
Kallikrein-Related Peptidase 6 (KLK6) as a Contributor toward an Aggressive Cancer Cell Phenotype: A Potential Role in Colon Cancer Peritoneal Metastasis

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Reverse Transcription Polymerase Chain Reaction (RT-PCR)
2.4. Enzyme-Linked Immunosorbent Assay (ELISA) for KLK6
2.5. Intracellular Calcium Measurement
2.6. Immunofluorescence Staining
2.7. Western Blot Analysis
2.8. Colony Formation Assay
2.9. Cell Adhesion Assay
2.10. Spheroid Formation
2.11. Tissue Immunohistochemistry
2.12. Peritoneal Fluid
2.13. Statistical Analysis
3. Results
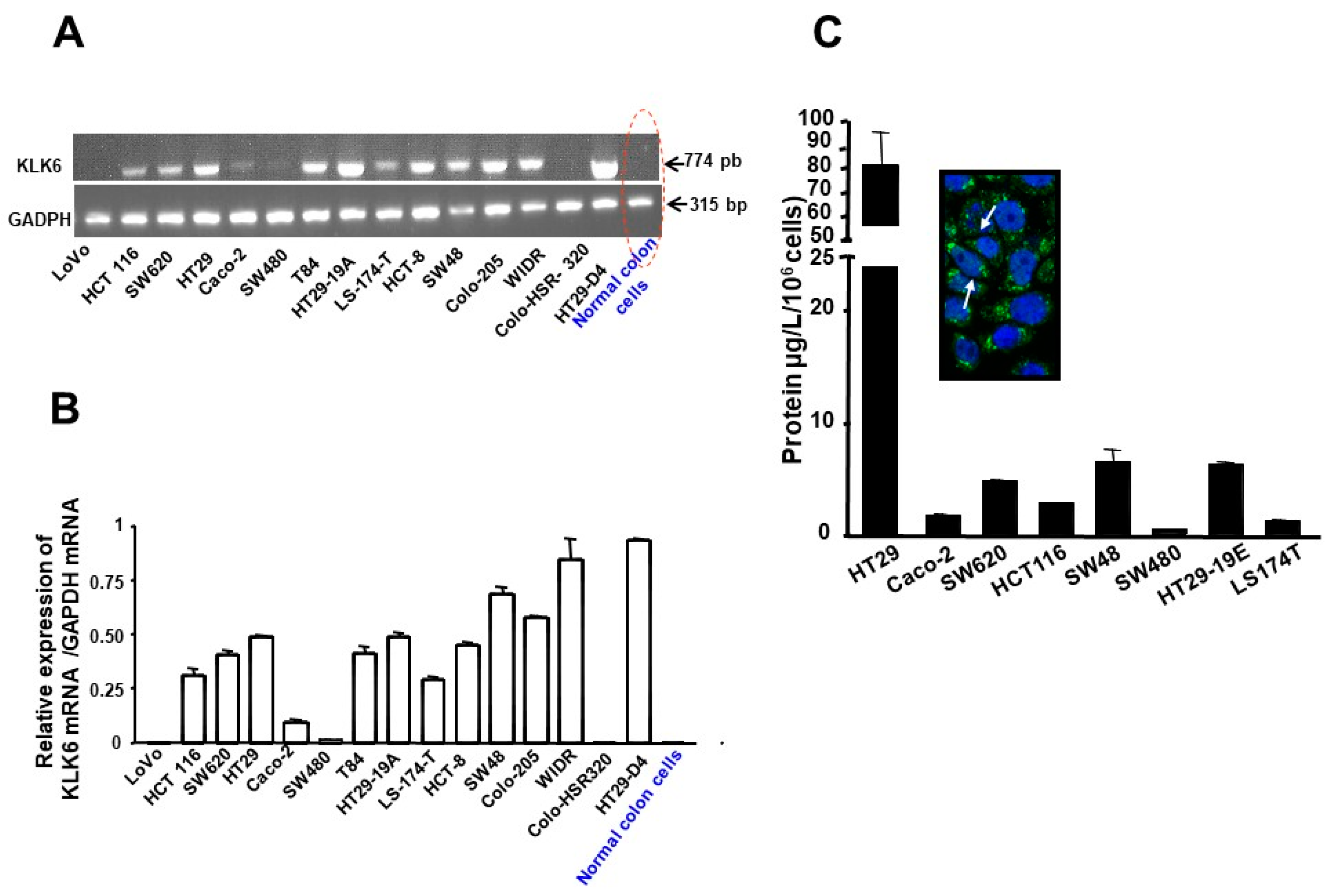
3.1. Expression Patterns of KLKs in Human Colon Cancer Cell Lines
3.2. Kallikrein 6 Expression in Human Colon Cancer Cell Lines
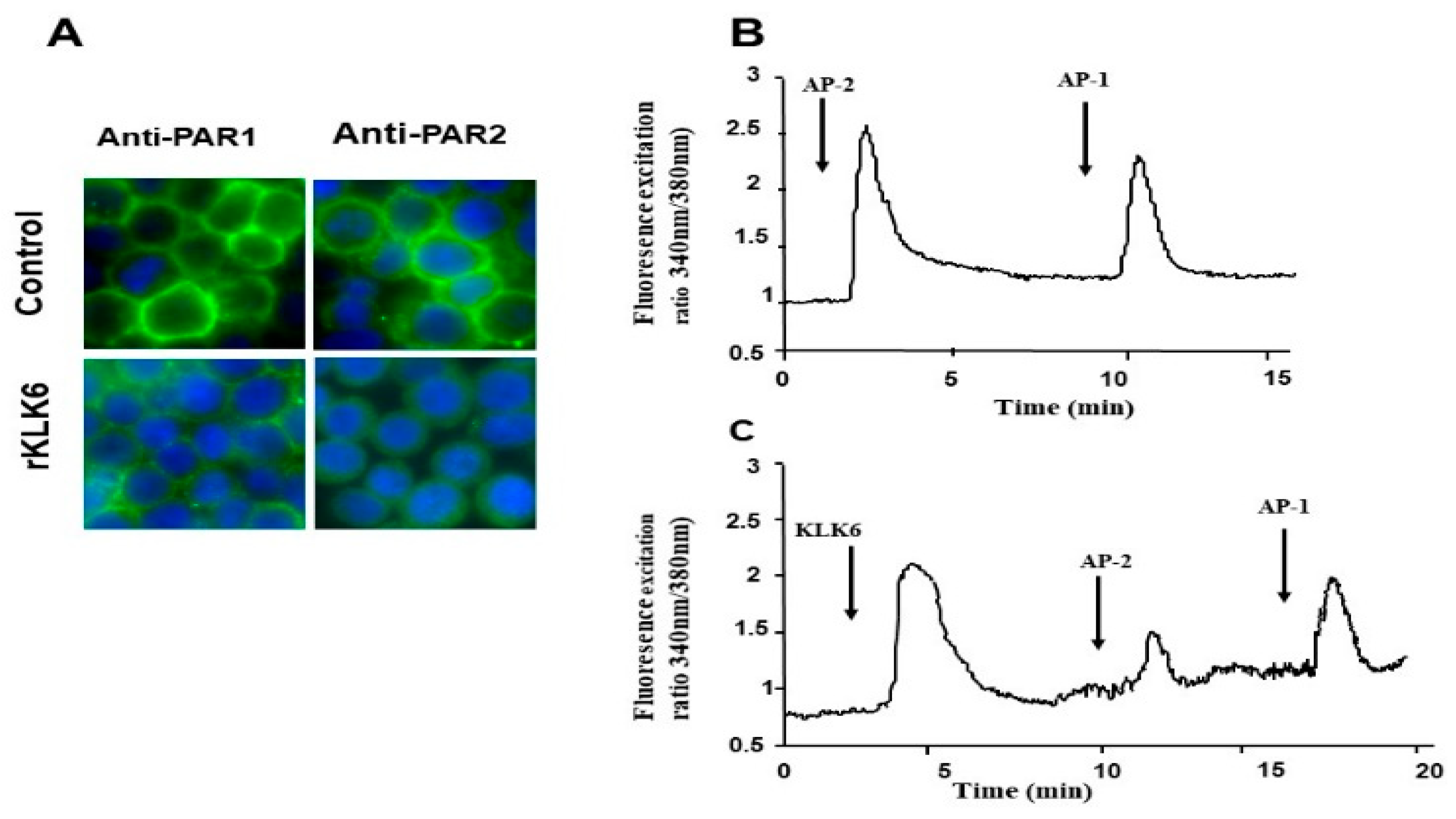
3.3. KLK6 Induced Loss of PAR1 and PAR2 from the Cell Surface of Colon Cancer-Derived Cell Line HT29
3.4. Calcium Signaling Triggered by KLK6 Is Mediated by PAR2 but Not by PAR1
3.5. KLK6 Protein Induces ERK1/2 Phosphorylation in Human Colon Cancer Cells
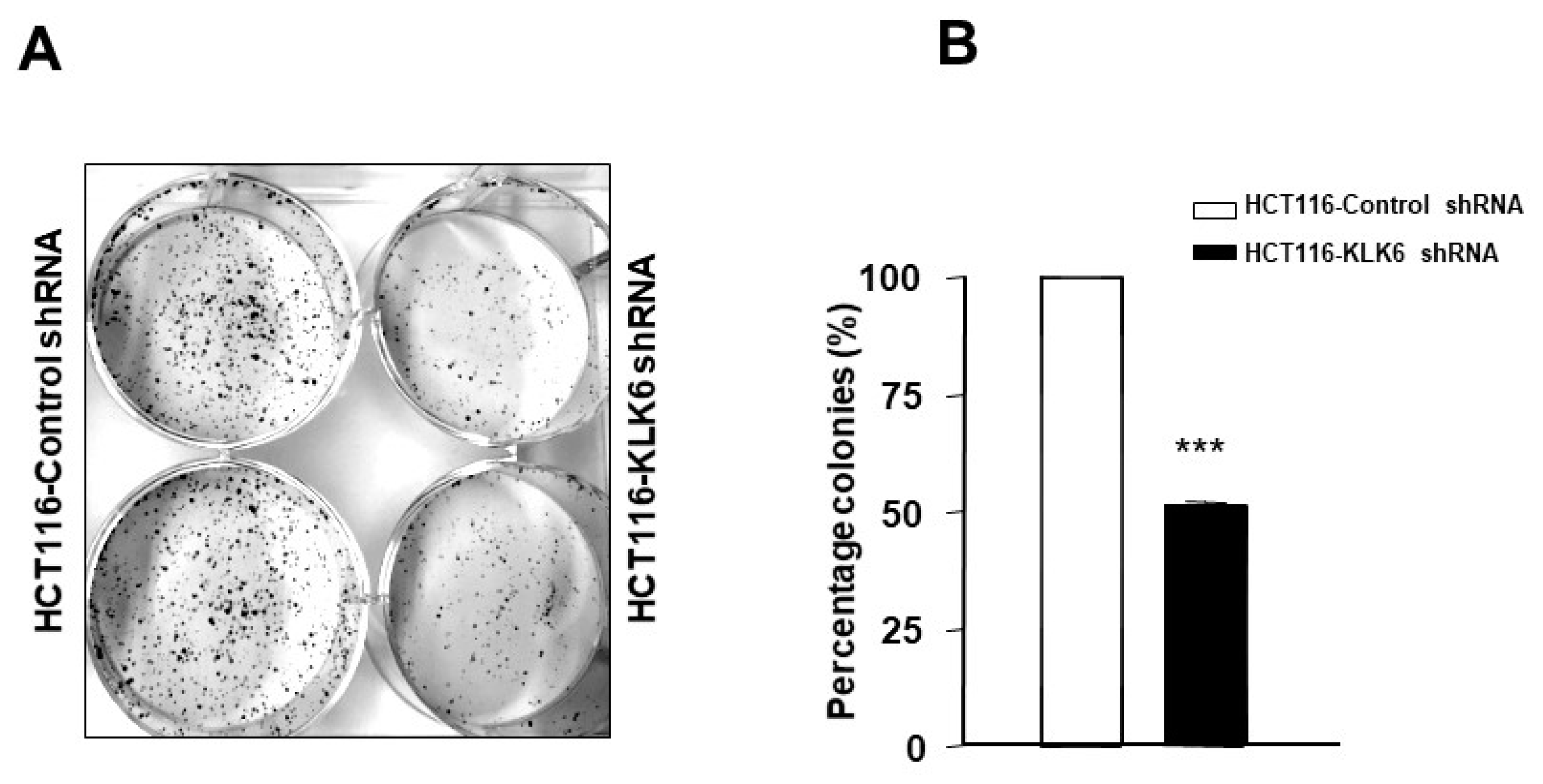
3.6. KLK6 Silencing in HCT116 Cells Reduced Colony Formation
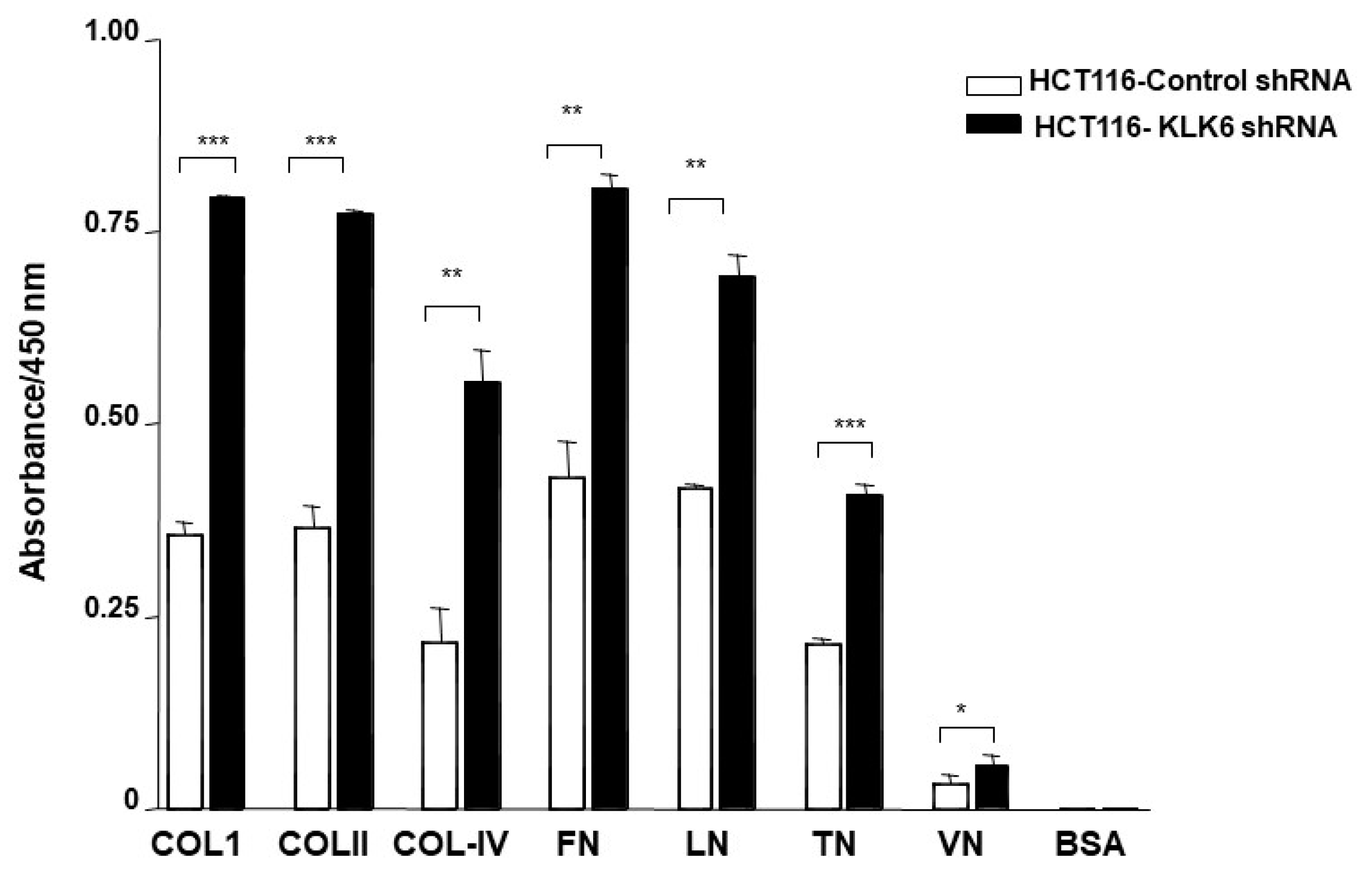
3.7. KLK6 Silencing in HCT116 Cells Alters Cell Adhesion
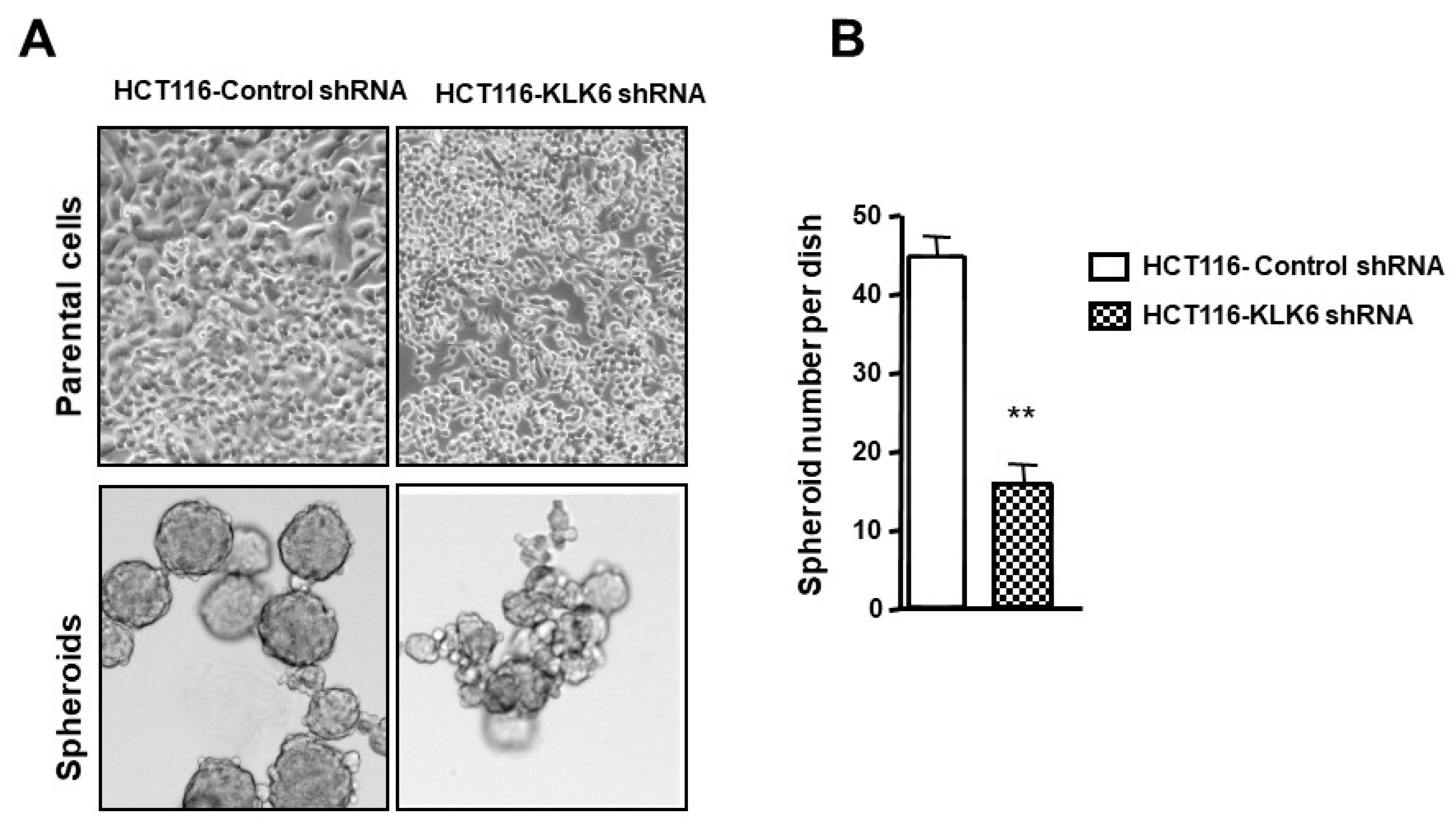
3.8. KLK6 Effect on Spheroid Forming Ability of Colon Cancer Cells
3.9. KLK6 Is Expressed in Colon Cancer Tumors In Vivo

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cases | Age | Gender | Tumor Site | Histological Type | Tumors Stage | Differentiation Grade | Percent of Stained Cells | Staining Intensity | Scores |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 77 | F | Cecum | Adenocarcinoma | T3N0 MX | 3 | 50% | 2 | 100 |
| 2 | 69 | F | Left colon | Adenocarcinoma | T4N0MX | 2 | 0% | 0 | 0 |
| 3 | 70 | F | Sigmoid | Adenocarcinoma | T3N0MX | 3 | 80% | 2 | 160 |
