
Innate Immunity: A Balance between Disease and Adaption to Stress



{kind=link}
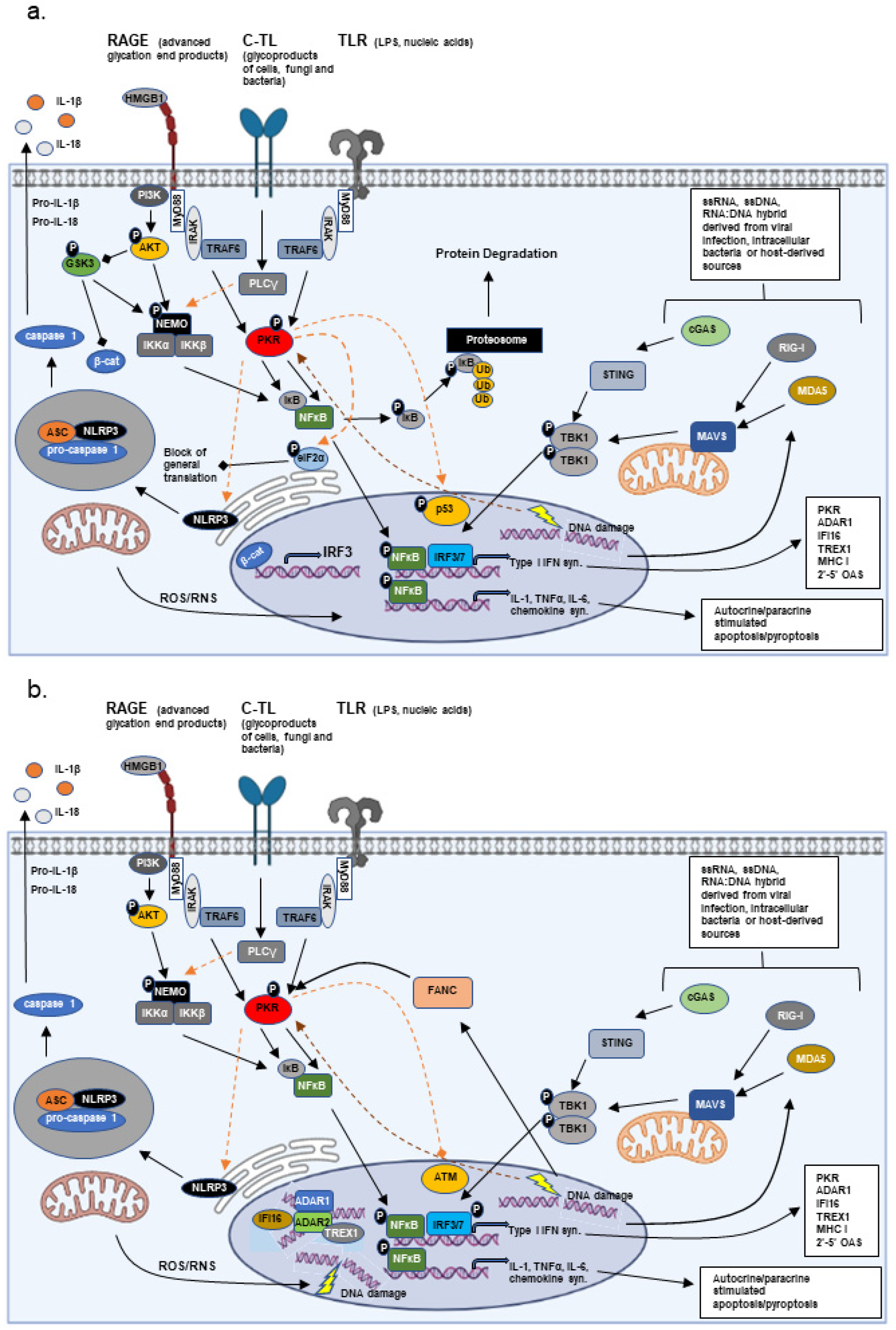
Abstract
1. Introduction
2. Innate Immunity vs. Adaptive Immunity
3. Chronic Inflammation and Disease
3.1. Fanconi Anemia (A Genetic Alteration Stimulating an Innate Immune Response)
3.2. Complement (A Genetic Alteration of an Immune System Component)
3.3. Environmental/Behavioral Factors
3.3.1. Pathogenic Organisms
3.3.2. Environmental Toxins
3.3.3. Behavioral Factors
4. Adaption vs. Disease in Chronic Inflammation
4.1. Bone Marrow Failure Disorders and Acute Myelogenous Leukemia
4.2. Hemoglobin Beta
4.3. Gastro-Intestinal Microbiome
4.4. Innate Immune Control of DNA Damage Repair and Epigentic Modifications
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Granger, D.N.; Senchenkova, E. Inflammation and the Microcirculation; Morgan & Claypool Life Sciences Publisher: San Rafael, CA, USA, 2010. [Google Scholar]
- Gasteiger, G.; D’Osualdo, A.; Schubert, D.A.; Weber, A.; Bruscia, E.M.; Hartl, D. Cellular Innate Immunity: An Old Game with New Players. J. Innate Immun. 2017, 9, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Prantner, D.; Nallar, S.; Vogel, S.N. The role of RAGE in host pathology and crosstalk between RAGE and TLR4 in innate immune signal transduction pathways. FASEB J. 2020, 34, 15659–15674. [Google Scholar] [CrossRef] [PubMed]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Gan, Y.; Li, X.; Han, S.; Liang, Q.; Ma, X.; Rong, P.; Wang, W.; Li, W. The cGAS/STING Pathway: A Novel Target for Cancer Therapy. Front. Immunol. 2021, 12, 795401. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Gal-Ben-Ari, S.; Barrera, I.; Ehrlich, M.; Rosenblum, K. PKR: A Kinase to Remember. Front. Mol. Neurosci. 2018, 11, 480. [Google Scholar] [CrossRef]
- Ito, T.; Yang, M.; May, W.S. RAX, a cellular activator for double-stranded RNA-dependent protein kinase during stress signaling. J. Biol. Chem. 1999, 274, 15427–15432. [Google Scholar] [CrossRef]
- Patel, R.C.; Sen, G.C. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 1998, 17, 4379–4390. [Google Scholar] [CrossRef]
- Blalock, W.L. Opposing forces fight over the same ground to regulate interferon signaling. Biochem. J. 2021, 478, 1853–1859. [Google Scholar] [CrossRef]
- Piazzi, M.; Bavelloni, A.; Faenza, I.; Blalock, W. Glycogen synthase kinase (GSK)-3 and the double-strand RNA-dependent kinase, PKR: When two kinases for the common good turn bad. Biochim. Et Biophys. Acta. Mol. Cell Res. 2020, 1867, 118769. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Nakamura, T.; Inouye, K.; Li, J.; Tang, Y.; Lundback, P.; Valdes-Ferrer, S.I.; Olofsson, P.S.; Kalb, T.; Roth, J.; et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 2012, 488, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- Kok, K.H.; Lui, P.Y.; Ng, M.H.; Siu, K.L.; Au, S.W.; Jin, D.Y. The double-stranded RNA-binding protein PACT functions as a cellular activator of RIG-I to facilitate innate antiviral response. Cell Host Microbe 2011, 9, 299–309. [Google Scholar] [CrossRef]
- Bennett, R.L.; Blalock, W.L.; Abtahi, D.M.; Pan, Y.; Moyer, S.A.; May, W.S. RAX, the PKR activator, sensitizes cells to inflammatory cytokines, serum withdrawal, chemotherapy, and viral infection. Blood 2006, 108, 821–829. [Google Scholar] [CrossRef]
- Sokol, C.L.; Luster, A.D. The chemokine system in innate immunity. Cold Spring Harb. Perspect. Biol. 2015, 7, a016303. [Google Scholar] [CrossRef]
- Eiz-Vesper, B.; Schmetzer, H.M. Antigen-Presenting Cells: Potential of Proven und New Players in Immune Therapies. Transfus. Med. Hemother. 2020, 47, 429–431. [Google Scholar] [CrossRef]
- Kambayashi, T.; Laufer, T.M. Atypical MHC class II-expressing antigen-presenting cells: Can anything replace a dendritic cell? Nat. Rev. Immunol. 2014, 14, 719–730. [Google Scholar] [CrossRef]
- Dong, C.; Flavell, R.A. Cell fate decision: T-helper 1 and 2 subsets in immune responses. Arthritis Res. 2000, 2, 179–188. [Google Scholar] [CrossRef]
- Uribe-Querol, E.; Rosales, C. Phagocytosis: Our Current Understanding of a Universal Biological Process. Front. Immunol. 2020, 11, 1066. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, e17023. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhou, Y.; Ko, K.S.; Yang, H. Interactions between Myc and Mediators of Inflammation in Chronic Liver Diseases. Mediat. Inflamm. 2015, 2015, 276850. [Google Scholar] [CrossRef]
