
Non-Equilibrium Protein Folding and Activation by ATP-Driven Chaperones


Abstract
:1. Introduction
Assumptions and Notations
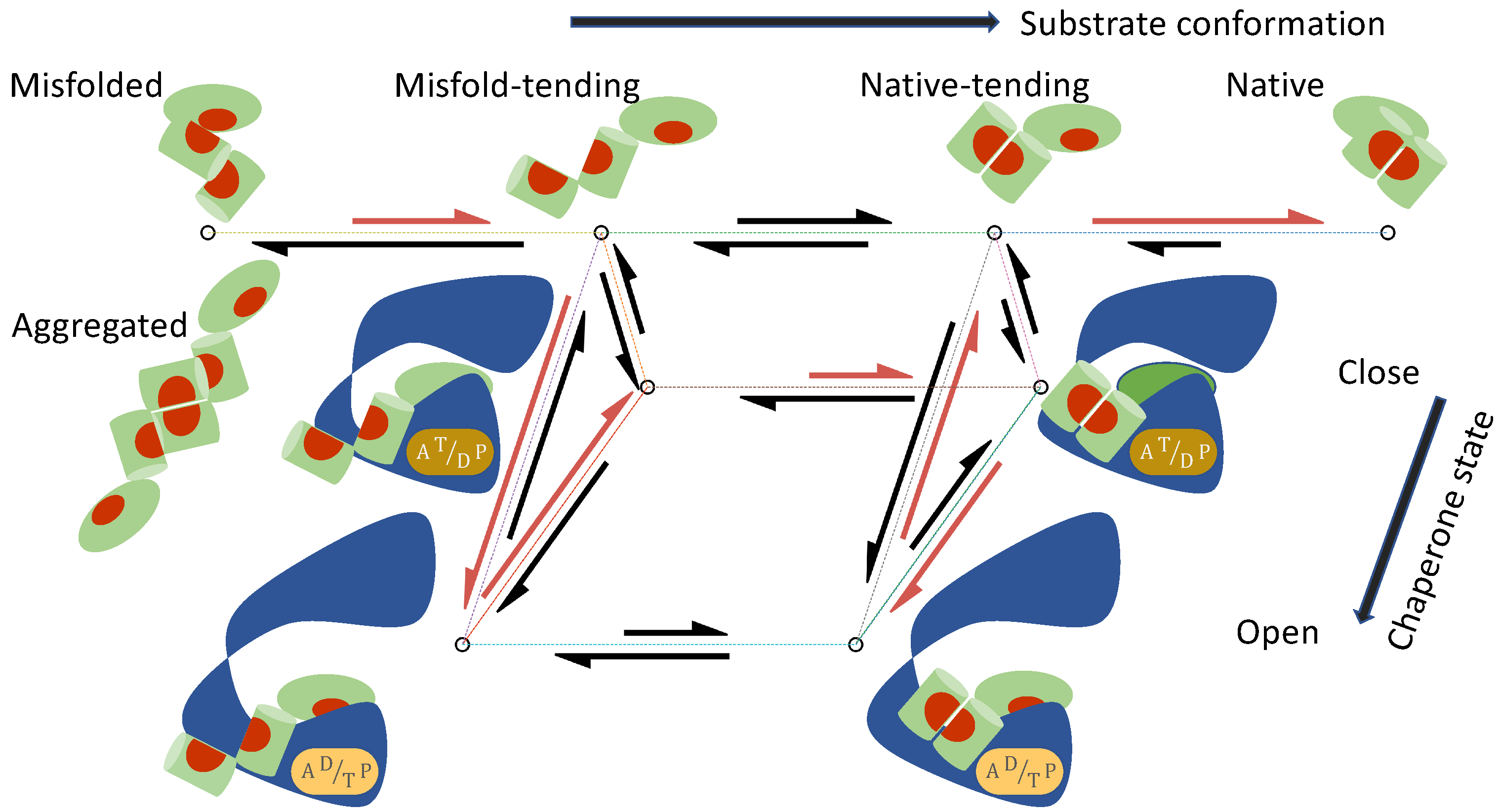
- The substrate protein can convert among a set of conformations , both when it is free in solution and when it is bound to the chaperone. I will use M to denote the misfolded/aggregated conformation and N the native conformation. In addition, I will consider two classes of intermediate conformations: the unfolded and misfold-tending (or aggregation-tending) conformation U, and the non-native but native-tending conformation F. To avoid a proliferation of symbols and to underscore the mechanistic commonality shared by protein folding and activation, in the discussion of kinase activation, I will use M to denote the inactive conformation, N the active conformation, U the inactive-tending conformation, and F the active-tending conformation.
- The chaperone can transition among a set of states , each state i characterized by its conformational state (e.g., open or closed) and the numbers and the types (ATP vs ADP) of bound nucleotides.
- The substrate in conformation S binds to the chaperone in state i with the association rate constant and the dissociation rate constant :
- The free substrate in solution converts between conformation S and conformation :The corresponding conformational equilibrium constant is
- The substrate bound to the chaperone in state i converts between conformation S and conformation :
- The chaperone transitions between state i and state j when it is bound to a substrate in conformation S:
2. Materials and Methods
2.1. Proof That Symmetry Breaking Is Required for Non-Equilibrium Protein Folding
2.2. Derivation of the Upper Bound of the Native Concentration at the Steady State of Non-Equilibrium Folding
3. Results
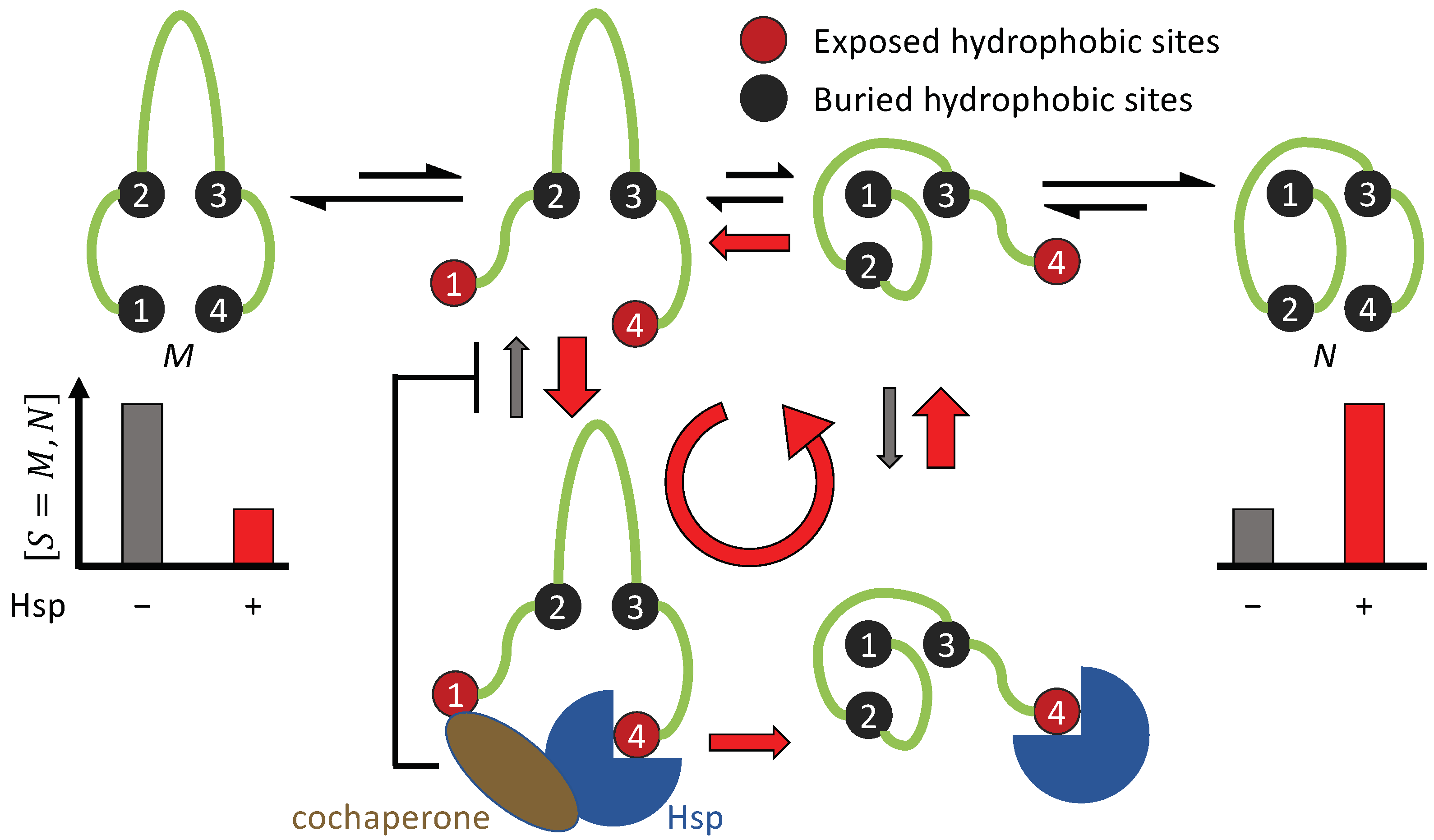
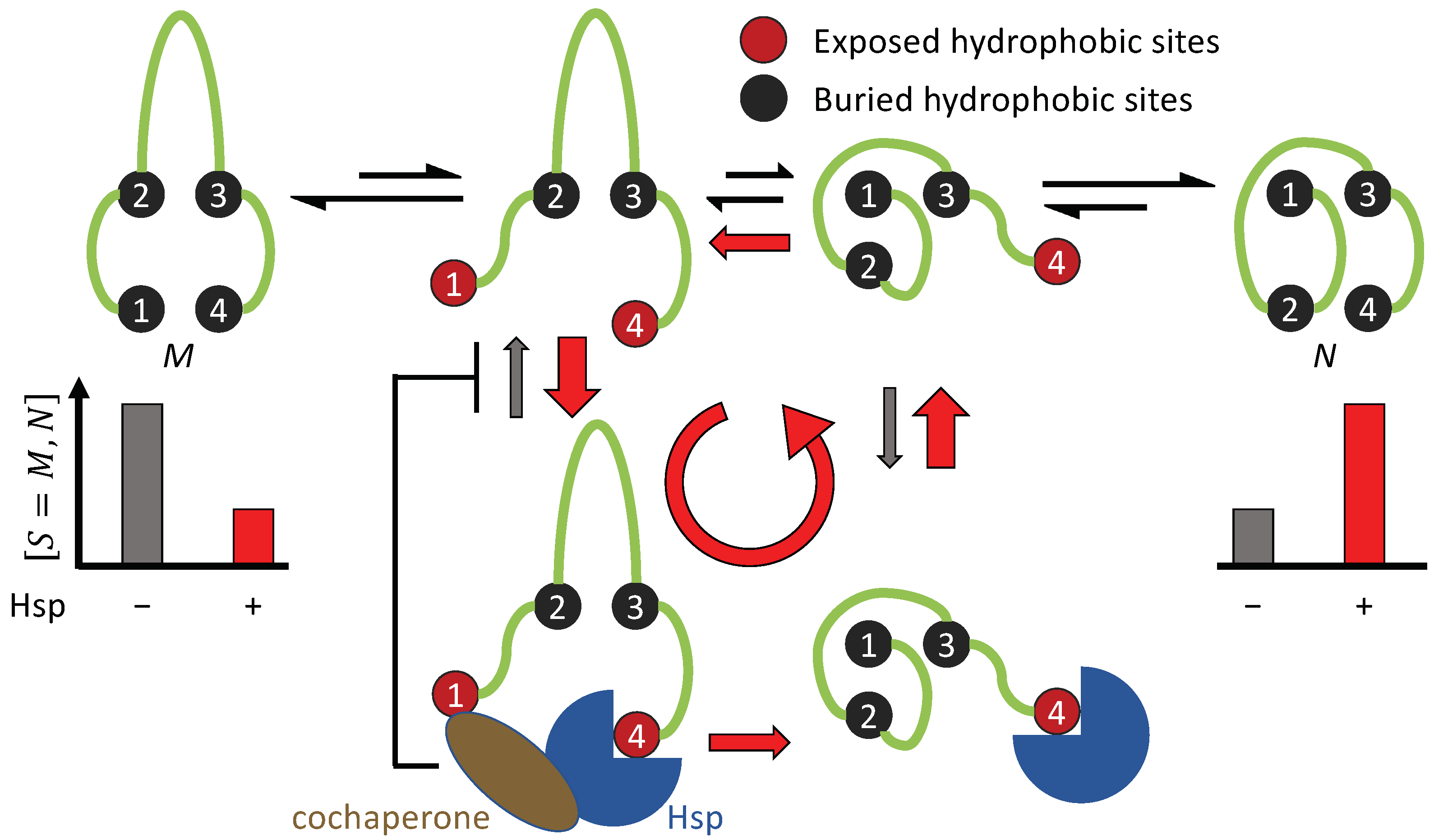
3.1. Non-Equilibrium Folding Requires Kinetic Symmetry Breaking
- The ratio of association rate constants does not depend on the chaperone state i for all pair of substrate conformations S and and for all open state i.
- The dissociation rate constant does not depend on the substrate conformation S, i.e.,for all open state i and for all conformation S.
- The transition rates between chaperone states are independent of the conformation of the bound substrate, i.e., does not depend on S for all pair .
- For every closed state i of the chaperone, there is an open state j, such that the chaperone can reversibly transition between states j and i without consuming chemical energy.
3.1.1. Requisites for Breaking the Binding and Unbinding Symmetries (Conditions 1 and 2)
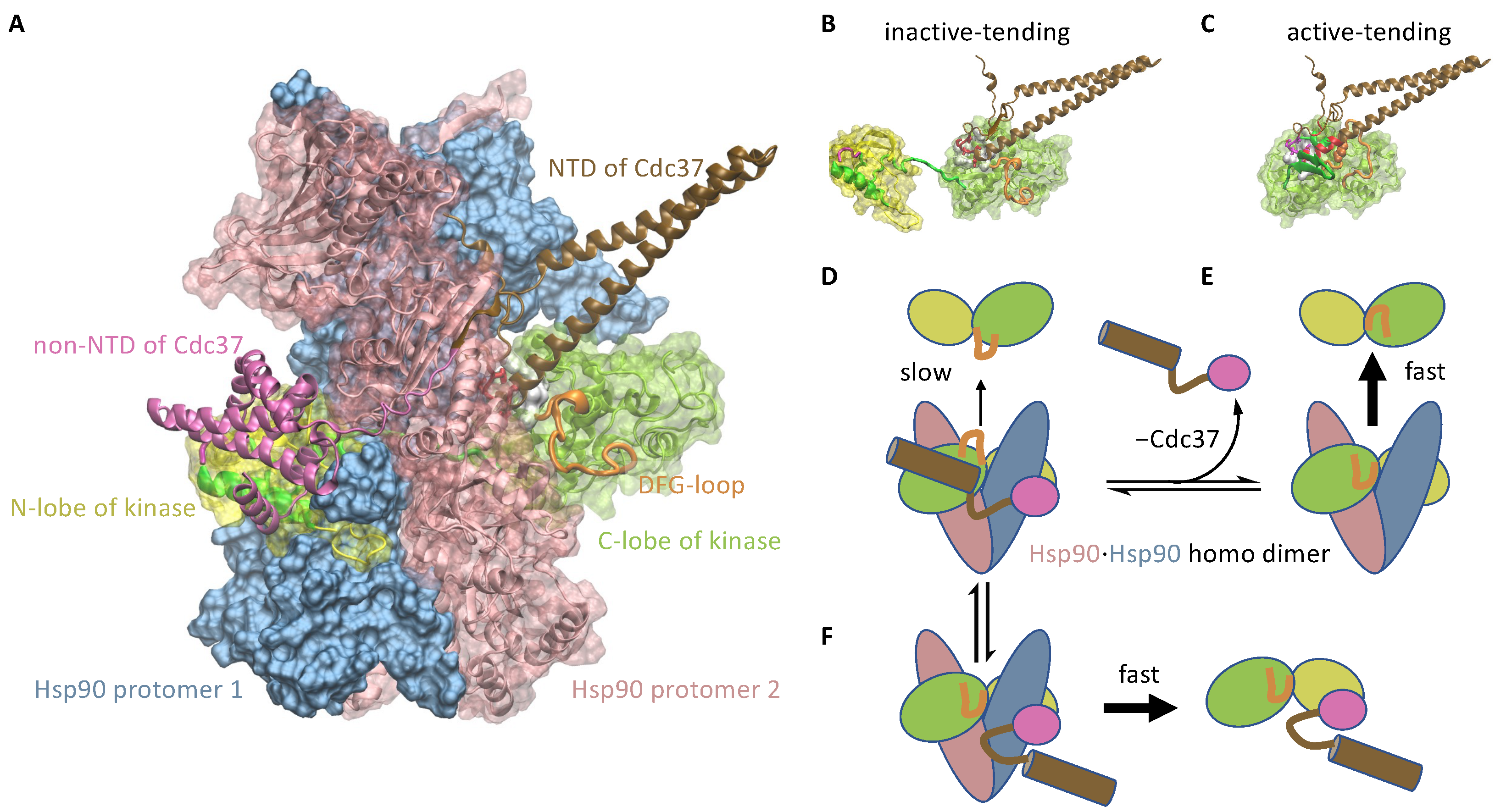
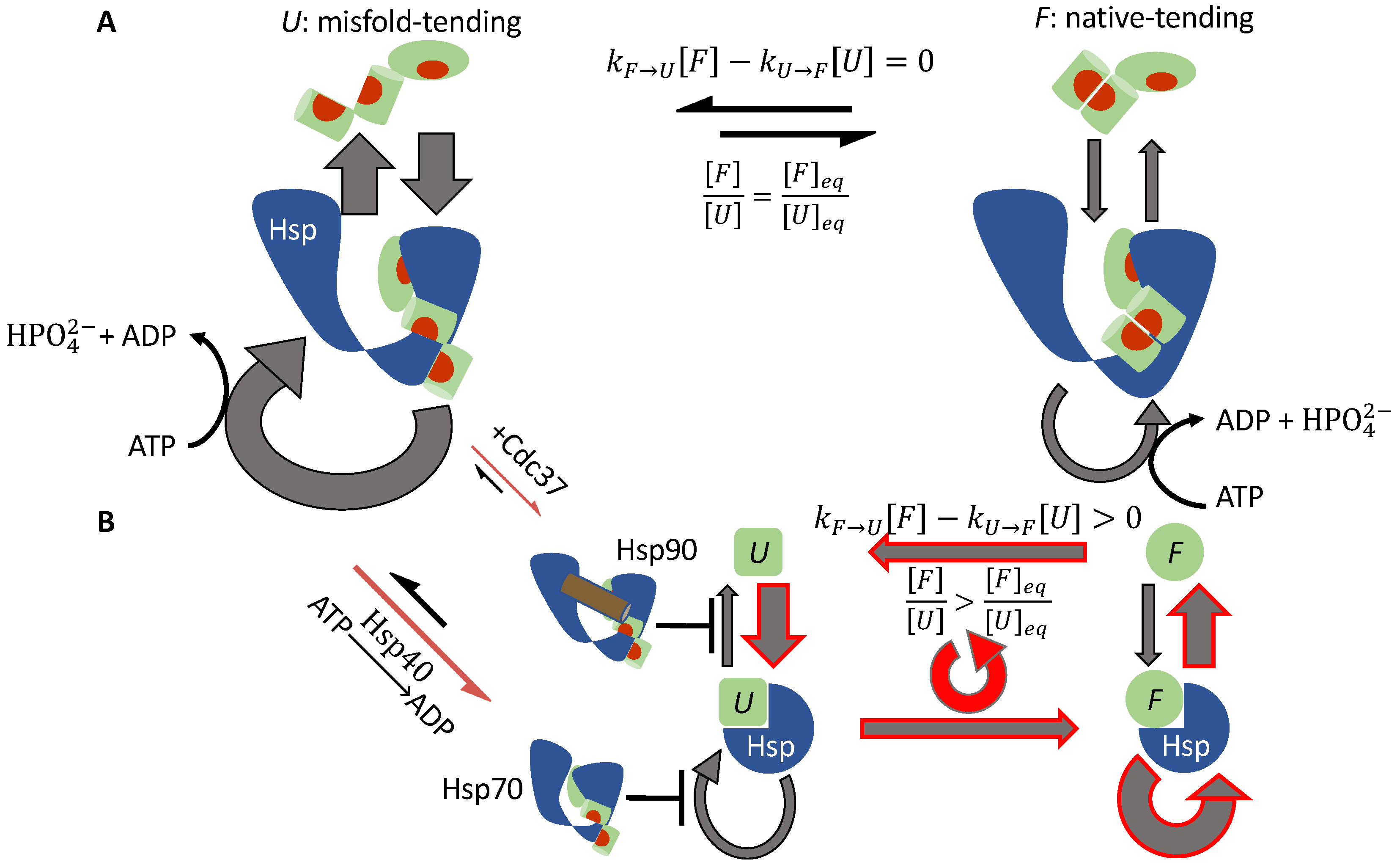
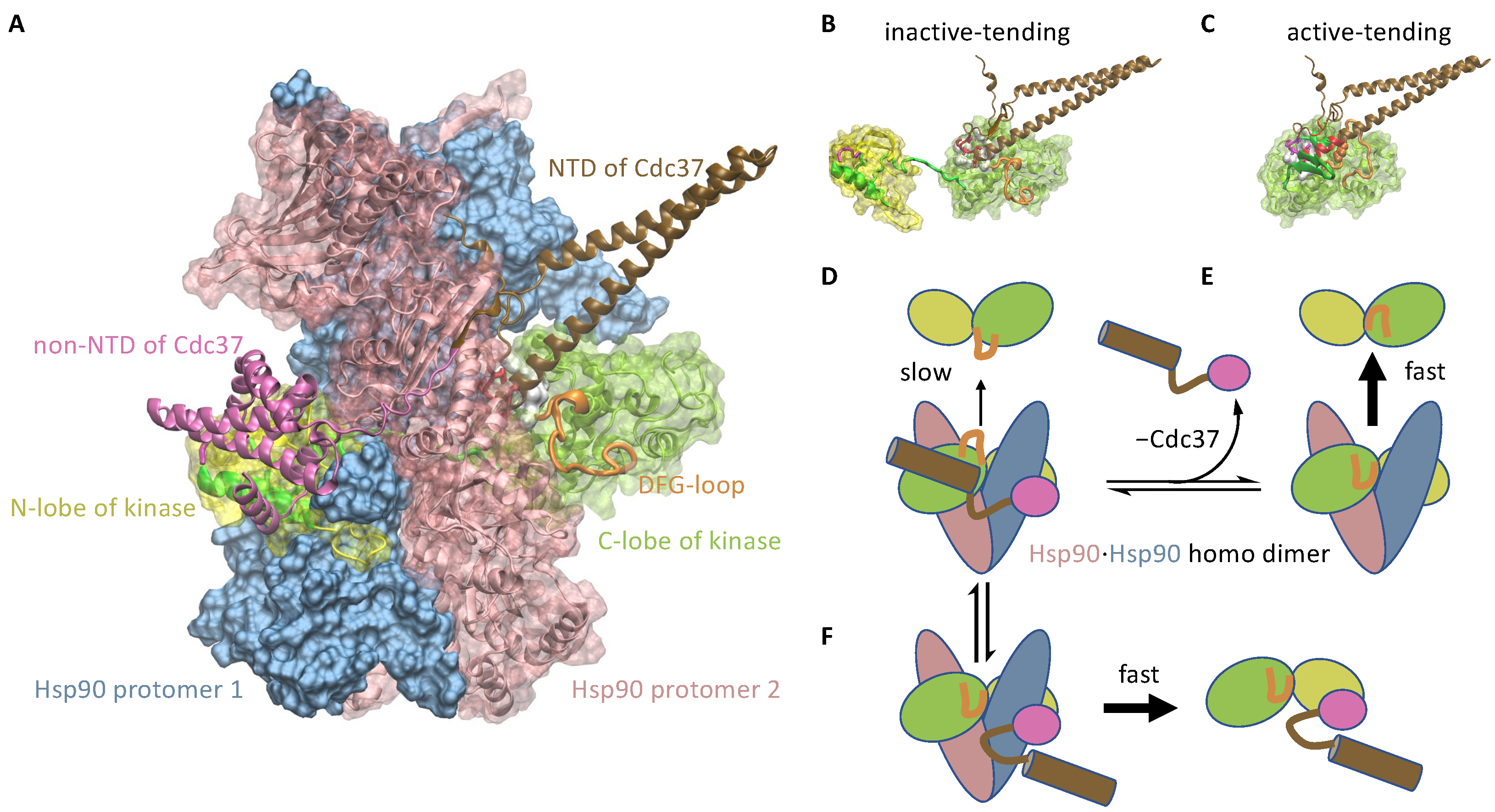
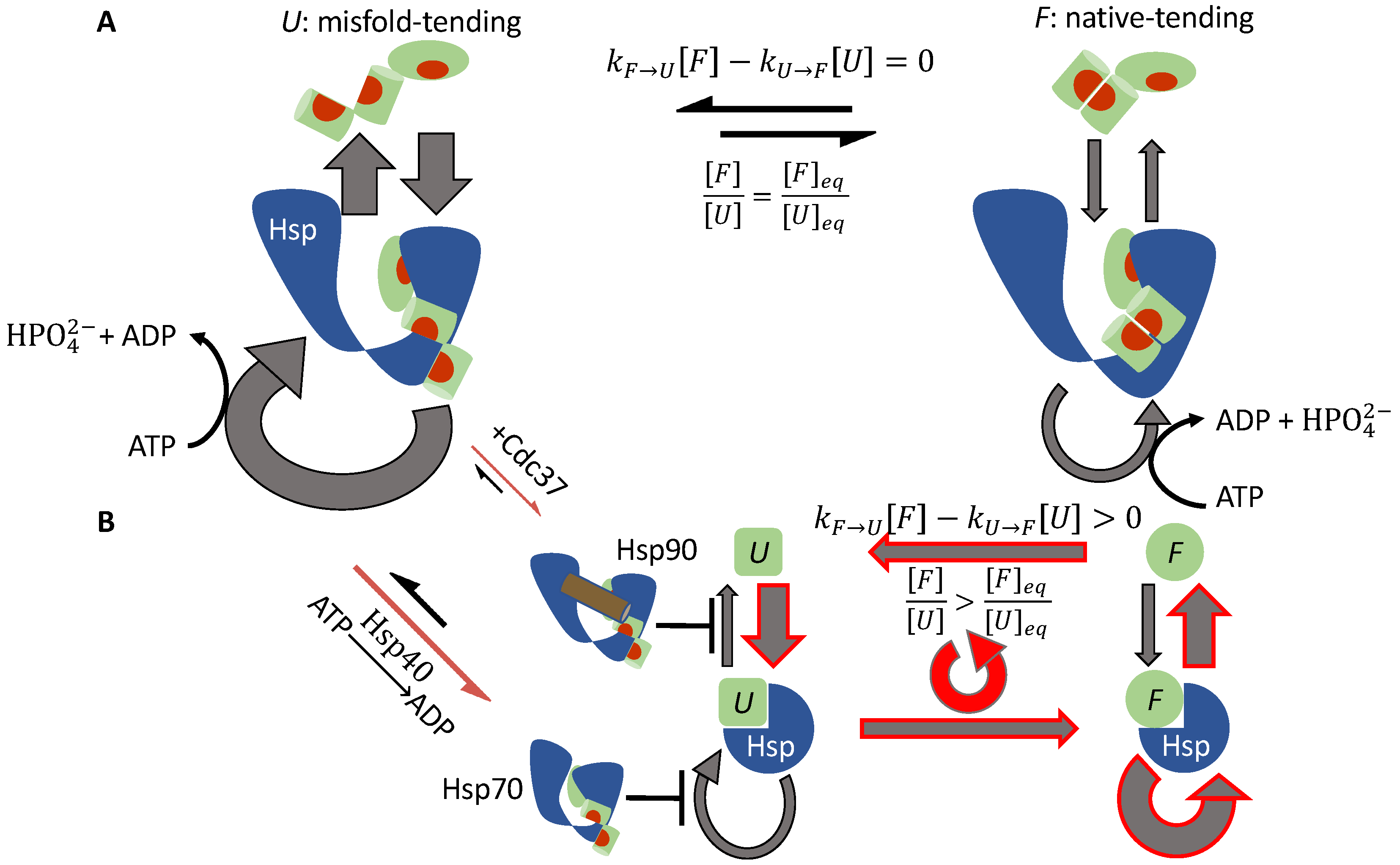
3.1.2. Cdc37 Enables Hsp90 to Differentiate between the Active-Tending and Inactive-Tending Conformations of a Client Kinase
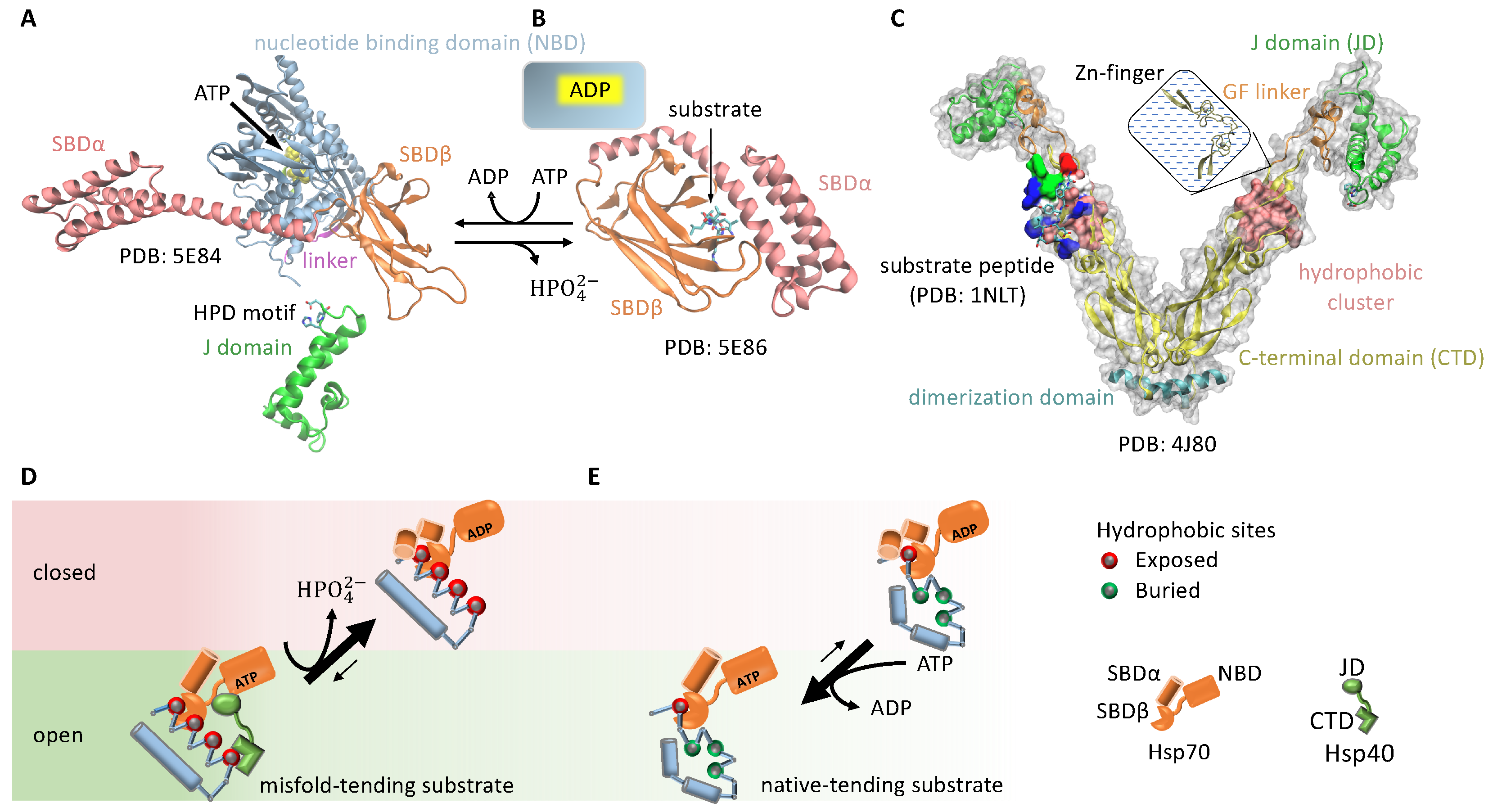
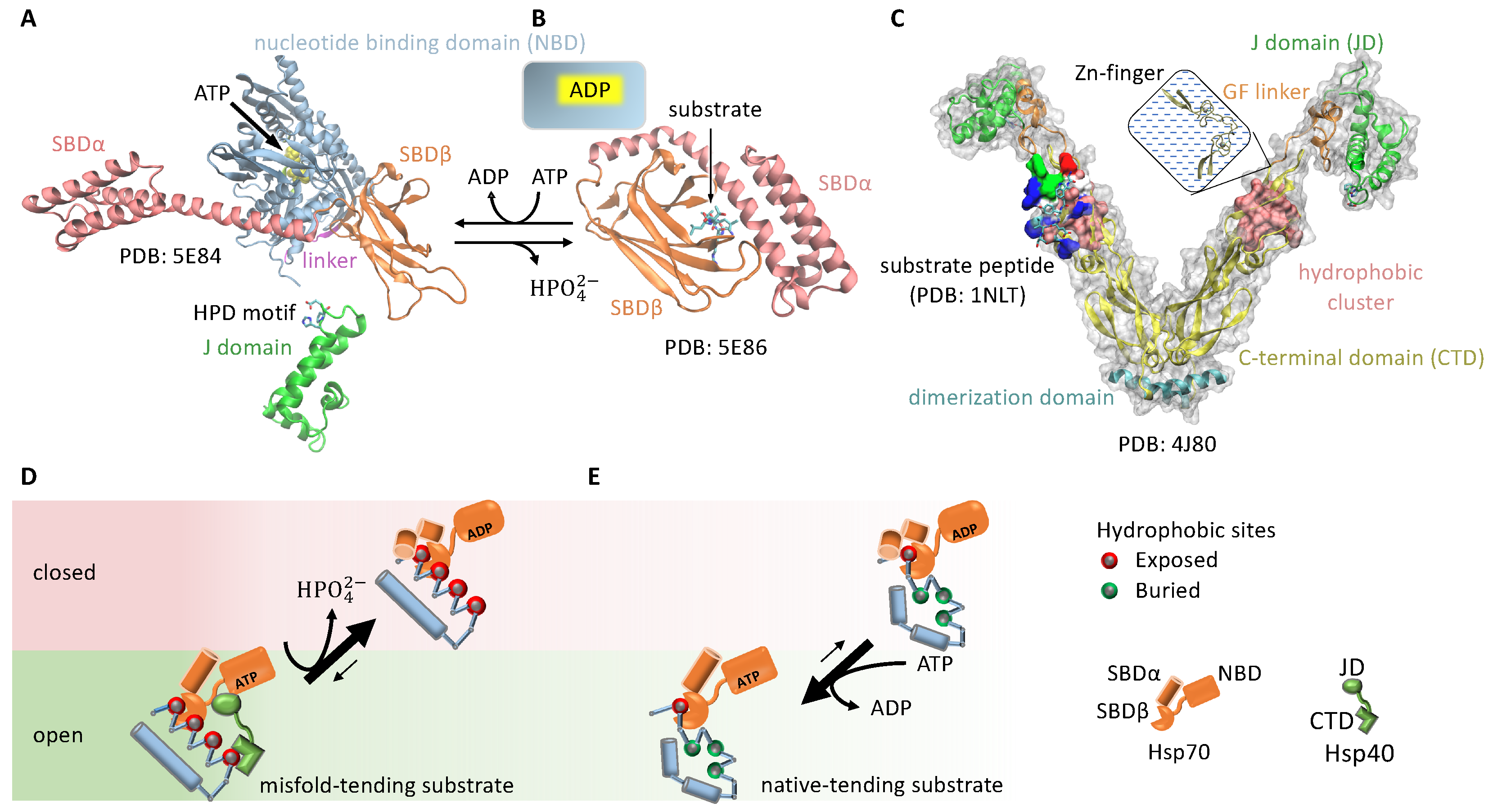
3.1.3. Cochaperone Hsp40 Enables Differential ATP Hydrolysis by Hsp70 Bound to a Substrate in Different Conformations
3.1.4. Hsp70 and Hsp90 Perform Non-Equilibrium Folding by Preferentially Releasing Substrate Proteins in Native-Tending Conformations