| 4 | 63 | M | Sigmoid | Adenocarcinoma | T3N0MX | 3 | 0% | 0 | 0 |
| 5 | 60 | M | Right colon | Adenocarcinoma | T3N0MX | 1 | 0% | 0 | 0 |
| 6 | 66 | M | Sigmoid | Adenocarcinoma | T3N0MX | 3 | 0% | 0 | 0 |
| 7 | 52 | F | Sigmoid | Adenocarcinoma | T3N1MX | 3 | 70% | 2 | 140 |
| 8 | 50 | F | Right colon | Adenocarcinoma | T3N0MX | 3 | 90% | 3 | 270 |
| 9 | 49 | M | Right colon | Adenocarcinoma | T3N1MX | 3 | 100% | 1 | 100 |
| 10 | 54 | F | Left colon | Adenocarcinoma | T3N0MX | 2 | 90% | 3 | 270 |
| 11 | 86 | F | Sigmoid | Adenocarcinoma | T4N0MX | 2 | 100% | 2 | 200 |
| 12 | 60 | M | Rectum | Adenocarcinoma | T3N0MX | 3 | 0% | 0 | 0 |
| 13 | 57 | M | Cecum | Adenocarcinoma | T3N0MX | 3 | 100% | 2 | 200 |
| 14 | 78 | M | Right colon | Adenocarcinoma | T3N1MX | 3 | 90% | 2 | 180 |
| 15 | 75 | M | Sigmoid | Tubulo-villous adenoma | T1N0MX | 3 | 10% | 1 | 10 |
| 16 | 91 | F | Cecum | Mucinous adenocarcinoma | T3N0M0 | 2 | 60% | 1 | 60 |
| 17 | 77 | M | Sigmoid | Mucinous adenocarcinoma | T3N2Mx | 2 | 10% | 1 | 10 |
| 18 | 84 | F | Cecum | Adenocarcinoma | T3N0Mx | 3 | 40% | 1 | 40 |
| 19 | 85 | F | Right colon | Adenocarcinoma | T3N2Mx | 2 | 40% | 1 | 40 |
| 20 | 90 | F | Cecum | Adenocarcinoma | T2N0Mx | 3 | 60% | 2 | 120 |
| 21 | 76 | M | Right colon | Adenocarcinoma | T3N1M1 | 2 | 10% | 3 | 30 |
| 22 | 81 | M | Right colon | Tubulo-villous adenoma | TisN0MX | 3 | 40% | 2 | 80 |
| 23 | 74 | F | Right colon | Adenocarcinoma | T3N1MX | 1 | 90% | 3 | 270 |
| 24 | 67 | M | Right colon | Adenocarcinoma | T3N2Mx | 2 | 0% | 0 | 0 |
| 25 | 79 | M | Right colon | Adenocarcinoma | T4N1MX | 2 | 30% | 2 | 60 |
| 26 | 42 | F | Sigmoid | Adenocarcinoma | T4N2M1 | 3 | 10% | 2 | 20 |
| 27 | 71 | M | Right colon | Adenocarcinoma | T4N1Mx | 2 | 50% | 1 | 50 |
| 28 | 89 | M | Sigmoid | Adenocarcinoma | T1N0Mx | 3 | 95% | 3 | 285 |