- Bae, S.; Park, P.S.U.; Lee, Y.; Mun, S.H.; Giannopoulou, E.; Fujii, T.; Lee, K.P.; Violante, S.N.; Cross, J.R.; Park-Min, K.H. MYC-mediated early glycolysis negatively regulates proinflammatory responses by controlling IRF4 in inflammatory macrophages. Cell Rep. 2021, 35, 109264. [Google Scholar] [CrossRef]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Et Biophys. Acta 2014, 1843, 2563–2582. [Google Scholar] [CrossRef]
- Cheroni, C.; Manganaro, L.; Donnici, L.; Bevilacqua, V.; Bonnal, R.J.P.; Rossi, R.L.; De Francesco, R. Novel interferon-sensitive genes unveiled by correlation-driven gene selection and systems biology. Sci. Rep. 2021, 11, 18043. [Google Scholar] [CrossRef]
- Uzhachenko, R.V.; Shanker, A. CD8(+) T Lymphocyte and NK Cell Network: Circuitry in the Cytotoxic Domain of Immunity. Front. Immunol. 2019, 10, 1906. [Google Scholar] [CrossRef]
- Muntjewerff, E.M.; Meesters, L.D.; van den Bogaart, G. Antigen Cross-Presentation by Macrophages. Front. Immunol. 2020, 11, 1276. [Google Scholar] [CrossRef]
- Belizario, J.E.; Brandao, W.; Rossato, C.; Peron, J.P. Thymic and Postthymic Regulation of Naive CD4(+) T-Cell Lineage Fates in Humans and Mice Models. Mediat. Inflamm. 2016, 2016, 9523628. [Google Scholar] [CrossRef]
- Tuzlak, S.; Dejean, A.S.; Iannacone, M.; Quintana, F.J.; Waisman, A.; Ginhoux, F.; Korn, T.; Becher, B. Repositioning TH cell polarization from single cytokines to complex help. Nat. Immunol. 2021, 22, 1210–1217. [Google Scholar] [CrossRef] [PubMed]
- Duncan, D.D.; Swain, S.L. Role of antigen-presenting cells in the polarized development of helper T cell subsets: Evidence for differential cytokine production by Th0 cells in response to antigen presentation by B cells and macrophages. Eur. J. Immunol. 1994, 24, 2506–2514. [Google Scholar] [CrossRef] [PubMed]
- Tsai, D.Y.; Hung, K.H.; Chang, C.W.; Lin, K.I. Regulatory mechanisms of B cell responses and the implication in B cell-related diseases. J. Biomed. Sci. 2019, 26, 64. [Google Scholar] [CrossRef] [PubMed]
- Hampe, C.S. B Cell in Autoimmune Diseases. Scientifica 2012, 2012, 215308. [Google Scholar] [CrossRef]
- Zambrano-Zaragoza, J.F.; Romo-Martinez, E.J.; Duran-Avelar Mde, J.; Garcia-Magallanes, N.; Vibanco-Perez, N. Th17 cells in autoimmune and infectious diseases. Int. J. Inflamm. 2014, 2014, 651503. [Google Scholar] [CrossRef]
- Esplugues, E.; Huber, S.; Gagliani, N.; Hauser, A.E.; Town, T.; Wan, Y.Y.; O’Connor, W., Jr.; Rongvaux, A.; Van Rooijen, N.; Haberman, A.M.; et al. Control of TH17 cells occurs in the small intestine. Nature 2011, 475, 514–518. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Cabelof, D.C.; Raffoul, J.J.; Ge, Y.; Van Remmen, H.; Matherly, L.H.; Heydari, A.R. Age-related loss of the DNA repair response following exposure to oxidative stress. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2006, 61, 427–434. [Google Scholar] [CrossRef]
- Schumpert, C.A.; Anderson, C.; Dudycha, J.L.; Patel, R.C. Involvement of Daphnia pulicaria Sir2 in regulating stress response and lifespan. Aging 2016, 8, 402–417. [Google Scholar] [CrossRef][Green Version]
- Yousefzadeh, M.; Henpita, C.; Vyas, R.; Soto-Palma, C.; Robbins, P.; Niedernhofer, L. DNA damage-how and why we age? eLife 2021, 10, e62852. [Google Scholar] [CrossRef]
- Zhu, X.; Chen, Z.; Shen, W.; Huang, G.; Sedivy, J.M.; Wang, H.; Ju, Z. Inflammation, epigenetics, and metabolism converge to cell senescence and ageing: The regulation and intervention. Signal Transduct. Target. Ther. 2021, 6, 245. [Google Scholar] [CrossRef] [PubMed]
- Hayman, T.J.; Glazer, P.M. Regulation of the Cell-Intrinsic DNA Damage Response by the Innate Immune Machinery. Int. J. Mol. Sci. 2021, 22, 12761. [Google Scholar] [CrossRef] [PubMed]
- Nakad, R.; Schumacher, B. DNA Damage Response and Immune Defense: Links and Mechanisms. Front. Genet. 2016, 7, 147. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, J.; Sejas, D.P.; Rathbun, K.R.; Bagby, G.C.; Pang, Q. The Fanconi anemia proteins functionally interact with the protein kinase regulated by RNA (PKR). J. Biol. Chem. 2004, 279, 43910–43919. [Google Scholar] [CrossRef]
