
Passive Immunization in Alpha-Synuclein Preclinical Animal Models

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. αsyn and Its Role in the Pathogenesis of Synucleinopathies
3. Prion-like Behavior and Gut-to-Brain Propagation
4. Subtypes of α-Synucleinopathies
5. Therapeutic Strategies Targeting αsyn Pathology
6. Passive Immunization Strategies in Animal Models and Clinical Trials
6.1. C-Terminal Targeting Approaches
6.2. N-Terminal and NAC Targeting Approaches
6.3. Conformational Targeting Approaches
6.4. Passive Candidates Translated into Clinical Trials
7. Towards Personalized Immunotherapy
8. Future Perspectives and Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Chartier-Harlin, M.-C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [Green Version]
- Visanji, N.P.; Lang, A.E.; Kovacs, G.G. Beyond the synucleinopathies: Alpha synuclein as a driving force in neurodegenerative comorbidities. Transl. Neurodegener. 2019, 8, 28. [Google Scholar] [CrossRef] [Green Version]
- Papp, M.I.; Kahn, J.E.; Lantos, P.L. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome). J. Neurol. Sci. 1989, 94, 79–100. [Google Scholar] [CrossRef]
- Jellinger, K.A.; Lantos, P.L. Papp-Lantos inclusions and the pathogenesis of multiple system atrophy: An update. Acta Neuropathol. 2010, 119, 657–667. [Google Scholar] [CrossRef]
- Kovacs, G.G. Molecular Pathological Classification of Neurodegenerative Diseases: Turning towards Precision Medicine. Int. J. Mol. Sci. 2016, 17, 189. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Emre, M. Dementia with Lewy bodies and Parkinson disease dementia: More different than similar? Neurology 2020, 94, 858–859. [Google Scholar] [CrossRef]
- Borghammer, P. How does parkinson’s disease begin? Perspectives on neuroanatomical pathways, prions, and histology. Mov. Disord. 2018, 33, 48–57. [Google Scholar] [CrossRef]
- Beach, T.G.; Adler, C.H.; Sue, L.I.; Vedders, L.; Lue, L.; White Iii, C.L.; Akiyama, H.; Caviness, J.N.; Shill, H.A.; Sabbagh, M.N.; et al. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2010, 119, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendoza-Velásquez, J.J.; Flores-Vázquez, J.F.; Barrón-Velázquez, E.; Sosa-Ortiz, A.L.; Illigens, B.-M.W.; Siepmann, T. Autonomic Dysfunction in α-Synucleinopathies. Front. Neurol. 2019, 10, 363. [Google Scholar] [CrossRef] [Green Version]
- Thaisetthawatkul, P. Pure Autonomic Failure. Curr. Neurol. Neurosci. Rep. 2016, 16, 74. [Google Scholar] [CrossRef] [PubMed]
- Palma, J.-A.; Norcliffe-Kaufmann, L.; Kaufmann, H. Diagnosis of multiple system atrophy. Auton. Neurosci. 2018, 211, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Borghammer, P. The α-Synuclein Origin and Connectome Model (SOC Model) of Parkinson’s Disease: Explaining Motor Asymmetry, Non-Motor Phenotypes, and Cognitive Decline. J. Parkinsons. Dis. 2021, 11, 455–474. [Google Scholar] [CrossRef]
- Borghammer, P.; Horsager, J.; Andersen, K.; Van Den Berge, N.; Raunio, A.; Murayama, S.; Parkkinen, L.; Myllykangas, L. Neuropathological evidence of body-first vs. brain-first Lewy body disease. Neurobiol. Dis. 2021, 161, 105557. [Google Scholar] [CrossRef] [PubMed]
- Albus, A.; Jördens, M.; Möller, M.; Dodel, R. Encoding the Sequence of Specific Autoantibodies Against beta-Amyloid and alpha-Synuclein in Neurodegenerative Diseases. Front. Immunol. 2019, 10, 2033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Games, D.; Valera, E.; Spencer, B.; Rockenstein, E.; Mante, M.; Adame, A.; Patrick, C.; Ubhi, K.; Nuber, S.; Sacayon, P.; et al. Reducing C-terminal-truncated α-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J. Neurosci. 2014, 34, 9441–9454. [Google Scholar] [CrossRef]
- Bergström, A.L.; Kallunki, P.; Fog, K. Development of Passive Immunotherapies for Synucleinopathies. Mov. Disord. 2015, 31, 16–21. [Google Scholar] [CrossRef]
- Antonini, A.; Bravi, D.; Sandre, M.; Bubacco, L. Immunization therapies for Parkinson’s disease: State of the art and considerations for future clinical trials. Expert Opin. Investig. Drugs 2020, 29, 685–695. [Google Scholar] [CrossRef]
- Li, X.; Koudstaal, W.; Fletcher, L.; Costa, M.; van Winsen, M.; Siregar, B.; Inganäs, H.; Kim, J.; Keogh, E.; Macedo, J.; et al. Naturally occurring antibodies isolated from PD patients inhibit synuclein seeding in vitro and recognize Lewy pathology. Acta Neuropathol. 2019, 137, 825–836. [Google Scholar] [CrossRef] [Green Version]
- Scott, K.M.; Kouli, A.; Yeoh, S.L.; Clatworthy, M.R.; Williams-Gray, C.H. A Systematic Review and Meta-Analysis of Alpha Synuclein Auto-Antibodies in Parkinson’s Disease. Front. Neurol. 2018, 9, 815. [Google Scholar] [CrossRef] [Green Version]
- Brudek, T.; Winge, K.; Folke, J.; Christensen, S.; Fog, K.; Pakkenberg, B.; Pedersen, L.O. Autoimmune antibody decline in Parkinson’s disease and Multiple System Atrophy; a step towards immunotherapeutic strategies. Mol. Neurodegener. 2017, 12, 44. [Google Scholar] [CrossRef]
- Folke, J.; Rydbirk, R.; Løkkegaard, A.; Salvesen, L.; Hejl, A.-M.; Starhof, C.; Bech, S.; Winge, K.; Christensen, S.; Pedersen, L.Ø.; et al. Distinct Autoimmune Anti-α-Synuclein Antibody Patterns in Multiple System Atrophy and Parkinson’s Disease. Front. Immunol. 2019, 10, 2253. [Google Scholar] [CrossRef]
- Folke, J.; Rydbirk, R.; Løkkegaard, A.; Hejl, A.-M.; Winge, K.; Starhof, C.; Salvesen, L.; Pedersen, L.Ø.; Aznar, S.; Pakkenberg, B.; et al. Cerebrospinal fluid and plasma distribution of anti-α-synuclein IgMs and IgGs in multiple system atrophy and Parkinson’s disease. Parkinsonism Relat. Disord. 2021, 87, 98–104. [Google Scholar] [CrossRef]
