
A Comparative Evaluation of the Structural and Dynamic Properties of Insect Odorant Binding Proteins


Abstract
1. Introduction
- are expressed not only in the insect olfactory organs but also in other body parts [14]. For example, it was shown in proteomic studies that of the 66 genes encoding OBPs in Anopheles gambiae only 18 are detectable in olfactory tissues [15]. These results raise questions regarding the actual number of OBPs that may have a purely olfactory function.
2. Materials and Methods
3. Results
3.1. Sequence Analysis
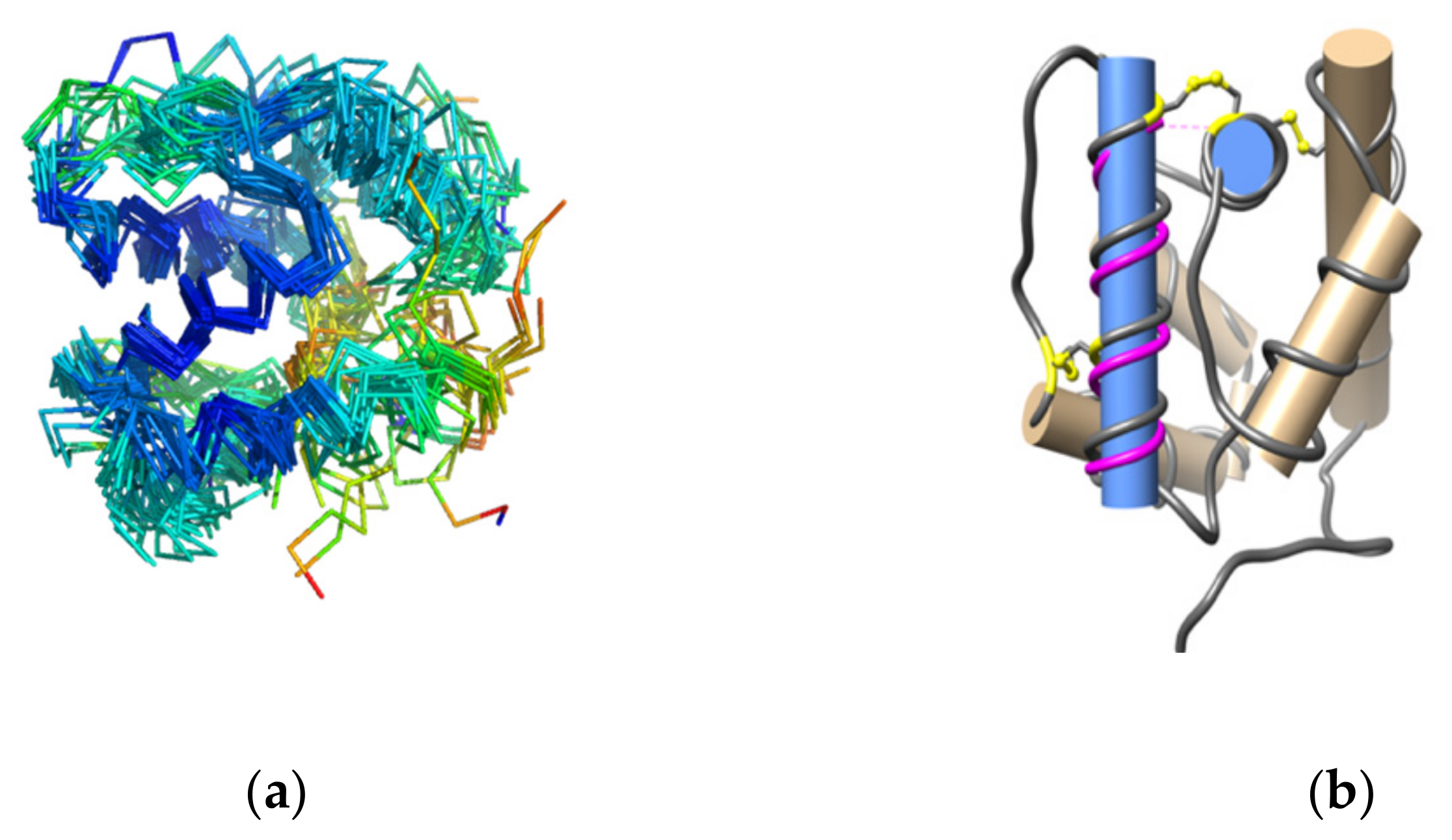
3.2. Structure Analysis
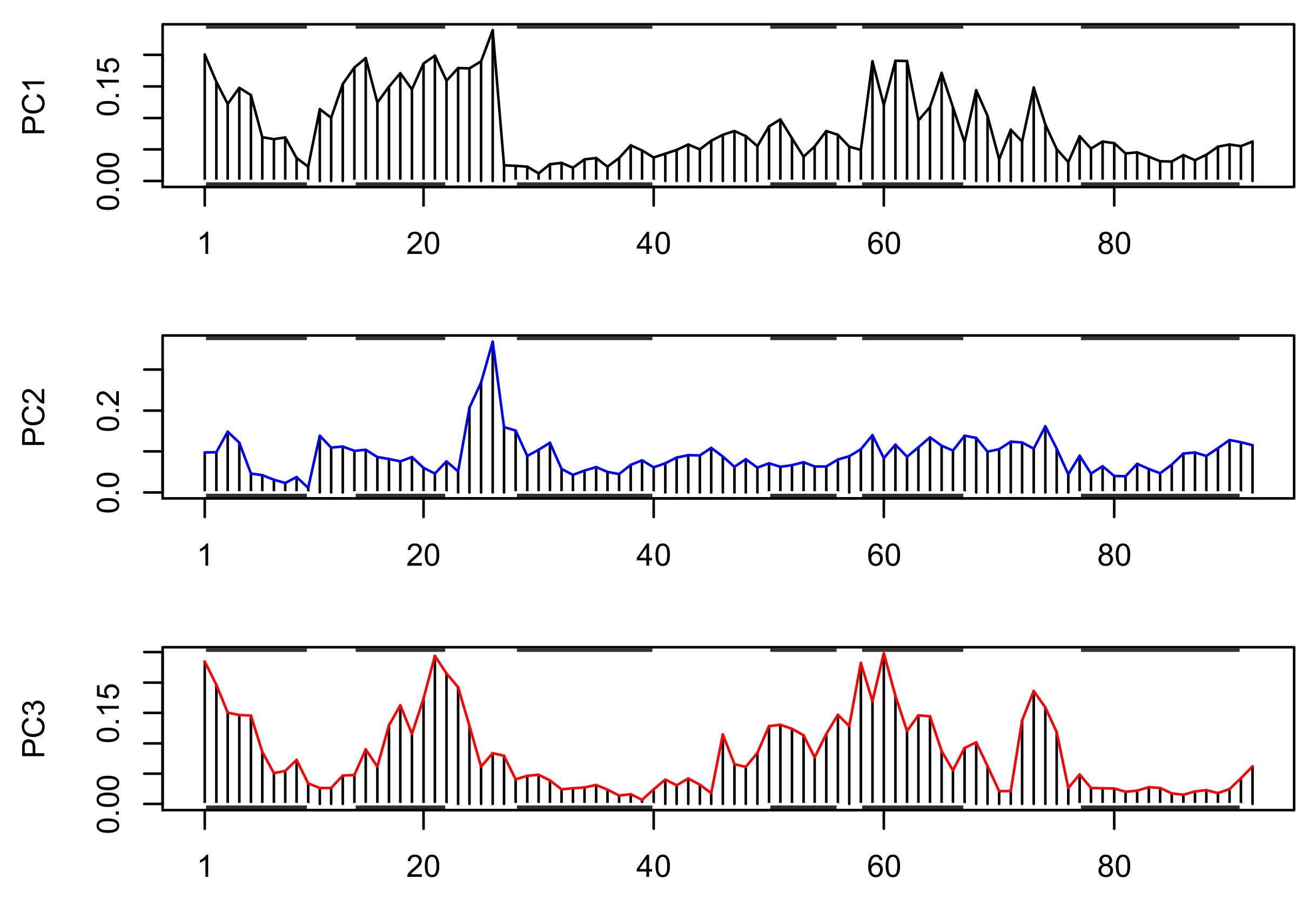
3.3. Principal Component Analysis (PCA)
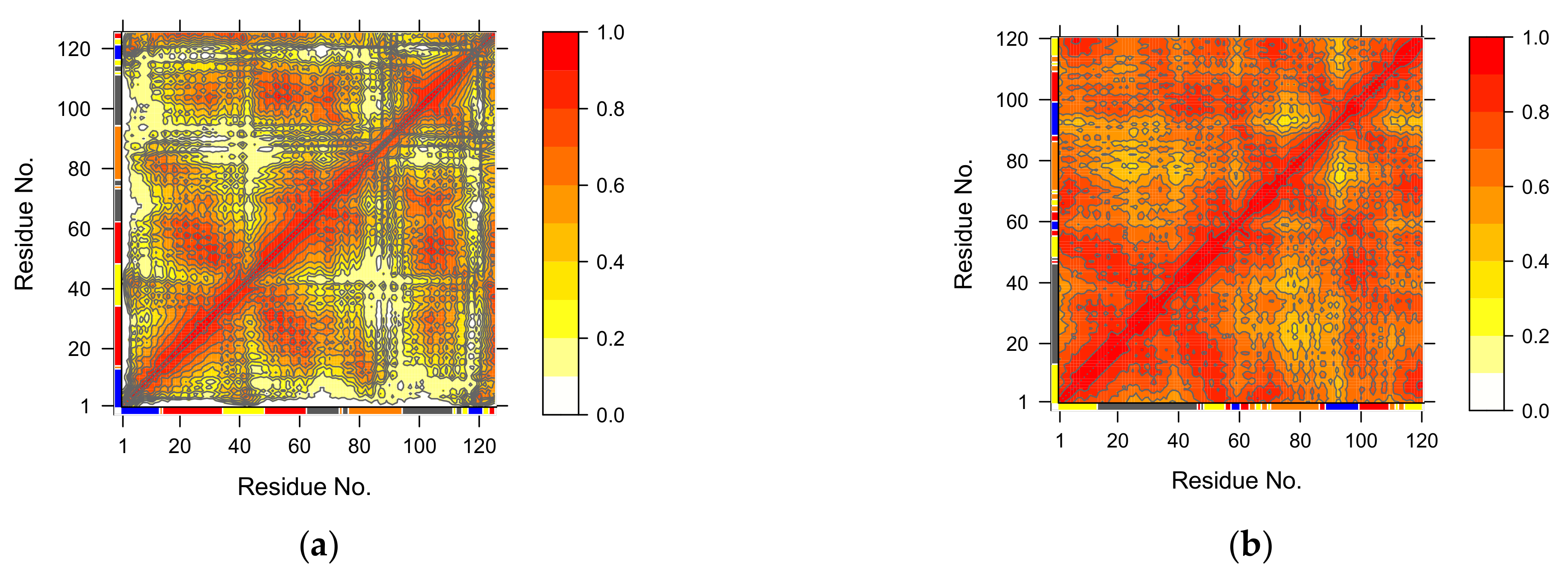
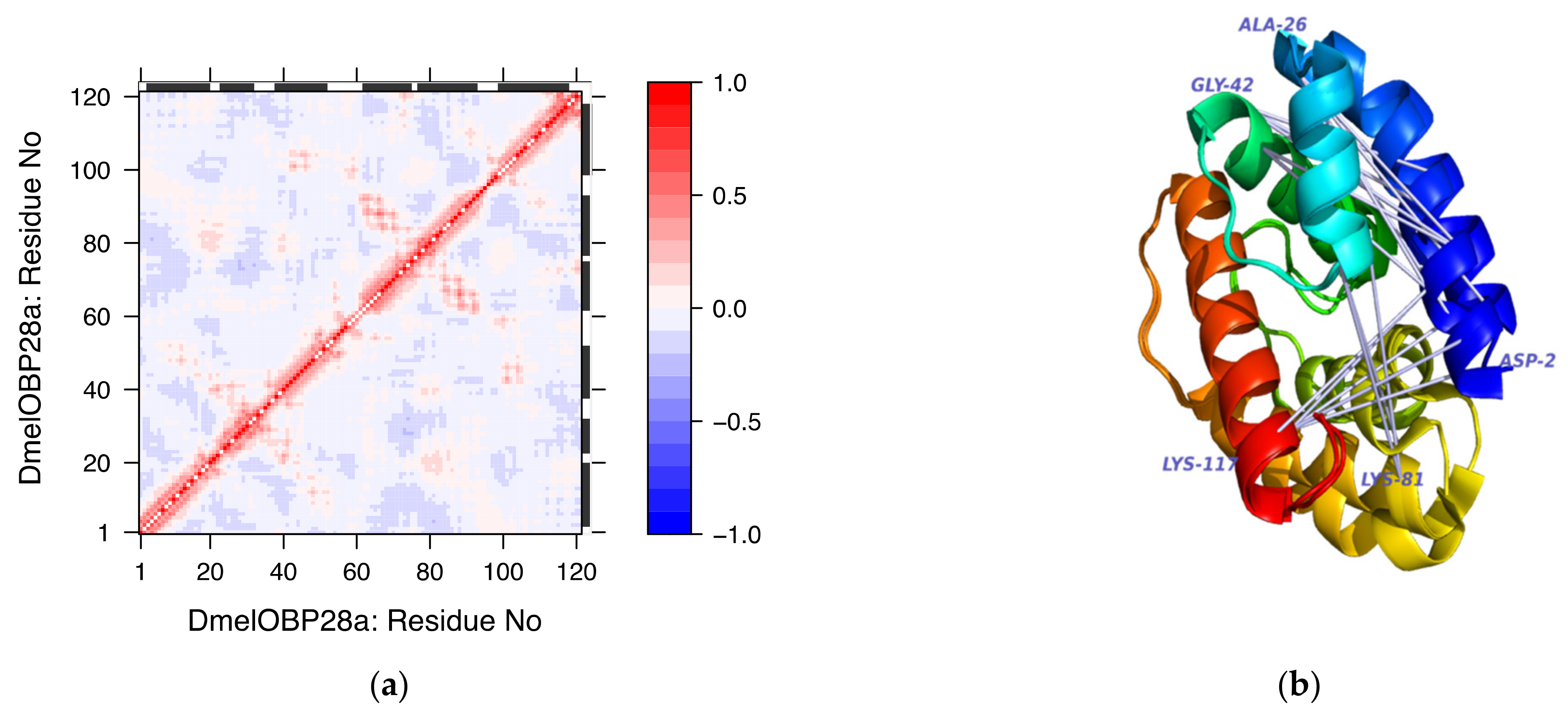
3.4. Normal Mode Analysis (NMA)
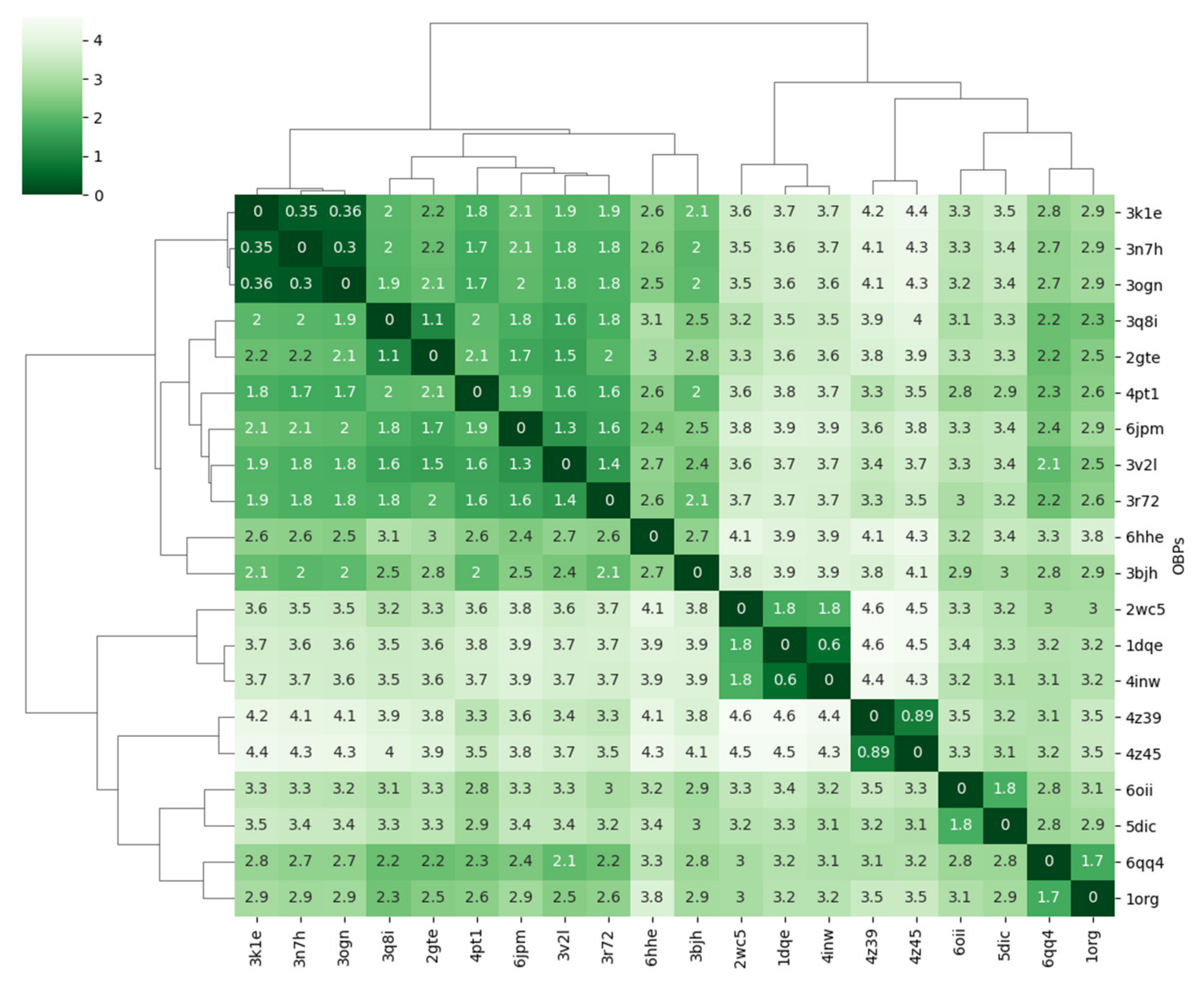
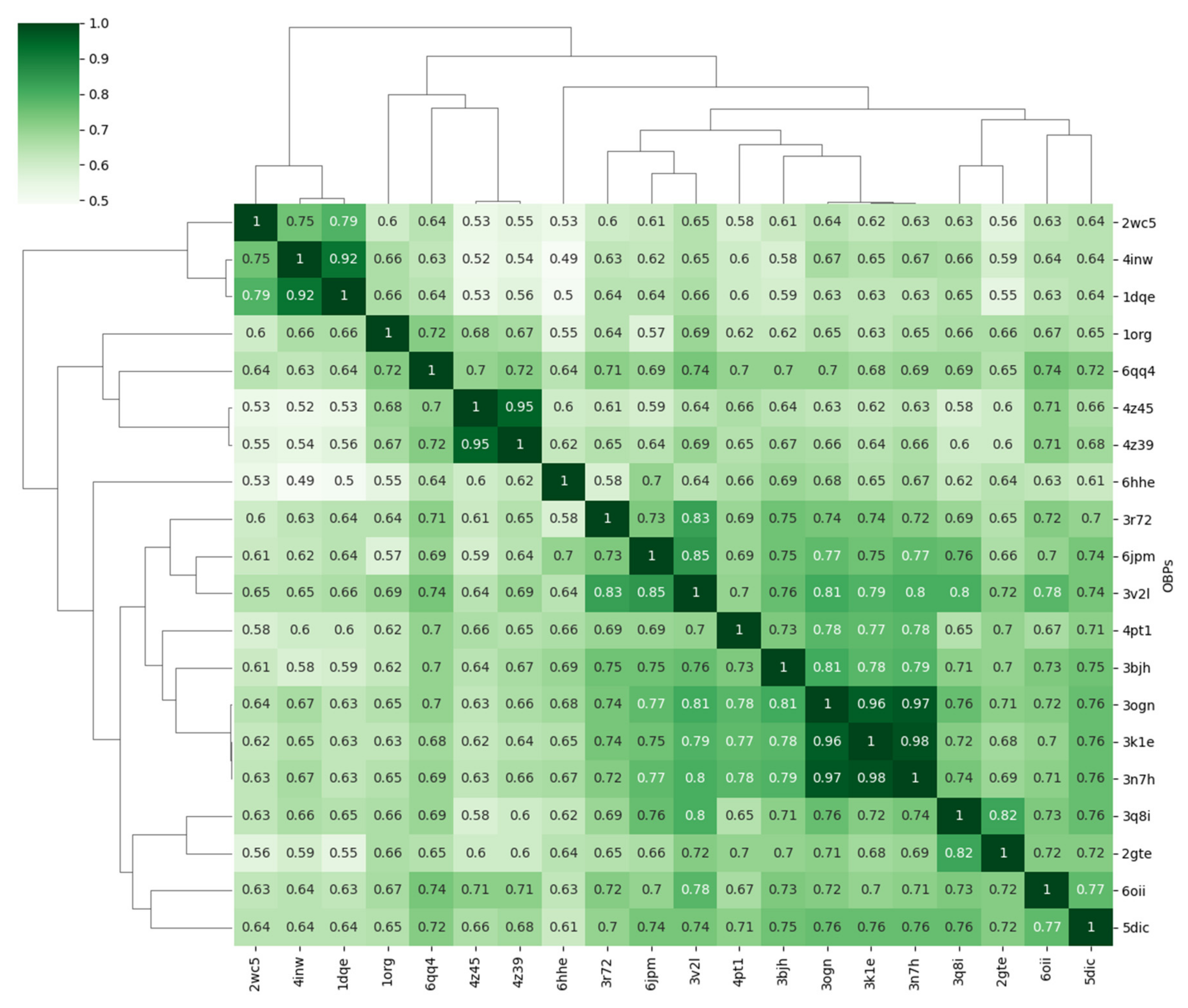
3.4.1. Covariance Similarity Analysis
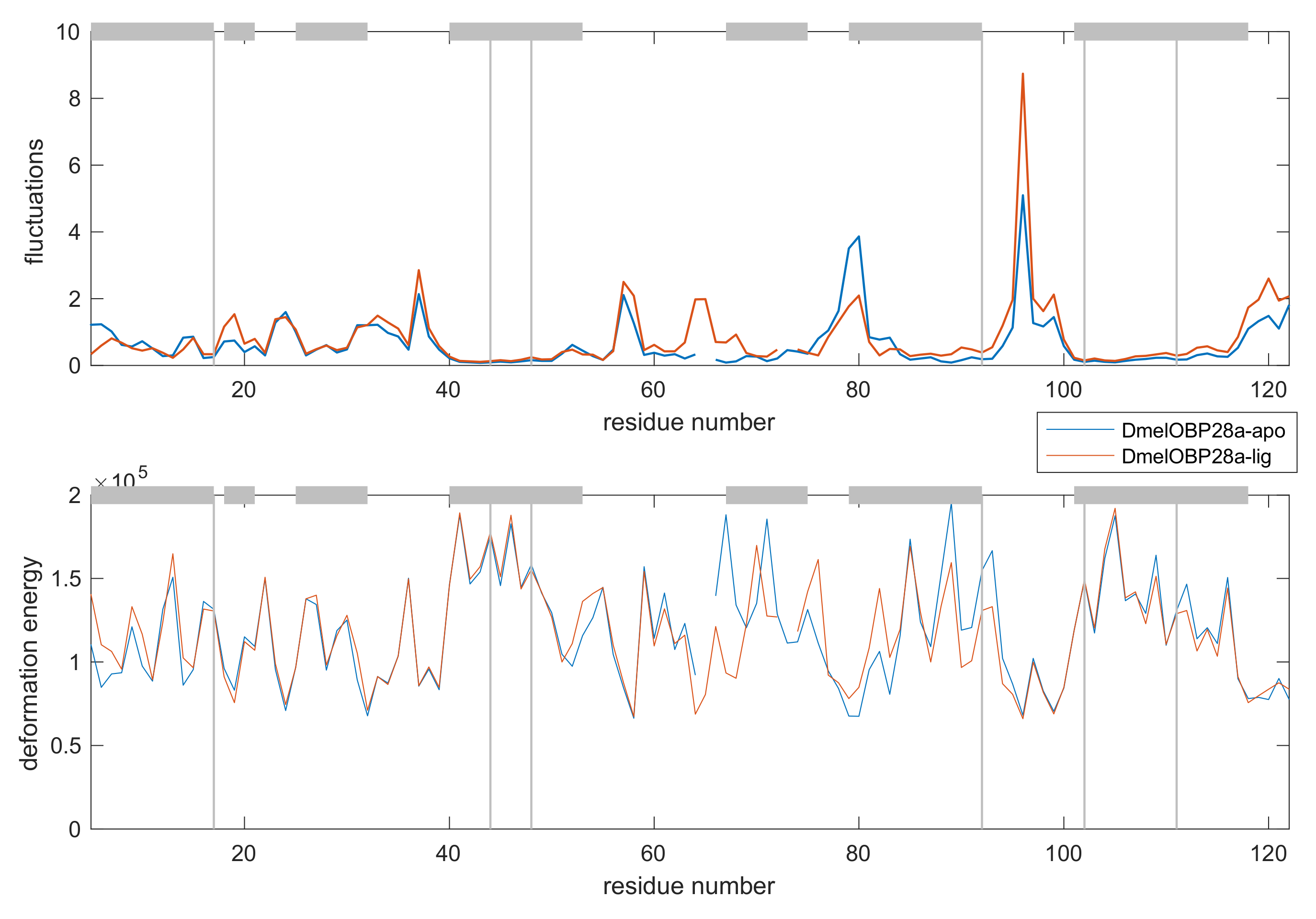
3.4.2. Comparative Fluctuation and Deformation Analysis
3.4.3. Single Analysis on Selected Examples
4. Discussion
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Multiple Structural Alignment of OBPs


Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Title 1 | PDB ID | Angle (Degrees) | Distance (Å) |
|---|---|---|---|
| 1 | 1dqe | 83.4 | 7.5 |
| 2 | 1org | 85.7 | 8.1 |
| 3 | 2gte | 77.9 | 7.8 |
| 4 | 2wc5 | 83.0 | 6.8 |
| 5 | 3bjh | 88.5 | 7.4 |
| 6 | 3k1e | 87.3 | 7.6 |
| 7 | 3n7h | 88.0 | 7.8 |
| 8 | 3ogn | 88.1 | 7.8 |
| 9 | 3q8i | 85.0 | 7.4 |
| 10 | 3r72 | 88.2 | 7.4 |
| 11 | 3v2l | 81.0 | 7.7 |
| 12 | 4inw | 81.5 | 7.3 |
| 13 | 4pt1 | 84.2 | 7.5 |
| 14 | 4z39 | 54.4 | 7.9 |
| 15 | 4z45 | 66.0 | 7.6 |
| 16 | 5dic | 75.4 | 8.1 |
| 17 | 6hhe | 83.8 | 7.8 |
| 18 | 6jpm | 81.4 | 7.3 |
| 19 | 6oii | 81.8 | 7.7 |
| 20 | 6qq4 | 78.9 | 7.7 |
Appendix C


Appendix D
Normal Modes Analysis Using Elastic Network Models



