
Population Dynamics of Epithelial-Mesenchymal Heterogeneity in Cancer Cells


Abstract
:1. Introduction
2. Methods
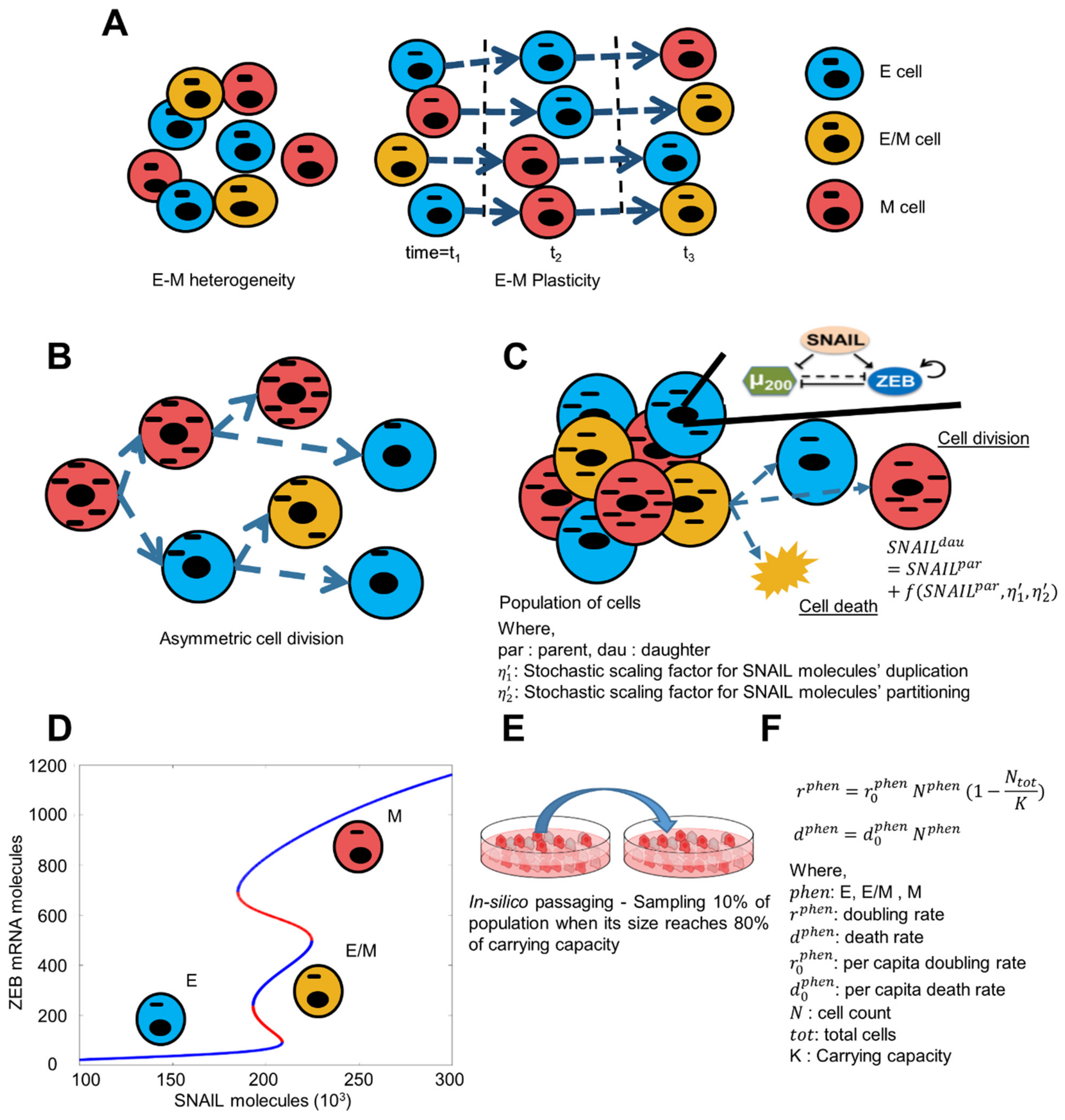
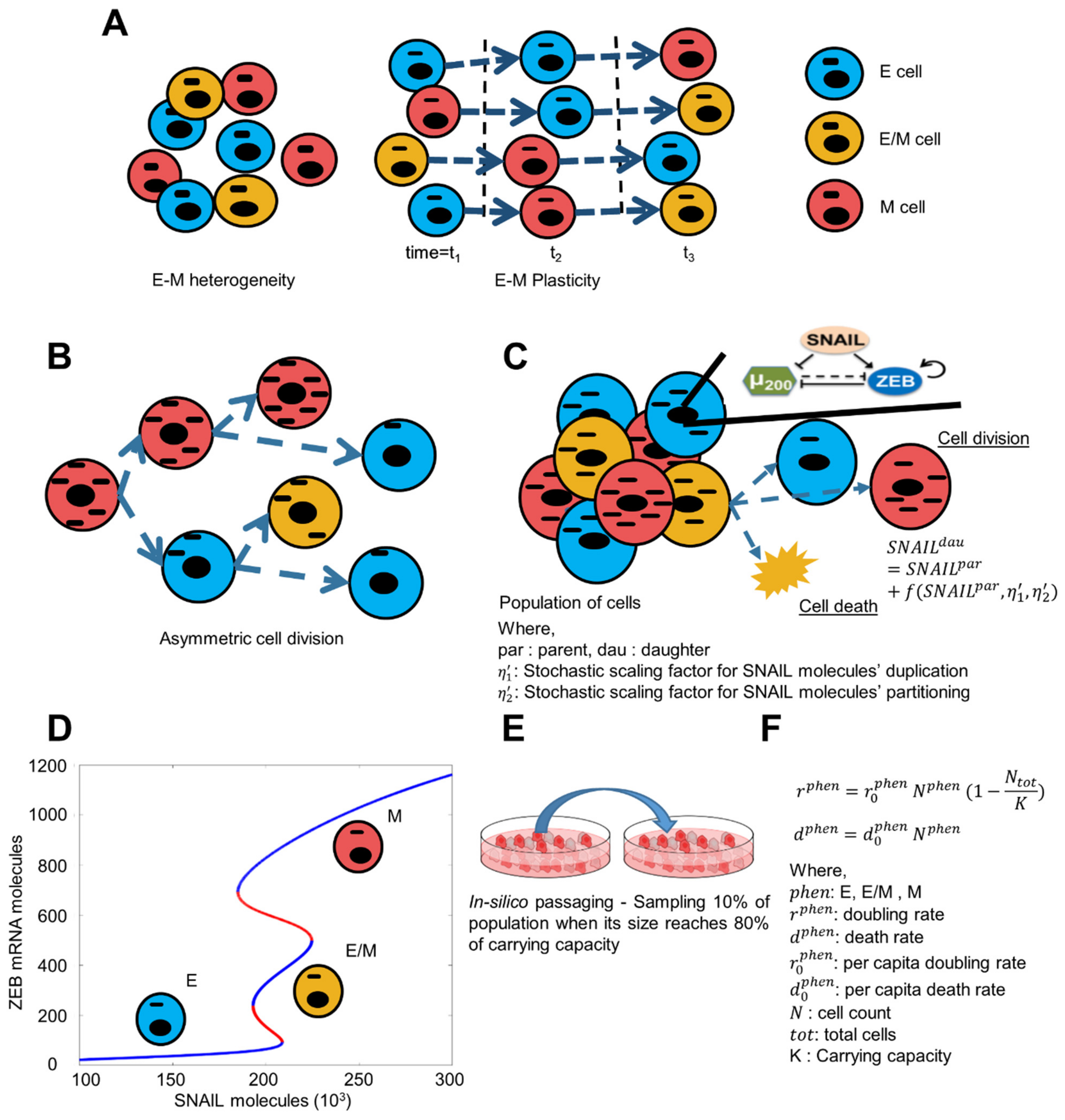
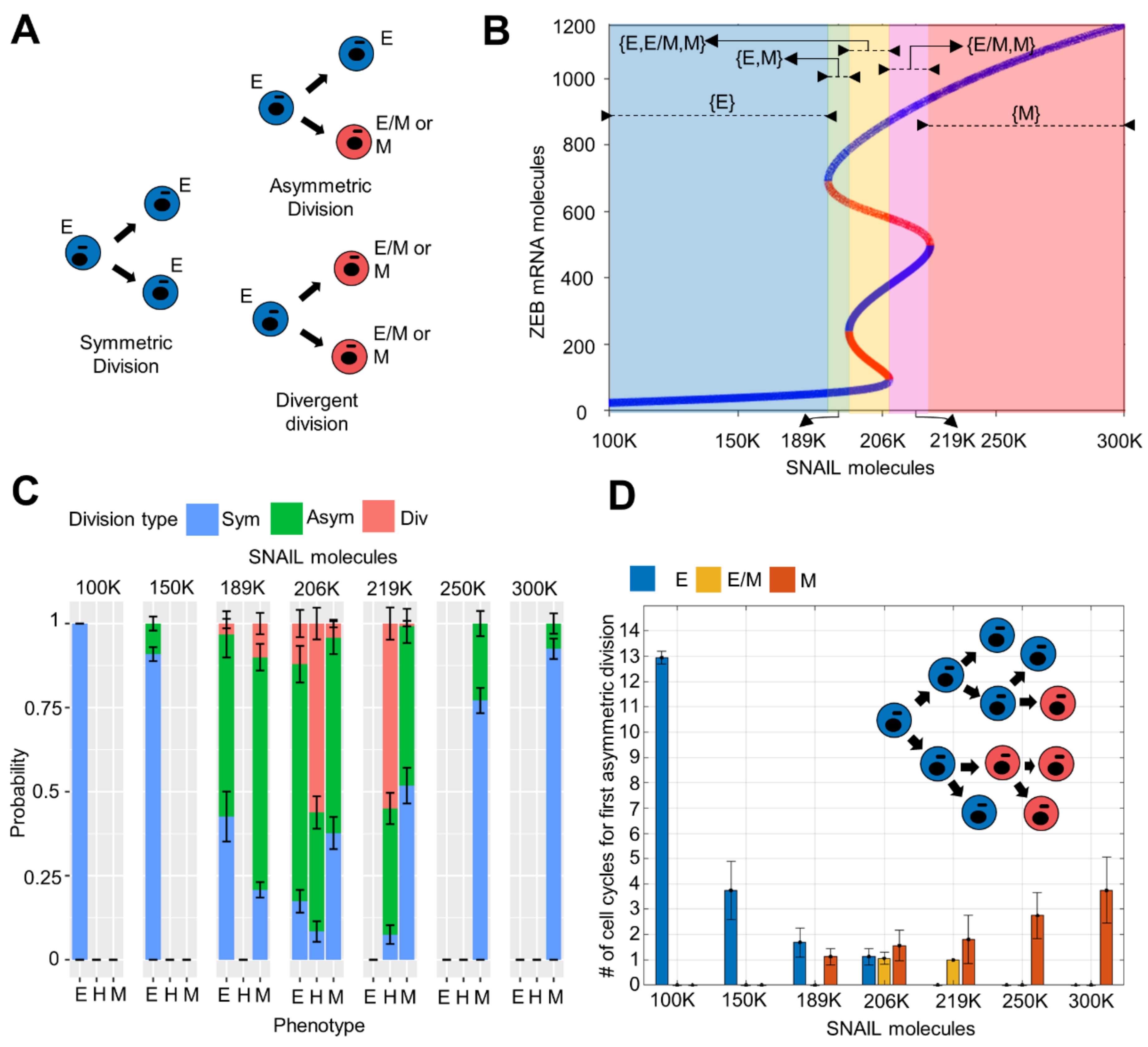
2.1. Asymmetric Distribution of Molecular Content on Cell Division
2.2. Dynamics of Core EMT Regulatory Network
2.3. Simulation of Population Dynamics
2.3.1. Generation of Population as per Initial Phenotypic Fraction
2.3.2. Avg. Birth and Death Rate of Cells
2.3.3. Population Growth
2.4. Cell Doubling Quantification
3. Results
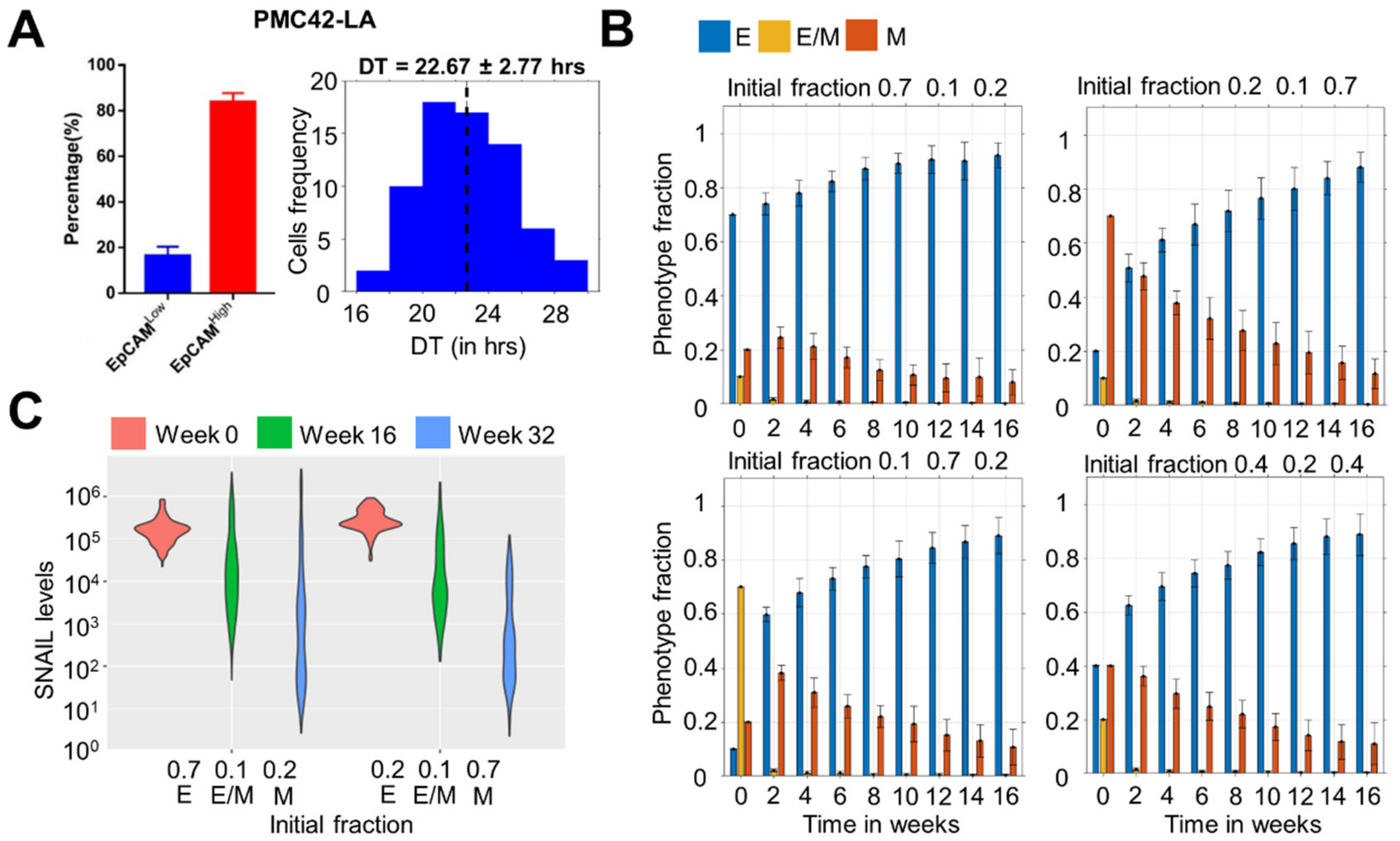
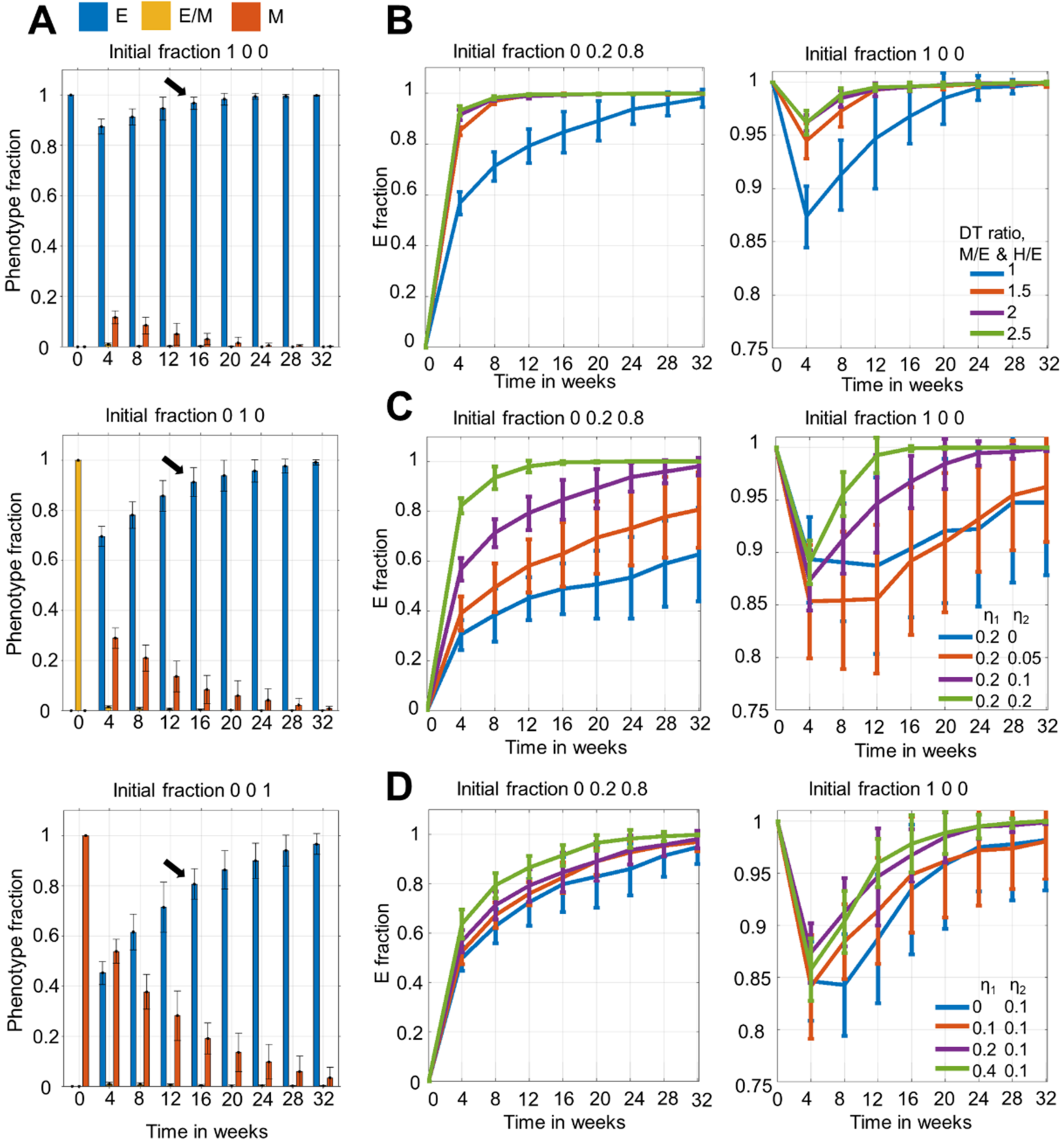
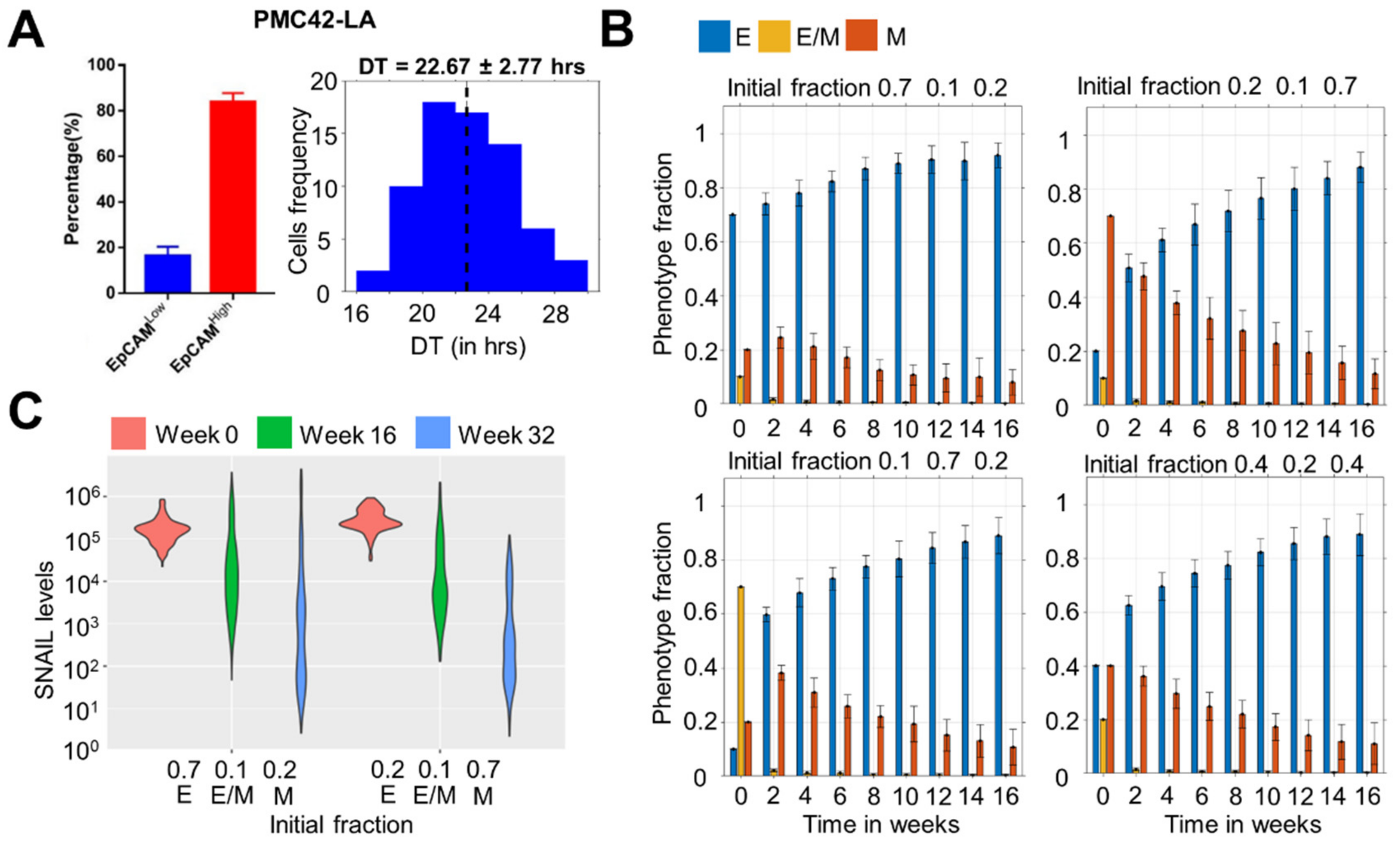
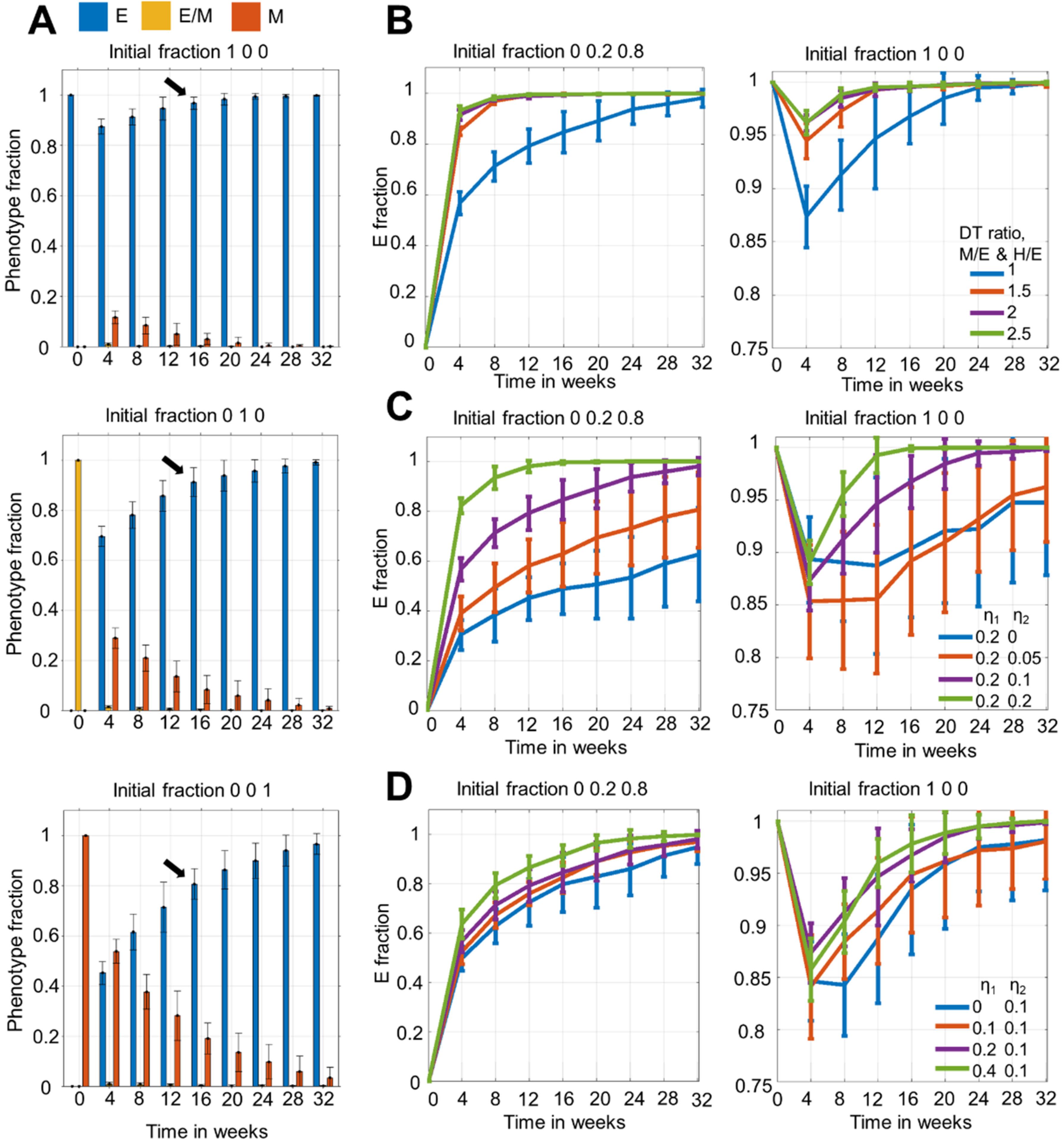
3.1. Dominance of Epithelial Cells in the Population over Time Irrespective of Initial Distribution
3.2. Time to Attain Epithelial Dominance Depends on Initial Fractions, Doubling Times of Phenotypes and the Extent of Stochastic Fluctuations during Cell Division
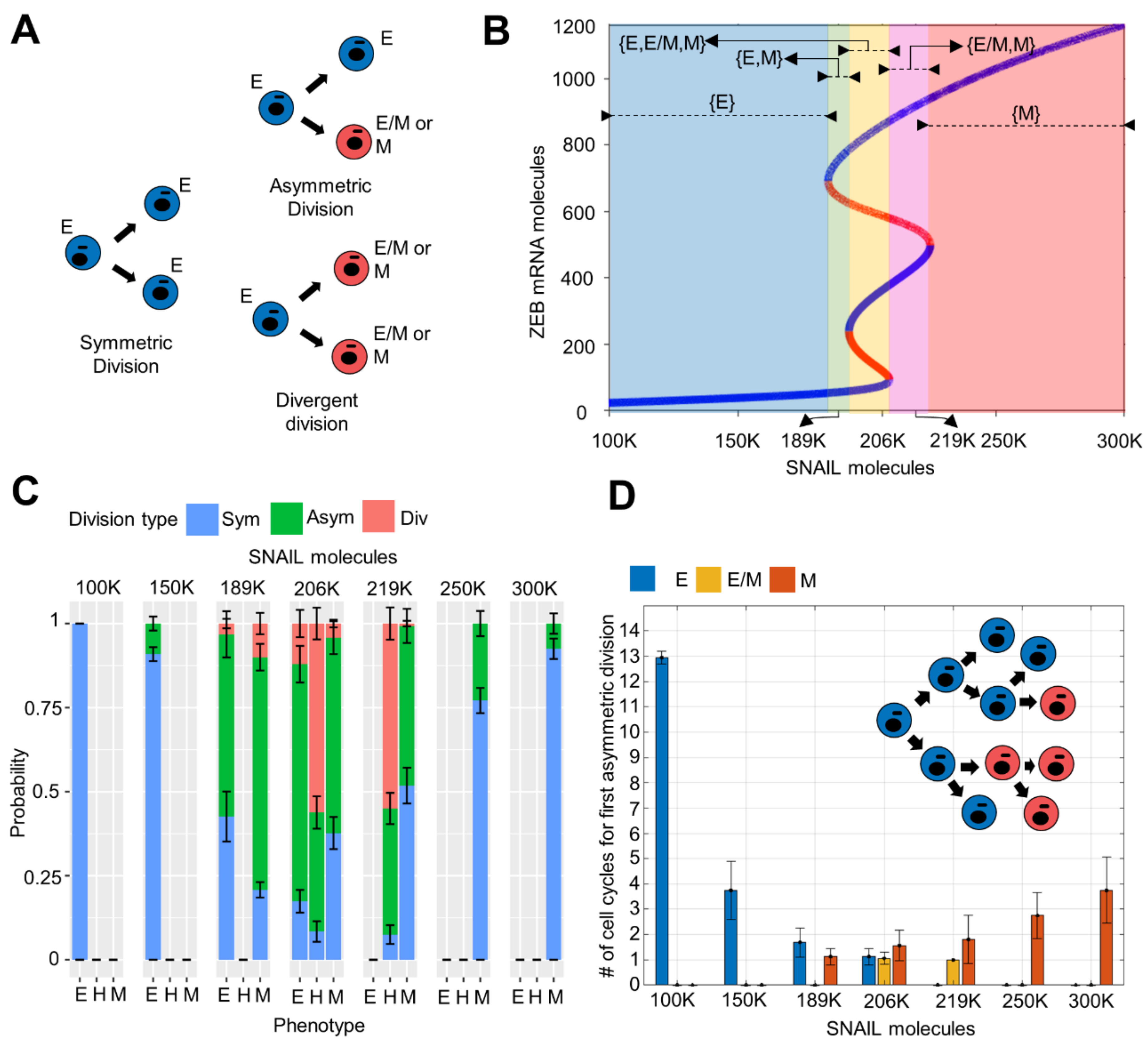
3.3. Phenotypic Switching Probability and Rate in Cell Division Events Depends on the Cells’ Location on E-M Axis
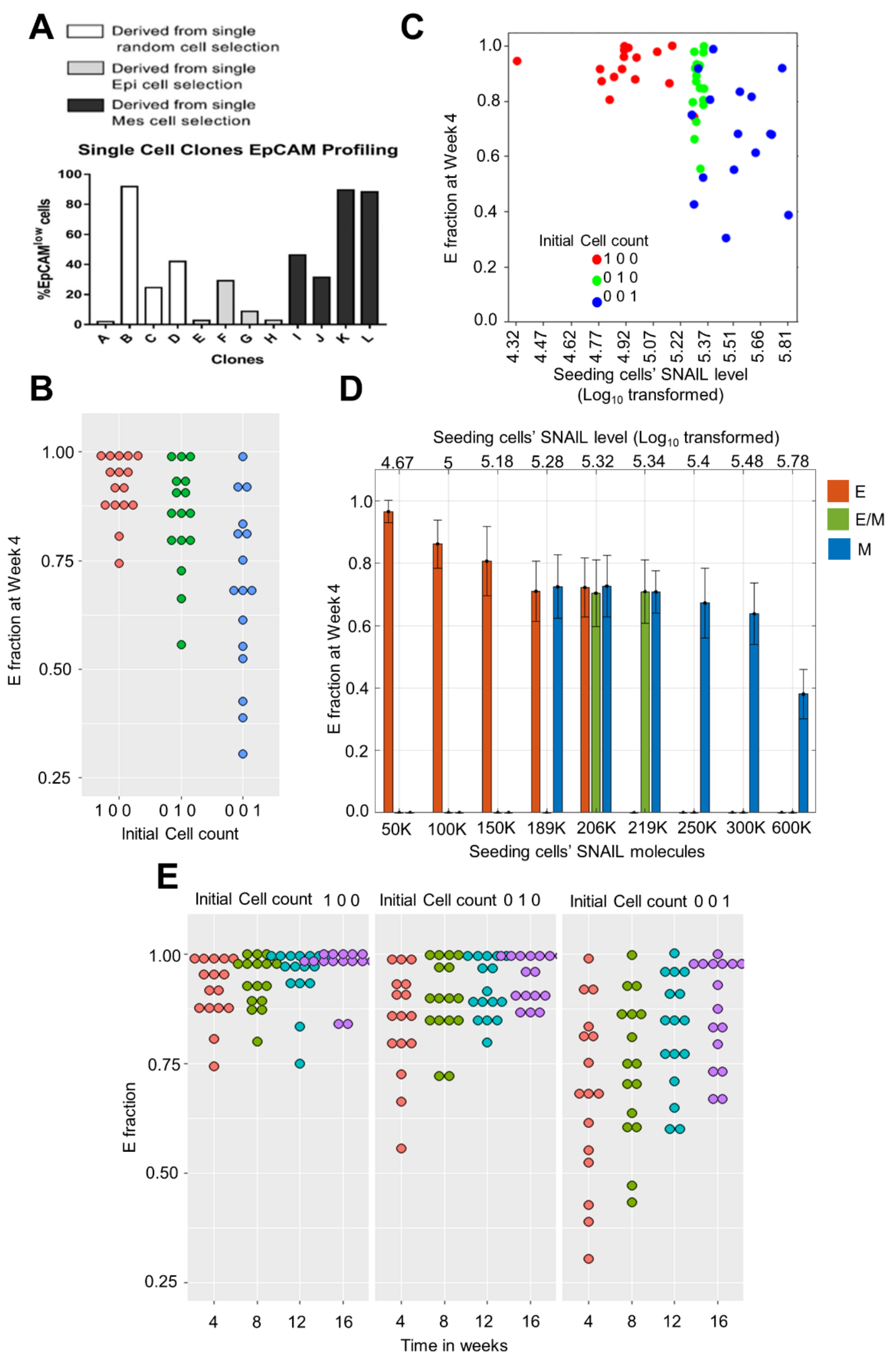
3.4. Heterogeneity in E Fraction at Initial Time Points among Single Cell Clones
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Biswas, A.; De, S. Drivers of dynamic intratumor heterogeneity and phenotypic plasticity. Am. J. Physiol.-Cell Physiol. 2021, 320, C750–C760. [Google Scholar] [CrossRef]
- Shlyakhtina, Y.; Moran, K.L.; Portal, M.M. Genetic and Non-Genetic Mechanisms Underlying Cancer Evolution. Cancers 2021, 13, 1380. [Google Scholar] [CrossRef]