- Pang, Q.; Keeble, W.; Diaz, J.; Christianson, T.A.; Fagerlie, S.; Rathbun, K.; Faulkner, G.R.; O’Dwyer, M.; Bagby, G.C., Jr. Role of double-stranded RNA-dependent protein kinase in mediating hypersensitivity of Fanconi anemia complementation group C cells to interferon gamma, tumor necrosis factor-alpha, and double-stranded RNA. Blood 2001, 97, 1644–1652. [Google Scholar] [CrossRef]
- Landelouci, K.; Sinha, S.; Pepin, G. Type-I Interferon Signaling in Fanconi Anemia. Front. Cell. Infect. Microbiol. 2022, 12, 820273. [Google Scholar] [CrossRef]
- Sumpter, R., Jr.; Levine, B. Emerging functions of the Fanconi anemia pathway at a glance. J. Cell Sci. 2017, 130, 2657–2662. [Google Scholar] [CrossRef]
- Gueiderikh, A.; Maczkowiak-Chartois, F.; Rosselli, F. A new frontier in Fanconi anemia: From DNA repair to ribosome biogenesis. Blood Rev. 2022, 52, 100904. [Google Scholar] [CrossRef]
- Garcia-Muse, T.; Aguilera, A. R Loops: From Physiological to Pathological Roles. Cell 2019, 179, 604–618. [Google Scholar] [CrossRef]
- Pang, Q.; Keeble, W.; Christianson, T.A.; Faulkner, G.R.; Bagby, G.C. FANCC interacts with Hsp70 to protect hematopoietic cells from IFN-gamma/TNF-alpha-mediated cytotoxicity. EMBO J. 2001, 20, 4478–4489. [Google Scholar] [CrossRef]
- Pang, Q.; Christianson, T.A.; Keeble, W.; Koretsky, T.; Bagby, G.C. The anti-apoptotic function of Hsp70 in the interferon-inducible double-stranded RNA-dependent protein kinase-mediated death signaling pathway requires the Fanconi anemia protein, FANCC. J. Biol. Chem. 2002, 277, 49638–49643. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, J.; Benlimame, N.; Zeng-Rong, N.; Xiao, D.; Scrivens, P.J.; Koromilas, A.E.; Alaoui-Jamali, M.A. Identification of the interferon-inducible double-stranded RNA-dependent protein kinase as a regulator of cellular response to bulky adducts. Cancer Res. 2000, 60, 6800–6804. [Google Scholar] [PubMed]
- Hao, C.; Shao, R.; Raju, U.; Fang, B.; Swisher, S.G.; Pataer, A. Accumulation of RNA-dependent protein kinase (PKR) in the nuclei of lung cancer cells mediates radiation resistance. Oncotarget 2016, 7, 38235–38242. [Google Scholar] [CrossRef] [PubMed]
- Shiromoto, Y.; Sakurai, M.; Minakuchi, M.; Ariyoshi, K.; Nishikura, K. ADAR1 RNA editing enzyme regulates R-loop formation and genome stability at telomeres in cancer cells. Nat. Commun. 2021, 12, 1654. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, S.; Prados-Carvajal, R.; Fernandez-Avila, M.J.; Silva, S.; Silvestris, D.A.; Endara-Coll, M.; Rodriguez-Real, G.; Domingo-Prim, J.; Mejias-Navarro, F.; Romero-Franco, A.; et al. ADAR-mediated RNA editing of DNA:RNA hybrids is required for DNA double strand break repair. Nat. Commun. 2021, 12, 5512. [Google Scholar] [CrossRef] [PubMed]
- Furquim, C.P.; Pivovar, A.; Amenabar, J.M.; Bonfim, C.; Torres-Pereira, C.C. Oral cancer in Fanconi anemia: Review of 121 cases. Crit. Rev. Oncol. Hematol. 2018, 125, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Grein Cavalcanti, L.; Fuentes Araujo, R.L.; Bonfim, C.; Torres-Pereira, C.C. Oral manifestations compatible with chronic graft-versus-host disease in patients with Fanconi anemia. Biol. Blood Marrow Transplant. 2015, 21, 275–280. [Google Scholar] [CrossRef]
- Nonaka, M.; Kimura, A. Genomic view of the evolution of the complement system. Immunogenetics 2006, 58, 701–713. [Google Scholar] [CrossRef]
- Ikinciogullari, A.; Tekin, M.; Dogu, F.; Reisli, I.; Tanir, G.; Yi, Z.; Garrison, N.; Brilliant, M.H.; Babacan, E. Meningococccal meningitis and complement component 6 deficiency associated with oculocutaneous albinism. Eur. J. Pediatrics 2005, 164, 177–179. [Google Scholar] [CrossRef]
- Fernie, B.A.; Hobart, M.J. Complement C7 deficiency: Seven further molecular defects and their associated marker haplotypes. Hum. Genet. 1998, 103, 513–519. [Google Scholar] [CrossRef]
- Witzel-Schlomp, K.; Hobart, M.J.; Fernie, B.A.; Orren, A.; Wurzner, R.; Rittner, C.; Kaufmann, T.; Schneider, P.M. Heterogeneity in the genetic basis of human complement C9 deficiency. Immunogenetics 1998, 48, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Seddon, J.M.; Yu, Y.; Miller, E.C.; Reynolds, R.; Tan, P.L.; Gowrisankar, S.; Goldstein, J.I.; Triebwasser, M.; Anderson, H.E.; Zerbib, J.; et al. Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat. Genet. 2013, 45, 1366–1370. [Google Scholar] [CrossRef] [PubMed]