Appendix E
Principal Components Analysis
References
- Kaupp, U.B. Olfactory signalling in vertebrates and insects: Differences and commonalities. Nat. Rev. Neurosci. 2010, 11, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Leal, W.S. Odorant Reception in Insects: Roles of Receptors, Binding Proteins, and Degrading Enzymes. Annu. Rev. Entomol. 2013, 58, 373–391. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Ha, T.S.; Smith, D.P. SNMP is a signaling component required for pheromone sensitivity in Drosophila. Proc. Natl. Acad. Sci. USA 2008, 105, 10996–11001. [Google Scholar] [CrossRef] [PubMed]
- Bohbot, J.D.; Pitts, R.J. The narrowing olfactory landscape of insect odorant receptors. Front. Ecol. Evol. 2015, 3, 39. [Google Scholar] [CrossRef]
- Vogt, R.G.; Riddiford, L.M. Pheromone binding and inactivation by moth antennae. Nature 1981, 293, 161–163. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Sun, J.S.; Xiao, S.; Carlson, J.R. The diverse small proteins called odorant-binding proteins. Open Biol. 2018, 8, 180208. [Google Scholar] [CrossRef]
- Rihani, K.; Ferveur, J.F.; Briand, L. The 40-Year Mystery of Insect Odorant-Binding Proteins. Biomolecules 2021, 11, 509. [Google Scholar] [CrossRef]
- Pelosi, P.; Zhou, J.J.; Ban, L.P.; Calvello, M. Soluble proteins in insect chemical communication. Cell Mol. Life Sci. 2006, 63, 1658–1676. [Google Scholar] [CrossRef]
- Sandler, B.H.; Nikonova, L.; Leal, W.S.; Clardy, J. Sexual attraction in the silkworm moth: Structure of the pheromone-binding-protein-bombykol complex. Chem. Biol. 2000, 7, 143–151. [Google Scholar] [CrossRef]
- Maida, R.; Ziegelberger, G.; Kaissling, K.E. Ligand binding to six recombinant pheromone-binding proteins of Antheraea polyphemus and Antheraea pernyi. J. Comp. Physiol. B 2003, 173, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Lautenschlager, C.; Leal, W.S.; Clardy, J. Bombyx mori pheromone-binding protein binding nonpheromone ligands: Implications for pheromone recognition. Structure 2007, 15, 1148–1154. [Google Scholar] [CrossRef] [PubMed]
