2610. [Google Scholar] [CrossRef]
- Ishola, A.A.; Adewole, K.E. In Silico Screening Reveals Histone Deacetylase 7 and ERK1/2 as Potential Targets for Artemisinin Dimer and Artemisinin Dimer Hemisuccinate. Curr. Drug Discov. Technol. 2019, 17, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Pathania, S.; Singh, P.K.; Narang, R.K.; Rawal, R.K. Identifying novel putative ERK1/2 inhibitors via hybrid scaffold hopping–FBDD approach. J. Biomol. Struct. Dyn. 2021, 39, 1–16. [Google Scholar] [CrossRef]
- Xi, D.; Niu, Y.; Li, H.; Noha, S.M.; Temml, V.; Schuster, D.; Wang, C.; Xu, F.; Xu, P. Discovery of carbazole derivatives as novel allosteric MEK inhibitors by pharmacophore modeling and virtual screening. Eur. J. Med. Chem. 2019, 178, 802–817. [Google Scholar] [CrossRef]
- Ashtekar, S.S.; Bhatia, N.M.; Bhatia, M.S. Exploration of Leads from Natural Domain Targeting HER2 in Breast Cancer: An In-Silico Approach. Int. J. Pept. Res. Ther. 2019, 25, 659–667. [Google Scholar] [CrossRef]
- Pasha, M.K.; Jabeen, I.; Samarasinghe, S. 3D QSAR and pharmacophore studies on inhibitors of insuline like growth factor 1 receptor (IGF-1R) and insulin receptor (IR) as potential anti-cancer agents. Curr. Res. Chem. Biol. 2022, 2, 100019. [Google Scholar] [CrossRef]
- Muthumanickam, S.; Indhumathi, T.; Boomi, P.; Balajee, R.; Jeyakanthan, J.; Anand, K.; Ravikumar, S.; Kumar, P.; Sudha, A.; Jiang, Z. In silico approach of naringin as potent phosphatase and tensin homolog (PTEN) protein agonist against prostate cancer. J. Biomol. Struct. Dyn. 2020, 40, 1629–1638. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.-H.; Cheng, T.-C.; Leu, Y.-L.; Chuang, K.-H.; Tzou, S.-C.; Chen, C.-S. Discovery of Akt kinase inhibitors through structure-based virtual screening and their evaluation as potential anticancer agents. Int. J. Mol. Sci. 2015, 16, 3202–3212. [Google Scholar] [CrossRef] [PubMed]
- Saidel, M.É.; dos Santos, K.C.; Nagano, L.F.P.; Montanari, C.A.; Leitão, A. Novel anti-prostate cancer scaffold identified by the combination of in silico and cell-based assays targeting the PI3K-AKT-mTOR pathway. Bioorganic Med. Chem. Lett. 2017, 27, 4001–4006. [Google Scholar] [CrossRef]
- Peddi, S.R.; Sivan, S.K.; Manga, V. Discovery and design of new PI3K inhibitors through pharmacophore-based virtual screening, molecular docking, and binding free energy analysis. Struct. Chem. 2018, 29, 1753–1766. [Google Scholar] [CrossRef]
- Zahler, S.; Tietze, S.; Totzke, F.; Kubbutat, M.; Meijer, L.; Vollmar, A.M.; Apostolakis, J. Inverse In Silico Screening for Identification of Kinase Inhibitor Targets. Chem. Biol. 2007, 14, 1207–1214. [Google Scholar] [CrossRef]
- Yang, W.; AbdulHameed, M.D.M.; Hamza, A.; Zhan, C.-G. New inhibitor of 3-phosphoinositide dependent protein kinase-1 identified from virtual screening. Bioorganic Med. Chem. Lett. 2012, 22, 1629–1632. [Google Scholar] [CrossRef][Green Version]
- Xiao, Z.; Riccardi, D.; Velazquez, H.A.; Chin, A.L.; Yates, C.R.; Carrick, J.D.; Smith, J.C.; Baudry, J.; Quarles, L.D. A computationally identified compound antagonizes excess FGF-23 signaling in renal tubules and a mouse model of hypophosphatemia. Sci. Signal. 2016, 9, ra113. [Google Scholar] [CrossRef]
- Velazquez, H.A.; Riccardi, D.; Xiao, Z.; Quarles, L.D.; Yates, C.R.; Baudry, J.; Smith, J.C. Ensemble docking to difficult targets in early-stage drug discovery: Methodology and application to fibroblast growth factor 23. Chem. Biol. Drug Des. 2018, 91, 491–504. [Google Scholar] [CrossRef]
- Wahlberg, S.S.; Schmeits, J.; Thomas, G.; Loda, M.; Garber, J.; Syngal, S.; Kolodner, R.D.; Fox, E. Evaluation of microsatellite instability and immunohistochemistry for the prediction of germ-line MSH2 and MLH1 mutations in hereditary nonpolyposis colon cancer families. Cancer Res. 2002, 62, 3485–3492. [Google Scholar] [PubMed]
- Evans, D.G.; Walsh, S.; Hill, J.; McMahon, R.T. Strategies for identifying hereditary nonpolyposis colon cancer. Semin. Oncol. 2007, 34, 411–417. [Google Scholar]
- Kawasaki, T.; Ohnishi, M.; Nosho, K.; Suemoto, Y.; Kirkner, G.J.; Meyerhardt, J.A.; Fuchs, C.S.; Ogino, S. CpG island methylator phenotype-low (CIMP-low) colorectal cancer shows not only few methylated CIMP-high-specific CpG islands, but also low-level methylation at individual loci. Mod. Pathol. 2008, 21, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Brieger, A.; Adryan, B.; Wolpert, F.; Passmann, S.; Zeuzem, S.; Trojan, J. Cytoskeletal scaffolding proteins interact with Lynch-Syndrome associated mismatch repair protein MLH1. Proteomics 2010, 10, 3343–3355. [Google Scholar] [CrossRef] [PubMed]
- Hinrichsen, I.; Ernst, B.P.; Nuber, F.; Passmann, S.; Schäfer, D.; Steinke, V.; Friedrichs, N.; Plotz, G.; Zeuzem, S.; Brieger, A. Reduced migration of MLH1 deficient colon cancer cells depends on SPTAN1. Mol. Cancer 2014, 13, 1–12. [Google Scholar] [CrossRef]