- Chartier, S.; Duyckaerts, C. Is Lewy pathology in the human nervous system chiefly an indicator of neuronal protection or of toxicity? Cell Tissue Res. 2018, 373, 149–160. [Google Scholar] [CrossRef]
- Ingelsson, M. Alpha-Synuclein Oligomers-Neurotoxic Molecules in Parkinson’s Disease and Other Lewy Body Disorders. Front. Neurosci. 2016, 10, 408. [Google Scholar] [CrossRef] [Green Version]
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [CrossRef] [Green Version]
- George, J.M. The synucleins. Genome Biol. 2002, 3, reviews3002. [Google Scholar] [CrossRef]
- Stefanis, L. α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [Green Version]
- Vargas, K.J.; Makani, S.; Davis, T.; Westphal, C.H.; Castillo, P.E.; Chandra, S.S. Synucleins regulate the kinetics of synaptic vesicle endocytosis. J. Neurosci. 2014, 34, 9364–9376. [Google Scholar] [CrossRef]
- Logan, T.; Bendor, J.; Toupin, C.; Thorn, K.; Edwards, R.H. α-Synuclein promotes dilation of the exocytotic fusion pore. Nat. Neurosci. 2017, 20, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Danzer, K.M.; Haasen, D.; Karow, A.R.; Moussaud, S.; Habeck, M.; Giese, A.; Kretzschmar, H.; Hengerer, B.; Kostka, M. Different species of alpha-synuclein oligomers induce calcium influx and seeding. J. Neurosci. 2007, 27, 9220–9232. [Google Scholar] [CrossRef] [PubMed]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Perovic, I.; Chittuluru, J.; Kaganovich, A.; Nguyen, L.T.T.; Liao, J.; Auclair, J.R.; Johnson, D.; Landeru, A.; Simorellis, A.K.; et al. A soluble α-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. USA 2011, 108, 17797–17802. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Hoffmann, M.; Li, X.; Wang, M.; Garen, C.R.; Petersen, N.O.; Woodside, M.T. Structural characteristics and membrane interactions of tandem α-synuclein oligomers. Sci. Rep. 2018, 8, 6755. [Google Scholar] [CrossRef]
- Trexler, A.J.; Rhoades, E. N-Terminal acetylation is critical for forming α-helical oligomer of α-synuclein. Protein Sci. 2012, 21, 601–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, A.; Roeters, S.J.; Schilderink, N.; Hommersom, B.; Heeren, R.M.A.; Woutersen, S.; Claessens, M.M.A.E.; Subramaniam, V. The Impact of N-terminal Acetylation of α-Synuclein on Phospholipid Membrane Binding and Fibril Structure. J. Biol. Chem. 2016, 291, 21110–21122. [Google Scholar] [CrossRef] [Green Version]
- Rossetti, G.; Musiani, F.; Abad, E.; Dibenedetto, D.; Mouhib, H.; Fernandez, C.O.; Carloni, P. Conformational ensemble of human α-synuclein physiological form predicted by molecular simulations. Phys. Chem. Chem. Phys. 2016, 18, 5702–5706. [Google Scholar] [CrossRef] [Green Version]
- Giasson, B.I.; Murray, I.V.; Trojanowski, J.Q.; Lee, V.M. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J. Biol. Chem. 2001, 276, 2380–2386. [Google Scholar] [CrossRef] [Green Version]
- Kara, E.; Lewis, P.A.; Ling, H.; Proukakis, C.; Houlden, H.; Hardy, J. α-Synuclein mutations cluster around a putative protein loop. Neurosci. Lett. 2013, 546, 67–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lázaro, D.F.; Rodrigues, E.F.; Langohr, R.; Shahpasandzadeh, H.; Ribeiro, T.; Guerreiro, P.; Gerhardt, E.; Kröhnert, K.; Klucken, J.; Pereira, M.D.; et al. Systematic comparison of the effects of alpha-synuclein mutations on its oligomerization and aggregation. PLoS Genet. 2014, 10, e1004741. [Google Scholar] [CrossRef]
- Bengoa-Vergniory, N.; Roberts, R.F.; Wade-Martins, R.; Alegre-Abarrategui, J. Alpha-synuclein oligomers: A new hope. Acta Neuropathol. 2017, 134, 819–838. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Jellinger, K.A. A critical evaluation of current staging of alpha-synuclein pathology in Lewy body disorders. Biochim. Biophys. Acta 2009, 1792, 730–740. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Ghebremedhin, E.; Rüb, U.; Bratzke, H.; Del Tredici, K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004, 318, 121–134. [Google Scholar] [CrossRef]
- Hawkes, C.H.; Del Tredici, K.; Braak, H. Parkinson’s disease: A dual-hit hypothesis. Neuropathol. Appl. Neurobiol. 2007, 33, 599–614. [Google Scholar] [CrossRef]
- Dickson, D.W.; Fujishiro, H.; DelleDonne, A.; Menke, J.; Ahmed, Z.; Klos, K.J.; Josephs, K.A.; Frigerio, R.; Burnett, M.; Parisi, J.E.; et al. Evidence that incidental Lewy body disease is pre-symptomatic Parkinson’s disease. Acta Neuropathol. 2008, 115, 437–444. [Google Scholar] [CrossRef]
- Tsuboi, Y.; Uchikado, H.; Dickson, D.W. Neuropathology of Parkinson’s disease dementia and dementia with Lewy bodies with reference to striatal pathology. Parkinsonism Relat. Disord. 2007, 13 (Suppl. 3), S221–S224. [Google Scholar] [CrossRef]
- McKeith, I.G.; Dickson, D.W.; Lowe, J.; Emre, M.; O’Brien, J.T.; Feldman, H.; Cummings, J.; Duda, J.E.; Lippa, C.; Perry, E.K.; et al. Diagnosis and management of dementia with Lewy bodies: Third report of the DLB Consortium. Neurology 2005, 65, 1863–1872. [Google Scholar] [CrossRef] [Green Version]
- Zaccai, J.; Brayne, C.; McKeith, I.; Matthews, F.; Ince, P.G. Patterns and stages of alpha-synucleinopathy: Relevance in a population-based cohort. Neurology 2008, 70, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Cairns, N.J.; Lantos, P.L.; Goedert, M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci. Lett. 1998, 251, 205–208. [Google Scholar] [CrossRef]