3.1.5. The Potential Role of Sequential Hydrolyses of Multiple ATPs in the Chaperone Cycle
3.2. An Upper Bound of Non-Equilibrium Protein Folding and Its Implications
3.2.1. Chaperones Bind to Unstable Intermediate Conformations of Substrates to Drive Non-Equilibrium Folding
3.2.2. Chaperones Stabilize the Native Structures of Slow-Folding Proteins
3.2.3. ATP-Driven Chaperones Buffer Destabilizing Mutations
4. Discussion
Implications for Protein Native Structures and Their Folding Pathways
5. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| Hsp40 | Heat shock protein 40 |
| Hsp70 | Heat shock protein 70 |
| Hsp90 | Heat shock protein 90 |
| Cdc37 | Cell division cycle 37 |
| cryo-EM | Cryogenic electron microscopy |
| PDB | Protein data bank |
References
- Anfinsen, C.B. Principles that Govern the Folding of Protein Chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Dill, K.A. Dominant forces in protein folding. Biochemistry 1990, 29, 7133–7155. [Google Scholar] [CrossRef]
- Bukau, B.; Weissman, J.; Horwich, A. Molecular Chaperones and Protein Quality Control. Cell 2006, 125, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Balchin, D.; Hayer-Hartl, M.; Hartl, U.F. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef]
- Thirumalai, D.; Lorimer, G.H. Chaperonin-Mediated Protein Folding. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 245–269. [Google Scholar] [CrossRef]
- Lin, Z.; Rye, H.S. GroEL-Mediated Protein Folding: Making the Impossible, Possible. Crit. Rev. Biochem. Mol. Biol. 2008, 41, 211–239. [Google Scholar] [CrossRef] [Green Version]
- Horwich, A.L.; Fenton, W.A. Chaperonin-mediated protein folding: Using a central cavity to kinetically assist polypeptide chain folding. Q. Rev. Biophys. 2009, 42. [Google Scholar] [CrossRef]
- Mayer, M.P. Hsp70 chaperone dynamics and molecular mechanism. Trends Biochem. Sci. 2013, 38, 507–514. [Google Scholar] [CrossRef]
- Mayer, M.P.; Gierasch, L.M. Recent advances in the structural and mechanistic aspects of Hsp70 molecular chaperones. J. Biol. Chem. 2019, 294, 2085–2097. [Google Scholar] [CrossRef] [Green Version]
- Pearl, L.H.; Prodromou, C. Structure and Mechanism of the Hsp90 Molecular Chaperone Machinery. Annu. Rev. Biochem. 2006, 75, 271–294. [Google Scholar] [CrossRef]
- Pearl, L.H. Review: The HSP90 molecular chaperone—An enigmatic ATPase. Biopolymers 2016, 105, 594–607. [Google Scholar] [CrossRef] [Green Version]
- Radli, M.; Rüdiger, S.G. Dancing with the Diva: Hsp90-Client Interactions. J. Mol. Biol. 2018, 430, 3029–3040. [Google Scholar] [CrossRef]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Chiosis, G.; Dickey, C.A.; Johnson, J.L. A global view of Hsp90 functions. Nat. Struct. Mol. Biol. 2013, 20, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef]
- Prodromou, C.; Bjorklund, D.M. Advances towards Understanding the Mechanism of Action of the Hsp90 Complex. Biomolecules 2022, 12, 600. [Google Scholar] [CrossRef]
- Ranson, N.A.; Farr, G.W.; Roseman, A.M.; Gowen, B.; Fenton, W.A.; Horwich, A.L.; Saibil, H.R. ATP-Bound States of GroEL Captured by Cryo-Electron Microscopy. Cell 2001, 107, 869–879. [Google Scholar] [CrossRef]
- Krukenberg, K.A.; Street, T.O.; Lavery, L.A.; Agard, D.A. Conformational dynamics of the molecular chaperone Hsp90. Q. Rev. Biophys. 2011, 44, 229–255. [Google Scholar] [CrossRef] [Green Version]