| 29 | 72 | F | Cecum | Adenocarcinoma | T2N0Mx | 2 | 100% | 4 | 400 |
| 30 | 72 | F | Left colon | Adenocarcinoma | T2N0Mx | 2 | 100% | 3 | 300 |
| 31 | 65 | M | Right colon | Tubulo-villous adenoma | T1N0Mx | 3 | 20% | 2 | 40 |
| 32 | 69 | F | Left colon | Adenocarcinoma | T3N1Mx | 2 | 10% | 1 | 10 |
| 33 | 45 | F | Left colon | Adenocarcinoma | T3N1Mx | 3 | 100% | 3 | 300 |
| 34 | 47 | F | Cecum | Adenocarcinoma | T3N1Mx | 2 | 80% | 3 | 240 |
| 35 | 60 | F | Left colon | Adenocarcinoma | T3N0Mx | 3 | 100% | 4 | 400 |
| 36 | 70 | M | Right colon | Adenocarcinoma | T3N1Mx | 2 | 10% | 2 | 20 |
| 37 | 64 | F | Sigmoid | Adenocarcinoma | T3N0Mx | 2 | 5% | 1 | 5 |
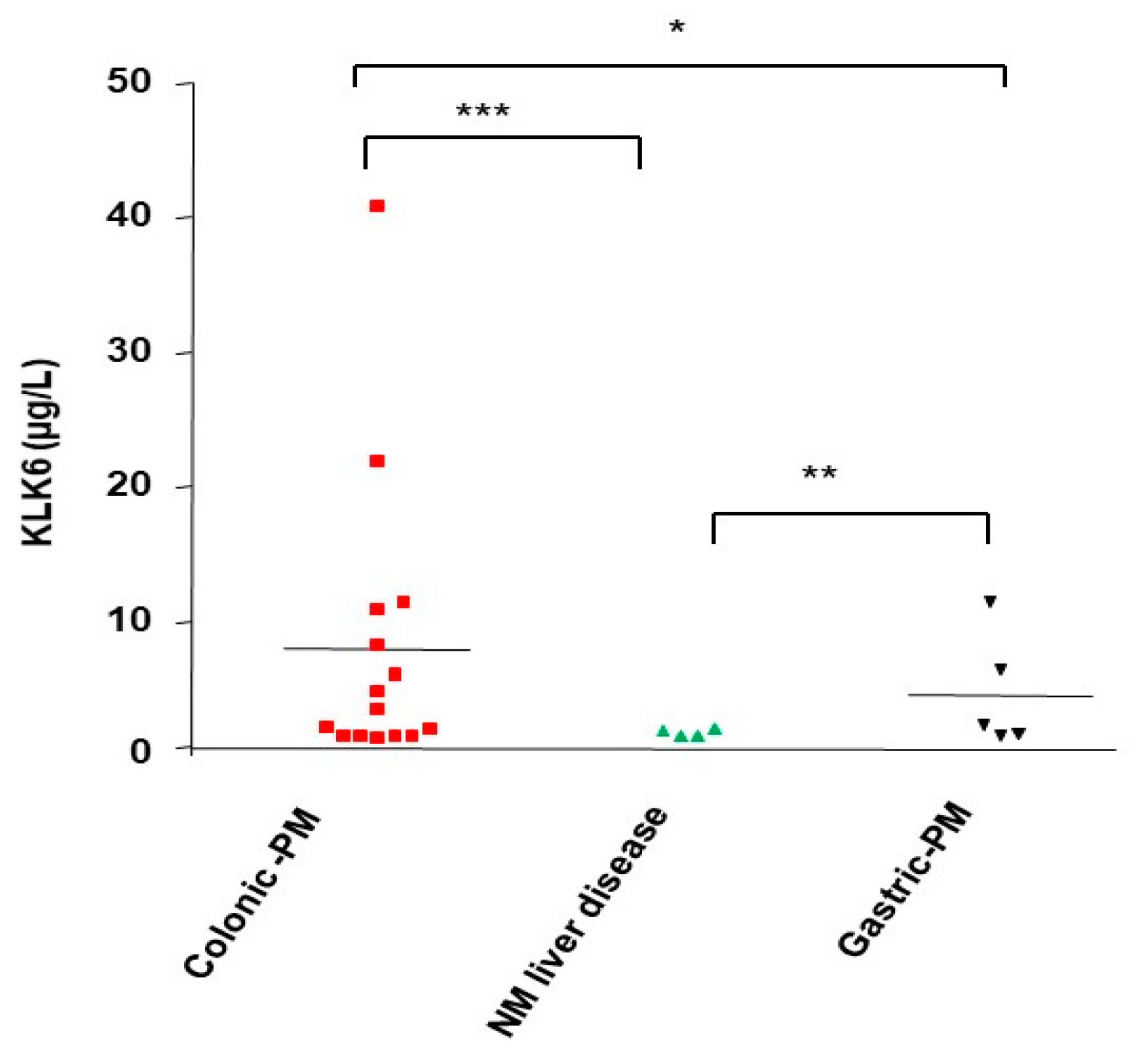
3.10. KLK6 Identification in Malignant Ascitic Fluids from Peritoneal Metastasis of Colon Cancers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Lemoine, L.; Sugarbaker, P.; Van der Speeten, K. Pathophysiology of colorectal peritoneal carcinomatosis: Role of the peritoneum. World J. Gastroenterol. 2016, 22, 7692–7707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pretzsch, E.; Bosch, F.; Neumann, J.; Ganschow, P.; Bazhin, A.; Guba, M.; Werner, J.; Angele, M. Mechanisms of Metastasis in Colorectal Cancer and Metastatic Organotropism: Hematogenous versus Peritoneal Spread. J. Oncol. 2019, 2019, 7407190. [Google Scholar] [CrossRef] [PubMed]
- Sevenich, L.; Joyce, J.A. Pericellular proteolysis in cancer. Genes Dev. 2014, 28, 2331–2347. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Chen, L.; Lv, X.; Hou, G.; Wang, Y.; Jiang, C.; Zhu, H.; Xu, N.; Wu, L.; Lou, X.; et al. The levels of serine proteases in colon tissue interstitial fluid and serum serve as an indicator of colorectal cancer progression. Oncotarget 2016, 7, 32592–32606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrabalan, A.; Ramachandran, R. Molecular mechanisms regulating Proteinase-Activated Receptors (PARs). Febs J. 2021, 288, 2697–2726. [Google Scholar] [CrossRef] [PubMed]