- Gilman, S.; Wenning, G.K.; Low, P.A.; Brooks, D.J.; Mathias, C.J.; Trojanowski, J.Q.; Wood, N.W.; Colosimo, C.; Durr, A.; Fowler, C.J.; et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008, 71, 670–676. [Google Scholar] [CrossRef]
- Kaji, S.; Maki, T.; Ishimoto, T.; Yamakado, H.; Takahashi, R. Insights into the pathogenesis of multiple system atrophy: Focus on glial cytoplasmic inclusions. Transl. Neurodegener. 2020, 9, 7. [Google Scholar] [CrossRef] [Green Version]
- Cykowski, M.D.; Coon, E.A.; Powell, S.Z.; Jenkins, S.M.; Benarroch, E.E.; Low, P.A.; Schmeichel, A.M.; Parisi, J.E. Expanding the spectrum of neuronal pathology in multiple system atrophy. Brain 2015, 138, 2293–2309. [Google Scholar] [CrossRef] [PubMed]
- Duda, J.E.; Giasson, B.I.; Gur, T.L.; Montine, T.J.; Robertson, D.; Biaggioni, I.; Hurtig, H.I.; Stern, M.B.; Gollomp, S.M.; Grossman, M.; et al. Immunohistochemical and biochemical studies demonstrate a distinct profile of alpha-synuclein permutations in multiple system atrophy. J. Neuropathol. Exp. Neurol. 2000, 59, 830–841. [Google Scholar] [CrossRef] [Green Version]
- Brettschneider, J.; Irwin, D.J.; Boluda, S.; Byrne, M.D.; Fang, L.; Lee, E.B.; Robinson, J.L.; Suh, E.; Van Deerlin, V.M.; Toledo, J.B.; et al. Progression of alpha-synuclein pathology in multiple system atrophy of the cerebellar type. Neuropathol. Appl. Neurobiol. 2017, 43, 315–329. [Google Scholar] [CrossRef]
- Fujishiro, H.; Ahn, T.-B.; Frigerio, R.; DelleDonne, A.; Josephs, K.A.; Parisi, J.E.; Eric Ahlskog, J.; Dickson, D.W. Glial cytoplasmic inclusions in neurologically normal elderly: Prodromal multiple system atrophy? Acta Neuropathol. 2008, 116, 269–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koga, S.; Li, F.; Zhao, N.; Roemer, S.F.; Ferman, T.J.; Wernick, A.I.; Walton, R.L.; Faroqi, A.H.; Graff-Radford, N.R.; Cheshire, W.P.; et al. Clinicopathologic and genetic features of multiple system atrophy with Lewy body disease. Brain Pathol. 2020, 30, 766–778. [Google Scholar] [CrossRef]
- Hague, K.; Lento, P.; Morgello, S.; Caro, S.; Kaufmann, H. The distribution of Lewy bodies in pure autonomic failure: Autopsy findings and review of the literature. Acta Neuropathol. 1997, 94, 192–196. [Google Scholar] [CrossRef]
- Li, J.-Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Björklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Brundin, P. Lewy body pathology in long-term fetal nigral transplants: Is Parkinson’s disease transmitted from one neural system to another? Neuropsychopharmacology 2009, 34, 254. [Google Scholar] [CrossRef]
- Uchihara, T.; Giasson, B.I. Propagation of alpha-synuclein pathology: Hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. 2016, 131, 49–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedert, M.; Masuda-Suzukake, M.; Falcon, B. Like prions: The propagation of aggregated tau and α-synuclein in neurodegeneration. Brain 2017, 140, 266–278. [Google Scholar] [CrossRef] [Green Version]
- Stokholm, M.G.; Danielsen, E.H.; Hamilton-Dutoit, S.J.; Borghammer, P. Pathological α-synuclein in gastrointestinal tissues from prodromal Parkinson disease patients. Ann. Neurol. 2016, 79, 940–949. [Google Scholar] [CrossRef]
- Chiang, H.-L.; Lin, C.-H. Altered Gut Microbiome and Intestinal Pathology in Parkinson’s Disease. J. Mov. Disord. 2019, 12, 67–83. [Google Scholar] [CrossRef]
- Skjærbæk, C.; Knudsen, K.; Horsager, J.; Borghammer, P. Gastrointestinal Dysfunction in Parkinson’s Disease. J. Clin. Med. 2021, 10, 493. [Google Scholar] [CrossRef] [PubMed]
- Orimo, S.; Uchihara, T.; Nakamura, A.; Mori, F.; Kakita, A.; Wakabayashi, K.; Takahashi, H. Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008, 131, 642–650. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Illigens, B.M.; McCormick, M.P.; Wang, N.; Gibbons, C.H. Alpha-Synuclein in Skin Nerve Fibers as a Biomarker for Alpha-Synucleinopathies. J. Clin. Neurol. 2019, 15, 135–142. [Google Scholar] [CrossRef]
- Donadio, V.; Incensi, A.; Rizzo, G.; De Micco, R.; Tessitore, A.; Devigili, G.; Del Sorbo, F.; Bonvegna, S.; Infante, R.; Magnani, M.; et al. Skin Biopsy May Help to Distinguish Multiple System Atrophy-Parkinsonism from Parkinson’s Disease With Orthostatic Hypotension. Mov. Disord. 2020, 35, 1649–1657. [Google Scholar] [CrossRef]
- Brumberg, J.; Kuzkina, A.; Lapa, C.; Mammadova, S.; Buck, A.; Volkmann, J.; Sommer, C.; Isaias, I.U.; Doppler, K. Dermal and cardiac autonomic fiber involvement in Parkinson’s disease and multiple system atrophy. Neurobiol. Dis. 2021, 153, 105332. [Google Scholar] [CrossRef]
- Leclair-Visonneau, L.; Clairembault, T.; Coron, E.; Le Dily, S.; Vavasseur, F.; Dalichampt, M.; Péréon, Y.; Neunlist, M.; Derkinderen, P. REM sleep behavior disorder is related to enteric neuropathology in Parkinson disease. Neurology 2017, 89, 1612–1618. [Google Scholar] [CrossRef] [PubMed]
- Donadio, V.; Incensi, A.; Del Sorbo, F.; Rizzo, G.; Infante, R.; Scaglione, C.; Modugno, N.; Fileccia, E.; Elia, A.E.; Cencini, F.; et al. Skin Nerve Phosphorylated α-Synuclein Deposits in Parkinson Disease with Orthostatic Hypotension. J. Neuropathol. Exp. Neurol. 2018, 77, 942–949. [Google Scholar] [CrossRef] [Green Version]
- Hirayama, M. Sweating dysfunctions in Parkinson’s disease. J. Neurol. 2006, 253, vii42–vii47. [Google Scholar] [CrossRef]
- Cheon, S.-M.; Ha, M.-S.; Park, M.J.; Kim, J.W. Nonmotor symptoms of Parkinson’s disease: Prevalence and awareness of patients and families. Parkinsonism Relat. Disord. 2008, 14, 286–290. [Google Scholar] [CrossRef]