- Dragon-Durey, M.A.; Quartier, P.; Fremeaux-Bacchi, V.; Blouin, J.; de Barace, C.; Prieur, A.M.; Weiss, L.; Fridman, W.H. Molecular basis of a selective C1s deficiency associated with early onset multiple autoimmune diseases. J. Immunol. 2001, 166, 7612–7616. [Google Scholar] [CrossRef] [PubMed]
- Slingsby, J.H.; Norsworthy, P.; Pearce, G.; Vaishnaw, A.K.; Issler, H.; Morley, B.J.; Walport, M.J. Homozygous hereditary C1q deficiency and systemic lupus erythematosus. A new family and the molecular basis of C1q deficiency in three families. Arthritis Rheum. 1996, 39, 663–670. [Google Scholar] [CrossRef]
- Petry, F.; Hauptmann, G.; Goetz, J.; Grosshans, E.; Loos, M. Molecular basis of a new type of C1q-deficiency associated with a non-functional low molecular weight (LMW) C1q: Parallels and differences to other known genetic C1q-defects. Immunopharmacology 1997, 38, 189–201. [Google Scholar] [CrossRef]
- Wetsel, R.A.; Kulics, J.; Lokki, M.L.; Kiepiela, P.; Akama, H.; Johnson, C.A.; Densen, P.; Colten, H.R. Type II human complement C2 deficiency. Allele-specific amino acid substitutions (Ser189 --> Phe; Gly444 --> Arg) cause impaired C2 secretion. J. Biol. Chem. 1996, 271, 5824–5831. [Google Scholar] [CrossRef]
- Singer, L.; Whitehead, W.T.; Akama, H.; Katz, Y.; Fishelson, Z.; Wetsel, R.A. Inherited human complement C3 deficiency. An amino acid substitution in the beta-chain (ASP549 to ASN) impairs C3 secretion. J. Biol. Chem. 1994, 269, 28494–28499. [Google Scholar] [CrossRef]
- Barba, G.; Rittner, C.; Schneider, P.M. Genetic basis of human complement C4A deficiency. Detection of a point mutation leading to nonexpression. J. Clin. Investig. 1993, 91, 1681–1686. [Google Scholar] [CrossRef]
- Wilson, W.A.; Perez, M.C. Complete C4B deficiency in black Americans with systemic lupus erythematosus. J. Rheumatol. 1988, 15, 1855–1858. [Google Scholar]
- Yang, Y.; Chung, E.K.; Wu, Y.L.; Savelli, S.L.; Nagaraja, H.N.; Zhou, B.; Hebert, M.; Jones, K.N.; Shu, Y.; Kitzmiller, K.; et al. Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): Low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am. J. Hum. Genet. 2007, 80, 1037–1054. [Google Scholar] [CrossRef]
- Macedo, A.C.; Isaac, L. Systemic Lupus Erythematosus and Deficiencies of Early Components of the Complement Classical Pathway. Front. Immunol. 2016, 7, 55. [Google Scholar] [CrossRef] [PubMed]
- Gold, B.; Merriam, J.E.; Zernant, J.; Hancox, L.S.; Taiber, A.J.; Gehrs, K.; Cramer, K.; Neel, J.; Bergeron, J.; Barile, G.R.; et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat. Genet. 2006, 38, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Goicoechea de Jorge, E.; Harris, C.L.; Esparza-Gordillo, J.; Carreras, L.; Arranz, E.A.; Garrido, C.A.; Lopez-Trascasa, M.; Sanchez-Corral, P.; Morgan, B.P.; Rodriguez de Cordoba, S. Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Sprong, T.; Roos, D.; Weemaes, C.; Neeleman, C.; Geesing, C.L.; Mollnes, T.E.; van Deuren, M. Deficient alternative complement pathway activation due to factor D deficiency by 2 novel mutations in the complement factor D gene in a family with meningococcal infections. Blood 2006, 107, 4865–4870. [Google Scholar] [CrossRef] [PubMed]
- Maga, T.K.; Nishimura, C.J.; Weaver, A.E.; Frees, K.L.; Smith, R.J. Mutations in alternative pathway complement proteins in American patients with atypical hemolytic uremic syndrome. Hum. Mutat. 2010, 31, E1445–E1460. [Google Scholar] [CrossRef]
- Servais, A.; Fremeaux-Bacchi, V.; Lequintrec, M.; Salomon, R.; Blouin, J.; Knebelmann, B.; Grunfeld, J.P.; Lesavre, P.; Noel, L.H.; Fakhouri, F. Primary glomerulonephritis with isolated C3 deposits: A new entity which shares common genetic risk factors with haemolytic uraemic syndrome. J. Med. Genet. 2007, 44, 193–199. [Google Scholar] [CrossRef]
- Van de Ven, J.P.; Nilsson, S.C.; Tan, P.L.; Buitendijk, G.H.; Ristau, T.; Mohlin, F.C.; Nabuurs, S.B.; Schoenmaker-Koller, F.E.; Smailhodzic, D.; Campochiaro, P.A.; et al. A functional variant in the CFI gene confers a high risk of age-related macular degeneration. Nat. Genet. 2013, 45, 813–817. [Google Scholar] [CrossRef]