- Ackermann, A.; Schrecker, C.; Bon, D.; Friedrichs, N.; Bankov, K.; Wild, P.; Plotz, G.; Zeuzem, S.; Herrmann, E.; Hansmann, M.-L. Downregulation of SPTAN1 is related to MLH1 deficiency and metastasis in colorectal cancer. PLoS ONE 2019, 14, e0213411. [Google Scholar]
- Ahuja, N.; Mohan, A.L.; Li, Q.; Stolker, J.M.; Herman, J.G.; Hamilton, S.R.; Baylin, S.B.; Issa, J.-P.J. Association between CpG island methylation and microsatellite instability in colorectal cancer. Cancer Res. 1997, 57, 3370–3374. [Google Scholar]
- Shibata, D.M.; Sato, F.; Mori, Y.; Perry, K.; Yin, J.; Wang, S.; Xu, Y.; Olaru, A.; Selaru, F.; Spring, K. Hypermethylation of HPP1 is associated with hMLH1 hypermethylation in gastric adenocarcinomas. Cancer Res. 2002, 62, 5637–5640. [Google Scholar]
- Wallace, S.S.; Murphy, D.L.; Sweasy, J.B. Base excision repair and cancer. Cancer Lett. 2012, 327, 73–89. [Google Scholar] [CrossRef]
- Hazra, T.K.; Hill, J.W.; Izumi, T.; Mitra, S. Multiple DNA glycosylases for repair of 8-oxoguanine and their potential in vivo functions. Prog. Nucleic Acid Res. Mol. Biol. 2001, 68, 193–205. [Google Scholar]
- Godwin, R.C.; Melvin, R.; Salsbury, F.R. Molecular dynamics simulations and computer-aided drug discovery. In Computer-aided Drug Discovery; Springer: Berlin/Heidelberg, Germany, 2015; pp. 1–30. [Google Scholar]
- Negureanu, L.; Salsbury, F.R., Jr. The molecular origin of the MMR-dependent apoptosis pathway from dynamics analysis of MutSα-DNA complexes. J. Biomol. Struct. Dyn. 2012, 30, 347–361. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Screening Type | Ligands | Receptor/PDB ID | Summaries | Ref. |
|---|---|---|---|---|
| A set of docking methods followed by molecular dynamic simulations | ZINC13, NCI, and Maybridge databases | APC-Asef/3NMZ MAI peptides/PDB: 5IZA, 5IZ6, 5B6G, 5IZ9, and 5IZ8 | The main target was to prevent APC-Asef interaction that spreads CRC to the entire colon. The induced fit was performed on compounds with a variety of chemical scaffolds and direct interaction with Arg549 and other active site residues. Because of the strong interactions with Arg549, visible conformational changes occur, allowing for proper positioning inside the peptide binding region. The top hit inside the APC-Asef binding region was subjected to specific MD simulations, which revealed substantial interactions necessary for biochemical recognition in a dynamic microenvironment. | [59] |
| Structure-based virtual screening by rigid and flexible docking followed by in vitro assays | 13.3 million drug-like and 89.4 natural product compounds | TNKS-1/2RF5 TNKS-2/3KR8 | This study targets the WNT/β-catenin pathway by developing inhibitors against tankyrase 1/2. Out of 11 structurally representative top hits, one compound was selected for experimental analysis | [60] |
| Structure-based virtual screening followed by biological assays | 500,000 structurally diverse compounds | Homology modeling of the closely related Smoothened receptor (PDB ID: 4JVK) | The study’s aim was to screen ligands targeting the transmembrane domain of frizzled protein-7 Fzd7. Fzd7 inhibitors have been identified in six small molecule drugs. With IC50 values in the sub-micromolar range, the strongest hit, SRI37892, effectively suppressed Wnt/Fzd7 signaling. | [61] |
| High-throughput, and ligand docking-based virtual screening | 20,000 natural products | Human Telomeric DNA/1KF1 | Using the X-ray crystal structure of the intramolecular human telomeric G-quadruplex DNA, a model of the intramolecular G-quadruplex loop isomer of NHE III1 was created. The aim of this study is to stabilize the c-myc G-quadruplex. The naphthopyrone fonsecin B was found the top candidate. | [62] |
| Binding site identification, drug design, and large-scale virtual screening | 4.7 million compounds from ZINC12 drug-like subset | Myc-Max recognizing DNA/1NKP | A binding site on the structurally organized Myc-Max complex’s DNA-binding domain was discovered. Computer-aided drug design was employed to identify a small molecule that can inhibit Myc-Max functionality. In vitro analysis found a chemically different scaffold inhibitor than the previously identified Myc inhibitor. | [63] |
| A comprehensive molecular docking and bioinformatics analysis followed by in vitro assays | NSC765600 and NSC765691, derived from diflunisal and fostamatinib respectively | CCND1/6P8G CDK4/4O9W PLK1/2W9F and CD44/1UUH | CCND1/CDK4/PLK1/CD44 were identified as target genes for NSC765600 and NSC765691 compounds by target prediction tools. In numerous cancer types, the mRNA levels of CCND1/CDK4/PLK1/CD44 were greater in tumor tissues than in normal tissues. Protein-protein interaction networks among those genes have been shown after taking into account the gene neighborhood, gene fusion, gene co-occurrence, and the coexpression of CDK4 with CCND1, CD44, and PLK1, and CCND1 with PLK1 have been illustrated. The antiproliferative and cytotoxic effects of the 2 compounds against a panel of NCI-60 cancer cell lines have been illustrated. | [64] |