- Clare, D.K.; Vasishtan, D.; Stagg, S.; Quispe, J.; Farr, G.W.; Topf, M.; Horwich, A.L.; Saibil, H.R. ATP-Triggered Conformational Changes Delineate Substrate-Binding and -Folding Mechanics of the GroEL Chaperonin. Cell 2012, 149, 113–123. [Google Scholar] [CrossRef] [Green Version]
- Kityk, R.; Vogel, M.; Schlecht, R.; Bukau, B.; Mayer, M.P. Pathways of allosteric regulation in Hsp70 chaperones. Nat. Commun. 2015, 6, 8308. [Google Scholar] [CrossRef] [Green Version]
- Alderson, T.; Kim, J.; Markley, J. Dynamical Structures of Hsp70 and Hsp70-Hsp40 Complexes. Structure 2016, 24, 1014–1030. [Google Scholar] [CrossRef] [Green Version]
- Weissman, J.S.; Kashi, Y.; Fenton, W.A.; Horwich, A.L. GroEL-mediated protein folding proceeds by multiple rounds of binding and release of nonnative forms. Cell 1994, 78, 693–702. [Google Scholar] [CrossRef]
- Szabo, A.; Langer, T.; Schröder, H.; Flanagan, J.; Bukau, B.; Hartl, F.U. The ATP hydrolysis-dependent reaction cycle of the Escherichia coli Hsp70 system DnaK, DnaJ, and GrpE. Proc. Natl. Acad. Sci. USA 1994, 91, 10345–10349. [Google Scholar] [CrossRef] [Green Version]
- Doyle, S.M.; Genest, O.; Wickner, S. Protein rescue from aggregates by powerful molecular chaperone machines. Nat. Rev. Mol. Cell Biol. 2013, 14, 617–629. [Google Scholar] [CrossRef]
- Imamoglu, R.; Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. Bacterial Hsp70 resolves misfolded states and accelerates productive folding of a multi-domain protein. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Rios, P.D.L.; Goloubinoff, P. Hsp70 chaperones use ATP to remodel native protein oligomers and stable aggregates by entropic pulling. Nat. Struct. Mol. Biol. 2016, 23, 766–769. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Hyeon, C.; Ye, X.; Lorimer, G.H.; Thirumalai, D. Molecular chaperones maximize the native state yield on biological times by driving substrates out of equilibrium. Proc. Natl. Acad. Sci. USA 2017, 114, E10919–E10927. [Google Scholar] [CrossRef] [Green Version]
- Goloubinoff, P.; Sassi, A.S.; Fauvet, B.; Barducci, A.; Rios, P. Chaperones convert the energy from ATP into the nonequilibrium stabilization of native proteins. Nat. Chem. Biol. 2018, 14. [Google Scholar] [CrossRef] [Green Version]
- Xu, H. Cochaperones enable Hsp70 to use ATP energy to stabilize native proteins out of the folding equilibrium. Sci. Rep. 2018, 8, 13213. [Google Scholar] [CrossRef] [Green Version]
- Xu, H. ATP-driven Non-equilibrium Activation of Kinase Clients by the Molecular Chaperone Hsp90. Biophys. J. 2020, 119, 1538–1549. [Google Scholar] [CrossRef]
- Verba, K.A.; Agard, D.A. How Hsp90 and Cdc37 Lubricate Kinase Molecular Switches. Trends Biochem. Sci. 2017, 42, 799–811. [Google Scholar] [CrossRef]
- Kirschke, E.; Goswami, D.; Southworth, D.; Griffin, P.; Agard, D. Glucocorticoid Receptor Function Regulated by Coordinated Action of the Hsp90 and Hsp70 Chaperone Cycles. Cell 2014, 157, 1685–1697. [Google Scholar] [CrossRef] [Green Version]
- Todd, M.J.; Lorimer, G.H.; Thirumalai, D. Chaperonin-facilitated protein folding: Optimization of rate and yield by an iterative annealing mechanism. Proc. Natl. Acad. Sci. USA 1996, 93, 4030–4035. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Mayer, M.P.; Tomita, M. Modeling Hsp70-Mediated Protein Folding. Biophys. J. 2006, 91, 496–507. [Google Scholar] [CrossRef] [Green Version]