- Palma, J.-A.; Kaufmann, H. Orthostatic Hypotension in Parkinson Disease. Clin. Geriatr. Med. 2020, 36, 53–67. [Google Scholar] [CrossRef]
- Angot, E.; Steiner, J.A.; Hansen, C.; Li, J.-Y.; Brundin, P. Are synucleinopathies prion-like disorders? Lancet Neurol. 2010, 9, 1128–1138. [Google Scholar] [CrossRef]
- Jan, A.; Gonçalves, N.P.; Vaegter, C.B.; Jensen, P.H.; Ferreira, N. The Prion-Like Spreading of Alpha-Synuclein in Parkinson’s Disease: Update on Models and Hypotheses. Int. J. Mol. Sci. 2021, 22, 8338. [Google Scholar] [CrossRef]
- Ferreira, N.; Gonçalves, N.P.; Jan, A.; Jensen, N.M.; van der Laan, A.; Mohseni, S.; Vægter, C.B.; Jensen, P.H. Trans-synaptic spreading of alpha-synuclein pathology through sensory afferents leads to sensory nerve degeneration and neuropathic pain. Acta Neuropathol. Commun. 2021, 9, 31. [Google Scholar] [CrossRef]
- Braak, H.; Rüb, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef]
- Pan-Montojo, F.; Anichtchik, O.; Dening, Y.; Knels, L.; Pursche, S.; Jung, R.; Jackson, S.; Gille, G.; Spillantini, M.G.; Reichmann, H.; et al. Progression of Parkinson’s disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS ONE 2010, 5, e8762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Den Berge, N.; Ferreira, N.; Gram, H.; Mikkelsen, T.W.; Alstrup, A.K.O.; Casadei, N.; Tsung-Pin, P.; Riess, O.; Nyengaard, J.R.; Tamgüney, G.; et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019, 138, 535–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Den Berge, N.; Ferreira, N.; Mikkelsen, T.W.; Alstrup, A.K.O.; Tamgüney, G.; Karlsson, P.; Terkelsen, A.J.; Nyengaard, J.R.; Jensen, P.H.; Borghammer, P. Ageing promotes pathological alpha-synuclein propagation and autonomic dysfunction in wild-type rats. Brain 2021, 144, 1853–1868. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kwon, S.H.; Kam, T.I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641.e7. [Google Scholar] [CrossRef]
- Svensson, E.; Horváth-Puhó, E.; Thomsen, R.W.; Djurhuus, J.C.; Pedersen, L.; Borghammer, P.; Sørensen, H.T. Vagotomy and subsequent risk of Parkinson’s disease. Ann. Neurol. 2015, 78, 522–529. [Google Scholar] [CrossRef]
- Liu, B.; Fang, F.; Pedersen, N.L.; Tillander, A.; Ludvigsson, J.F.; Ekbom, A.; Svenningsson, P.; Chen, H.; Wirdefeldt, K. Vagotomy and Parkinson disease: A Swedish register-based matched-cohort study. Neurology 2017, 88, 1996–2002. [Google Scholar] [CrossRef] [Green Version]
- Parkkinen, L.; Pirttilä, T.; Alafuzoff, I. Applicability of current staging/categorization of alpha-synuclein pathology and their clinical relevance. Acta Neuropathol. 2008, 115, 399–407. [Google Scholar] [CrossRef] [Green Version]
- Jellinger, K.A. Is Braak staging valid for all types of Parkinson’s disease? J. Neural Transm. 2019, 126, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Raunio, A.; Kaivola, K.; Tuimala, J.; Kero, M.; Oinas, M.; Polvikoski, T.; Paetau, A.; Tienari, P.J.; Myllykangas, L. Lewy-related pathology exhibits two anatomically and genetically distinct progression patterns: A population-based study of Finns aged 85. Acta Neuropathol. 2019, 138, 771–782. [Google Scholar] [CrossRef] [Green Version]
- Borghammer, P.; Van Den Berge, N. Brain-First versus Gut-First Parkinson’s Disease: A Hypothesis. J. Parkinsons. Dis. 2019, 9, S281–S295. [Google Scholar] [CrossRef] [Green Version]
- Horsager, J.; Andersen, K.B.; Knudsen, K.; Skjærbæk, C.; Fedorova, T.D.; Okkels, N.; Schaeffer, E.; Bonkat, S.K.; Geday, J.; Otto, M.; et al. Brain-first versus body-first Parkinson’s disease: A multimodal imaging case-control study. Brain 2020, 143, 3077–3088. [Google Scholar] [CrossRef]
- Högl, B.; Stefani, A.; Videnovic, A. Idiopathic REM sleep behaviour disorder and neurodegeneration—An update. Nat. Rev. Neurol. 2018, 14, 40–55. [Google Scholar] [CrossRef]
- McCarter, S.J.; Gehrking, T.L.; St Louis, E.K.; Suarez, M.D.; Boeve, B.F.; Silber, M.H.; Low, P.A.; Singer, W. Autonomic dysfunction and phenoconversion in idiopathic REM sleep behavior disorder. Clin. Auton. Res. Off. J. Clin. Auton. Res. Soc. 2020, 30, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.B.; Hansen, A.K.; Sommerauer, M.; Fedorova, T.D.; Knudsen, K.; Vang, K.; Van Den Berge, N.; Kinnerup, M.; Nahimi, A.; Pavese, N.; et al. Altered sensorimotor cortex noradrenergic function in idiopathic REM sleep behaviour disorder—A PET study. Parkinsonism Relat. Disord. 2020, 75, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, K.; Fedorova, T.D.; Horsager, J.; Andersen, K.B.; Skjærbæk, C.; Berg, D.; Schaeffer, E.; Brooks, D.J.; Pavese, N.; Van Den Berge, N.; et al. Asymmetric Dopaminergic Dysfunction in Brain-First versus Body-First Parkinson’s Disease Subtypes. J. Parkinsons. Dis. 2021, 11, 1677–1687. [Google Scholar] [CrossRef]
- Van Den Berge, N.; Ulusoy, A. Animal models of brain-first and body-first Parkinson’s disease. Neurobiol. Dis. 2021, 163, 105599. [Google Scholar] [CrossRef]
- Fleming, S.M.; Davis, A.; Simons, E. Targeting alpha-synuclein via the immune system in Parkinson’s disease: Current vaccine therapies. Neuropharmacology 2022, 202, 108870. [Google Scholar] [CrossRef]
- Takahashi, M.; Suzuki, M.; Fukuoka, M.; Fujikake, N.; Watanabe, S.; Murata, M.; Wada, K.; Nagai, Y.; Hohjoh, H. Normalization of Overexpressed α-Synuclein Causing Parkinson’s Disease by a Moderate Gene Silencing with RNA Interference. Mol. Ther. Nucleic Acids 2015, 4, e241. [Google Scholar] [CrossRef]