- Jolly, M.K.; Celià-Terrassa, T. Dynamics of Phenotypic Heterogeneity Associated with EMT and Stemness during Cancer Progression. J. Clin. Med. 2019, 8, 1542. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Merritt, E.; Hu, X.; Cruz, A.; Jiang, C.; Sarkodie, H.; Zhou, Z.; Malhotra, J.; Riedlinger, G.M.; De, S. Non-Genetic Intra-Tumor Heterogeneity Is a Major Predictor of Phenotypic Heterogeneity and Ongoing Evolutionary Dynamics in Lung Tumors. Cell Rep. 2019, 29, 2164–2174. [Google Scholar] [CrossRef] [Green Version]
- Lewis, A.C.; Kats, L.M. Non-genetic heterogeneity, altered cell fate and differentiation therapy. EMBO Mol. Med. 2021, 13, e12670. [Google Scholar] [CrossRef]
- Su, Y.; Wei, W.; Robert, L.; Xue, M.; Tsoi, J.; Garcia-diaz, A.; Homet, B.M.; Kim, J.; Ng, R.H.; Lee, J.W.; et al. Single-cell analysis resolves the cell state transition and signaling dynamics associated with melanoma drug-induced resistance. Proc. Natl. Acad. Sci. USA 2017, 114, 13679–13684. [Google Scholar] [CrossRef] [Green Version]
- Jolly, M.K.; Kulkarni, P.; Weninger, K.; Orban, J.; Levine, H. Phenotypic plasticity, bet-hedging, and androgen independence in prostate cancer: Role of non-genetic heterogeneity. Front. Oncol. 2018, 8, 50. [Google Scholar] [CrossRef]
- Ruscetti, M.; Dadashian, E.L.; Guo, W.; Quach, B.; Mulholland, D.J.; Park, J.W.; Tran, L.M.; Kobayashi, N.; Bianchi-Frias, D.; Xing, Y.; et al. HDAC inhibition impedes epithelial-mesenchymal plasticity and suppresses metastatic, castration-resistant prostate cancer. Oncogene 2016, 35, 3781–3795. [Google Scholar] [CrossRef] [Green Version]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Rios, A.C.; Capaldo, B.D.; Vaillant, F.; Pal, B.; van Ineveld, R.; Dawson, C.A.; Chen, Y.; Nolan, E.; Fu, N.Y.; Jackling, F.C.; et al. Intraclonal Plasticity in Mammary Tumors Revealed through Large-Scale Single-Cell Resolution 3D Imaging. Cancer Cell 2019, 35, 618–632. [Google Scholar] [CrossRef] [Green Version]