- Drakou, C.E.; Tsitsanou, K.E.; Potamitis, C.; Fessas, D.; Zervou, M.; Zographos, S.E. The crystal structure of the AgamOBP1*Icaridin complex reveals alternative binding modes and stereo-selective repellent recognition. Cell. Mol. Life Sci. 2017, 74, 319–338. [Google Scholar] [CrossRef] [PubMed]
- Biessmann, H.; Nguyen, Q.K.; Le, D.; Walter, M.F. Microarray-based survey of a subset of putative olfactory genes in the mosquito Anopheles gambiae. Insect Mol. Biol. 2005, 14, 575–589. [Google Scholar] [CrossRef]
- Mastrobuoni, G.; Qiao, H.; Iovinella, I.; Sagona, S.; Niccolini, A.; Boscaro, F.; Caputo, B.; Orejuela, M.R.; Torre, A.d.; Kempa, S.; et al. A Proteomic Investigation of Soluble Olfactory Proteins in Anopheles gambiae. PLoS ONE 2013, 8, e75162. [Google Scholar] [CrossRef]
- Tegoni, M.; Campanacci, V.; Cambillau, C. Structural aspects of sexual attraction and chemical communication in insects. Trends Biochem. Sci. 2004, 29, 257–264. [Google Scholar] [CrossRef]
- Fox, N.K.; Brenner, S.E.; Chandonia, J.M. SCOPe: Structural Classification of Proteins--extended, integrating SCOP and ASTRAL data and classification of new structures. Nucleic Acids Res. 2014, 42, D304–D309. [Google Scholar] [CrossRef]
- Zhou, J.J. Odorant-binding proteins in insects. Vitam. Horm. 2010, 83, 241–272. [Google Scholar] [CrossRef]
- Brito, N.F.; Moreira, M.F.; Melo, A.C.A. A look inside odorant-binding proteins in insect chemoreception. J. Insect Physiol. 2016, 95, 51–65. [Google Scholar] [CrossRef]
- Venthur, H.; Zhou, J.J. Odorant Receptors and Odorant-Binding Proteins as Insect Pest Control Targets: A Comparative Analysis. Front. Physiol. 2018, 9, 1163. [Google Scholar] [CrossRef]