- Richards, A.; Buddles, M.R.; Donne, R.L.; Kaplan, B.S.; Kirk, E.; Venning, M.C.; Tielemans, C.L.; Goodship, J.A.; Goodship, T.H. Factor H mutations in hemolytic uremic syndrome cluster in exons 18-20, a domain important for host cell recognition. Am. J. Hum. Genet. 2001, 68, 485–490. [Google Scholar] [CrossRef]
- Raychaudhuri, S.; Iartchouk, O.; Chin, K.; Tan, P.L.; Tai, A.K.; Ripke, S.; Gowrisankar, S.; Vemuri, S.; Montgomery, K.; Yu, Y.; et al. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat. Genet. 2011, 43, 1232–1236. [Google Scholar] [CrossRef]
- Xu, Y.Y.; Zhi, Y.X.; Yin, J.; Wang, L.L.; Wen, L.P.; Gu, J.Q.; Guan, K.; Craig, T.; Zhang, H.Y. Mutational spectrum and geno-phenotype correlation in Chinese families with hereditary angioedema. Allergy 2012, 67, 1430–1436. [Google Scholar] [CrossRef]
- Bafunno, V.; Bova, M.; Loffredo, S.; Divella, C.; Petraroli, A.; Marone, G.; Montinaro, V.; Margaglione, M.; Triggiani, M. Mutational spectrum of the c1 inhibitor gene in a cohort of Italian patients with hereditary angioedema: Description of nine novel mutations. Ann. Hum. Genet. 2014, 78, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Boackle, S.A.; Hanvivadhanakul, P.; Ulgiati, D.; Grossman, J.M.; Lee, Y.; Shen, N.; Abraham, L.J.; Mercer, T.R.; Park, E.; et al. Association of a common complement receptor 2 haplotype with increased risk of systemic lupus erythematosus. Proc. Natl. Acad. Sci. USA 2007, 104, 3961–3966. [Google Scholar] [CrossRef] [PubMed]
- Thiel, J.; Kimmig, L.; Salzer, U.; Grudzien, M.; Lebrecht, D.; Hagena, T.; Draeger, R.; Voelxen, N.; Bergbreiter, A.; Jennings, S.; et al. Genetic CD21 deficiency is associated with hypogammaglobulinemia. J. Allergy Clin. Immunol. 2012, 129, 801–810.e806. [Google Scholar] [CrossRef] [PubMed]
- Feichtinger, R.G.; Olahova, M.; Kishita, Y.; Garone, C.; Kremer, L.S.; Yagi, M.; Uchiumi, T.; Jourdain, A.A.; Thompson, K.; D’Souza, A.R.; et al. Biallelic C1QBP Mutations Cause Severe Neonatal-, Childhood-, or Later-Onset Cardiomyopathy Associated with Combined Respiratory-Chain Deficiencies. Am. J. Hum. Genet. 2017, 101, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.J.; Canete, P.F.; Wang, H.; Medhavy, A.; Bones, J.; Roco, J.A.; He, Y.; Qin, Y.; Cappello, J.; Ellyard, J.I.; et al. TLR7 gain-of-function genetic variation causes human lupus. Nature 2022, 605, 349–356. [Google Scholar] [CrossRef]
- Mao, D.; Reuter, C.M.; Ruzhnikov, M.R.Z.; Beck, A.E.; Farrow, E.G.; Emrick, L.T.; Rosenfeld, J.A.; Mackenzie, K.M.; Robak, L.; Wheeler, M.T.; et al. De novo EIF2AK1 and EIF2AK2 Variants Are Associated with Developmental Delay, Leukoencephalopathy, and Neurologic Decompensation. Am. J. Hum. Genet. 2020, 106, 570–583. [Google Scholar] [CrossRef]
- Kuipers, D.J.S.; Mandemakers, W.; Lu, C.S.; Olgiati, S.; Breedveld, G.J.; Fevga, C.; Tadic, V.; Carecchio, M.; Osterman, B.; Sagi-Dain, L.; et al. EIF2AK2 Missense Variants Associated with Early Onset Generalized Dystonia. Ann. Neurol. 2021, 89, 485–497. [Google Scholar] [CrossRef]
- Burnett, S.B.; Vaughn, L.S.; Sharma, N.; Kulkarni, R.; Patel, R.C. Dystonia 16 (DYT16) mutations in PACT cause dysregulated PKR activation and eIF2alpha signaling leading to a compromised stress response. Neurobiol. Dis. 2020, 146, 105135. [Google Scholar] [CrossRef]
- Shah, A.; Kishore, U.; Shastri, A. Complement System in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 13647. [Google Scholar] [CrossRef]
- Ajjan, R.A.; Schroeder, V. Role of complement in diabetes. Mol. Immunol. 2019, 114, 270–277. [Google Scholar] [CrossRef]
- Sturfelt, G.; Truedsson, L. Complement in the immunopathogenesis of rheumatic disease. Nat. Rev. Rheumatol. 2012, 8, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Esposito, J.; Brown, Z.; Stevens, W.; Sahhar, J.; Rabusa, C.; Zochling, J.; Roddy, J.; Walker, J.; Proudman, S.M.; Nikpour, M. The association of low complement with disease activity in systemic sclerosis: A prospective cohort study. Arthritis Res. Ther. 2016, 18, 246. [Google Scholar] [CrossRef] [PubMed]
- Pellicano, C.; Miglionico, M.; Romaggioli, L.; Colalillo, A.; Vantaggio, L.; Napodano, C.; Calla, C.; Gulli, F.; Marino, M.; Basile, U.; et al. Increased Complement Activation in Systemic Sclerosis Patients with Skin and Lung Fibrosis. J. Pers. Med. 2022, 12, 284. [Google Scholar] [CrossRef]
- Kourtzelis, I.; Rafail, S. The dual role of complement in cancer and its implication in anti-tumor therapy. Ann. Transl. Med. 2016, 4, 265. [Google Scholar] [CrossRef] [PubMed]