- Darmoul, D.; Marie, J.C.; Devaud, H.; Gratio, V.; Laburthe, M. Initiation of human colon cancer cell proliferation by trypsin acting at protease-activated receptor-2. Br. J. Cancer 2001, 85, 772–779. [Google Scholar] [CrossRef] [Green Version]
- Darmoul, D.; Gratio, V.; Devaud, H.; Lehy, T.; Laburthe, M. Aberrant expression and activation of the thrombin receptor protease-activated receptor-1 induces cell proliferation and motility in human colon cancer cells. Am. J. Pathol. 2003, 162, 1503–1513. [Google Scholar] [CrossRef] [Green Version]
- Darmoul, D.; Gratio, V.; Devaud, H.; Laburthe, M. Protease-activated receptor 2 in colon cancer: Trypsin-induced MAPK phosphorylation and cell proliferation are mediated by epidermal growth factor receptor transactivation. J. Biol. Chem. 2004, 279, 20927–20934. [Google Scholar] [CrossRef] [Green Version]
- Darmoul, D.; Gratio, V.; Devaud, H.; Peiretti, F.; Laburthe, M. Activation of proteinase-activated receptor 1 promotes human colon cancer cell proliferation through epidermal growth factor receptor transactivation. Mol. Cancer Res. 2004, 2, 514–522. [Google Scholar] [CrossRef]
- Kryza, T.; Silva, M.L.; Loessner, D.; Heuze-Vourc’h, N.; Clements, J.A. The kallikrein-related peptidase family: Dysregulation and functions during cancer progression. Biochimie 2016, 122, 283–299. [Google Scholar] [CrossRef]
- Kontos, C.K.; Mavridis, K.; Talieri, M.; Scorilas, A. Kallikrein-related peptidases (KLKs) in gastrointestinal cancer: Mechanistic and clinical aspects. Thromb. Haemost. 2012, 110, 450–457. [Google Scholar] [CrossRef]
- Filippou, P.S.; Karagiannis, G.S.; Musrap, N.; Diamandis, E.P. Kallikrein-related peptidases (KLKs) and the hallmarks of cancer. Crit. Rev. Clin. Lab. Sci. 2016, 53, 277–291. [Google Scholar] [CrossRef]
