
Exogenous Lactogenic Signaling Stimulates Beta Cell Replication In Vivo and In Vitro


Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. In Vivo Pseudopregnancy Model: Pellet Implantation and PL Injections
2.3. Histology
2.4. Histology Quantification
2.5. Cell Culture
In Vitro Pseudopregnancy Proliferation Assay
2.6. Statistical Analysis
3. Results
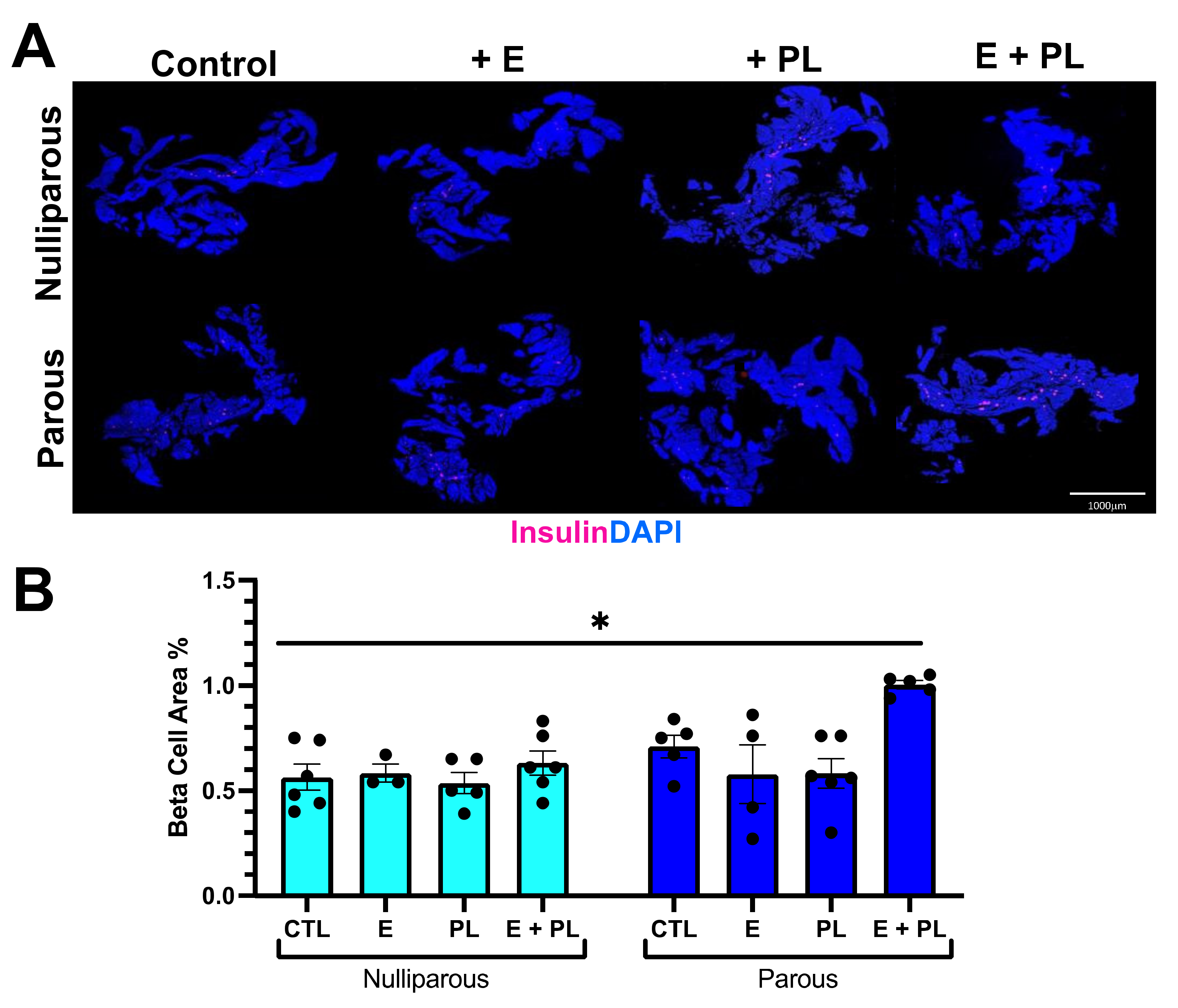
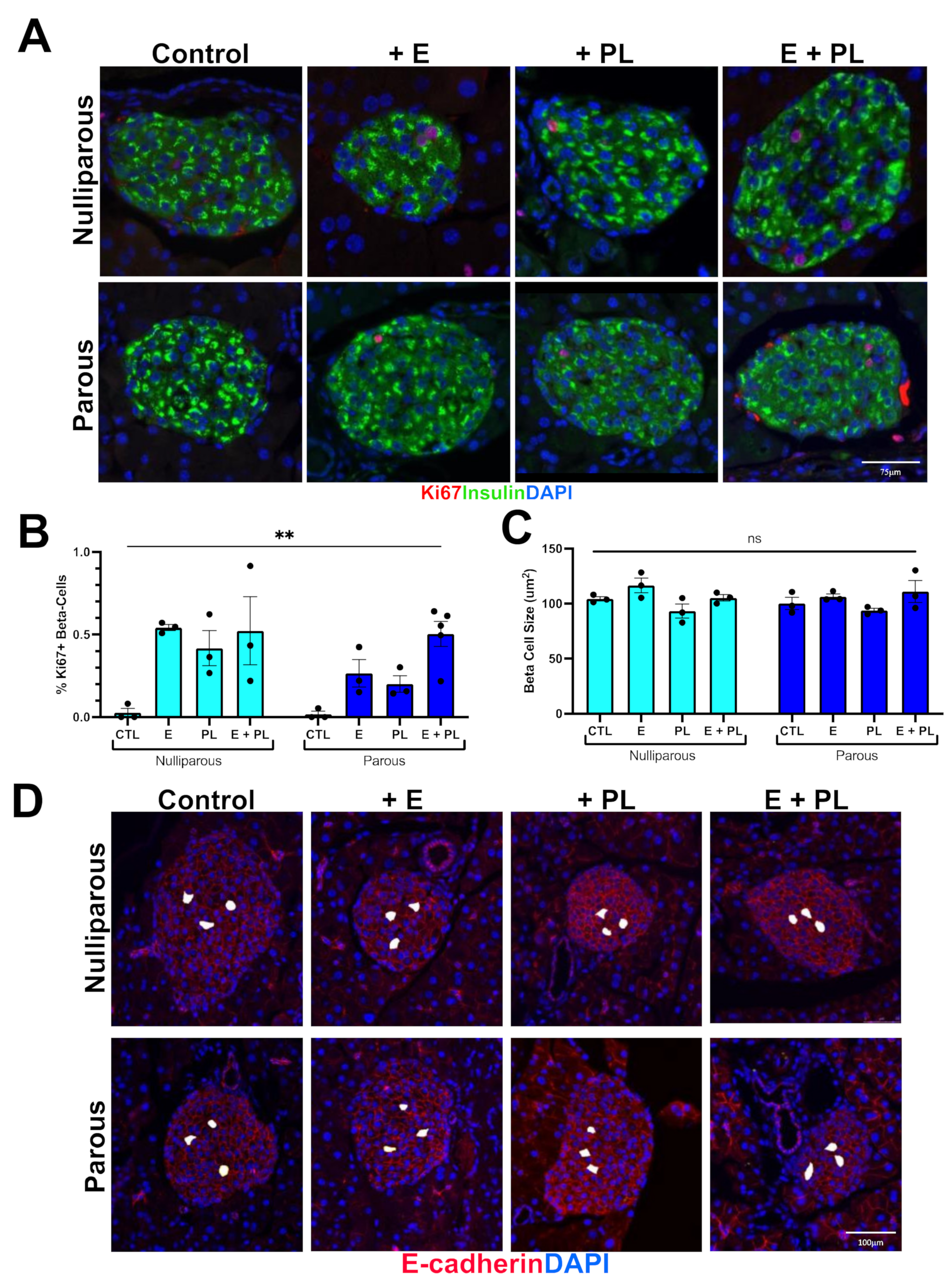
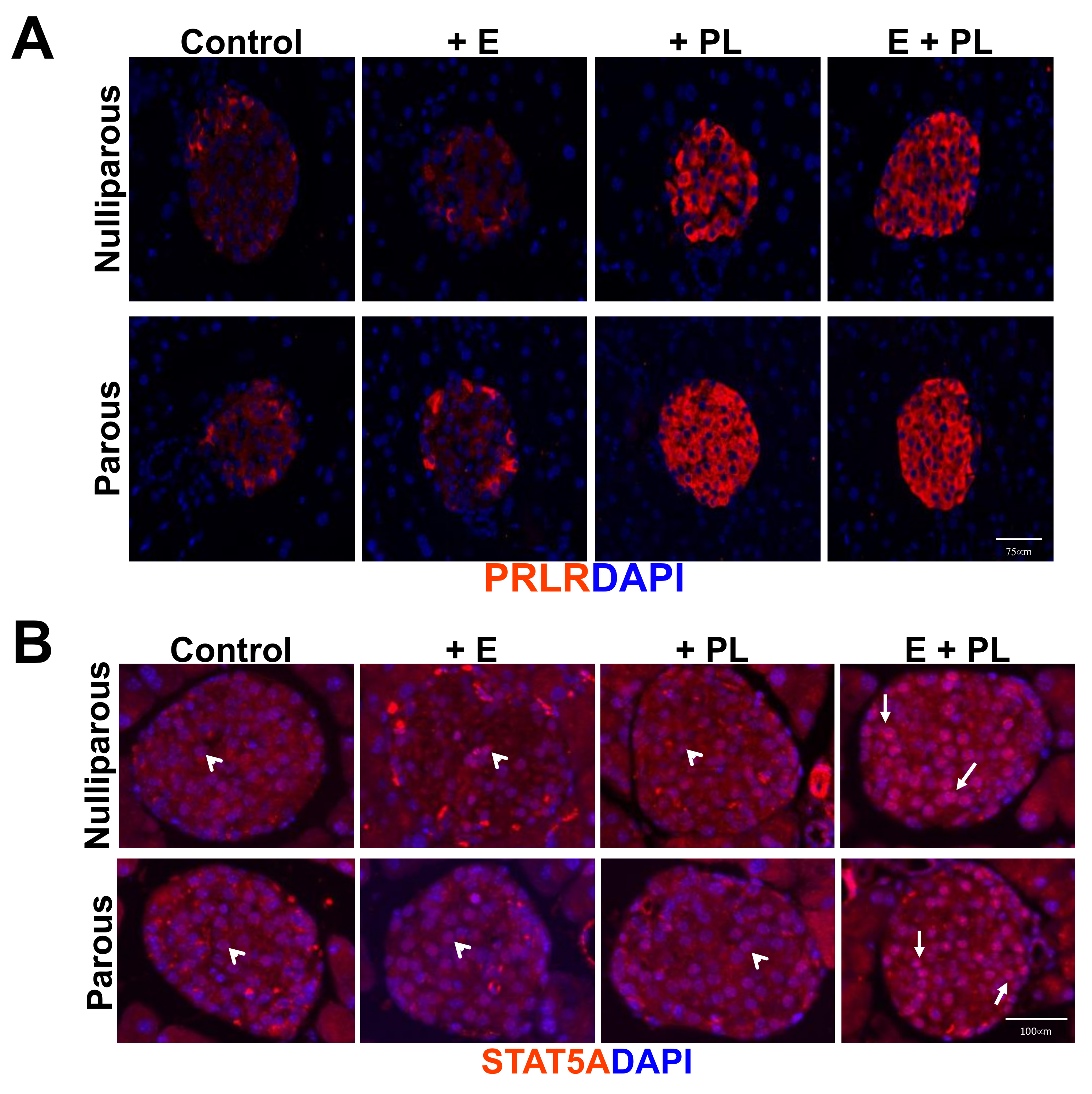
3.1. Pseudopregnancy Promotes Beta Cell Proliferation in Multiparous Mice In-Vivo
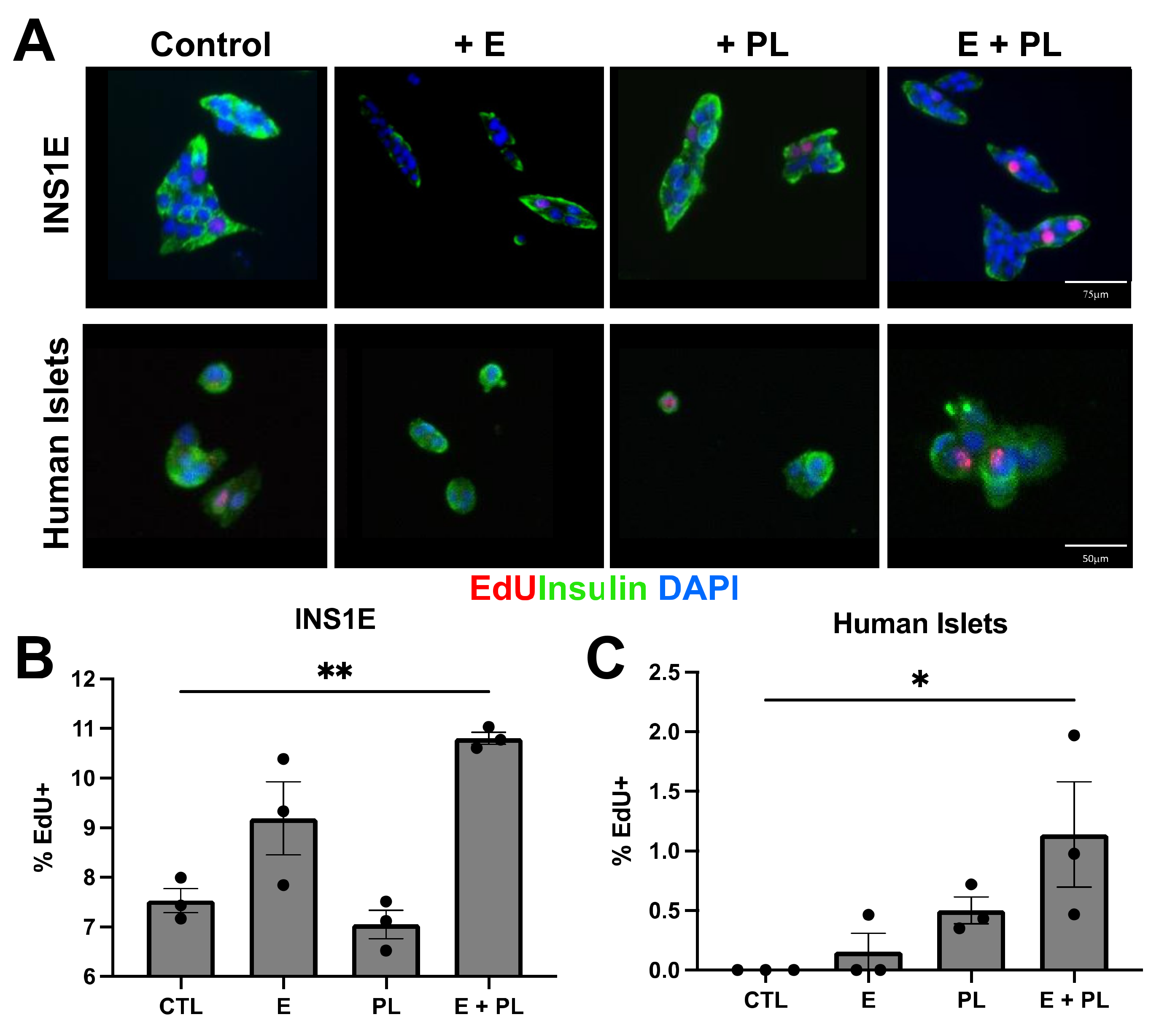
3.2. Pseudopregnancy Induces Beta Cell Replication In Vitro
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Gorvin, C.M. The prolactin receptor: Diverse and emerging roles in pathophysiology. J. Clin. Transl. Endocrinol. 2015, 2, 85–91. [Google Scholar] [CrossRef] [PubMed][Green Version]
- King, H. Epidemiology of glucose intolerance and gestational diabetes in women of childbearing age. Diabetes Care 1998, 21 (Suppl. S2), B9–B13. [Google Scholar] [PubMed]
- Gaudier, F.L.; Hauth, J.C.; Poist, M.; Corbett, D.; Cliver, S.P. Recurrence of gestational diabetes mellitus. Obstet. Gynecol. 1992, 80, 755–758. [Google Scholar] [PubMed]