- Laval, K.; Enquist, L.W. The Potential Role of Herpes Simplex Virus Type 1 and Neuroinflammation in the Pathogenesis of Alzheimer’s Disease. Front. Neurol. 2021, 12, 658695. [Google Scholar] [CrossRef]
- Canet, G.; Dias, C.; Gabelle, A.; Simonin, Y.; Gosselet, F.; Marchi, N.; Makinson, A.; Tuaillon, E.; Van de Perre, P.; Givalois, L.; et al. HIV Neuroinfection and Alzheimer’s Disease: Similarities and Potential Links? Front. Cell. Neurosci. 2018, 12, 307. [Google Scholar] [CrossRef]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64, S84–S101. [Google Scholar] [CrossRef]
- Yi, Z.; Yuan, Z. Hepatitis C Virus-Associated Cancers. Adv. Exp. Med. Biol. 2017, 1018, 129–146. [Google Scholar] [CrossRef]
- Wroblewski, L.E.; Peek, R.M., Jr.; Wilson, K.T. Helicobacter pylori and gastric cancer: Factors that modulate disease risk. Clin. Microbiol. Rev. 2010, 23, 713–739. [Google Scholar] [CrossRef]
- Singh, S.K.; Girschick, H.J. Lyme borreliosis: From infection to autoimmunity. Clin. Microbiol. Infect. 2004, 10, 598–614. [Google Scholar] [CrossRef]
- Kubelkova, K.; Macela, A. Innate Immune Recognition: An Issue More Complex Than Expected. Front. Cell. Infect. Microbiol. 2019, 9, 241. [Google Scholar] [CrossRef] [PubMed]
- Al Bander, Z.; Nitert, M.D.; Mousa, A.; Naderpoor, N. The Gut Microbiota and Inflammation: An Overview. Int. J. Environ. Res. Public Health 2020, 17, 7618. [Google Scholar] [CrossRef] [PubMed]
- Lopetuso, L.R.; Ianiro, G.; Allegretti, J.R.; Bibbo, S.; Gasbarrini, A.; Scaldaferri, F.; Cammarota, G. Fecal transplantation for ulcerative colitis: Current evidence and future applications. Expert Opin. Biol. Ther. 2020, 20, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Zhou, Y.; Liao, B.; Liao, M.; Shi, Y.; Wei, Y.; Huang, Y.; Zhou, X.; Cheng, L.; Ren, B. The Interaction Between the Microbiome and Tumors. Front. Cell. Infect. Microbiol. 2021, 11, 673724. [Google Scholar] [CrossRef]
- Benedetti, S.; Nuvoli, B.; Catalani, S.; Galati, R. Reactive oxygen species a double-edged sword for mesothelioma. Oncotarget 2015, 6, 16848–16865. [Google Scholar] [CrossRef]
- Bartrip, P.W. History of asbestos related disease. Postgrad. Med. J. 2004, 80, 72–76. [Google Scholar] [CrossRef]
- Bunderson-Schelvan, M.; Pfau, J.C.; Crouch, R.; Holian, A. Nonpulmonary outcomes of asbestos exposure. J. Toxicol. Environ. Health B Crit. Rev. 2011, 14, 122–152. [Google Scholar] [CrossRef]
- Rosenthal, G.J.; Simeonova, P.; Corsini, E. Asbestos toxicity: An immunologic perspective. Rev. Environ. Health 1999, 14, 11–20. [Google Scholar] [CrossRef]
- Brinkmann, V.; Ale-Agha, N.; Haendeler, J.; Ventura, N. The Aryl Hydrocarbon Receptor (AhR) in the Aging Process: Another Puzzling Role for This Highly Conserved Transcription Factor. Front. Physiol. 2019, 10, 1561. [Google Scholar] [CrossRef]
- Sekine, H.; Mimura, J.; Oshima, M.; Okawa, H.; Kanno, J.; Igarashi, K.; Gonzalez, F.J.; Ikuta, T.; Kawajiri, K.; Fujii-Kuriyama, Y. Hypersensitivity of aryl hydrocarbon receptor-deficient mice to lipopolysaccharide-induced septic shock. Mol. Cell. Biol. 2009, 29, 6391–6400. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Hanieh, H.; Nakahama, T.; Kishimoto, T. The roles of aryl hydrocarbon receptor in immune responses. Int. Immunol. 2013, 25, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Does Alcohol Cause Cancer? Available online: https://cancerresearchuk.org/about-cancer/causes-of-cancer/alcohol-and-cancer/does-alcohol-cause-cancer (accessed on 28 March 2022).
- Seitz, H.K.; Stickel, F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat. Rev. Cancer 2007, 7, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Garaycoechea, J.I.; Crossan, G.P.; Langevin, F.; Mulderrig, L.; Louzada, S.; Yang, F.; Guilbaud, G.; Park, N.; Roerink, S.; Nik-Zainal, S.; et al. Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 2018, 553, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Salaspuro, M. Local Acetaldehyde: Its Key Role in Alcohol-Related Oropharyngeal Cancer. Visc. Med. 2020, 36, 167–173. [Google Scholar] [CrossRef]
- What’s in a Cigarette? Available online: https://www.cancerresearchuk.or/about-cancer/causes-of-cancer/smoking-and-cancer/whats-in-a-cigarette-0 (accessed on 28 March 2022).