- Biddle, A.; Liang, X.; Gammon, L.; Fazil, B.; Harper, L.J.; Emich, H.; Costea, D.E.; Mackenzie, I.C. Cancer stem cells in squamous cell carcinoma switch between two distinct phenotypes that are preferentially migratory or proliferative. Cancer Res. 2011, 71, 5317–5326. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [Green Version]
- Deshmukh, A.P.; Vasaikar, S.V.; Tomczak, K.; Tripathi, S.; Den Hollander, P.; Arslan, E.; Chakraborty, P.; Soundararajan, R.; Jolly, M.K.; Rai, K.; et al. Identification of EMT signaling cross-talk and gene regulatory networks by single-cell RNA sequencing. Proc. Natl. Acad. Sci. USA 2021, 118, e2102050118. [Google Scholar] [CrossRef]
- Karacosta, L.G.; Anchang, B.; Ignatiadis, N.; Kimmey, S.C.; Benson, J.A.; Shrager, J.B.; Tibshirani, R.; Bendall, S.C.; Plevritis, S.K. Mapping Lung Cancer Epithelial-Mesenchymal Transition States and Trajectories with Single-Cell Resolution. Nat. Commun. 2019, 10, 5587. [Google Scholar] [CrossRef] [Green Version]
- George, J.T.; Jolly, M.K.; Xu, S.; Somarelli, J.A.; Levine, H. Survival outcomes in cancer patients predicted by a partial EMT gene expression scoring metric. Cancer Res. 2017, 77, 6415–6428. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, S.; Monkman, J.; Blick, T.; Duijf, P.H.; Nagaraj, S.H.; Thompson, E.W. Multi-Omics Characterization of the Spontaneous Mesenchymal–Epithelial Transition in the PMC42 Breast Cancer Cell Lines. J. Clin. Med. 2019, 8, 1253. [Google Scholar] [CrossRef] [Green Version]
- Jolly, M.K.; Somarelli, J.A.; Sheth, M.; Biddle, A.; Tripathi, S.C.; Armstrong, A.J.; Hanash, S.M.; Bapat, S.A.; Rangarajan, A.; Levine, H. Hybrid epithelial/mesenchymal phenotypes promote metastasis and therapy resistance across carcinomas. Pharmacol. Ther. 2019, 194, 161–184. [Google Scholar] [CrossRef]