- Srinivasan, S.; Kryza, T.; Batra, J.; Clements, J. Remodelling of the tumour microenvironment by the kallikrein-related peptidases. Nat. Rev. Cancer 2022, 22, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Gratio, V.; Beaufort, N.; Seiz, L.; Maier, J.; Virca, G.D.; Debela, M.; Grebenchtchikov, N.; Magdolen, V.; Darmoul, D. Kallikrein-related peptidase 4: A new activator of the aberrantly expressed protease-activated receptor 1 in colon cancer cells. Am. J. Pathol. 2010, 176, 1452–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gratio, V.; Loriot, C.; Virca, G.D.; Oikonomopoulou, K.; Walker, F.; Diamandis, E.P.; Hollenberg, M.D.; Darmoul, D. Kallikrein-related peptidase 14 acts on proteinase-activated receptor 2 to induce signaling pathway in colon cancer cells. Am. J. Pathol. 2011, 179, 2625–2636. [Google Scholar] [CrossRef]
- Oikonomopoulou, K.; Diamandis, E.P.; Hollenberg, M.D. Kallikrein-related peptidases: Proteolysis and signaling in cancer, the new frontier. Biol. Chem. 2010, 391, 299–310. [Google Scholar] [CrossRef]
- Bayani, J.; Diamandis, E.P. The physiology and pathobiology of human kallikrein-related peptidase 6 (KLK6). Clin. Chem. Lab. Med. 2012, 50, 211–233. [Google Scholar] [CrossRef] [PubMed]
- Yousef, G.M.; Borgono, C.A.; White, N.M.; Robb, J.D.; Michael, I.P.; Oikonomopoulou, K.; Khan, S.; Diamandis, E.P. In silico analysis of the human kallikrein gene 6. Tumor Biol. 2004, 25, 282–289. [Google Scholar] [CrossRef]
- Kim, J.T.; Song, E.Y.; Chung, K.S.; Kang, M.A.; Kim, J.W.; Kim, S.J.; Yeom, Y.I.; Kim, J.H.; Kim, K.H.; Lee, H.G. Upregulation and clinical significance of serine protease kallikrein 6 in colon cancer. Cancer 2011, 117, 2608–2619. [Google Scholar] [CrossRef]