| A comprehensive molecular docking and bioinformatics analysis followed by in vitro assays | Sulfasalazine | KRAS/6BP1, MMP7/2Y6C and CD44/1UUH | The molecular docking revealed a unique interaction between sulfasalazine and KRAS, MMP&, and CD44. Bioinformatic analysis identified overexpression of those oncogenes in CRC cells. The synergistic effects of the sulfasalazine and cisplatin were successful in reducing cell viability, colony, and sphere formation in CRC cell lines. Sulfasalazine therapy reduced KRAS/MMP7/CD44 expression in CRC cell lines in a dose-dependent fashion. | [65] |
| Molecular docking and virtual screening followed by in vitro and in vivo assays | 13,000 diverse small molecules from the ZINC database | ND | 68 compounds were identified from the screening to interact with the binding site of α5β1-integrin. By inhibiting the urokinase receptor/integrins interaction, 2-(Pyridin-2-ylamino)-quinolin-8-ol and 2,2′-(methylimino)di (8-quinolinol) suppressed ERK activation. In vivo, these two drugs suppressed ERK activation, tumor development, and metastasis in a model head and neck cancer. | [66] |
| Protein binding pocket prediction and structure-based virtual screening | 5000 chemical compounds collected from ZINC were chosen based on structural similarity indices to the four ligand probes | GSK3β/3DU8 | A protein binding pocket screening was done on an X-ray model of human GSK3 beta using the geometric analysis via the Voronoi tessellation algorithm. Pocket geometry is the most important factor in ligand binding. Using molecular docking to find probable binding sites yielded comparable results to protein pocket prediction. | [67] |
| Computational drug-repositioning approach for identifying novel anti-cancer agents | 973,296 chemical–gene interactions from Comparative Toxicogenomics Database including 7570 chemicals/drugs and 20,116 genes | ND | DrugPredict platform was employed to repurpose chemicals and drugs for endothelial ovarian cancer. Indomethacin decreases cell viability and promotes apoptosis in patients with primary high grade severe cancer-derived cell lines. Because it inhibits β-catenin and represses multiple Wnt signaling targets, such as Lgr5, TCF7, and Axin2, it proved effective against platinum-resistant ovarian cancer cells. | [68] |
| Virtual screening by molecular docking followed by in vitro assays. | 1990 small molecules from the National Cancer Institute database | β-catenin/Tcf4 complex (PDB/1JPW chain A) | Site A hotspot on beta-catenin was chosen as a virtual screening pharmacophore. The top-ranked molecule has effectively reduced the β-catenin/Tcf4 driven activity in the CRC cell line. It prevents β-catenin from directly binding to Tcf4 and suppresses the expression and activity of Wnt/β-catenin target genes and gene products. | [69] |
| New binding pockets detection, structure- and ligand-based virtual screening, molecular dynamics simulations, and binding free energy calculations | 1880 structures from diversity Set II were obtained from the ZINC database. 50 structures from the above were selected for similarity screening from the ZINC15 database | Domain 1 and 2 of LRP6/4DG6, domains 3 and 4 of LRP6/4A0P | After applying Lipinski’s rule of five and flexible molecular docking, ten candidate compounds were found, five of which were for each binding pocket. It has been concluded that ZINC03954520, ZINC01729523, ZINC03898665, ZINC13152226, ZINC26730911, and ZINC01069082 are possibly appropriate compounds for inhibiting LRP6 using RMSD, RMSF, the radius of gyration, and MMPBSA binding free energy calculations. | [70] |
| Ensemble docking-based virtual screening | 3520 natural products | Tp53/1TSR | Natural products were screened to identify a ligand that stabilizes the function of the wild type p53 by targeting its Loop1/Sheet3 pocket. Due to the flexibility of Loop1, ensemble docking for 7 conformations was performed. Compound torilin not only enhanced p53 activity but also p21 protein production, which is downstream of p53. | [71] |
| The Nanoluc/YFP-based bioluminescence resonance energy transfer (BRET) test was combined with structure-based virtual screening and followed by | Commercially available protein-protein interaction small molecules from ChemDiv | Bcl-xL/2YXJ | The purpose of this study is to find inhibitors of Bax/Bcl-xL and Bak/Bcl-xL interactions. Based on BRET techniques, a screening platform for Bak/Bcl-xL and Bax/Bcl-xL interactions were developed and identified inhibitors of both interactions. ABT-737, an inhibitor for Bcl-xL, was employed as a positive control drug to identify more inhibitors. 50 Compounds were selected via virtual screening that targeted the ABT-737 binding site and only BIP-A1001 and BIP-A2001 showed dose-response inhibition for the Bax and Bcl-xL interactions within low micromolar concentration | [72] |