- Kim, Y.-C.; Miller, A.; Lins, L.C.R.F.; Han, S.-W.; Keiser, M.S.; Boudreau, R.L.; Davidson, B.L.; Narayanan, N.S. RNA Interference of Human α-Synuclein in Mouse. Front. Neurol. 2017, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. Autophagy and Alpha-Synuclein: Relevance to Parkinson’s Disease and Related Synucleopathies. Mov. Disord. 2016, 31, 178–192. [Google Scholar] [CrossRef]
- Pena-Diaz, S.; Pujols, J.; Ventura, S. Small molecules to prevent the neurodegeneration caused by α-synuclein aggregation. Neural Regen. Res. 2020, 15, 2260–2261. [Google Scholar] [CrossRef]
- Meissner, W.G.; Traon, A.P.-L.; Foubert-Samier, A.; Galabova, G.; Galitzky, M.; Kutzelnigg, A.; Laurens, B.; Lührs, P.; Medori, R.; Péran, P.; et al. A Phase 1 Randomized Trial of Specific Active α-Synuclein Immunotherapies PD01A and PD03A in Multiple System Atrophy. Mov. Disord. 2020, 35, 1957–1965. [Google Scholar] [CrossRef]
- Nimmo, J.T.; Smith, H.; Wang, C.Y.; Teeling, J.L.; Nicoll, J.A.R.; Verma, A.; Dodart, J.-C.; Liu, Z.; Lin, F.; Carare, R.O. Immunisation with UB-312 in the Thy1SNCA mouse prevents motor performance deficits and oligomeric α-synuclein accumulation in the brain and gut. Acta Neuropathol. 2021, 143, 55–73. [Google Scholar] [CrossRef]
- Sulzer, D.; Alcalay, R.N.; Garretti, F.; Cote, L.; Kanter, E.; Agin-Liebes, J.; Liong, C.; McMurtrey, C.; Hildebrand, W.H.; Mao, X.; et al. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 2017, 546, 656–661. [Google Scholar] [CrossRef] [Green Version]
- Lindestam Arlehamn, C.S.; Dhanwani, R.; Pham, J.; Kuan, R.; Frazier, A.; Rezende Dutra, J.; Phillips, E.; Mallal, S.; Roederer, M.; Marder, K.S.; et al. α-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat. Commun. 2020, 11, 1875. [Google Scholar] [CrossRef] [Green Version]
- Masliah, E.; Rockenstein, E.; Mante, M.; Crews, L.; Spencer, B.; Adame, A.; Patrick, C.; Trejo, M.; Ubhi, K.; Rohn, T.T.; et al. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of lewy body disease. PLoS ONE 2011, 6, e19338. [Google Scholar] [CrossRef] [Green Version]
- Bae, E.-J.; Lee, H.-J.; Rockenstein, E.; Ho, D.-H.; Park, E.-B.; Yang, N.-Y.; Desplats, P.; Masliah, E.; Lee, S.-J. Antibody-aided clearance of extracellular α-synuclein prevents cell-to-cell aggregate transmission. J. Neurosci. 2012, 32, 13454–13469. [Google Scholar] [CrossRef]
- Tran, H.T.; Chung, C.H.-Y.; Iba, M.; Zhang, B.; Trojanowski, J.Q.; Luk, K.C.; Lee, V.M.Y. A-synuclein immunotherapy blocks uptake and templated propagation of misfolded α-synuclein and neurodegeneration. Cell Rep. 2014, 7, 2054–2065. [Google Scholar] [CrossRef] [Green Version]
- Lindström, V.; Fagerqvist, T.; Nordström, E.; Eriksson, F.; Lord, A.; Tucker, S.; Andersson, J.; Johannesson, M.; Schell, H.; Kahle, P.J.; et al. Immunotherapy targeting α-synuclein protofibrils reduced pathology in (Thy-1)-h[A30P] α-synuclein mice. Neurobiol. Dis. 2014, 69, 134–143. [Google Scholar] [CrossRef]
- Shahaduzzaman, M.; Nash, K.; Hudson, C.; Sharif, M.; Grimmig, B.; Lin, X.; Bai, G.; Liu, H.; Ugen, K.E.; Cao, C.; et al. Anti-human α-synuclein N-terminal peptide antibody protects against dopaminergic cell death and ameliorates behavioral deficits in an AAV-α-synuclein rat model of Parkinson’s disease. PLoS ONE 2015, 10, e0116841. [Google Scholar] [CrossRef]
- El-Agnaf, O.; Overk, C.; Rockenstein, E.; Mante, M.; Florio, J.; Adame, A.; Vaikath, N.; Majbour, N.; Lee, S.-J.; Kim, C.; et al. Differential effects of immunotherapy with antibodies targeting α-synuclein oligomers and fibrils in a transgenic model of synucleinopathy. Neurobiol. Dis. 2017, 104, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Spencer, B.; Valera, E.; Rockenstein, E.; Overk, C.; Mante, M.; Adame, A.; Zago, W.; Seubert, P.; Barbour, R.; Schenk, D.; et al. Anti-α-synuclein immunotherapy reduces α-synuclein propagation in the axon and degeneration in a combined viral vector and transgenic model of synucleinopathy. Acta Neuropathol. Commun. 2017, 5, 7. [Google Scholar] [CrossRef] [Green Version]
- Kallab, M.; Herrera-Vaquero, M.; Johannesson, M.; Eriksson, F.; Sigvardson, J.; Poewe, W.; Wenning, G.K.; Nordström, E.; Stefanova, N. Region-Specific Effects of Immunotherapy with Antibodies Targeting α-synuclein in a Transgenic Model of Synucleinopathy. Front. Neurosci. 2018, 12, 452. [Google Scholar] [CrossRef] [Green Version]
- Schofield, D.J.; Irving, L.; Calo, L.; Bogstedt, A.; Rees, G.; Nuccitelli, A.; Narwal, R.; Petrone, M.; Roberts, J.; Brown, L.; et al. Preclinical development of a high affinity α-synuclein antibody, MEDI1341, that can enter the brain, sequester extracellular α-synuclein and attenuate α-synuclein spreading in vivo. Neurobiol. Dis. 2019, 132, 104582. [Google Scholar] [CrossRef]
- Huang, Y.-R.; Xie, X.-X.; Ji, M.; Yu, X.-L.; Zhu, J.; Zhang, L.-X.; Liu, X.-G.; Wei, C.; Li, G.; Liu, R.-T. Naturally occurring autoantibodies against α-synuclein rescues memory and motor deficits and attenuates α-synuclein pathology in mouse model of Parkinson’s disease. Neurobiol. Dis. 2019, 124, 202–217. [Google Scholar] [CrossRef]
- Henderson, M.X.; Covell, D.J.; Chung, C.H.-Y.; Pitkin, R.M.; Sandler, R.M.; Decker, S.C.; Riddle, D.M.; Zhang, B.; Gathagan, R.J.; James, M.J.; et al. Characterization of novel conformation-selective α-synuclein antibodies as potential immunotherapeutic agents for Parkinson’s disease. Neurobiol. Dis. 2020, 136, 104712. [Google Scholar] [CrossRef]