- Christodoulou, S.; Alexopoulou, D.K.; Kontos, C.K.; Scorilas, A.; Papadopoulos, I.N. Kallikrein-related peptidase-6 (KLK6) mRNA expression is an independent prognostic tissue biomarker of poor disease-free and overall survival in colorectal adenocarcinoma. Tumor Biol. 2014, 35, 4673–4685. [Google Scholar] [CrossRef]
- Ohlsson, L.; Lindmark, G.; Israelsson, A.; Palmqvist, R.; Oberg, A.; Hammarstrom, M.L.; Hammarstrom, S. Lymph node tissue kallikrein-related peptidase 6 mRNA: A progression marker for colorectal cancer. Br. J. Cancer. 2012, 107, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Tailor, P.D.; Kodeboyina, S.K.; Bai, S.; Patel, N.; Sharma, S.; Ratnani, A.; Copland, J.A.; She, J.X.; Sharma, A. Diagnostic and prognostic biomarker potential of kallikrein family genes in different cancer types. Oncotarget 2018, 9, 17876–17888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Du, J.; Gu, J.; Jin, L.; Pu, Y.; Fei, B. A 65--gene signature for prognostic prediction in colon adenocarcinoma. Int. J. Mol. Med. 2018, 41, 2021–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Sells, E.; Pandey, R.; Abril, E.R.; Hsu, C.H.; Krouse, R.S.; Nagle, R.B.; Pampalakis, G.; Sotiropoulou, G.; Ignatenko, N.A. Kallikrein 6 protease advances colon tumorigenesis via induction of the high mobility group A2 protein. Oncotarget 2019, 10, 6062–6078. [Google Scholar] [CrossRef] [Green Version]
- Pandey, R.; Zhou, M.; Chen, Y.; Darmoul, D.; Kisiel, C.C.; Nfonsam, V.N.; Ignatenko, N.A. Molecular Pathways Associated with Kallikrein 6 Overexpression in Colorectal Cancer. Genes 2021, 12, 749. [Google Scholar] [CrossRef]
- Sells, E.; Pandey, R.; Chen, H.; Skovan, B.A.; Cui, H.; Ignatenko, N.A. Specific microRNA-mRNA Regulatory Network of Colon Cancer Invasion Mediated by Tissue Kallikrein-Related Peptidase 6. Neoplasia 2017, 19, 396–411. [Google Scholar] [CrossRef]
- Recommendations for cryopreservation of cells tumor tissues to be used for molecular analyses. Ann. Pathol. 2001, 21, 184–201.