| Pharmacophore- and structure-based virtual screening | 582,474 compounds from TimTec Compound Libraries | MDM2/3JZK | Based on a conventional Mdm2 inhibitor, a set of pharmacophoric characteristics was developed and utilized to screen a ligand library, and the potential inhibitors were docked into the receptor to check their potential to stop MDM2-p53 interaction. Triazolopyrimidine was among top 5 compounds that bind to the MDM2 active site. | [73] |
| Pharmacophore virtual screening and molecular dynamic simulations. | National Cancer Institute and ZINC Libraries | Caspase-9/1JXQ | Due to a substantial missing section of the crystallographic structure, the caspase-9 structure was refined. Four structures were employed with PDB IDs of 4DGE, 4DGA, 2PBj, and 1Z9H to build the missing part. For evaluating the ligands’ forms of interaction in the protein binding pocket, a pharmacophore model approach was applied. The compound selected from pharmacophore screening and rigid docking was further checked for binding pose stability through MDS with stable hydrogen bonds. | [74] |
| Structure- and ligand-based 3D pharmacophore models followed by in vitro assays | 50,000 compounds from Maybridge database | Caspase-3/1pau | Using 25 various compounds, a ligand-based pharmacophore model was generated. Further docking experiments on known inhibitors revealed that the amino acids Arg207, Ser209, and Trp214 found in the active region of caspase-3 are critical for ligand binding. From this study, methyl piperazine was identified as a non-peptide inhibitor against Caspase-3. | [75] |
| Homology modeling for predicting target protein sequence and virtual screening for finding inhibitors | Mcule database was used for small molecule virtual screening | TNFRSF10B/2ZB9, 3NKE, 3NKD | TNFRSF10B best model was built by using 2ZB9 template and assessed by 3 different software with high scores. An evolutionary tool was employed to construct a neighbor-joining tree of the target gene based on TNFRSF10A, TNFRSF10D and TNFRSF10B genes. Virtual screening revealed 4 lead compounds with inhibitory activities against the mutated TNFRSF10B activity. To investigate the highly interacting proteins of the target protein, a functional partner network of the TNFRSF10B protein was created. TNFSF10 was utilized as a ligand-protein in protein-protein docking because it had the greatest interaction. | [76] |
| Virtual screening (pharmacophoric molecular identification), molecular docking, followed by molecular dynamics and experimental assays | 8 million compounds from a clean and drug-like subset of the ZINC database, and 260,071 compounds from the NCI-2003 library | The crystal structure of TGF-b3 in complex with the extracellular domain of TßRII/1KTZ | The main purpose of this study was to discover drugs that antagonize TGF-b signaling by protein-protein competitively inhibiting TGF-b binding to TßRII. Two compounds were found with a quite good binding affinity (26 and 18 µM). Three compounds were found to bind to SS1 on TßRII over the duration of the simulations, according to molecular dynamics trajectories. The 3 compounds share the chemical property of being aromatic and fairly flat | [77] |
| Shape-based virtual screening followed by experimental work and X-ray crystallization study for TGFb-1 inhibitor | 200,000 Compounds in the multi-conformational Catalyst database | The pharmacophoric query was constructed using SB203580′s conformation as shown in the X-ray combination with p38 (PDB: 1a9u). | The pharmacophore features were chosen based on a derived alignment of p38-SB203580 (a triarylimidazole) with TβRI’s ATP site. 87 compounds were identified satisfying both the shape constraint and pharmacophore features. With IC50 of 60 nM, HTS466284 was found to be a strong, non-toxic inhibitor of TßRI in vitro and in cell culture. The aromatic contacts of the HTS466284 indicated by the shape question are satisfied by the quinoline, pyrazole, and pyridyl rings. | [78] |
| De novo synthesis of caspase-6 inhibitors using neural network, and molecular docking-based ligand screening | 2.4 million molecules were retrieved from PubMed to train the RNN model | caspase-6/3OD5 | For de novo molecular design of caspase-6 inhibitors, a gated recurrent unit (GRU)-based RNN network was merged with transfer learning and classical machine learning. A prediction model was trained on known caspase-6 inhibitors and decoys. The 6927 synthesized inhibitors that were developed share the same chemical space as the known caspase-6 inhibitors. The synthesized inhibitors are predicted to have comparable binding mechanisms to the known 577 caspase-6 inhibitors. | [79] |
| VEGFR2 Inhibitor | PDB ID | Resolution | Comments | Inhibitor Type/other RTKs Inhibition |
|---|---|---|---|---|
| Sorafenib | 4ASD | 2.03 Å | Mutated | Type IIA, also inhibits VEGFR2/3, BRaf, CRaf, mutated BRaf, Kit, Flt3, RET and PDGFRB |
| Axitinib | 4AG8 | 1.95 Å | Mutated | Type IIA, also inhibits VEGFR2/3, PDGFRB |
| Sunitinib | 4AGD | 2.81 Å | Mutated | Type I, also inhibits PDGFRB/alpha, VEGFR2/3, Kit, Flt3, CSF-1R, and RET |