- Weihofen, A.; Liu, Y.; Arndt, J.W.; Huy, C.; Quan, C.; Smith, B.A.; Baeriswyl, J.-L.; Cavegn, N.; Senn, L.; Su, L.; et al. Development of an aggregate-selective, human-derived α-synuclein antibody BIIB054 that ameliorates disease phenotypes in Parkinson’s disease models. Neurobiol. Dis. 2019, 124, 276–288. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Wu, K.-J.; Hsieh, W.; Harvey, B.K.; Hoffer, B.J.; Wang, Y.; Yu, S.-J. Administration of AAV-Alpha Synuclein NAC Antibody Improves Locomotor Behavior in Rats Overexpressing Alpha Synuclein. Genes 2021, 12, 948. [Google Scholar] [CrossRef]
- Nordström, E.; Eriksson, F.; Sigvardson, J.; Johannesson, M.; Kasrayan, A.; Jones-Kostalla, M.; Appelkvist, P.; Söderberg, L.; Nygren, P.; Blom, M.; et al. ABBV-0805, a novel antibody selective for soluble aggregated α-synuclein, prolongs lifespan and prevents buildup of α-synuclein pathology in mouse models of Parkinson’s disease. Neurobiol. Dis. 2021, 161, 105543. [Google Scholar] [CrossRef]
- Masliah, E.; Rockenstein, E.; Adame, A.; Alford, M.; Crews, L.; Hashimoto, M.; Seubert, P.; Lee, M.; Goldstein, J.; Chilcote, T.; et al. Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron 2005, 46, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Gorbatyuk, O.S.; Li, S.; Nash, K.; Gorbatyuk, M.; Lewin, A.S.; Sullivan, L.F.; Mandel, R.J.; Chen, W.; Meyers, C.; Manfredsson, F.P.; et al. In vivo RNAi-mediated alpha-synuclein silencing induces nigrostriatal degeneration. Mol. Ther. 2010, 18, 1450–1457. [Google Scholar] [CrossRef]
- Kahle, P.J.; Neumann, M.; Ozmen, L.; Muller, V.; Jacobsen, H.; Schindzielorz, A.; Okochi, M.; Leimer, U.; van Der Putten, H.; Probst, A.; et al. Subcellular localization of wild-type and Parkinson’s disease-associated mutant alpha -synuclein in human and transgenic mouse brain. J. Neurosci. 2000, 20, 6365–6373. [Google Scholar] [CrossRef] [Green Version]
- Kuo, Y.-M.; Li, Z.; Jiao, Y.; Gaborit, N.; Pani, A.K.; Orrison, B.M.; Bruneau, B.G.; Giasson, B.I.; Smeyne, R.J.; Gershon, M.D.; et al. Extensive enteric nervous system abnormalities in mice transgenic for artificial chromosomes containing Parkinson disease-associated alpha-synuclein gene mutations precede central nervous system changes. Hum. Mol. Genet. 2010, 19, 1633–1650. [Google Scholar] [CrossRef]
- Ghosh, S.S.; Wang, J.; Yannie, P.J.; Ghosh, S. Intestinal Barrier Dysfunction, LPS Translocation, and Disease Development. J. Endocr. Soc. 2020, 4, bvz039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van IJzendoorn, S.C.D.; Derkinderen, P. The Intestinal Barrier in Parkinson’s Disease: Current State of Knowledge. J. Parkinsons. Dis. 2019, 9, S323–S329. [Google Scholar] [CrossRef] [Green Version]
- Boertien, J.M.; Pereira, P.A.B.; Aho, V.T.E.; Scheperjans, F. Increasing Comparability and Utility of Gut Microbiome Studies in Parkinson’s Disease: A Systematic Review. J. Parkinsons. Dis. 2019, 9, S297–S312. [Google Scholar] [CrossRef] [Green Version]
- Brudek, T. Inflammatory Bowel Diseases and Parkinson’s Disease. J. Parkinsons. Dis. 2019, 9, S331–S344. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-S.; Lobbestael, E.; Vermeire, S.; Sabino, J.; Cleynen, I. Inflammatory bowel disease and Parkinson’s disease: Common pathophysiological links. Gut 2021, 70, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Knudsen, K.; Haase, A.-M.; Fedorova, T.D.; Bekker, A.C.; Østergaard, K.; Krogh, K.; Borghammer, P. Gastrointestinal Transit Time in Parkinson’s Disease Using a Magnetic Tracking System. J. Parkinsons. Dis. 2017, 7, 471–479. [Google Scholar] [CrossRef]
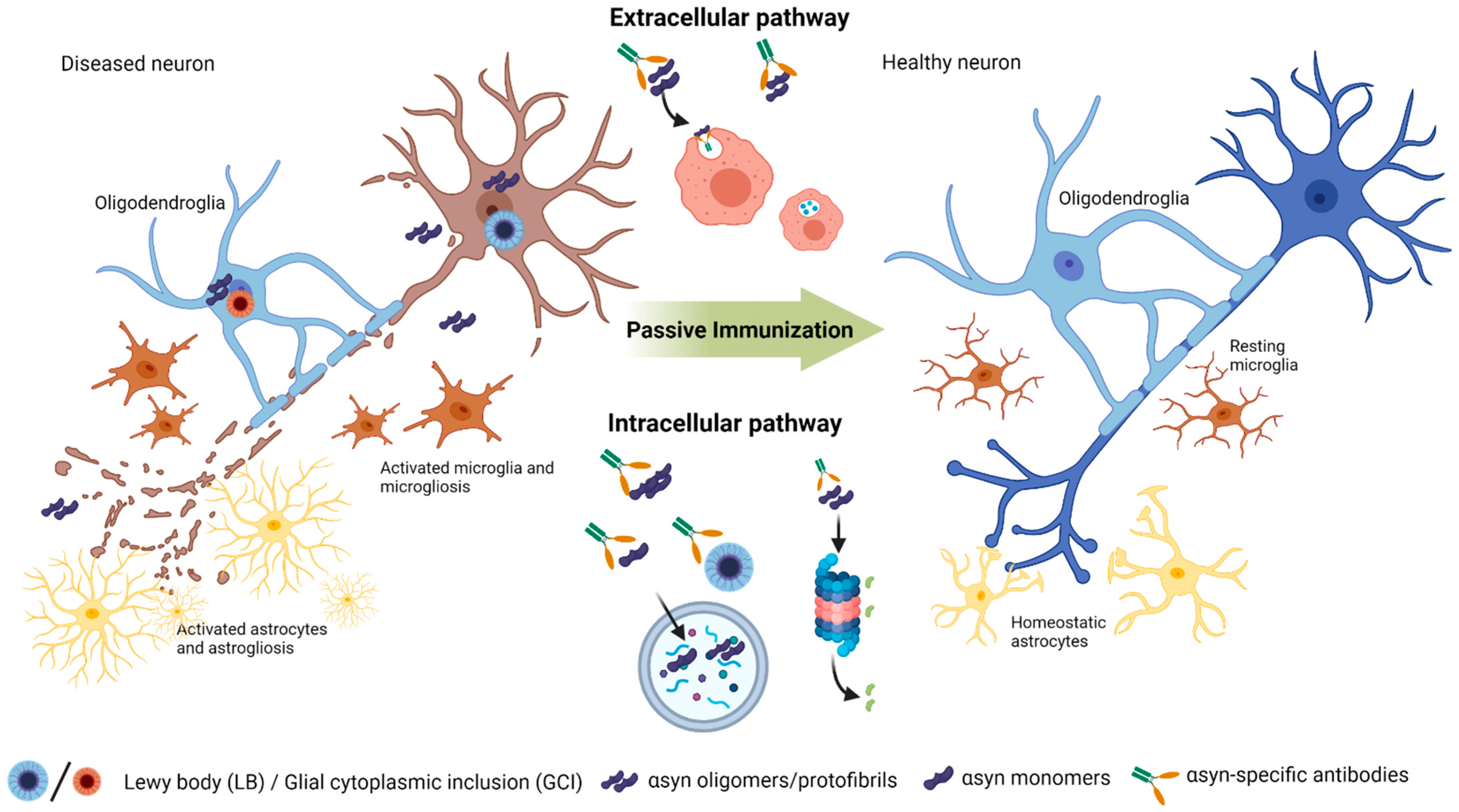

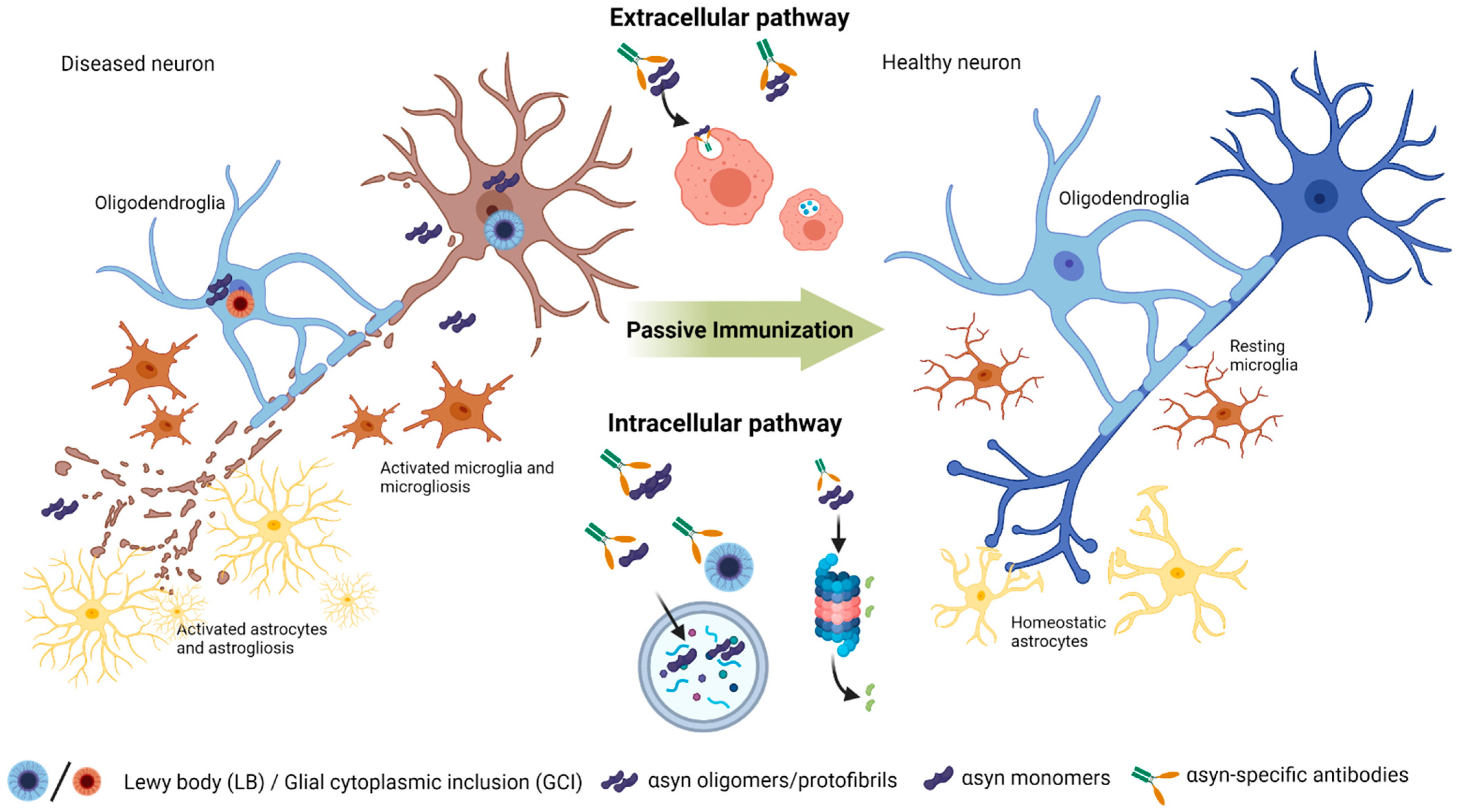{kind=link}
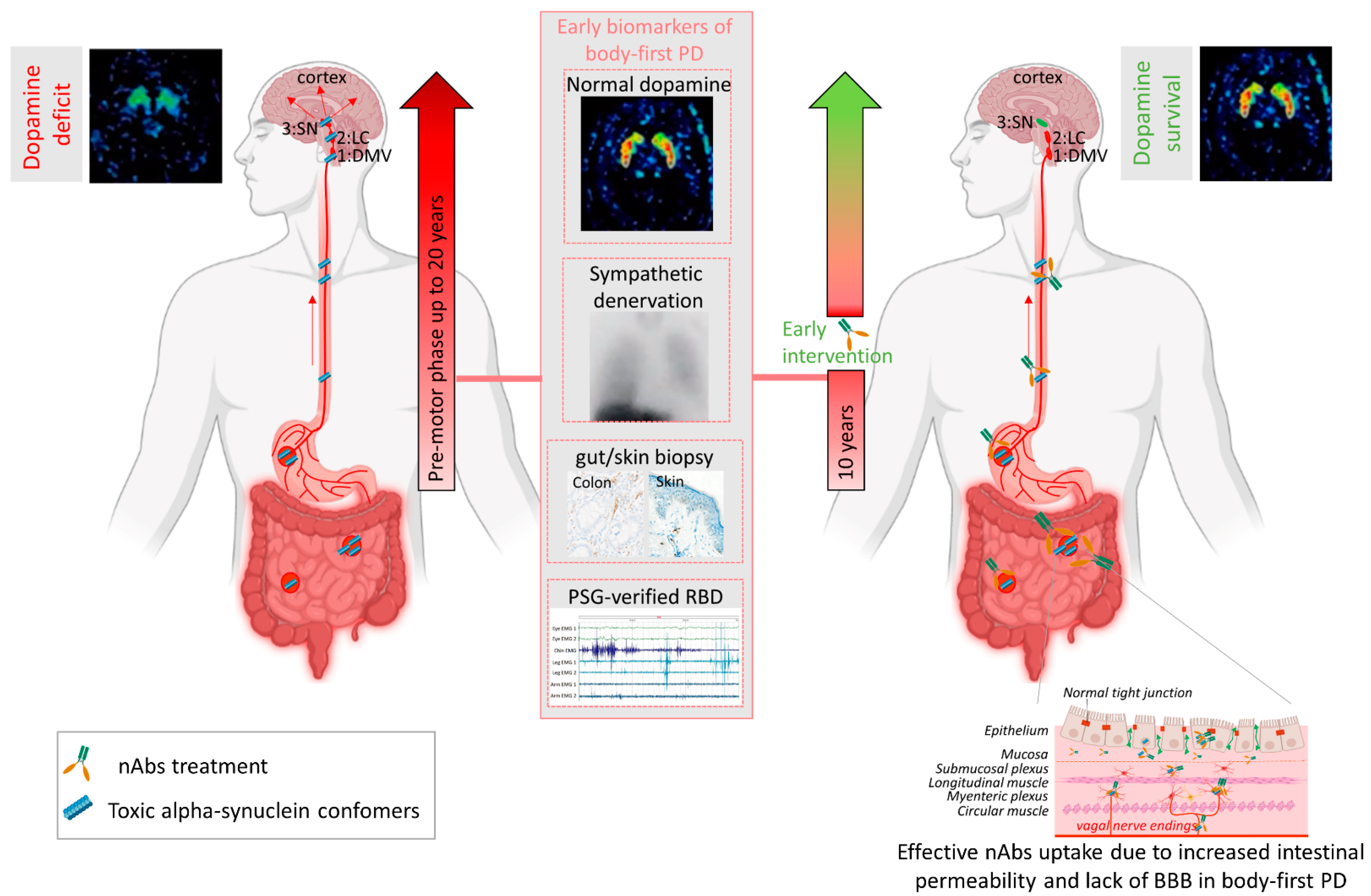
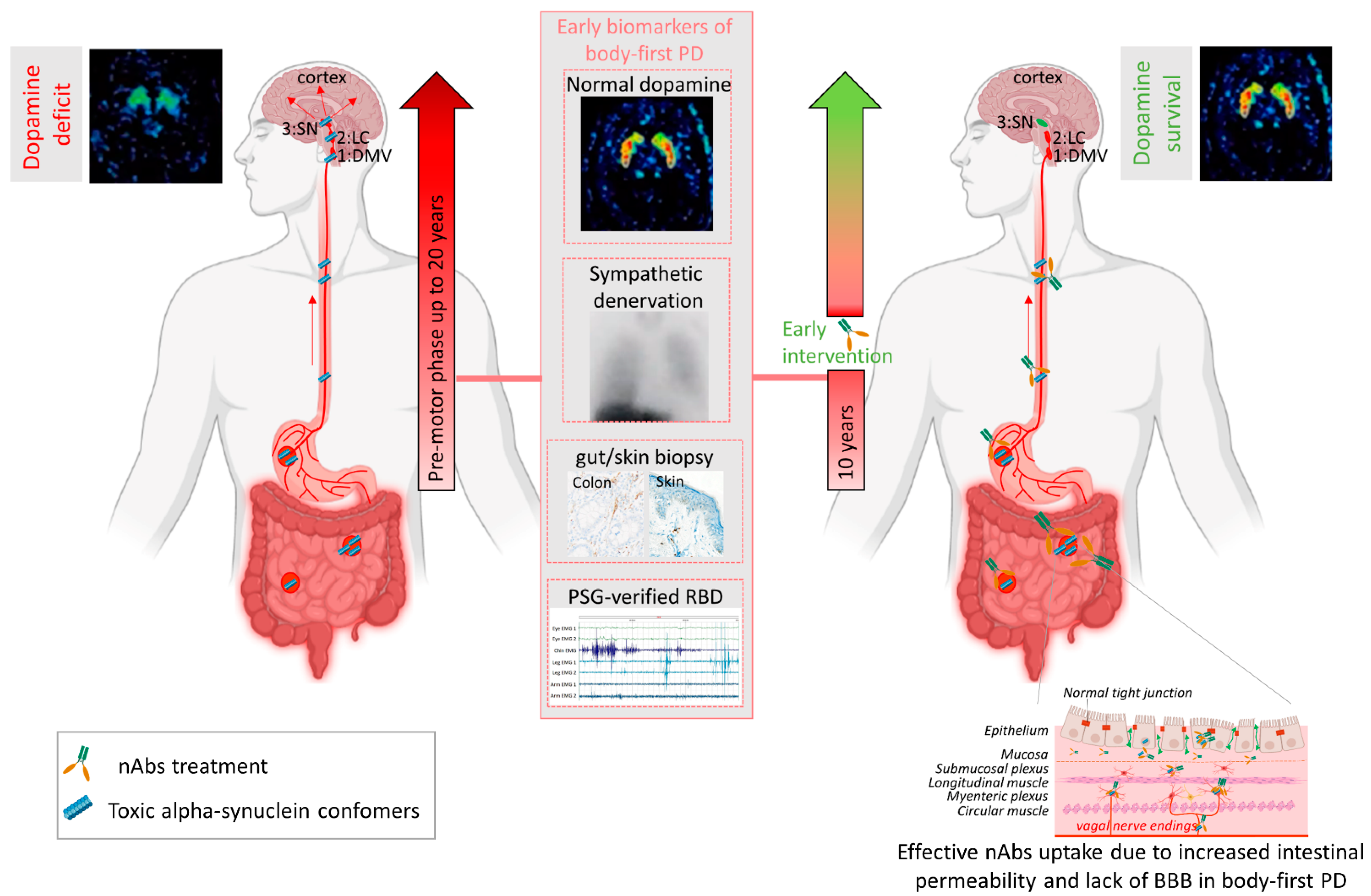{kind=link}
| Target (αsyn) | Antibody/Clone | Binding Site (aa) | Ab Origin Immunization Method | Injection Frequency Duration Amount | Animal Model | αsyn Pathological Effects | Neuronal Effects | Other Non-Neuronal Effects | Behavioral Effects | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| C-term. | 9E4 (IgG1) | C-term. 118–126 | human Full-length (FL) αsyn (h-αsyn) | i.p., weekly 6 m 10 mg/kg b.w. | PD/DLB: PDGFb αsyn mice (line D) | ↓ FL αsyn in neocortex neuropils; ↓ Reduced CC-αsyn in neocortex (intraneuronal and neuropil), Hippocampus (intraneuronal/neuropil); ↓ Reduced insoluble-FL-αsyn oligo.; ↓ soluble-CC-αsyn mono./oligo.; ↓ insoluble-CC-αsyn mono./oligo. | ↑ synaptic densities; ↑ Pre-synaptic terminals; ↑ PSD95; ↑ Synapsin. | ↓ Astrogliosis in PFC. | ↑ Rotarod time; ↓ Path in Morris water maze. | [106] |
| C-term. | Ab274 (IgG2a) | C-term. 120–140 | h-αsyn | i.p., weekly 4 wks 1 mg/mL hippocampal injection | PD: PDGFb αsyn mice (line M) | ↓ αsyn (70–80% in cortex and hippocampus; ↓ αsyn (30–35% in striatum); ↓ αsyn in brain homogenates; ↓ αsyn in neurons and glial cells (total) (neocortex, Hippocampus, Striatum); ↑ Cathepsin-D and αsyn coloc.; ↑ αsyn clearance by microglia. | ↓ NeuN cell loss; ↓ NeuN cell loss (hipp); ↑ increased synaptophysin (Hippocampus). | ↓ TNF-a and IL-6; ↑ Iba-1 in hippocampus; ↓ Astrogliosis. | ↓ latency to turn (Pole test); ↓ Total activity in open field. | [107] |
| NAC to C-term. | 1H7 (IgG1) | 91–99 (NAC to C-terminal) | FL h-αsyn | i.p., weekly 6 m 10 mg/kg b.w. | PD: Thy1 αsyn (line 61) mice | ↓ αsyn and αsyn aggre. (temporal and striatal neuropil) ↓ axonal αsyn (striatum) | ↓ TH loss in striatum; ↑ synapto-physin + MAP2 (neocortex and striatum) | ↓ Astrogliosis ↓ Microgliosis | ↓ Memory and learning deficits; ↓ error on transversal beam) | [18] |