- Henzler-Wildman, K.; Kern, D. Dynamic personalities of proteins. Nature 2007, 450, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Bahar, I.; Lezon, T.R.; Bakan, A.; Shrivastava, I.H. Normal mode analysis of biomolecular structures: Functional mechanisms of membrane proteins. Chem. Rev. 2010, 110, 1463–1497. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, H.; Krieger, J.M.; Bahar, I. Shared Signature Dynamics Tempered by Local Fluctuations Enables Fold Adaptability and Specificity. Mol. Biol. Evol. 2019, 36, 2053–2068. [Google Scholar] [CrossRef] [PubMed]
- Hinsen, K. Analysis of domain motions by approximate normal mode calculations. Proteins 1998, 33, 417–429. [Google Scholar] [CrossRef]
- Na, H.; Jernigan, R.L.; Song, G. Bridging between NMA and Elastic Network Models: Preserving All-Atom Accuracy in Coarse-Grained Models. PLoS Comput. Biol. 2015, 11, e1004542. [Google Scholar] [CrossRef]
- Tiwari, S.P.; Reuter, N. Similarity in Shape Dictates Signature Intrinsic Dynamics Despite No Functional Conservation in TIM Barrel Enzymes. PLoS Comput. Biol. 2016, 12, e1004834. [Google Scholar] [CrossRef]
- Skjaerven, L.; Yao, X.Q.; Scarabelli, G.; Grant, B.J. Integrating protein structural dynamics and evolutionary analysis with Bio3D. BMC Bioinform. 2014, 15, 399. [Google Scholar] [CrossRef]
- Grant, B.J.; Rodrigues, A.P.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef]
- Grant, B.J.; Skjaerven, L.; Yao, X.Q. The Bio3D packages for structural bioinformatics. Protein Sci. 2021, 30, 20–30. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Marks, D.S.; Colwell, L.J.; Sheridan, R.; Hopf, T.A.; Pagnani, A.; Zecchina, R.; Sander, C. Protein 3D structure computed from evolutionary sequence variation. PLoS ONE 2011, 6, e28766. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, R.; Fieldhouse, R.J.; Hayat, S.; Sun, Y.; Antipin, Y.; Yang, L.; Hopf, T.; Marks, D.S.; Sander, C. EVfold.org: Evolutionary Couplings and Protein 3D Structure Prediction. bioRxiv 2015, 021022. [Google Scholar] [CrossRef]