| Pazopanib | 3CJG | 2.25 Å | Not mutated | Type I, also inhibits PDGFRB/alpha, VEGFR2/3, FGFR1/3, Kit, Lck, Fms, Itk. |
| Lenvatinib | 3WZD | 1.57 Å | Mutated | Type I1/2A, also inhibits PDGFR, VEGFR2/3, FGFR, Kit, RET |
| PF-00337210 | 2XIR | 1.50 Å | Mutated | Type II inhibitor |
| CHEMBL272198 | 3B8R | 2.70 Å | Mutated | Type I, also inhibits Aurora B, ABL1, c-MET, Tie2, Lck, Lyn |
| CHEMBL194911 | 1YWN | 1.71 Å | Mutated | Tie-2 and VEGFR2 dual inhibitors |
| 2-Anilino-5-aryloxazole | 1Y6A | 2.10 Å | Not mutated | |
| LENVATINIB | 3WZD | 1.57 Å | Mutated | Also inhibits VEGFR2/3, PDGFR, FGFR, Kit, RET |
| TIVOZANIB | 4ASE | 1.83 Å | Mutated | Pan-inhibitor of VEGF receptors |
| MOTESANIB | 3EFL | 2.20 Å | Mutated | Inhibitor of VEGF, PDGF, and Kit receptors |
| Screening Method | Database Size | Summaries | Ref. |
|---|---|---|---|
| High throughput virtual screening for EGFR inhibitors | 400,000 compound library of tyrosine kinase inhibitors from ChemBioBase | Indenopyrazole framework was reported as cyclin-dependent kinase inhibitor. The framework was discovered to be one of the most prevalent structures among the top 100 scoring compounds, prompting the development of a series of indenopyrazoles. Interestingly, some of the synthesized compounds suppressed VEGFR-2 tyrosine kinase at 1 micromolar. | [97] |
| Molecular docking, multicomplex pharmacophore and fingerprint-based 2D similarity in an individual and a combined manner. | 409 actives and 24,680 decoys | In a retrospective comparison, the three combined approaches outperformed 43 of 45 previously published articles. The results showed that the 2D fingerprint ECFP 4 outperformed the multicomplex pharmacophore Glide SP. In self- and cross-docking studies, Glide SP docking with PDB ID: 3EWH was shown to be the best choice for molecular docking-based screening. | [98] |
| Molecular flexible docking followed by virtual screening, pharmacophore and ligand energy inspection | 284 compounds from the PubChem database were found with the highest similarity with the best active compound. | Among 23 inhibitors, compound CHEMBL346631 (Pubchem CID: 9936664) was identified as the highest efficient ligand interaction with VEGFR2. The greatest affinity against Renal Cell Carcinoma was found in the dicarboxamide (SCHEMBL469307) from the PubChem database. The original inhibitor chemical is more stable in the receptor protein than the virtually screened one. | [99] |
| Virtual screening followed by molecular dynamics and binding free energy decomposition calculations | 30,792 natural derivatives from the ZINC 15 database | Three 1-azabicyclo [2.2.2] octane-3-carboxamide derivatives with excellent affinity were discovered using the VEGFR2 inhibitor as a reference to uncover more inhibitors from natural resources. These potential molecules might be VEGFR-2 inhibitors, according to the RMSD study of each VEGFR-2–inhibitor combination, in addition, they showed low binding free energy and decomposition energy for each VEGFR-2–inhibitor interaction. | [100] |
| Virtual screening by using homology models, pharmacophore modeling and in vitro studies | 46 derivatives of 2-anilino-5-phenyloxazoles | As VEGFR2 inhibitors, two 2-anilino-5-phenyloxazole derivatives were shown to be effective. Because the crystal structure of VEGFR2 was not available at the time of this work, homology models were employed instead. At the ATP-binding region, the compounds shared a pharmacophore and established hydrogen bonds with the backbone’s Cys919. The activation loop was disordered between residues 1046 and 1065 in both crystal structures, indicating that residues beyond this region were not directly contributing to the binding affinity. | [101] |
| Structure-based pharmacophore models followed by virtual screening of several commercial databases. | Key Organics (48,768), Maybridge (94,448), Otava (69,700), Life Chemicals (248,445), Asinex (358,126) | Following pharmacophore modeling, 16,000 and 19,000 compounds were identified as type I and type II inhibitors respectively. A total of 100 compounds were taken to biological testing after the flexible docking. Three compounds with excellent binding and drug-like characteristics were discovered. The 3-membered ring of the triazinoindole derivative (IC50 = 1.6 micromolar) establishes two standard hydrogen bonds with the backbone NH and the carbonyl oxygen of Cys917 in the kinase motif (type II). | [102] |
| De novo structure-based identification methods followed by in vitro assays | A range of pyrazole-based compounds was designed to be employed. | Using a structure-based de novo design, the researchers discovered a new VEGFR2 inhibitor scaffold. As a multi-tyrosine kinase inhibitor, this resulted in the development of a pyrazole-based molecule (JK-P3) that targets VEGFR2 kinase activity and angiogenesis while also inhibiting FGFR kinases in vitro. | [103] |
| Screening Type | Ligands | Receptor/PDB ID | Findings | Ref. |
|---|---|---|---|---|