- Le Comité consultatif national d’éthique pour les sciences de la vie et de la santé. French Bioethics Law, No. 2004-800; Art. L. 1232-1, Art. L. 1235-2, Art. L. 1245-2. J.O.R.F.; 2004; July 8th.
- Fearon, E.R. Molecular Genetics of Colorectal Cancer. Annu. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef]
- Chung, H.; Hamza, M.; Oikonomopoulou, K.; Gratio, V.; Saifeddine, M.; Virca, G.D.; Diamandis, E.P.; Hollenberg, M.D.; Darmoul, D. Kallikrein-related peptidase signaling in colon carcinoma cells: Targeting proteinase-activated receptors. Biol. Chem. 2012, 393, 413–420. [Google Scholar] [CrossRef]
- Loessner, D.; Quent, V.M.; Kraemer, J.; Weber, E.C.; Hutmacher, D.W.; Magdolen, V.; Clements, J.A. Combined expression of KLK4, KLK5, KLK6, and KLK7 by ovarian cancer cells leads to decreased adhesion and paclitaxel-induced chemoresistance. Gynecol. Oncol. 2012, 127, 569–578. [Google Scholar] [CrossRef] [Green Version]
- Haddada, M.; Draoui, H.; Deschamps, L.; Walker, F.; Delaunay, T.; Brattsand, M.; Magdolen, V.; Darmoul, D. Kallikrein-related peptidase 7 overexpression in melanoma cells modulates cell adhesion leading to a malignant phenotype. Biol. Chem. 2018, 399, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Delaunay, T.; Deschamps, L.; Haddada, M.; Walker, F.; Soosaipillai, A.; Soualmia, F.; El Amri, C.; Diamandis, E.P.; Brattsand, M.; Magdolen, V.; et al. Aberrant expression of kallikrein-related peptidase 7 is correlated with human melanoma aggressiveness by stimulating cell migration and invasion. Mol. Oncol. 2017, 11, 1330–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tieng, F.Y.F.; Abu, N.; Sukor, S.; Mohd Azman, Z.A.; Mahamad Nadzir, N.; Lee, L.H.; Mutalib, N.S.A. L1CAM, CA9, KLK6, HPN, and ALDH1A1 as Potential Serum Markers in Primary and Metastatic Colorectal Cancer Screening. Diagnostics 2020, 10, 444. [Google Scholar] [CrossRef] [PubMed]
- Vakrakou, A.; Devetzi, M.; Papachristopoulou, G.; Malachias, A.; Scorilas, A.; Xynopoulos, D.; Talieri, M. Kallikrein-related peptidase 6 (KLK6) expression in the progression of colon adenoma to carcinoma. Biol. Chem. 2014, 395, 1105–1117. [Google Scholar] [CrossRef]
- Ogawa, K.; Utsunomiya, T.; Mimori, K.; Tanaka, F.; Inoue, H.; Nagahara, H.; Murayama, S.; Mori, M. Clinical significance of human kallikrein gene 6 messenger RNA expression in colorectal cancer. Clin. Cancer Res. 2005, 11, 2889–2893. [Google Scholar] [CrossRef] [Green Version]
- Petraki, C.; Dubinski, W.; Scorilas, A.; Saleh, C.; Pasic, M.D.; Komborozos, V.; Khalil, B.; Gabril, M.Y.; Streutker, C.; Diamandis, E.P.; et al. Evaluation and prognostic significance of human tissue kallikrein-related peptidase 6 (KLK6) in colorectal cancer. Pathol. Res. Pract. 2012, 208, 104–108. [Google Scholar] [CrossRef]
- Adamopoulos, P.G.; Kontos, C.K.; Scorilas, A. Molecular cloning of novel transcripts of human kallikrein-related peptidases 5, 6, 7, 8 and 9 (KLK5–KLK9), using Next-generation sequencing. Sci. Rep. 2017, 7, 17299. [Google Scholar] [CrossRef] [Green Version]
- Candido, J.B.; Maiques, O.; Boxberg, M.; Kast, V.; Peerani, E.; Tomás-Bort, E.; Weichert, W.; Sananes, A.; Papo, N.; Magdolen, V.; et al. Kallikrein-Related Peptidase 6 Is Associated with the Tumour Microenvironment of Pancreatic Ductal Adenocarcinoma. Cancers 2021, 13, 3969. [Google Scholar] [CrossRef]
- Henkhaus, R.S.; Gerner, E.W.; Ignatenko, N.A. Kallikrein 6 is a mediator of K-RAS-dependent migration of colon carcinoma cells. Biol. Chem. 2008, 389, 757–764. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Ding, Z.; Zhang, H.; Chen, Q. Identification of Prognostic Biomarkers and Drugs Targeting Them in Colon Adenocarcinoma: A Bioinformatic Analysis. Integr. Cancer Ther. 2019, 18, 1534735419864434. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Wang, L.; Ma, Q.; Yang, Y.; Yang, Z.; Wang, B.; He, N. Let7i5p inhibits the proliferation and metastasis of colon cancer cells by targeting kallikreinrelated peptidase 6. Oncol. Rep. 2018, 40, 1459–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pampalakis, G.; Zingkou, E.; Sidiropoulos, K.G.; Diamandis, E.P.; Zoumpourlis, V.; Yousef, G.M.; Sotiropoulou, G. Biochemical pathways mediated by KLK6 protease in breast cancer. Mol. Oncol. 2019, 13, 2329–2343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugihara, S.; Sugimoto, S.; Tachibana, K.; Kobashi, M.; Nomura, H.; Miyake, T.; Hirai, Y.; Yamasaki, O.; Morizane, S. TNF-alpha and IL-17A induce the expression of lympho-epithelial Kazal-type inhibitor in epidermal keratinocytes. J. Dermatol. Sci. 2019, 96, 26–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, Y.; Ozawa, S.; Miyamoto, C.; Maehata, Y.; Suzuki, A.; Maeda, T.; Baba, Y. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013, 13, 89. [Google Scholar] [CrossRef] [Green Version]