- Strzelak, A.; Ratajczak, A.; Adamiec, A.; Feleszko, W. Tobacco Smoke Induces and Alters Immune Responses in the Lung Triggering Inflammation, Allergy, Asthma and Other Lung Diseases: A Mechanistic Review. Int. J. Environ. Res. Public Health 2018, 15, 1033. [Google Scholar] [CrossRef]
- How Does Smoking Cause Cancer? Available online: https://www.cancerresearchuk.org/about-cancer/causes-of-cancer/smoking-and-cancer/how-does-smoking-cause-cancer (accessed on 28 March 2022).
- Fu, X.; Shi, H.; Qi, Y.; Zhang, W.; Dong, P. M2 polarized macrophages induced by CSE promote proliferation, migration, and invasion of alveolar basal epithelial cells. Int. Immunopharmacol. 2015, 28, 666–674. [Google Scholar] [CrossRef]
- Rovina, N.; Koutsoukou, A.; Koulouris, N.G. Inflammation and immune response in COPD: Where do we stand? Mediat. Inflamm. 2013, 2013, 413735. [Google Scholar] [CrossRef]
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef]
- Koonin, E.V.; Wolf, Y.I. Is evolution Darwinian or/and Lamarckian? Biol. Direct 2009, 4, 42. [Google Scholar] [CrossRef]
- Monroe, J.G.; Srikant, T.; Carbonell-Bejerano, P.; Becker, C.; Lensink, M.; Exposito-Alonso, M.; Klein, M.; Hildebrandt, J.; Neumann, M.; Kliebenstein, D.; et al. Mutation bias reflects natural selection in Arabidopsis thaliana. Nature 2022, 602, 101–105. [Google Scholar] [CrossRef]
- Matic, I. Mutation Rate Heterogeneity Increases Odds of Survival in Unpredictable Environments. Mol. Cell 2019, 75, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Sharma, U. Paternal Contributions to Offspring Health: Role of Sperm Small RNAs in Intergenerational Transmission of Epigenetic Information. Front. Cell Dev. Biol. 2019, 7, 215. [Google Scholar] [CrossRef] [PubMed]
- Donkin, I.; Barres, R. Sperm epigenetics and influence of environmental factors. Mol. Metab. 2018, 14, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Van Niekerk, G.; Davids, L.M.; Hattingh, S.M.; Engelbrecht, A.M. Cancer stem cells: A product of clonal evolution? Int. J. Cancer 2017, 140, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Savage, S.A.; Dufour, C. Classical inherited bone marrow failure syndromes with high risk for myelodysplastic syndrome and acute myelogenous leukemia. Semin. Hematol. 2017, 54, 105–114. [Google Scholar] [CrossRef]
- Blalock, W.L.; Bavelloni, A.; Piazzi, M.; Faenza, I.; Cocco, L. A role for PKR in hematologic malignancies. J. Cell. Physiol. 2010, 223, 572–591. [Google Scholar] [CrossRef]
- Piazzi, M.; Bavelloni, A.; Gallo, A.; Faenza, I.; Blalock, W.L. Signal Transduction in Ribosome Biogenesis: A Recipe to Avoid Disaster. Int. J. Mol. Sci. 2019, 20, 2718. [Google Scholar] [CrossRef]
- Fisher, D.A.C.; Fowles, J.S.; Zhou, A.; Oh, S.T. Inflammatory Pathophysiology as a Contributor to Myeloproliferative Neoplasms. Front. Immunol. 2021, 12, 683401. [Google Scholar] [CrossRef]
- Follo, M.Y.; Finelli, C.; Mongiorgi, S.; Clissa, C.; Bosi, C.; Martinelli, G.; Blalock, W.L.; Cocco, L.; Martelli, A.M. PKR is activated in MDS patients and its subcellular localization depends on disease severity. Leukemia 2008, 22, 2267–2269. [Google Scholar] [CrossRef]
- Blalock, W.L.; Grimaldi, C.; Fala, F.; Follo, M.; Horn, S.; Basecke, J.; Martinelli, G.; Cocco, L.; Martelli, A.M. PKR activity is required for acute leukemic cell maintenance and growth: A role for PKR-mediated phosphatase activity to regulate GSK-3 phosphorylation. J. Cell. Physiol. 2009, 221, 232–241. [Google Scholar] [CrossRef]
- Blalock, W.L.; Bavelloni, A.; Piazzi, M.; Tagliavini, F.; Faenza, I.; Martelli, A.M.; Follo, M.Y.; Cocco, L. Multiple forms of PKR present in the nuclei of acute leukemia cells represent an active kinase that is responsive to stress. Leukemia 2011, 25, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Byrne, M.; Brown, K.D.; Konopleva, M.Y.; Kornblau, S.M.; Bennett, R.L.; May, W.S. PKR inhibits the DNA damage response, and is associated with poor survival in AML and accelerated leukemia in NHD13 mice. Blood 2015, 126, 1585–1594. [Google Scholar] [CrossRef] [PubMed]
- Melamed, D.; Nov, Y.; Malik, A.; Yakass, M.B.; Bolotin, E.; Shemer, R.; Hiadzi, E.K.; Skorecki, K.L.; Livnat, A. De novo mutation rates at the single-mutation resolution in a human HBB gene region associated with adaptation and genetic disease. Genome Res. 2022, 32, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef] [PubMed]
- Suppli, M.P.; Bagger, J.I.; Lelouvier, B.; Broha, A.; Demant, M.; Konig, M.J.; Strandberg, C.; Lund, A.; Vilsboll, T.; Knop, F.K. Hepatic microbiome in healthy lean and obese humans. JHEP Rep. 2021, 3, 100299. [Google Scholar] [CrossRef]