| Structure-based screening | Curcumin, litreol, triterpene | EGFR/3POZ | The predicted pharmacological features of curcumin were found to be better than litreol and triterpene. | [126] |
| Pharmacophore and docking screening for Korean P. ginseng active compounds | 128 ginsenosides | EGFR/1M17 | Molecular docking studies identified 14 hit molecules based on scoring function and suitable binding orientation with critical active site amino acids. | [127] |
| The combination of docking and molecular dynamics simulation had been carried out to design new quinazoline derivatives compounds | Erlotinib, Afatinib, and WZ4002 were optimized into A1, B1, and C1 lead compounds. | EGFR/1M17 | Molecular docking was successful in designing new potential compounds using the pharmacophore model of lead compounds. The interaction between lead compounds and the receptor was evaluated by MMGBSA. A1 is a potential compound as an EGFR inhibitor. | [128] |
| Structure-based virtual screening | 615,462 compounds were obtained from the ZINC database | EGFR/1M17 | Six compounds displayed good effects when compared with erlotinib at 30 μM. At 2 μM, one compound showed inhibiting effects close to those from erlotinib. | [129] |
| Structure-based virtual screening for non-small cell lung cancer (NSCLC) | 93 million compounds obtained from the PubChem database | AKT/3AOX | The virtual screening showed that (PubChem CID123449015) is more efficient to be a better prospective candidate for NSCLC treatment having better pharmacological profile than the pre-established compound PubChem CID71721648 with low toxicity and cytotoxicity | [130] |
| Structure-based screening for repurposing of an antifungal drug against gastrointestinal stromal tumors | A docking with 36 antifungal drugs and 5 antineoplastic drugs. | PDGFRA/5K5X | Itraconazole was predicted as a better PDGFRA inhibitor among all the computationally tested drugs. The binding affinity of Imatinib was close to that of Itraconazole. | [131] |
| Structure-based virtual screening toward the experimental DNA G-quadruplex (G4s) structures of c-myc and c-Kit | 693,000 commercial compounds obtained from Asinex | c-myc/1XAV and 2L7V c-Kit/4WO2, 4WO3 and 2O3M | Ensemble docking simulations resulted in 442 for c-myc and 634 molecules for c-Kit G4s. The 76 shared hits in complex with both receptors investigated for their thermodynamic behavior. Three N-(4-piperidinylmethyl)amine derivatives effectively stabilized both G-quadruplex oncogene promoter structures | [132] |
| Machine learning-based virtual screening with multiple PI3Kγ protein structures. | 87 crystallographic structures of PI3Kγ-inhibitor complexes | PI3Kγ/4wwo, 5g2n, 3r7q, 3ml8, 2a5u, 4flh, 4fjy, 4ps7, 2v4l, 3dbs | The developed NBC model integrating ten PI3Kγ proteins showed a satisfactory prediction power against PI3Kγ inhibitors. JN-KI3 ligand exhibits the most potent selective inhibitory bioactivity. The results of molecular docking, MD simulation, and free energy calculations reveal that JN-KI3 contains the highest binding free energy against PI3Kγ than Class IA isoforms. | [133] |
| A support vector machine as a virtual screening tool for searching Abl inhibitors from large compound libraries | 13 and a half Million PubChem, 168K MDDR, and 6 638 MDDR molecules | Similarity screening with known Abl inhibitors | The model shows substantial capability in identifying Abl inhibitors at substantially lower false-hit rate. 29 072 inhibitors (0.21%) of 13.5 M PubChem lib. 659 inhibitors (0.39%) of 168K MDDR lib. 330 (5.0%) of 6 638 MDDR lib. | [134] |
| A structure- and ligand-based virtual screening were involved to investigate ligands targeting the allosteric site of Abl kinase | 1424 compounds from DrugBank database v3.0 | Abl/3K5V | A series of in silico techniques like virtual screening, molecular dynamics, and steered molecular dynamic simulations were employed. Gefitinib was identified as an inhibitor for over-expressing Bcr-Abl protein in the K562 CML cell line. It was found that the combination of imatinib and gefitinib produced a synergistic antiproliferative effect in such a cell line. | [135] |
| High Throughput Virtual Screening, Standard Precision, and Extra Precision docking, followed by molecular dynamic simulations. | Natural product libraries of ZINC database and Drug bank database | Abl1/3QRJ | Comparative docking analysis was also carried out on the active site of the ABL tyrosine kinase receptor with a reported reference inhibitor. The purpose was to identify inhibitors for mutated BCR-ABL protein. Six inhibitors were further validated and analyzed through pharmacokinetics properties and a series of ADMET parameters by in-silico methods | [136] |
| Structure-based pharmacophore modeling, virtual screening, and molecular docking simulations | 200,000 commercially compounds | 14-3-3σ isoform/1YWT | The purpose was to design a small molecule able to inhibit protein–protein interactions between 14-3-3 and c-Abl. BV02 which was designed by in silico process is a terephthalic acid derivative and was found as an anti-proliferative on human leukemia cells either sensitive or resistant to Imatinib due to the T315I mutation. It also mediates c-Abl release from 14-3-3 protein. | [137] |