- Kwak, S.H.; Kim, H.S.; Choi, S.H.; Lim, S.; Cho, Y.M.; Park, K.S.; Jang, H.C.; Kim, M.Y.; Cho, N.H.; Metzger, B.E. Subsequent pregnancy after gestational diabetes mellitus: Frequency and risk factors for recurrence in Korean women. Diabetes Care 2008, 31, 1867–1871. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Liu, Y.; Li, C.; Lin, J.; Liu, X.M.; Sheng, J.Z.; Huang, H.F. Frequency and risk factors for recurrent gestational diabetes mellitus in primiparous women: A case control study. BMC Endocr. Disord. 2019, 19, 22. [Google Scholar] [CrossRef]
- Labriola, L.; Montor, W.R.; Krogh, K.; Lojudice, F.H.; Genzini, T.; Goldberg, A.C.; Eliaschewitz, F.G.; Sogayar, M.C. Beneficial effects of prolactin and laminin on human pancreatic islet-cell cultures. Mol. Cell. Endocrinol. 2007, 263, 120–133. [Google Scholar] [CrossRef]
- Le, T.N.; Elsea, S.H.; Romero, R.; Chaiworapongsa, T.; Francis, G.L. Prolactin receptor gene polymorphisms are associated with gestational diabetes. Genet. Test Mol. Biomark. 2013, 17, 567–571. [Google Scholar] [CrossRef]
- Huang, C.; Snider, F.; Cross, J.C. Prolactin receptor is required for normal glucose homeostasis and modulation of beta-cell mass during pregnancy. Endocrinology 2009, 150, 1618–1626. [Google Scholar] [CrossRef]
- Fujinaka, Y.; Takane, K.; Yamashita, H.; Vasavada, R.C. Lactogens promote beta cell survival through JAK2/STAT5 activation and Bcl-XL upregulation. J. Biol. Chem. 2007, 282, 30707–30717. [Google Scholar] [CrossRef]