- Rios, P.D.L.; Barducci, A. Hsp70 chaperones are non-equilibrium machines that achieve ultra-affinity by energy consumption. eLife 2014, 3, e02218. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.K.; Rios, P.; Christen, P.; Lustig, A.; Goloubinoff, P. The kinetic parameters and energy cost of the Hsp70 chaperone as a polypeptide unfoldase. Nat. Chem. Biol. 2010, 6, 914–920. [Google Scholar] [CrossRef]
- Burston, S.G.; Weissman, J.S.; Farr, G.W.; Fenton, W.A.; Norwich, A.L. Release of both native and non-native proteins from a cis-only GroEL ternary complex. Nature 1996, 383, 96–99. [Google Scholar] [CrossRef]
- Astumian, D.R. Microscopic reversibility as the organizing principle of molecular machines. Nat. Nanotechnol. 2012, 7, 684–688. [Google Scholar] [CrossRef]
- Sekhar, A.; Velyvis, A.; Zoltsman, G.; Rosenzweig, R.; Bouvignies, G.; Kay, L.E. Conserved conformational selection mechanism of Hsp70 chaperone-substrate interactions. eLife 2018, 7, e32764. [Google Scholar] [CrossRef]
- Park, S.J.; Borin, B.N.; Martinez-Yamout, M.A.; Dyson, H.J. The client protein p53 adopts a molten globule–like state in the presence of Hsp90. Nat. Struct. Mol. Biol. 2011, 18, 537–541. [Google Scholar] [CrossRef] [Green Version]
- Verba, K.A.; Wang, R.Y.R.; Arakawa, A.; Liu, Y.; Shirouzu, M.; Yokoyama, S.; Agard, D.A. Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 2016, 352, 1542–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayer-Hartl, M.; Bracher, A.; Hartl, F.U. The GroEL–GroES Chaperonin Machine: A Nano-Cage for Protein Folding. Trends Biochem. Sci. 2016, 41, 62–76. [Google Scholar] [CrossRef]
- Siligardi, G.; Panaretou, B.; Meyer, P.; Singh, S.; Woolfson, D.N.; Piper, P.W.; Pearl, L.H.; Prodromou, C. Regulation of Hsp90 ATPase activity by the co-chaperone Cdc37p/p50cdc37. J. Biol. Chem. 2002, 277, 20151–20159. [Google Scholar] [CrossRef] [Green Version]
- Caplan, A.J.; Mandal, A.K.; Theodoraki, M.A. Molecular chaperones and protein kinase quality control. Trends Cell Biol. 2007, 17, 87–92. [Google Scholar] [CrossRef]
- Keramisanou, D.; Aboalroub, A.; Zhang, Z.; Liu, W.; Marshall, D.; Diviney, A.; Larsen, R.; Landgraf, R.; Gelis, I. Molecular Mechanism of Protein Kinase Recognition and Sorting by the Hsp90 Kinome-Specific Cochaperone Cdc37. Mol. Cell 2016, 62, 260–271. [Google Scholar] [CrossRef] [Green Version]
- Sreeramulu, S.; Jonker, H.R.; Langer, T.; Richter, C.; Lancaster, R.C.; Schwalbe, H. The Human Cdc37·Hsp90 Complex Studied by Heteronuclear NMR Spectroscopy. J. Biol. Chem. 2009, 284, 3885–3896. [Google Scholar] [CrossRef] [Green Version]
- Eckl, J.M.; Scherr, M.J.; Freiburger, L.; Daake, M.A.; Sattler, M.; Richter, K. Hsp90·Cdc37 Complexes with Protein Kinases Form Cooperatively with Multiple Distinct Interaction Sites. J. Biol. Chem. 2015, 290, 30843–30854. [Google Scholar] [CrossRef] [Green Version]
- Keramisanou, D.; Kumar, M.V.; Boose, N.; Abzalimov, R.R.; Gelis, I. Assembly mechanism of early Hsp90-Cdc37-kinase complexes. Sci. Adv. 2022, 8, eabm9294. [Google Scholar] [CrossRef]
- Bjorklund, D.M.; Morgan, R.M.L.; Oberoi, J.; Day, K.L.I.M.; Galliou, P.A.; Prodromou, C. Recognition of BRAF by CDC37 and Reevaluation of the Activation mechanism for the Class 2 BRAF-L597R mutant. bioRxiv 2022. [Google Scholar] [CrossRef]