| 5C1 (IgG1) (9E4 analog) | C-term. 118–126 | GCC-VDPDNEAYE peptide | ↓ αsyn and αsyn aggr. (temporal and striatal neuropil) ↓ axonal as αsyn yn (striatum) | ↓ TH loss in striatum; ↑ synaptophysin + MAP2 (neocortex and striatum | ↓ Astrogliosis ↓ Microgliosis | ↓ Memory and learning deficits; | ||||
| 5D12 (IgG1) (9E4 analog) | C-term. 118–126 | VDPDNEAYE-GCC peptide | ↓ αsyn (neocortex) | - | - | - | ||||
| 9E4 | C-term. 118–126 | FL h-αsyn | ↓ αsyn and αsyn aggre. (temporal and striatal neuropil) ↓ axonal αsyn (striatum) | ↓ TH loss in striatum; ↑ synaptophysin + MAP2 (neocortex and striatum | ↓ Astrogliosis ↓ Microgliosis | ↓ Memory and learning deficits; ↓ error on transversal beam) | ||||
| N/C-term. | Syn303 | N-term. 1–5 | human phos./nitr. αsyn | i.p., weekly 180 days 30 mg/kg b. w. | PD: Intra-striatal injection of PFF in wt mice | ↓ insoluble αsyn aggre. and pS129-αsyn; ↓ Reduced αsyn spread in SNc (30%) and contra- and ipsilateral amygdala (40%). | ↓ neuron loss; ↓ PFF neuron entry and PFF transmission; ↓ TH cell loss. | - | ↑ latency to hang (Wirehang time) | [108] |
| Syn211 | C-term. 121–125 (mono./oligo./Fibrils) | h-αsyn positive for DNEAY-peptide | ↓ insoluble αsyn aggre. and pS129-αsyn | ↓ neuron loss; ↓ PFF neuron entry and PFF transmission. | - | - | ||||
| Proto-fibril | mAB47 (IgG1) | Conformational | h-αsyn oligomers (hybridoma) | i.p., weekly 14 wks 10 mg/kg b.w. | PD: Thy-1 H[A30P] mice | ↓ αsyn protofibrils in spinal cord | - | - | - | [109] |
| N/C-term. | AB1 | N-term. 16–35 | αsyn peptide (16–35aa) | i.p., 14 days 3 m, 1 mg/rat (2x first), then 0.5 mg/mL | PD: Nigral AVV-CBA-αsyn in wt rats | ↓ αsyn in SN | ↓ DA and NeuN cell loss. | ↓ Microgliosis | - | [110] |
| AB2 | C-term. 93–115 | αsyn peptide (93–115aa) | ↓ αsyn brain homogenate | - | ↓ Microgliosis | - | ||||
| Oligo and late aggre. | Syn-01 | Conformational (Oligo./aggre.) | αsyn -> hybridomas | i.p., weekly 3 m 30 mg/kg b.w. | PD/DLB: mThy1 αsyn (Line 61) Mice | ↓ αsyn (neocortex, hippocampus, striatum, SN); ↓ PK-resistant αsyn (neocortex, hippocampus, striatum); ↓ oligomeric αsyn; ↓ 5G4 aggregated αsyn | ↓ NeuN hippocampal loss (CA3); ↑ Synapsin I/Synaptophysin ratio;↓ αsyn/synaptophysin ratio | ↓ Astrogliosis; ↓ Microgliosis | ↓ beam breaks (total activity) | [111] |
| Syn-02 | ↓ αsyn (striatum);↓ PK-resistant αsyn (hippocampus, striatum); ↓ total αsyn; ↓ oligo. αsyn;↓ 5G4 aggregated αsyn. | - | - | - | ||||||
| Syn-04 | ↓ αsyn (neocortex, hippocampus, Striatum, SN);↓ PK-resistant αsyn (neocortex, hippocampus, striatum); ↓ total αsyn; ↓ oligomeric αsyn; ↓ 5G4 aggregated αsyn | ↓ NeuN hippocampal loss (CA3); ↑ Synapsin I/Synaptophysin ratio;↓ αsyn/synaptophysin ratio | ↓ Astrogliosis; ↓ Microgliosis | ↓ beam breaks (total activity) | ||||||
| Syn-F1 | Conformational (late aggre.) | ↓ αsyn (neocortex, hippocampus, striatum, SN); ↓ PK-resistant αsyn (hippocampus); ↓ oligomeric αsyn. | ↓ NeuN hippocampal loss (CA3); ↑ Synapsin I/Synaptophysin ratio;↓ αsyn/synaptophysin ratio | - | ↓ beam breaks (total activity) | |||||
| Syn-F2 | ↓ αsyn (neocortex, striatum, SN); ↓ PK-resistant αsyn (hippocampus); ↓ oligomeric αsyn. | ↓ αsyn/synaptophysin ratio | - | - | ||||||
| Aggre. | 1H7 | C-term. 91–99 | FL h-αsyn | i.p., weekly 3 m 30 mg/kg b.w. | PD: mThy1 αsyn (61) mice, intra-hippocampal inj. of LV-αsyn | ↓ axonal αsyn ↑ coloc. of αsyn and microglia | ↑ axonal integrity | - | ↓ water maze time to localization | [112] |
| Oligo | Rec47 (mAB47 as in [109]) | Conformational, Binding to C-terminal 121–127 | h-αsyn oligomers (hybridoma) | i.p., bi-weekly 3 m 20 mg/kg b.w. | MSA: PLP αsyn transgenic mice | ↓ soluble and insoluble αsyn (hippocampus) ↓ GCI’s in spinal cord; ↑ pS129 αsyn (SNpc, pontine nuclei and inferior olives) ↑ Co-localization of LCS (autophagy) and p-S129 αsyn. | ↓ Microgliosis; ↓ activated MG; ↑ Iba-1 and olig-αsyn co-localization | - | [113] | |
| Aggre. | MEDI1341 (IgG1) | C-term. | Human phage library cloned into IgG1 | i.p., weekly 13 wks 20 mg/kg b.w. | PD: mThy1 αsyn (Line 61) mice – intra-hippocampal injection of LV-αsyn | ↓ contralateral and ipsilateral αsyn (hippocampus); ↓ contralateral axonal αsyn ↓ αsyn (neocortex) ↓ interstitial fluid αsyn levels ↓ CSF fluid αsyn levels ↓ αsyn positive neurons (neocortex and hippocampus) | - | - | - | [114] |
| Mono. and Oligo. | nAb isolated from IViG | nAbs isolated from IViG using αsyn column chromatography | s.c., weekly 4 wks Low dosage: 0.8 mg/kg b.w. | PD: A53T tg mice | ↓ pS129-αsyn (brainstem) ↓ soluble αsyn (brainstem) | ↓ Astrogliosis(Striatum);↑ Microglia and αsyn coloc. | ↓ Pole test (time to descend/time to turn). | [115] | ||
| s.c., weekly 4 wks High dosage: 2.4 mg/kg b.w. | ↓ pS129-αsyn (brainstem and neocortex) ↓ soluble αsyn (brainstem) ↓ Reduced total soluble and insoluble h-αsyn (brainstem); ↓ fibrillary-oligo. αsyn; ↓ pS129-αsyn/NfL ratio. ↑ Microglia and αsyn co-localization. | ↑ PSD95 (brainstem), ↑ synaptophysin (brainstem); ↓ TH cell loss (striatum, brainstem) | ↓ astrogliosis(striatum) ↓ microgliosis(striatum); ↓ MCP-1(brainstem). | ↓ Pole test (time to descend/time to turn); ↑ Body suspension test (hanging); ↑ Y maze (duration in new arm/new entries). | ||||||