- Hopf, T.A.; Green, A.G.; Schubert, B.; Mersmann, S.; Scharfe, C.P.I.; Ingraham, J.B.; Toth-Petroczy, A.; Brock, K.; Riesselman, A.J.; Palmedo, P.; et al. The EVcouplings Python framework for coevolutionary sequence analysis. Bioinformatics 2019, 35, 1582–1584. [Google Scholar] [CrossRef]
- Fuglebakk, E.; Tiwari, S.P.; Reuter, N. Comparing the intrinsic dynamics of multiple protein structures using elastic network models. Biochim. Biophys. Acta 2015, 1850, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Wang, S. Chapter Five—Algorithms, Applications, and Challenges of Protein Structure Alignment. In Advances in Protein Chemistry and Structural Biology; Donev, R., Ed.; Academic Press: Cambridge, MA, USA, 2014; Volume 94, pp. 121–175. [Google Scholar]
- Konagurthu, A.S.; Whisstock, J.C.; Stuckey, P.J.; Lesk, A.M. MUSTANG: A multiple structural alignment algorithm. Proteins 2006, 64, 559–574. [Google Scholar] [CrossRef]
- Chung, S.Y.; Subbiah, S. A structural explanation for the twilight zone of protein sequence homology. Structure 1996, 4, 1123–1127. [Google Scholar] [CrossRef]
- Khor, B.Y.; Tye, G.J.; Lim, T.S.; Choong, Y.S. General overview on structure prediction of twilight-zone proteins. Theor. Biol. Med. Model. 2015, 12, 15. [Google Scholar] [CrossRef]
- Fuglebakk, E.; Echave, J.; Reuter, N. Measuring and comparing structural fluctuation patterns in large protein datasets. Bioinformatics 2012, 28, 2431–2440. [Google Scholar] [CrossRef]
- Ishida, Y.; Ishibashi, J.; Leal, W.S. Fatty acid solubilizer from the oral disk of the blowfly. PLoS ONE 2013, 8, e51779. [Google Scholar] [CrossRef]
- Wang, J.; Murphy, E.J.; Nix, J.C.; Jones, D.N.M. Aedes aegypti Odorant Binding Protein 22 selectively binds fatty acids through a conformational change in its C-terminal tail. Sci. Rep. 2020, 10, 3300. [Google Scholar] [CrossRef]