- Gupta, P.B.; Fillmore, C.M.; Jiang, G.; Shapira, S.D.; Tao, K.; Kuperwasser, C.; Lander, E.S. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 2011, 146, 633–644. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Sakane, K.; Tominaga, K.; Gotoh, N.; Niwa, T.; Kikuchi, Y.; Tada, K.; Goshima, N.; Semba, K.; Inoue, J.I. Intratumoral bidirectional transitions between epithelial and mesenchymal cells in triple-negative breast cancer. Cancer Sci. 2017, 108, 1210–1222. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, S.; Monkman, J.; Blick, T.; Pinto, C.; Waltham, A.; Nagaraj, S.H.; Thompson, E.W. Interrogation of Phenotypic Plasticity between Epithelial and Mesenchymal States in Breast Cancer. J. Clin. Med. 2019, 8, 893. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.S.; Abdollahi, B.; Wilkins, O.M.; Chakraborty, P.; Ognjenovic, N.B.; Muller, K.E.; Kumar Jolly, M.; Hassanpour, S.; Pattabiraman, D.R. Dynamic plasticity within the EMT spectrum, rather than static mesenchymal traits, drives tumor heterogeneity and metastatic progression of breast cancers. bioRxiv 2021. [Google Scholar] [CrossRef]
- Lu, M.; Jolly, M.K.; Levine, H.; Onuchic, J.N.; Ben-Jacob, E. MicroRNA-based regulation of epithelial-hybrid-mesenchymal fate determination. Proc. Natl. Acad. Sci. USA 2013, 110, 18144–18149. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, S.; Brabletz, T. The ZEB/miR-200 feedback loop--a motor of cellular plasticity in development and cancer? EMBO Rep. 2010, 11, 670–677. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, S.; Chakraborty, P.; Levine, H.; Jolly, M.K. A mechanism for epithelial-mesenchymal heterogeneity in a population of cancer cells. PLoS Comput. Biol. 2020, 16, e1007619. [Google Scholar] [CrossRef] [Green Version]
- In Proceedings of theGillespie, D.T. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 1977, 81, 2340–2361. [Google Scholar] [CrossRef]
- Sigal, A.; Milo, R.; Cohen, A.; Geva-Zatorsky, N.; Klein, Y.; Liron, Y.; Rosenfeld, N.; Danon, T.; Perzov, N.; Alon, U. Variability and memory of protein levels in human cells. Nature 2006, 444, 643–646. [Google Scholar] [CrossRef]