- Polier, S.; Samant, R.S.; Clarke, P.A.; Workman, P.; Prodromou, C.; Pearl, L.H. ATP-competitive inhibitors block protein kinase recruitment to the Hsp90-Cdc37 system. Nat. Chem. Biol. 2013, 9, 307–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiel, C.; Benisty, H.; Lloréns-Rico, V.; Serrano, L. The yin–yang of kinase activation and unfolding explains the peculiarity of Val600 in the activation segment of BRAF. eLife 2016, 5, e12814. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.P.; Schröder, H.; Rüdiger, S.; Paal, K.; Laufen, T.; Bukau, B. Multistep mechanism of substrate binding determines chaperone activity of Hsp70. Nat. Struct. Mol. Biol. 2000, 7, 586–593. [Google Scholar] [CrossRef]
- Yang, J.; Nune, M.; Zong, Y.; Zhou, L.; Liu, Q. Close and Allosteric Opening of the Polypeptide-Binding Site in a Human Hsp70 Chaperone BiP. Structure 2015, 23, 2191–2203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laufen, T.; Mayer, M.P.; Beisel, C.; Klostermeier, D.; Mogk, A.; Reinstein, J.; Bukau, B. Mechanism of regulation of Hsp70 chaperones by DnaJ cochaperones. Proc. Natl. Acad. Sci. USA 1999, 96, 5452–5457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittung-Stafshede, P.; Guidry, J.; Horne, B.E.; Landry, S.J. The J-Domain of Hsp40 Couples ATP Hydrolysis to Substrate Capture in Hsp70. Biochemistry 2003, 42, 4937–4944. [Google Scholar] [CrossRef]
- Rüdiger, S.; Schneider-Mergener, J.; Bukau, B. Its substrate specificity characterizes the DnaJ co-chaperone as a scanning factor for the DnaK chaperone. EMBO J. 2001, 20, 1042–1050. [Google Scholar] [CrossRef] [Green Version]
- Perales-Calvo, J.; Muga, A.; Moro, F. Role of DnaJ G/F-rich Domain in Conformational Recognition and Binding of Protein Substrates. J. Biol. Chem. 2010, 285, 34231–34239. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R.; Schäfer, U.; Seckler, R. Equilibrium Intermediates in the Reversible Unfolding of Firefly (Photinus pyralis) Luciferase. J. Biol. Chem. 1997, 272, 7099–7105. [Google Scholar] [CrossRef] [Green Version]
- Elnatan, D.; Betegon, M.; Liu, Y.; Ramelot, T.; Kennedy, M.A.; Agard, D.A. Symmetry broken and rebroken during the ATP hydrolysis cycle of the mitochondrial Hsp90 TRAP1. eLife 2017, 6, e25235. [Google Scholar] [CrossRef]
- Lopez, T.; Dalton, K.; Frydman, J. The Mechanism and Function of Group II Chaperonins. J. Mol. Biol. 2015, 427, 2919–2930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gestaut, D.; Limatola, A.; Joachimiak, L.; Frydman, J. The ATP-powered gymnastics of TRiC/CCT: An asymmetric protein folding machine with a symmetric origin story. Curr. Opin. Struct. Biol. 2019, 55, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, K.; Chatila, M.; Sinha, J.; Shi, Q.; Poschner, B.C.; Sikor, M.; Jiang, G.; Lamb, D.C.; Hartl, F.U.; Hayer-Hartl, M. Chaperonin-Catalyzed Rescue of Kinetically Trapped States in Protein Folding. Cell 2010, 142, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.C.; Chang, H.C.; Roeben, A.; Wischnewski, D.; Wischnewski, N.; Kerner, M.J.; Hartl, F.U.; Hayer-Hartl, M. Structural Features of the GroEL-GroES Nano-Cage Required for Rapid Folding of Encapsulated Protein. Cell 2006, 125, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.J.; Haldar, S.; Miličić, G.; Hartl, F.U.; Hayer-Hartl, M. Active Cage Mechanism of Chaperonin-Assisted Protein Folding Demonstrated at Single-Molecule Level. J. Mol. Biol. 2014, 426, 2739–2754. [Google Scholar] [CrossRef] [Green Version]