- Aono, S.; Nakagawa, S.; Reynolds, A.B.; Takeichi, M. p120(ctn) acts as an inhibitory regulator of cadherin function in colon carcinoma cells. J. Cell Biol. 1999, 145, 551–562. [Google Scholar] [CrossRef] [Green Version]
- Stadler, M.; Scherzer, M.; Walter, S.; Holzner, S.; Pudelko, K.; Riedl, A.; Unger, C.; Kramer, N.; Weil, B.; Neesen, J.; et al. Exclusion from spheroid formation identifies loss of essential cell–cell adhesion molecules in colon cancer cells. Sci. Rep. 2018, 8, 1151. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.K.; Yang, Y.; Byeon, S.; Jeong, Y.; Kwon, J.; Lee, K.H.; Son, S.M.; Han, H.S. E-Cadherin and Angiopoietin-2 as Potential Biomarkers for Colorectal Cancer With Peritoneal Carcinomatosis. Anticancer Res. 2021, 41, 4497–4504. [Google Scholar] [CrossRef]
- Dong, Y.; Loessner, D.; Irving-Rodgers, H.; Obermair, A.; Nicklin, J.L.; Clements, J.A. Metastasis of ovarian cancer is mediated by kallikrein related peptidases. Clin. Exp. Metastasis 2014, 31, 135–147. [Google Scholar] [CrossRef] [Green Version]
- Oikonomopoulou, K.; Batruch, I.; Smith, C.R.; Soosaipillai, A.; Diamandis, E.P.; Hollenberg, M.D. Functional proteomics of kallikrein-related peptidases in ovarian cancer ascites fluid. Biol. Chem. 2010, 391, 381–390. [Google Scholar] [CrossRef]
- Oikonomopoulou, K.; Hansen, K.K.; Baruch, A.; Hollenberg, M.D.; Diamandis, E.P. Immunofluorometric activity-based probe analysis of active KLK6 in biological fluids. Biol. Chem. 2008, 389, 747–756. [Google Scholar] [CrossRef]
- Berg, K.C.; Eide, P.W.; Eilertsen, I.A.; Johannessen, B.; Bruun, J.; Danielsen, S.A.; Bjørnslett, M.; Meza-Zepeda, M.A.; Eknæs, M.; Lind, G.E.; et al. Multi-omics of 34 colorectal cancer cell lines-a resource for biomedical studies. Mol. Cancer 2017, 16, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.J.; Routh, E.D.; Rubinas, T.; Peacock, J.; Martin, T.D.; Shen, X.J.; Sandler, R.S.; Kim, H.J.; Keku, T.; Der, C.J. KRAS/BRAF mutation status and ERK1/2 activation as biomarkers for MEK1/2 inhibitor therapy in colorectal cancer. Mol. Cancer Ther. 2009, 8, 834–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abajo, A.; Bitarte, N.; Zarate, R.; Boni, V.; Lopez, I.; Gonzalez-Huarriz, M.; Rodriguez, J.; Bandres, E.; Garcia-Foncillas, J. Identification of colorectal cancer metastasis markers by an angiogenesis-related cytokine-antibody array. World J. Gastroenterol. WJG 2012, 18, 637. Available online: https://www.wjgnet.com/1007-9327/full/v18/i7/637.htm (accessed on 12 May 2022). [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouzid, H.; Soualmia, F.; Oikonomopoulou, K.; Soosaipillai, A.; Walker, F.; Louati, K.; Lo Dico, R.; Pocard, M.; El Amri, C.; Ignatenko, N.A.; et al. Kallikrein-Related Peptidase 6 (KLK6) as a Contributor toward an Aggressive Cancer Cell Phenotype: A Potential Role in Colon Cancer Peritoneal Metastasis. Biomolecules 2022, 12, 1003. https://doi.org/10.3390/biom12071003
Bouzid H, Soualmia F, Oikonomopoulou K, Soosaipillai A, Walker F, Louati K, Lo Dico R, Pocard M, El Amri C, Ignatenko NA, et al. Kallikrein-Related Peptidase 6 (KLK6) as a Contributor toward an Aggressive Cancer Cell Phenotype: A Potential Role in Colon Cancer Peritoneal Metastasis. Biomolecules. 2022; 12(7):1003. https://doi.org/10.3390/biom12071003
Chicago/Turabian StyleBouzid, Hayet, Feryel Soualmia, Katerina Oikonomopoulou, Antoninus Soosaipillai, Francine Walker, Khaoula Louati, Rea Lo Dico, Marc Pocard, Chahrazade El Amri, Natalia A. Ignatenko, and et al. 2022. "Kallikrein-Related Peptidase 6 (KLK6) as a Contributor toward an Aggressive Cancer Cell Phenotype: A Potential Role in Colon Cancer Peritoneal Metastasis" Biomolecules 12, no. 7: 1003. https://doi.org/10.3390/biom12071003
APA StyleBouzid, H., Soualmia, F., Oikonomopoulou, K., Soosaipillai, A., Walker, F., Louati, K., Lo Dico, R., Pocard, M., El Amri, C., Ignatenko, N. A., & Darmoul, D. (2022). Kallikrein-Related Peptidase 6 (KLK6) as a Contributor toward an Aggressive Cancer Cell Phenotype: A Potential Role in Colon Cancer Peritoneal Metastasis. Biomolecules, 12(7), 1003. https://doi.org/10.3390/biom12071003