- Vega, S.; Morales, A.V.; Ocaña, O.H.; Valdés, F.; Fabregat, I.; Nieto, M.A. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004, 18, 1131–1143. [Google Scholar] [CrossRef] [Green Version]
- Jonckheere, S.; Adams, J.; De Groote, D.; Campbell, K.; Berx, G.; Goossens, S. Epithelial-Mesenchymal Transition (EMT) as a Therapeutic Target. Cells Tissues Organs 2021, in press. [Google Scholar] [CrossRef]
- Eichelberger, L.; Saini, M.; Moreno, H.D.; Klein, C.; Bartsch, J.M.; Falcone, M.; Reitberger, M.; Espinet, E.; Vogel, V.; Graf, E.; et al. Maintenance of epithelial traits and resistance to mesenchymal reprogramming promote proliferation in metastatic breast cancer. bioRxiv 2020. [Google Scholar] [CrossRef]
- Serresi, M.; Kertalli, S.; Li, L.; Schmitt, M.J.; Dramaretska, Y.; Wierikx, J.; Hulsman, D.; Gargiulo, G. Functional antagonism of chromatin modulators regulates epithelial-mesenchymal transition. Sci. Adv. 2021, 7, eabd7974. [Google Scholar] [CrossRef]
- Jia, W.; Tripathi, S.; Chakraborty, P.; Chedere, A.; Rangarajan, A.; Levine, H.; Jolly, M.K. Epigenetic feedback and stochastic partitioning during cell division can drive resistance to EMT. Oncotarget 2020, 11, 2611–2624. [Google Scholar] [CrossRef]
- Bocci, F.; Gearhart-Serna, L.; Boareto, M.; Riberio, M.; Ben-Jacob, E.; Devi, G.R.; Levine, H.; Onuchic, J.N.; Jolly, M.K. Toward understanding cancer stem cell heterogeneity in the tumor microenvironment. Proc. Natl. Acad. Sci. USA 2019, 116, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Scheel, C.; Eaton, E.N.; Li, S.H.J.; Chaffer, C.L.; Reinhardt, F.; Kah, K.J.; Bell, G.; Guo, W.; Rubin, J.; Richardson, A.L.; et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 2011, 145, 926–940. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Jolly, M.K.; George, J.T.; Pienta, K.J.; Levine, H. Computational Modeling of the Crosstalk Between Macrophage Polarization and Tumor Cell Plasticity in the Tumor Microenvironment. Front. Oncol. 2019, 9, 10. [Google Scholar] [CrossRef]