- Molinero, N.; Ruiz, L.; Milani, C.; Gutierrez-Diaz, I.; Sanchez, B.; Mangifesta, M.; Segura, J.; Cambero, I.; Campelo, A.B.; Garcia-Bernardo, C.M.; et al. The human gallbladder microbiome is related to the physiological state and the biliary metabolic profile. Microbiome 2019, 7, 100. [Google Scholar] [CrossRef]
- Ka, N.L.; Lim, G.Y.; Hwang, S.; Kim, S.S.; Lee, M.O. IFI16 inhibits DNA repair that potentiates type-I interferon-induced antitumor effects in triple negative breast cancer. Cell Rep. 2021, 37, 110138. [Google Scholar] [CrossRef]
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 1, 519–525. [Google Scholar] [CrossRef]
- Jimeno, S.; Balestra, F.R.; Huertas, P. The Emerging Role of RNA Modifications in DNA Double-Strand Break Repair. Front. Mol. Biosci. 2021, 8, 664872. [Google Scholar] [CrossRef]
- Tao, S.S.; Wu, G.C.; Zhang, Q.; Zhang, T.P.; Leng, R.X.; Pan, H.F.; Ye, D.Q. TREX1 As a Potential Therapeutic Target for Autoimmune and Inflammatory Diseases. Curr. Pharm. Des. 2019, 25, 3239–3247. [Google Scholar] [CrossRef]
- Tassinari, V.; Cesarini, V.; Tomaselli, S.; Ianniello, Z.; Silvestris, D.A.; Ginistrelli, L.C.; Martini, M.; De Angelis, B.; De Luca, G.; Vitiani, L.R.; et al. ADAR1 is a new target of METTL3 and plays a pro-oncogenic role in glioblastoma by an editing-independent mechanism. Genome Biol. 2021, 22, 51. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Hsu, P.J.; Xing, X.; Fang, J.; Lu, Z.; Zou, Q.; Zhang, K.J.; Zhang, X.; Zhou, Y.; Zhang, T.; et al. Mettl3-/Mettl14-mediated mRNA N(6)-methyladenosine modulates murine spermatogenesis. Cell Res. 2017, 27, 1216–1230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, L.; Peng, D.; Jiang, A.; He, Y.; Zeng, Y.; Xie, C.; Zhou, H.; Luo, X.; Liu, H.; et al. METTL3 and N6-Methyladenosine Promote Homologous Recombination-Mediated Repair of DSBs by Modulating DNA-RNA Hybrid Accumulation. Mol. Cell 2020, 79, 425–442.e427. [Google Scholar] [CrossRef] [PubMed]
- Geffroy, B.; Douhard, M. The Adaptive Sex in Stressful Environments. Trends Ecol. Evol. 2019, 34, 628–640. [Google Scholar] [CrossRef]
- Kishimoto, S.; Uno, M.; Okabe, E.; Nono, M.; Nishida, E. Environmental stresses induce transgenerationally inheritable survival advantages via germline-to-soma communication in Caenorhabditis elegans. Nat. Commun. 2017, 8, 14031. [Google Scholar] [CrossRef]
- Cheng, C.; Kirkpatrick, M. Environmental Plasticity in the Intersexual Correlation and Sex Bias of Gene Expression. J. Hered. 2017, 108, 754–758. [Google Scholar] [CrossRef]
- Marcho, C.; Oluwayiose, O.A.; Pilsner, J.R. The preconception environment and sperm epigenetics. Andrology 2020, 8, 924–942. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, G.; Zhang, F. Role of Peripheral Immune Cells-Mediated Inflammation on the Process of Neurodegenerative Diseases. Front. Immunol. 2020, 11, 582825. [Google Scholar] [CrossRef]
- Sim, K.Y.; Im, K.C.; Park, S.G. The Functional Roles and Applications of Immunoglobulins in Neurodegenerative Disease. Int. J. Mol. Sci. 2020, 21, 5295. [Google Scholar] [CrossRef]
- Piazzi, M.; Bavelloni, A.; Cenni, V.; Faenza, I.; Blalock, W.L. Revisiting the Role of GSK3, A Modulator of Innate Immunity, in Idiopathic Inclusion Body Myositis. Cells 2021, 10, 3255. [Google Scholar] [CrossRef]
- Lin, B.; Goldbach-Mansky, R. Pathogenic insights from genetic causes of autoinflammatory inflammasomopathies and interferonopathies. J. Allergy Clin. Immunol. 2022, 149, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Nazmi, A.; Field, R.H.; Griffin, E.W.; Haugh, O.; Hennessy, E.; Cox, D.; Reis, R.; Tortorelli, L.; Murray, C.L.; Lopez-Rodriguez, A.B.; et al. Chronic neurodegeneration induces type I interferon synthesis via STING, shaping microglial phenotype and accelerating disease progression. Glia 2019, 67, 1254–1276. [Google Scholar] [CrossRef] [PubMed]
- Long, H.Z.; Cheng, Y.; Zhou, Z.W.; Luo, H.Y.; Wen, D.D.; Gao, L.C. PI3K/AKT Signal Pathway: A Target of Natural Products in the Prevention and Treatment of Alzheimer’s Disease and Parkinson’s Disease. Front. Pharmacol. 2021, 12, 648636. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faenza, I.; Blalock, W.L. Innate Immunity: A Balance between Disease and Adaption to Stress. Biomolecules 2022, 12, 737. https://doi.org/10.3390/biom12050737
Faenza I, Blalock WL. Innate Immunity: A Balance between Disease and Adaption to Stress. Biomolecules. 2022; 12(5):737. https://doi.org/10.3390/biom12050737
Chicago/Turabian StyleFaenza, Irene, and William L. Blalock. 2022. "Innate Immunity: A Balance between Disease and Adaption to Stress" Biomolecules 12, no. 5: 737. https://doi.org/10.3390/biom12050737
APA StyleFaenza, I., & Blalock, W. L. (2022). Innate Immunity: A Balance between Disease and Adaption to Stress. Biomolecules, 12(5), 737. https://doi.org/10.3390/biom12050737