- Baumketner, A.; Jewett, A.; Shea, J. Effects of Confinement in Chaperonin Assisted Protein Folding: Rate Enhancement by Decreasing the Roughness of the Folding Energy Landscape. J. Mol. Biol. 2003, 332, 701–713. [Google Scholar] [CrossRef]
- England, J.L.; Lucent, D.; Pande, V.S. A Role for Confined Water in Chaperonin Function. J. Am. Chem. Soc. 2008, 130, 11838–11839. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, H.; Hillger, F.; Pfeil, S.H.; Hoffmann, A.; Streich, D.; Haenni, D.; Nettels, D.; Lipman, E.A.; Schuler, B. Single-molecule spectroscopy of protein folding in a chaperonin cage. Proc. Natl. Acad. Sci. USA 2010, 107, 11793–11798. [Google Scholar] [CrossRef] [Green Version]
- Rüdiger, S.; Freund, S.M.; Veprintsev, D.B.; Fersht, A.R. CRINEPT-TROSY NMR reveals p53 core domain bound in an unfolded form to the chaperone Hsp90. Proc. Natl. Acad. Sci. USA 2002, 99, 11085–11090. [Google Scholar] [CrossRef] [Green Version]
- Drummond, D.A.; Wilke, C.O. Mistranslation-Induced Protein Misfolding as a Dominant Constraint on Coding-Sequence Evolution. Cell 2008, 134, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Cagan, A.; Baez-Ortega, A.; Brzozowska, N.; Abascal, F.; Coorens, T.H.H.; Sanders, M.A.; Lawson, A.R.J.; Harvey, L.M.R.; Bhosle, S.; Jones, D.; et al. Somatic mutation rates scale with lifespan across mammals. Nature 2022, 604, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.H.; Choe, J.; Loeb, L.A. Protein tolerance to random amino acid change. Proc. Natl. Acad. Sci. USA 2004, 101, 9205–9210. [Google Scholar] [CrossRef] [Green Version]
- Markiewicz, P.; Kleina, L.; Cruz, C.; Ehret, S.; Miller, J. Genetic Studies of the lac Repressor. XIV. Analysis of 4000 Altered Escherichia coli lac Repressors Reveals Essential and Non-essential Residues, as well as “Spacers” which do not Require a Specific Sequence. J. Mol. Biol. 1994, 240, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Pakula, A.A.; Sauer, R.T. Genetic Analysis of Protein Stability and Function. Annu. Rev. Genet. 1989, 23, 289–310. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.D.; Labthavikul, S.T.; Otey, C.R.; Arnold, F.H. Protein stability promotes evolvability. Proc. Natl. Acad. Sci. USA 2006, 103, 5869–5874. [Google Scholar] [CrossRef] [Green Version]
- Queitsch, C.; Sangster, T.A.; Lindquist, S. Hsp90 as a capacitor of phenotypic variation. Nature 2002, 417, 618–624. [Google Scholar] [CrossRef]
- Aguilar-Rodríguez, J.; Sabater-Muñoz, B.; Montagud-Martínez, R.; Berlanga, V.; Alvarez-Ponce, D.; Wagner, A.; Fares, M.A. The Molecular Chaperone DnaK Is a Source of Mutational Robustness. Genome Biol. Evol. 2016, 8, 2979–2991. [Google Scholar] [CrossRef] [Green Version]
- Karras, G.I.; Yi, S.; Sahni, N.; Fischer, M.; Xie, J.; Vidal, M.; D’Andrea, A.D.; Whitesell, L.; Lindquist, S. HSP90 Shapes the Consequences of Human Genetic Variation. Cell 2017, 168, 856–866.e12. [Google Scholar] [CrossRef] [Green Version]
- Zierer, B.K.; Rübbelke, M.; Tippel, F.; Madl, T.; Schopf, F.H.; Rutz, D.A.; Richter, K.; Sattler, M.; Buchner, J. Importance of cycle timing for the function of the molecular chaperone Hsp90. Nat. Struct. Mol. Biol. 2016, 23, 1020–1028. [Google Scholar] [CrossRef]