- Kondegowda, N.G.; Mozar, A.; Chin, C.; Otero, A.; Garcia-Ocana, A.; Vasavada, R.C. Lactogens protect rodent and human beta cells against glucolipotoxicity-induced cell death through Janus kinase-2 (JAK2)/signal transducer and activator of transcription-5 (STAT5) signalling. Diabetologia 2012, 55, 1721–1732. [Google Scholar] [CrossRef]
- Brelje, T.C.; Scharp, D.W.; Lacy, P.E.; Ogren, L.; Talamantes, F.; Robertson, M.; Friesen, H.G.; Sorenson, R.L. Effect of homologous placental lactogens, prolactins, and growth hormones on islet B-cell division and insulin secretion in rat, mouse, and human islets: Implication for placental lactogen regulation of islet function during pregnancy. Endocrinology 1993, 132, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, R.; Fleenor, D.; Freemark, M. Knockdown of prolactin receptors in a pancreatic beta cell line: Effects on DNA synthesis, apoptosis, and gene expression. Endocrine 2014, 46, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Freemark, M.; Avril, I.; Fleenor, D.; Driscoll, P.; Petro, A.; Opara, E.; Kendall, W.; Oden, J.; Bridges, S.; Binart, N.; et al. Targeted deletion of the PRL receptor: Effects on islet development, insulin production, and glucose tolerance. Endocrinology 2002, 143, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Pepin, M.E.; Bickerton, H.H.; Bethea, M.; Hunter, C.S.; Wende, A.R.; Banerjee, R.R. Prolactin Receptor Signaling Regulates a Pregnancy-Specific Transcriptional Program in Mouse Islets. Endocrinology 2019, 160, 1150–1163. [Google Scholar] [CrossRef]
- Shrivastava, V.; Lee, M.; Lee, D.; Pretorius, M.; Radford, B.; Makkar, G.; Huang, C. Beta cell adaptation to pregnancy requires prolactin action on both beta and non-beta cells. Sci. Rep. 2021, 11, 10372. [Google Scholar] [CrossRef]
- Deroo, B.J.; Hewitt, S.C.; Collins, J.B.; Grissom, S.F.; Hamilton, K.J.; Korach, K.S. Profile of estrogen-responsive genes in an estrogen-specific mammary gland outgrowth model. Mol. Reprod. Dev. 2009, 76, 733–750. [Google Scholar] [CrossRef]
- Silberstein, G.B.; Van Horn, K.; Shyamala, G.; Daniel, C.W. Essential role of endogenous estrogen in directly stimulating mammary growth demonstrated by implants containing pure antiestrogens. Endocrinology 1994, 134, 84–90. [Google Scholar] [CrossRef]
- Parsons, J.A.; Brelje, T.C.; Sorenson, R.L. Adaptation of islets of Langerhans to pregnancy: Increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology 1992, 130, 1459–1466. [Google Scholar] [CrossRef]
- Soares, M.J.; Talamantes, F. Genetic and litter size effects on serum placental lactogen in the mouse. Biol. Reprod. 1983, 29, 165–171. [Google Scholar] [CrossRef]