- Watanabe, K.; Panchy, N.; Noguchi, S.; Suzuki, H.; Hong, T. Combinatorial perturbation analysis reveals divergent regulations of mesenchymal genes during epithelial-to-mesenchymal transition. NPJ Syst. Biol. Appl. 2019, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Steinway, S.N.; Zañudo, J.G.T.; Michel, P.J.; Feith, D.J.; Loughran, T.P.; Albert, R. Combinatorial interventions inhibit TGFβ-driven epithelial-to-mesenchymal transition and support hybrid cellular phenotypes. NPJ Syst. Biol. Appl. 2015, 1, 15014. [Google Scholar] [CrossRef] [Green Version]
- Cook, D.P.; Vanderhyden, B.C. Context specificity of the EMT transcriptional response. Nat. Commun. 2020, 11, 2142. [Google Scholar] [CrossRef]
- Hari, K.; Sabuwala, B.; Subramani, B.V.; La Porta, C.A.M.; Zapperi, S.; Font-Clos, F.; Jolly, M.K. Identifying inhibitors of epithelial–mesenchymal plasticity using a network topology-based approach. NPJ Syst. Biol. Appl. 2020, 6, 15. [Google Scholar] [CrossRef]
- Cook, D.P.; Vanderhyden, B.C. Transcriptional census of epithelial-mesenchymal plasticity in cancer. Sci. Adv. 2022, 8, eabi7640. [Google Scholar] [CrossRef]
- Xin, Y.; Cummins, B.; Gedeon, T. Multistability in the epithelial-mesenchymal transition network. BMC Bioinform. 2020, 21, 71. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Balazsi, G. A landscape view on the interplay between EMT and cancer metastasis. NPJ Syst. Biol. Appl. 2018, 4, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunchu, B.; Cabernard, C. Principles and mechanisms of asymmetric cell division. Development 2020, 147, dev167650. [Google Scholar] [CrossRef]