| High throughout virtual screening for calculating the binding score, hydrogen bonds, and hydrophobic complementarity, and free energy of binding. | 300,000 molecules from the SPECS subset from the Zinc. The database was filtered down to 90,000 for compounds with a logS value of greater than—4 for better solubility | BRaf/2FB8 | Identification of a series of purine-2,6-dione analogs that are selective for BRaf. The best lead compound inhibits the kinase activity of BRAF with an IC50 value of 1.7 μM and high selectivity compared to other protein and lipid kinases. | [138] |
| A virtual docking screening along with pharmacokinetics and drug-likeness predictions to find V600E-BRAF inhibitors. | Eleven derivatives of 4-(quinolin-2-yl) pyrimidin-2-amine. | V600E-BRAF/3OG7 | Two derivatives of 4-(quinolin-2-yl) pyrimidin-2-amine were found to have binding patterns similar to that of the vemurafenib the drug used against V600E-BRAF malignancies. It is also indicated that the compounds had more favorable ligand-protein interaction energy than vemurafenib at the binding site of V600E-BRAF | [139] |
| Computer-aided drug discovery including pharmacophore modeling, molecular docking, and molecular dynamic simulations for finding KRAS G12D potential inhibitors | More than 214,000 compounds from InterBioScreen and ZINC databases | KRAS G12D/6GJ8 | Firstly, a common pharmacophoric feature model was generated to extract the important criteria for KRAS inhibition. Ligands from databases were mapped on the model and mapped compounds were finally subjected to molecular docking and dynamic simulations. Four potential inhibitors displaying favorable stability with KRAS G12D were obtained, and only 2 of them showed better binding free energies. | [140] |
| Fragment-based drug design was conducted to inhibit KRAS-PDEδ protein–protein interactions | Quinazolinone and f benzimidazole fragments that are attached with PDE gamma | PDEδ/5×73 PDEδ/4JV6 | A combination of the two fragments produced novel quinazolinone-imidazole KRAS-PDEδ inhibitors. The experimental results approved the high binding affinity and antitumor activity of this compound. | [141] |
| Structure-based screening for molecular binding interactions binding affinities | 49 Artemisinin derivatives | HDAC2/3C0Z ERK1/4QTB ERK2/5NGU | It has been found that artemisinin dimer and artemisinin dimer hemisuccinate are promising anticancer drug agents, with better therapeutic efficacy than the standard inhibitors; ulixertinib and apicidin for the treatment of cancer via inhibition of ERK1, ERK2 and HDAC7. | [142] |
| Scaffold hopping, followed by fragment-based drug discovery and molecular dynamics simulations | The ERK2 inhibitor Ulixertinib was used for scaffold hopping. | ERK2/6GDQ | Initial hits retained from scaffold hopping usually are not enough for finding potential hits. FBDD can be employed for improving the binding potential of the hopped hits. The identified ligands showed good binding affinity similar to Ulixertinib | [143] |
| Structure-based pharmacophore study, followed by virtual screening | 200,158 compounds from the SPECS library | (MAP2K2) MEK2/3DV3 | The pharmacophore model of MEK1 inhibitors was constructed and used for a large-scale virtual screening. 13 virtual hits against MEK1 were obtained from the SPECS library. Then, a small library of carbazoles was synthesized based on one hit by bioisosteric replacement with IC50 at the micromolar level of allosteric inhibition of MEK2. | [144] |
| Docking analysis, and pharmacophore modeling study | 350 anticancer natural products. | HER2/3RCD | The hits were selected for the comparative study with the established HER2 inhibitors lapatinib and neratinib and interactions were studied. Finally, the pharmacophoric model was built. Eight natural products were obtained as hits by virtual screening and the comparative study. Results revealed that mostly anthocyanidins have the potential to target the kinase domain of HER2. | [145] |
| 2D, 3D quantitative structure–activity relationship (QSAR) and pharmacophore studies. | 725 hits World Drug Index (WDI) and 19,773 from ChemBridge. | IGF-1R/5HZN | Virtual screening of structurally diverse ligands of dual inhibitors of IGF-1R and insulin receptor. Alignment independent molecular descriptors were established for 3Dconformations. Dual potential inhibition of IGF-1R and IR was found for Tirofiban, Practolol, Edoxaban, Novobiocin | [146] |
| Structure-based virtual screening, molecular docking, molecular dynamics simulation and ADME prediction | A set of compounds from the NCI database in addition to naringin | PTEN/1D5R | Naringin was found to have better binding with PTEN among the 5 top-ranked compounds, docking scores and energy. The pharmacokinetic properties, Lipinski’s rule violations and binding stabilities of naringin have achieved the best results. | [147] |