- Friedrichsen, B.N.; Galsgaard, E.D.; Nielsen, J.H.; Moldrup, A. Growth hormone- and prolactin-induced proliferation of insulinoma cells, INS-1, depends on activation of STAT5 (signal transducer and activator of transcription 5). Mol. Endocrinol. 2001, 15, 136–148. [Google Scholar] [CrossRef][Green Version]
- Mussmann, R.; Geese, M.; Harder, F.; Kegel, S.; Andag, U.; Lomow, A.; Burk, U.; Onichtchouk, D.; Dohrmann, C.; Austen, M. Inhibition of GSK3 promotes replication and survival of pancreatic beta cells. J. Biol. Chem. 2007, 282, 12030–12037. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, C.O.; Dolzhenko, E.; Hodges, E.; Smith, A.D.; Hannon, G.J. An epigenetic memory of pregnancy in the mouse mammary gland. Cell Rep. 2015, 11, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Kleinberger, J.W.; Takane, K.K.; Salim, F.; Fiaschi-Taesch, N.; Pappas, K.; Parsons, R.; Jiang, J.; Zhang, Y.; Liu, H.; et al. Augmented Stat5 Signaling Bypasses Multiple Impediments to Lactogen-Mediated Proliferation in Human beta-Cells. Diabetes 2015, 64, 3784–3797. [Google Scholar] [CrossRef] [PubMed]
- Mortlock, R.D.; Georgia, S.K.; Finley, S.D. Dynamic Regulation of JAK-STAT Signaling Through the Prolactin Receptor Predicted by Computational Modeling. Cell. Mol. Bioeng. 2021, 14, 15–30. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nulliparous | Parous |