- Tiwari, S.P.; Fuglebakk, E.; Hollup, S.M.; Skjaerven, L.; Cragnolini, T.; Grindhaug, S.H.; Tekle, K.M.; Reuter, N. WEBnm@ v2.0: Web server and services for comparing protein flexibility. BMC Bioinform. 2014, 15, 427. [Google Scholar] [CrossRef] [PubMed]
- Hollup, S.M.; Salensminde, G.; Reuter, N. WEBnm@: A web application for normal mode analyses of proteins. BMC Bioinform. 2005, 6, 52. [Google Scholar] [CrossRef] [PubMed]
- Ichiye, T.; Karplus, M. Collective motions in proteins: A covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins 1991, 11, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Pesenti, M.E.; Spinelli, S.; Bezirard, V.; Briand, L.; Pernollet, J.-C.; Campanacci, V.; Tegoni, M.; Cambillau, C. Queen Bee Pheromone Binding Protein pH-Induced Domain Swapping Favors Pheromone Release. J. Mol. Biol. 2009, 390, 981–990. [Google Scholar] [CrossRef]
- Romanowska, J.; Nowinski, K.S.; Trylska, J. Determining Geometrically Stable Domains in Molecular Conformation Sets. J. Chem. Theory Comput. 2012, 8, 2588–2599. [Google Scholar] [CrossRef]
- Gonzalez, D.; Rihani, K.; Neiers, F.; Poirier, N.; Fraichard, S.; Gotthard, G.; Chertemps, T.; Maibeche, M.; Ferveur, J.F.; Briand, L. The Drosophila odorant-binding protein 28a is involved in the detection of the floral odour ss-ionone. Cell Mol. Life Sci. 2020, 77, 2565–2577. [Google Scholar] [CrossRef]
- Bakan, A.; Dutta, A.; Mao, W.; Liu, Y.; Chennubhotla, C.; Lezon, T.R.; Bahar, I. Evol and ProDy for bridging protein sequence evolution and structural dynamics. Bioinformatics 2014, 30, 2681–2683. [Google Scholar] [CrossRef]
- Bahar, I.; Lezon, T.R.; Yang, L.W.; Eyal, E. Global dynamics of proteins: Bridging between structure and function. Annu. Rev. Biophys. 2010, 39, 23–42. [Google Scholar] [CrossRef]