- Panaretou, B.; Siligardi, G.; Meyer, P.; Maloney, A.; Sullivan, J.K.; Singh, S.; Millson, S.H.; Clarke, P.A.; Naaby-Hansen, S.; Stein, R.; et al. Activation of the ATPase Activity of Hsp90 by the Stress-Regulated Cochaperone Aha1. Mol. Cell 2002, 10, 1307–1318. [Google Scholar] [CrossRef] [Green Version]
- Panaretou, B.; Prodromou, C.; Roe, S.M.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. ATP binding and hydrolysis are essential to the function of the Hsp90 molecular chaperone in vivo. EMBO J. 1998, 17, 4829–4836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obermann, W.M.; Sondermann, H.; Russo, A.A.; Pavletich, N.P.; Hartl, F.U. In Vivo Function of Hsp90 Is Dependent on ATP Binding and ATP Hydrolysis. J. Cell Biol. 1998, 143, 901–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backe, S.J.; Sager, R.A.; Woodford, M.R.; Makedon, A.M.; Mollapour, M. Post-translational modifications of Hsp90 and translating the chaperone code. J. Biol. Chem. 2020, 295, 11099–11117. [Google Scholar] [CrossRef] [PubMed]
- Nitika.; Porter, C.M.; Truman, A.W.; Truttmann, M.C. Post-translational modifications of Hsp70 family proteins: Expanding the chaperone code. J. Biol. Chem. 2020, 295, 10689–10708. [Google Scholar] [CrossRef] [PubMed]
- Wisén, S.; Bertelsen, E.B.; Thompson, A.D.; Patury, S.; Ung, P.; Chang, L.; Evans, C.G.; Walter, G.M.; Wipf, P.; Carlson, H.A.; et al. Binding of a Small Molecule at a Protein–Protein Interface Regulates the Chaperone Activity of Hsp70–Hsp40. ACS Chem. Biol. 2010, 5, 611–622. [Google Scholar] [CrossRef]
- Taverna, D.M.; Goldstein, R.A. Why are proteins marginally stable? Proteins Struct. Funct. Bioinform. 2002, 46, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, K.; Dill, K. Cellular Proteomes Have Broad Distributions of Protein Stability. Biophys. J. 2010, 99, 3996–4002. [Google Scholar] [CrossRef] [Green Version]
- Park, C.; Marqusee, S. Pulse proteolysis: A simple method for quantitative determination of protein stability and ligand binding. Nat. Methods 2005, 2, 207–212. [Google Scholar] [CrossRef]
- To, P.; Whitehead, B.; Tarbox, H.E.; Fried, S.D. Nonrefoldability is Pervasive Across the E. coli Proteome. J. Am. Chem. Soc. 2021, 143, 11435–11448. [Google Scholar] [CrossRef]
- Leu, J.I.J.; Zhang, P.; Murphy, M.E.; Marmorstein, R.; George, D.L. Structural Basis for the Inhibition of HSP70 and DnaK Chaperones by Small-Molecule Targeting of a C-Terminal Allosteric Pocket. ACS Chem. Biol. 2014, 9, 2508–2516. [Google Scholar] [CrossRef] [Green Version]
- Wen, W.; Liu, W.; Shao, Y.; Chen, L. VER-155008, a small molecule inhibitor of HSP70 with potent anti-cancer activity on lung cancer cell lines. Exp. Biol. Med. 2014, 239, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Jakob, U.; Lilie, H.; Meyer, I.; Buchner, J. Transient Interaction of Hsp90 with Early Unfolding Intermediates of Citrate Synthase Implications for Heat Shock In Vivo. J. Biol. Chem. 1995, 270, 7288–7294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekhar, A.; Rosenzweig, R.; Bouvignies, G.; Kay, L.E. Hsp70 biases the folding pathways of client proteins. Proc. Natl. Acad. Sci. USA 2016, 113, E2794–E2801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, C.K.; Stultz, C.M. Constructing ensembles for intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2011, 21, 426–431. [Google Scholar] [CrossRef] [Green Version]