|---|---|
| Nulliparous + Estrogen (D0-D11) | Parous + Estrogen (D0-D11) |
| Nulliparous + Placental Lactogen (D7-D11) | Parous + Placental Lactogen (D7-D11) |
| Nulliparous + Estrogen (D0-D11) + Placental lactogen (D-D11) | Parous + Estrogen (D0-D11) + Placental lactogen (D7-D11) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Millette, K.; Rodriguez, K.; Sheng, X.; Finley, S.D.; Georgia, S. Exogenous Lactogenic Signaling Stimulates Beta Cell Replication In Vivo and In Vitro. Biomolecules 2022, 12, 215. https://doi.org/10.3390/biom12020215
Millette K, Rodriguez K, Sheng X, Finley SD, Georgia S. Exogenous Lactogenic Signaling Stimulates Beta Cell Replication In Vivo and In Vitro. Biomolecules. 2022; 12(2):215. https://doi.org/10.3390/biom12020215
Chicago/Turabian StyleMillette, Katelyn, Keith Rodriguez, Xia Sheng, Stacey D. Finley, and Senta Georgia. 2022. "Exogenous Lactogenic Signaling Stimulates Beta Cell Replication In Vivo and In Vitro" Biomolecules 12, no. 2: 215. https://doi.org/10.3390/biom12020215
APA StyleMillette, K., Rodriguez, K., Sheng, X., Finley, S. D., & Georgia, S. (2022). Exogenous Lactogenic Signaling Stimulates Beta Cell Replication In Vivo and In Vitro. Biomolecules, 12(2), 215. https://doi.org/10.3390/biom12020215