- Huh, D.; Paulsson, J. Non-genetic heterogeneity from stochastic partitioning at cell division. Nat. Genet. 2011, 43, 95–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, P.; Wang, L.; Chen, K.Y.; Srinivasan, T.; Murthy, P.K.L.; Tung, K.L.; Varanko, A.K.; Chen, H.J.; Ai, Y.; King, S.; et al. A miR-34a-Numb Feedforward Loop Triggered by Inflammation Regulates Asymmetric Stem Cell Division in Intestine and Colon Cancer. Cell Stem Cell 2016, 18, 189–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lathia, J.D.; Hitomi, M.; Gallagher, J.; Gadani, S.P.; Adkins, J.; Vasanji, A.; Liu, L.; Eyler, C.E.; Heddleston, J.M.; Wu, Q.; et al. Distribution of CD133 reveals glioma stem cells self-renew through symmetric and asymmetric cell divisions. Cell Death Dis. 2011, 2, e200. [Google Scholar] [CrossRef]
- Izumi, H.; Kaneko, Y. Evidence of asymmetric cell division and centrosome inheritance in human neuroblastoma cells. Proc. Natl. Acad. Sci. USA 2012, 109, 18048–18053. [Google Scholar] [CrossRef] [Green Version]
- Topolewski, P.; Zakrzewska, K.E.; Walczak, J.; Towski, K.; Muller-Newen, G.; Singh, A.; Komorowski, M. Phenotypic variability, not noise, accounts for most of the cell-to-cell heterogeneity in IFN-γ and oncostatin M signaling responses. Sci. Signal. 2022, 15. [Google Scholar] [CrossRef]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Peruzzi, G.; Miotto, M.; Maggio, R.; Ruocco, G.; Gosti, G. Asymmetric binomial statistics explains organelle partitioning variance in cancer cell proliferation. Commun. Phys. 2021, 4, 188. [Google Scholar] [CrossRef]
- Döhla, J.; Kuuluvainen, E.; Gebert, N.; Amaral, A.; Englund, J.I.; Gopalakrishnan, S.; Konovalova, S.; Nieminen, A.I.; Salminen, E.S.; Torregrosa Muñumer, R.; et al. Metabolic determination of cell fate through selective inheritance of mitochondria. Nat. Cell Biol. 2022, 24, 148–154. [Google Scholar] [CrossRef]
- den Hollander, P.; Vasaikar, S.; Castaneda, M.; Joseph, R.; Deshmukh, A.P.; Zhou, T.; Pietila, M.; Fu, C.; Symmans, W.F.; Soundararajan, R.; et al. Acquisition of cancer stem cell properties during EMT requires cell division. bioRxiv 2021. [Google Scholar] [CrossRef]