- Tompa, P. Unstructural biology coming of age. Curr. Opin. Struct. Biol. 2011, 21, 419–425. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Dunker, A.K. Intrinsically Disordered Proteins and Intrinsically Disordered Protein Regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef]
- Chong, S.H.; Chatterjee, P.; Ham, S. Computer Simulations of Intrinsically Disordered Proteins. Annu. Rev. Phys. Chem. 2016, 68, 1–18. [Google Scholar] [CrossRef]
- Best, R.B. Computational and theoretical advances in studies of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2017, 42, 147–154. [Google Scholar] [CrossRef]
- Baker, D.; Agard, D.A. Kinetics versus Thermodynamics in Protein Folding. Biochemistry 1994, 33, 7505–7509. [Google Scholar] [CrossRef]
- Kimchi-Sarfaty, C.; Oh, J.M.; Kim, I.W.; Sauna, Z.E.; Calcagno, A.M.; Ambudkar, S.V.; Gottesman, M.M. A “Silent” Polymorphism in the MDR1 Gene Changes Substrate Specificity. Science 2007, 315, 525–528. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Yoon, J.S.; Shishido, H.; Yang, Z.; Rooney, L.A.; Barral, J.M.; Skach, W.R. Translational tuning optimizes nascent protein folding in cells. Science 2015, 348, 444–448. [Google Scholar] [CrossRef] [Green Version]
- Walsh, I.M.; Bowman, M.A.; Santarriaga, I.F.S.; Rodriguez, A.; Clark, P.L. Synonymous codon substitutions perturb cotranslational protein folding in vivo and impair cell fitness. Proc. Natl. Acad. Sci. USA 2020, 117, 3528–3534. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.A.; Marx, A.; Bronstein, A.M. Codon-specific Ramachandran plots show amino acid backbone conformation depends on identity of the translated codon. Nat. Commun. 2022, 13, 2815. [Google Scholar] [CrossRef] [PubMed]
- Sorokina, I.; Mushegian, A.R.; Koonin, E.V. Is Protein Folding a Thermodynamically Unfavorable, Active, Energy-Dependent Process? Int. J. Mol. Sci. 2022, 23, 521. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
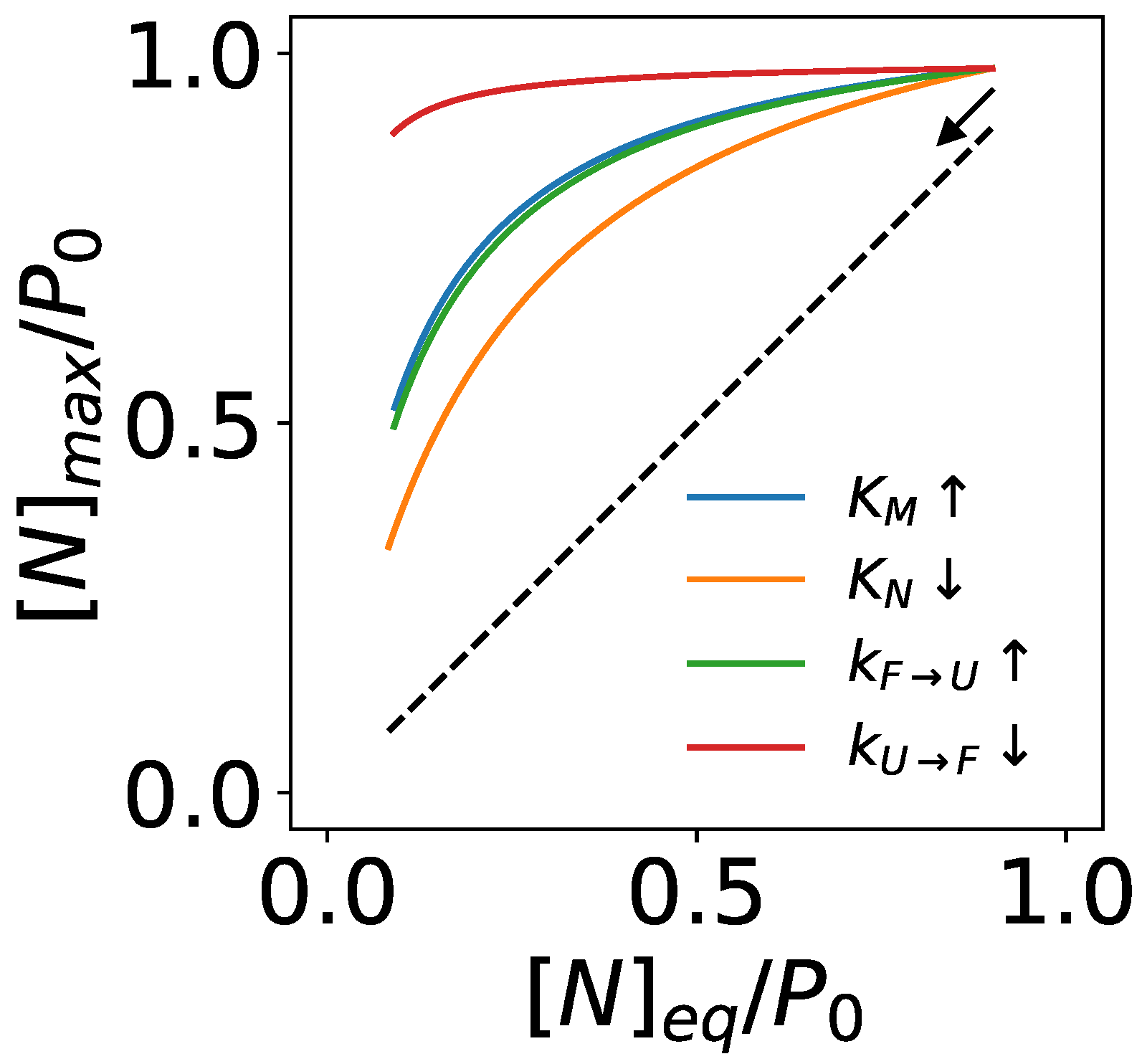{kind=link}
{kind=link}
| Misfolding and aggregation; | |
| Transition between intermediate conformations; | |
| Folding to native structure; | |
| Substrate in conformations binding to the open chaperone | |
| Transition of chaperone between open and closed states | |
| Conversion of protein bound to chaperone in states |
| (M) | (M) | |||||
|---|---|---|---|---|---|---|
| wild-type | 100 | 0.1 | 80 | 0.0092 | 0.07 | 1.0 |
| V600E | 10.24 | 0.4 | 80 | 0.0092 | 0.73 | 0.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, H. Non-Equilibrium Protein Folding and Activation by ATP-Driven Chaperones. Biomolecules 2022, 12, 832. https://doi.org/10.3390/biom12060832
Xu H. Non-Equilibrium Protein Folding and Activation by ATP-Driven Chaperones. Biomolecules. 2022; 12(6):832. https://doi.org/10.3390/biom12060832
Chicago/Turabian StyleXu, Huafeng. 2022. "Non-Equilibrium Protein Folding and Activation by ATP-Driven Chaperones" Biomolecules 12, no. 6: 832. https://doi.org/10.3390/biom12060832
APA StyleXu, H. (2022). Non-Equilibrium Protein Folding and Activation by ATP-Driven Chaperones. Biomolecules, 12(6), 832. https://doi.org/10.3390/biom12060832