- Aronson, H.E.; Royer, W.E., Jr.; Hendrickson, W.A. Quantification of tertiary structural conservation despite primary sequence drift in the globin fold. Protein Sci. 1994, 3, 1706–1711. [Google Scholar] [CrossRef]
- Maguid, S.; Fernandez-Alberti, S.; Ferrelli, L.; Echave, J. Exploring the common dynamics of homologous proteins. Application to the globin family. Biophys. J. 2005, 89, 3–13. [Google Scholar] [CrossRef]
- Togashi, Y.; Flechsig, H. Coarse-Grained Protein Dynamics Studies Using Elastic Network Models. Int. J. Mol. Sci. 2018, 19, 3899. [Google Scholar] [CrossRef] [PubMed]
- Yamato, T.; Laprevote, O. Normal mode analysis and beyond. Biophys. Physicobiol. 2019, 16, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Erickson, L.; Byrne, T.; Johansson, E.; Tygg, J.; Vikstrom, C. Multi- and Megavariate Data Analysis Basic Principles and Applications; Umetrics Academy: Malmö, Sweden, 2013; Volume 1, p. 521. [Google Scholar]
- Howe, P.W. Principal components analysis of protein structure ensembles calculated using NMR data. J. Biomol. NMR 2001, 20, 61–70. [Google Scholar] [CrossRef] [PubMed]
- David, C.C.; Jacobs, D.J. Principal component analysis: A method for determining the essential dynamics of proteins. Methods Mol. Biol. 2014, 1084, 193–226. [Google Scholar] [CrossRef] [PubMed]











| UniProt | Organism | Abbreviation | PDB ID | Resolution (Å) | No. Residues |
|---|---|---|---|---|---|
| Q8T6S0 | Anopheles gambiae | AgamOBP1 | 3n7h | 1.60 | 125 |
| Q8T6R7 | Anopheles gambiae | AgamOBP4 | 3q8i | 2.00 | 123 |
| Q7Q9J3 | Anopheles gambiae | AgamOBP20 | 3v2l | 1.80 | 120 |
| Q6Y2R8 | Aedes aegypti | AaegOBP1 | 3k1e | 1.85 | 124 |
| Q1HRL7 | Aedes aegypti | AaegOBP22 | 6oii | 1.85 | 120 |
| Q8T6I2 | Culex quinquefasciatus | CquiOBP1 | 3ogn | 1.30 | 124 |
| O02372 | Drosophila melanogaster | DmelOBP76α | 2gte | 1.40 | 124 |