- Hitomi, M.; Chumakova, A.P.; Silver, D.J.; Knudsen, A.M.; Pontius, W.D.; Murphy, S.; Anand, N.; Kristensen, B.W.; Lathia, J.D. Asymmetric cell division promotes therapeutic resistance in glioblastoma stem cells. JCI Insight 2021, 6, e130510. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bracken, C.P.; Smith, E.; Bert, A.G.; Wright, J.A.; Roslan, S.; Morris, M.; Wyatt, L.; Farshid, G.; Lim, Y.-Y.; et al. An autocrine TGF-beta/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol. Biol. Cell 2011, 22, 1686–1698. [Google Scholar] [CrossRef] [PubMed]
- Baptista, I.S.C.; Ribeiro, A.S. Stochastic models coupling gene expression and partitioning in cell division in Escherichia coli. BioSystems 2020, 193–194, 104154. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value | Parameter | Value |
|---|---|---|---|
| No. Of Mirna Binding Sites | 0 | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|---|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jain, P.; Bhatia, S.; Thompson, E.W.; Jolly, M.K. Population Dynamics of Epithelial-Mesenchymal Heterogeneity in Cancer Cells. Biomolecules 2022, 12, 348. https://doi.org/10.3390/biom12030348
Jain P, Bhatia S, Thompson EW, Jolly MK. Population Dynamics of Epithelial-Mesenchymal Heterogeneity in Cancer Cells. Biomolecules. 2022; 12(3):348. https://doi.org/10.3390/biom12030348
Chicago/Turabian StyleJain, Paras, Sugandha Bhatia, Erik W. Thompson, and Mohit Kumar Jolly. 2022. "Population Dynamics of Epithelial-Mesenchymal Heterogeneity in Cancer Cells" Biomolecules 12, no. 3: 348. https://doi.org/10.3390/biom12030348
APA StyleJain, P., Bhatia, S., Thompson, E. W., & Jolly, M. K. (2022). Population Dynamics of Epithelial-Mesenchymal Heterogeneity in Cancer Cells. Biomolecules, 12(3), 348. https://doi.org/10.3390/biom12030348