| Structure-based virtual screening followed by biological evaluation | 35,367 compounds from SPECS | AKT-1/3MVH | Two compounds were identified as AKT inhibitors with micromolar activity and high selectivity index against cancer cell lines. | [148] |
| bi- and three-dimensional physical-chemical filtrations followed by phenotypic assays. | 5.9 million compounds from eMolecules database | mTOR/4JT5 PI3Kα/4JPS | The aminopyridine scaffold was found to target the PI3K-AKT-mTOR pathway especially the mTOR and PI3Kα proteins. This kind of drug discovery produced soluble, stable, membrane-permeable and highly selective compounds. | [149] |
| Pharmacophore-based virtual screening, molecular docking, and binding free energy calculations study. The structural design of cyclic peptides also included | Three databases; TOS Lab 39,988 CPP 1411 and ASINEX 31,500 compounds | PI3Kα/4KYN | compounds having indole and benzothiazole moieties can act as potent inhibitors against PI3Kα. Linear and cyclic compounds were found to be effective for PI3Kα. 1, 3, 4-oxadiazole-based cyclic peptides with tryptophan showed that cyclic peptides can act as good inhibitors against PI3Kα | [150] |
| Virtual inverse screening followed by biological assays | Indirubin-3′-oxime (IOX) and three derivatives of bromo-indirubin-3′oxime; 5BIO, 6BIO, and 7BIO were screened against 6000 protein binding sites | 5 BIO: CDK2/1pxo 6 BIO: GSK3B/1q41 PDK1/1oky 7 BIO: RIFK/1nb9 IOX: CDK2/1pxp | The purpose is to identify kinase targets for three derivatives of indirubin; 5BIO, 6BIO, and 7BIO. 5BIO, 6BIO (EF = 16) and IOX (EF = 20) show significant enrichment of their well-known targets (CDK2, CDK5, GSK-3β) in the top 1%. This process has led to the identification of the kinase PDK1 as an unknown target of the indirubin derivative 6BIO. | [151] |
| Ligand-based screening, rigid and flexible receptor-based docking, molecular adynamic simulations and binding free energy calculations | 688,086 compounds from ZINC 15 were reduced to 157,623 compounds after the pre-screening process. | PDK1/2BIY | The compounds were first screened by using the ligand-based method, then rigid docking, followed by flexible molecular docking using, molecular dynamics simulation and molecular mechanics/Poisson–Boltzmann surface area (MM-PBSA) binding free energy calculations. The resulted compound inhibited many other cancer cell lines, such as multiple myeloma, non-small cell lung cancer, colon cancer, CNS cancer cells, Melanoma cell, Ovarian cancer cells, Renal cancer cells, Prostate cancer, and Breast cancer cell lines. | [152] |
| Ensemble docking to disrupt protein–protein interactions followed by rescoring with the molecular mechanics Poisson–Boltzmann surface area (MM/PBSA) | 84,589 compounds were studied by Xiao et al. [153] | FGF23/2P39 In addition to the homology of three crystal structures, two of FGF19/1PWA and 2P23 one of FGF12/1Q1U FGFR1/1FQ9 | The target selected has only a partial crystal structure and no a priori knowledge of small-molecule binding sites. Two putative binding sites for drug-like antagonist molecules binding to the hormone FGF23 were identified using a multicenter ensemble docking technique. The use of MM/PBSA rescoring to further enhance the MED results demonstrates the value of going from lower-resolution approaches to higher-resolution methods for refining a predicted binding mode. This study also reveals how the steric crowding of pockets by side-chain conformers might affect docking outcomes. Authors hypothesized that the protein–protein interface is being drugged and not a distal pocket that would indicate allosteric signaling | [154] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moshawih, S.; Lim, A.F.; Ardianto, C.; Goh, K.W.; Kifli, N.; Goh, H.P.; Jarrar, Q.; Ming, L.C. Target-Based Small Molecule Drug Discovery for Colorectal Cancer: A Review of Molecular Pathways and In Silico Studies. Biomolecules 2022, 12, 878. https://doi.org/10.3390/biom12070878
Moshawih S, Lim AF, Ardianto C, Goh KW, Kifli N, Goh HP, Jarrar Q, Ming LC. Target-Based Small Molecule Drug Discovery for Colorectal Cancer: A Review of Molecular Pathways and In Silico Studies. Biomolecules. 2022; 12(7):878. https://doi.org/10.3390/biom12070878
Chicago/Turabian StyleMoshawih, Said, Ai Fern Lim, Chrismawan Ardianto, Khang Wen Goh, Nurolaini Kifli, Hui Poh Goh, Qais Jarrar, and Long Chiau Ming. 2022. "Target-Based Small Molecule Drug Discovery for Colorectal Cancer: A Review of Molecular Pathways and In Silico Studies" Biomolecules 12, no. 7: 878. https://doi.org/10.3390/biom12070878
APA StyleMoshawih, S., Lim, A. F., Ardianto, C., Goh, K. W., Kifli, N., Goh, H. P., Jarrar, Q., & Ming, L. C. (2022). Target-Based Small Molecule Drug Discovery for Colorectal Cancer: A Review of Molecular Pathways and In Silico Studies. Biomolecules, 12(7), 878. https://doi.org/10.3390/biom12070878