| P54195 | Drosophila melanogaster | DmelOBP28a | 6qq4 | 2.00 | 121 |
| W8W3V2 | Ceratitis capitata | CcapOBP22 | 6hhe | 1.52 | 116 |
| P34174 | Bombyx mori | BmorPBP1 | 1dqe | 1.80 | 137 |
| P34170 | Bombyx mori | BmorGOBP2 | 2wc5 | 1.90 | 141 |
| D0E9M1 | Amyelois transitella | AtraPBP1 | 4inw | 1.14 | 140 |
| Q8WRW5 | Apis mellifera | AmelASP1 | 3bjh | 1.60 | 117 |
| Q8WRW2 | Apis mellifera | AmelASP5 | 3r72 | 1.15 | 122 |
| Q3HM32 | Locusta migratoria | LmigOBP1 | 4pt1 | 1.65 | 129 |
| Q8MTC1 | Leucophaea maderae | LmadPBP1 | 1org | 1.70 | 118 |
| A0A0S2E5N6 | Megoura viciae | MvicOBP3 | 4z39 | 1.30 | 121 |
| A0A0M4AUH6 | Nasonovia ribisnigri | NribOBP3 | 4z45 | 2.02 | 118 |
| L8B8J6 | Phormia regina | PregOBP56a | 5dic | 1.18 | 115 |
| A0A0R8PDN4 | Chrysopa pallens | CpalOBP4 | 6jpm | 2.10 | 119 |
| Minimum | 1st Quartile | Median | Mean | 3rd Quartile | Maximum |
|---|---|---|---|---|---|
| 0.090 | 0.151 | 0.188 | 0.244 | 0.221 | 1.000 |
| Minimum | 1st Quartile | Median | Mean | 3rd Quartile | Maximum |
|---|---|---|---|---|---|
| 0.000 | 2.117 | 2.876 | 2.656 | 3.235 | 4.446 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tzotzos, G. A Comparative Evaluation of the Structural and Dynamic Properties of Insect Odorant Binding Proteins. Biomolecules 2022, 12, 282. https://doi.org/10.3390/biom12020282
Tzotzos G. A Comparative Evaluation of the Structural and Dynamic Properties of Insect Odorant Binding Proteins. Biomolecules. 2022; 12(2):282. https://doi.org/10.3390/biom12020282
Chicago/Turabian StyleTzotzos, George. 2022. "A Comparative Evaluation of the Structural and Dynamic Properties of Insect Odorant Binding Proteins" Biomolecules 12, no. 2: 282. https://doi.org/10.3390/biom12020282
APA StyleTzotzos, G. (2022). A Comparative Evaluation of the Structural and Dynamic Properties of Insect Odorant Binding Proteins. Biomolecules, 12(2), 282. https://doi.org/10.3390/biom12020282