| Aggre. | Syn9048 (IgG1) | C-term. | hybridoma | i.p., weekly 6 m 30 mg/kg | PD: wt + αsyn PFF (5µg) unilateral inj. in dorsal striatum | ↓ αsyn ipsilateral SN; ↓ Contralateral amygdala. | ↓ DA cell loss; ↑ DOPAC | - | - | [116] |
| N-term. | Syn303 ([108]) | N-term. 1–5 | phos./nitr. h- αsyn | - | ↓ TH cell loss (ipsilateral) | - | - | |||
| Aggre. | BIIB054/cinpanemab | N-term: 1–10 (800-fold greater affinity to aggregated αsyn) | Healthy human memory B cells -> clones | i.p., weekly 60, 90 or 100 days 30 mg/kg b.w. | PD: wt C57BL/6JRccHsd mice + αsyn PFF intrastrial inj. | ↓ truncated αsyn 6kd (100d) | - | - | ↑ Hangwire (latency to fall, 60d) | [117] |
| PD: Tg αsyn A53T (M83) + αsyn PFF inoc. | - | - | - | ↓ paralysis (7 d) ↓ severe paralysis (5 d); ↓ weight loss (9 d). | ||||||
| PD: BAC αsyn A53T + αsyn PFF intrastrial inj. | - | ↑ contralateral DAT levels (striatum, 90d) | - | - | ||||||
| NAC-region | NAC32 | 53–87 | Yeast surface display library of an entire naïve repertoire of human scFV antibodies | Stereotaxis (AAV-NAC32) post 12 wks after αsyn inj. Beh. 4,8 and 12 wks after NAC32 inj. | PD: DAT-Cre rats + AAV-DIO- αsyn in SNpc. | ↓ αsyn (25%) (SNpc dorsal). | ↓ TH cell loss (SNpc dorsal) | - | ↓ Horizontal activity; ↓ Total distance travelled; ↓ Movement number; ↓ Movement time; ↑ Rest time; ↓ Vertical activity | [118] |
| Aggre. (Oligo/Proto-fibrils) | ABBV-0805/mAB47 for murine experiments | Humanized mAB47, binding to C-term. 121–127 | h-αsyn oligo. ->hybridoma, same as prior | i.v., bolus, starting at 2 m old, sampled multiple times. 0.1, 1, 10 mg/kg | wt C57BL/6 mice (pharmacokinetics) | - | 0.3% in the brain dose-dependent plasma content | - | - | [119] |
| i.p., weekly starting at age 12 m, 10mg/kg | PD: Thy-1-h[A30P] αsyn tg mice | - | - | - | ↑ Mean survival from 84 days to 160 days | |||||
| i.p., weekly starting at age 12 m, 20 mg/kg | PD: Thy-1-h[A30P] αsyn tg + 10 µg gastrocnemius i.m PFF inj., after mab treatment | - | - | - | ↑ Mean survival from 84 days to 95 days | |||||
| Starting 4 wks prior to PFF inj.; weekly mab inj. Prophylactic: 2–4 m, until severe motor deficits, 20 mg/kg | PD: Thy-1-h[A30P] αsyn tg + gastrocnemius PFF inj. 1 µg i.m. | ↓ soluble and insoluble αsyn (brain); ↓ insoluble pS129-αsyn; ↓ CSF pS129-αsyn; ↓ LB-509 αsyn inclusions (reticular nucleus); ↓ pS129-αsyn inclusion (midbrain). | - | - | - | |||||
| Post 2 wks after PFF inj.; weekly mab inj. Therapeutic: 2–4 m. until severe motor deficits, 20 mg/kg | PD: Thy-1-h[A30P] αsyn tg + gastrocnemius PFF inj. 1 µg i.m. | ↓ soluble and insoluble αsyn (brain); ↓ insoluble pS129-αsyn ↓ CSF pS129-αsyn; ↓ Dose-dependent soluble and insoluble αsyn (brain); ↓ soluble αsyn at low mab administration (0.25 mg/kg); ↓ insoluble αsyn (5 mg/kg); ↓ LB-509 αsyn inclusions in (reticular nucleus); ↓ pS129-αsyn inclusion (midbrain). | - | - | - | |||||
| weekly 16 wks 20 mg/kg | PD: A53T+/− mice (83) + i.c. (anterior olfactory nucleus) PFF inj. | ↓ pS129- αsyn pathology spreading to the contralateral hippocampus (CA1) (58%). | - | - | - |
| Target (αsyn) | Name | Companies | Antibody/Clone | Binding Site (aa) | Clinical Groups | Current Clinical Phase | Clinical Trial ID |
|---|---|---|---|---|---|---|---|
| Aggre. | PRX002/(Prasinezumab)–PASADENA study | Hoffman-La Roche; Prothena Biosciences Limited. | Humanized IgG1 mab version of murine 9E4 | Preferable aggregated αsyn within the C-terminal at aa 118–126 (VDPDNEAYE) | PD patients (H&Y < 2) | Phase II; active; recruitment completed. | NCT03100149 |
| Aggre. (Oligo/proto-fibrils) | ABBV-0805 | AbbVie; BioArctic Neuroscience AB | Humanized mAB47 mab | Preferable aggregated αsyn within the C-terminal at aa 121–127 (DNEAYEM) | PD patients (<5 years from diagnosis and H&Y < 3) | Phase I; recruiting. | NCT04127695 |
| Aggre. | MEDI1341 | Astra Zeneca; Takeda Pharmaceuticals | Humanized IgG1 mab | Preferable aggregated αsyn within the C-terminal (within the aa 103–129 region) | Healthy individuals (MEDI1341 vs. placebo) | Phase I; recruitment completed. | NCT03272165 |
| Aggre. | BIIB054 (Cinpanemab)–SPARK study | Biogen; Neuroimmune | Healthy human memory B cells derived mab | Preferable aggregated αsyn, oxidized at N-terminal aa: 4–10 (FMKGLSK) | PD patients (<3 years from diagnosis and H&Y < 2.5) | Phase II; Terminated | NCT03318523 |
| Aggre. | Lu AF82422–AMULET study | H. Lundbeck A/S; Genmab A/S | Humanized IgG1 mab | Preferable aggregated αsyn within the C-terminal at aa 112–117 (ILEDMP) | MSA-P and MSA-C patients (<5 years from diagnosis, UMSARS ≤ 16, MoCA ≥ 22) | Phase II; recruiting | NCT05104476 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Folke, J.; Ferreira, N.; Brudek, T.; Borghammer, P.; Van Den Berge, N. Passive Immunization in Alpha-Synuclein Preclinical Animal Models. Biomolecules 2022, 12, 168. https://doi.org/10.3390/biom12020168
Folke J, Ferreira N, Brudek T, Borghammer P, Van Den Berge N. Passive Immunization in Alpha-Synuclein Preclinical Animal Models. Biomolecules. 2022; 12(2):168. https://doi.org/10.3390/biom12020168
Chicago/Turabian StyleFolke, Jonas, Nelson Ferreira, Tomasz Brudek, Per Borghammer, and Nathalie Van Den Berge. 2022. "Passive Immunization in Alpha-Synuclein Preclinical Animal Models" Biomolecules 12, no. 2: 168. https://doi.org/10.3390/biom12020168
APA StyleFolke, J., Ferreira, N., Brudek, T., Borghammer, P., & Van Den Berge, N. (2022). Passive Immunization in Alpha-Synuclein Preclinical Animal Models. Biomolecules, 12(2), 168. https://doi.org/10.3390/biom12020168