
The Urokinase Plasminogen Activation System in Pancreatic Cancer: Prospective Diagnostic and Therapeutic Targets


Abstract
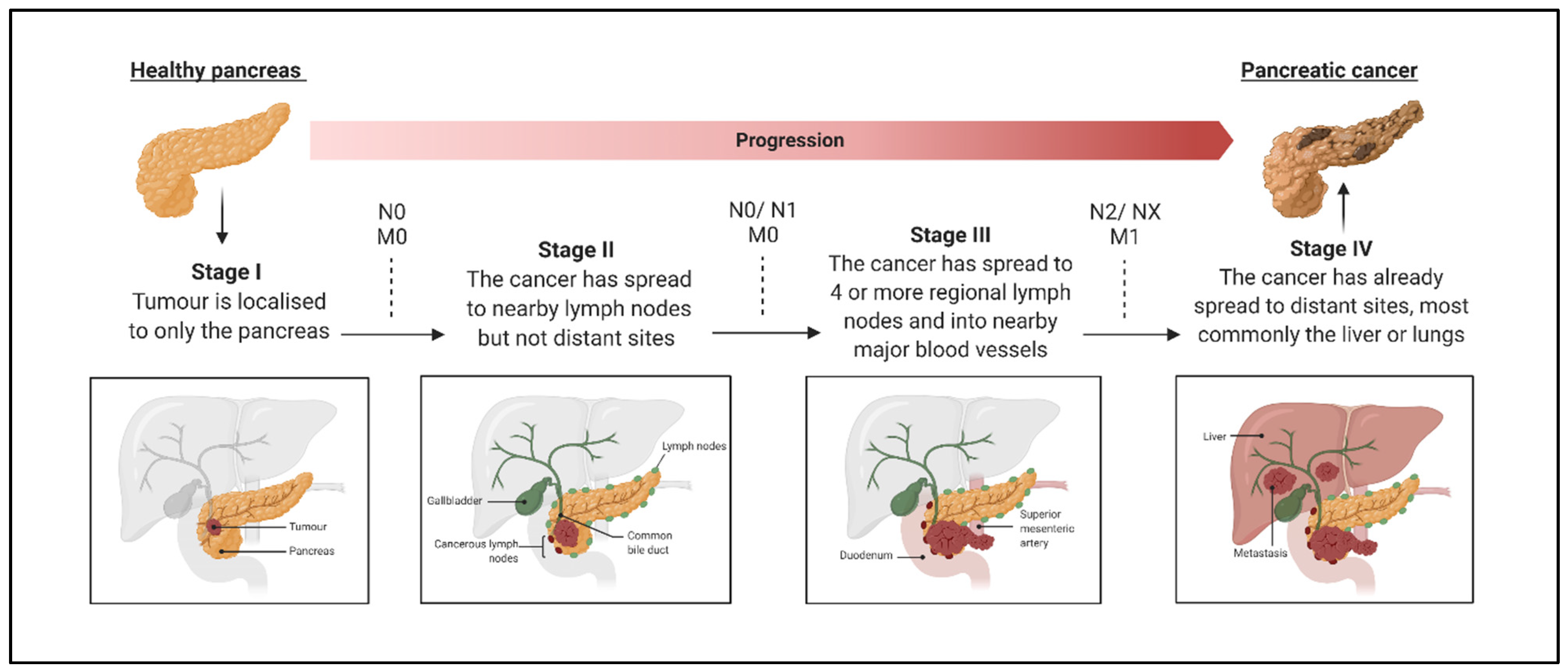
1. Introduction
2. The Plasminogen Activator System and Clinical Evidence for Its Role in PDAC
An Overview of the Urokinase Plasminogen Activator System

3. Clinical Evidence Supporting uPAS Overexpression in Pancreatic Cancer

{kind=link}
{kind=link}
{kind=link}
| Marker | Source | n * | Method | Findings | References |
|---|---|---|---|---|---|
| tPA, uPA, PAI-1, PAI-2 | Tumour | 97 | IHC | Elevated (↑) uPA in 76 samples (78.4%), ↑ tPA in 8 samples (8.2%), ↑ PAI-1 in 80 samples (82.5%), ↑ PAI-2 in 79 samples (81.4%) relative to healthy controls. ↑ PAI-2 associated with improved survival (p < 0.05). | [119] |
| uPA, uPAR | Tumour | 30 | IHC, Northern blot analysis | uPA ↑ 6-fold, uPAR ↑ 4-fold, relative to healthy controls. ↑ uPA and ↑ uPAR, together, associated with reduced (↓) median postoperative survival (median 9 months) compared with non-expression or singular expression of uPA or uPAR (median 18 months) (p < 0.006). | [33] |
| uPA, uPAR, MMP-9 | Tumour | 27 | IHC, ISH | ↑ uPA 93% of PDAC tissue. ↑ uPA, ↑ uPAR or ↑ MMP-9 associated with ↓ OS versus non-expression. ↑ uPA mRNA present in cytoplasm of tumour cells and adjacent pancreatic ducts. | [120] |
| uPAR | Tumour | 137 | IHC | 66% (n = 81) of patients with PDAC showed ↑ uPAR in neoplastic cells, 82% (n = 100) in tumour-associated stromal cells and 62% (n = 75) in both cell types (p < 0.001). ↑ uPAR in stromal cells associated with development of liver metastases. ↑ uPAR associated with ↓ OS and DFS. | [121] |
| uPA/PLAU, PAI-2/SERPINB2 | - | 109 | Genomic analysis | Frequent deletion of gene SERPINB2 in PDAC cohorts. ↑ uPA mRNA expression (PLAU) associated with ↓ DFS in PDAC resected patients (p = 0.00019) | [122] |
| uPA, uPAR | Tumour | 50 | IHC, ELISA, in situ hybridisation, PCR | ↑ uPA in 48 of 50 (96%) invasive PDAC tumour samples. Amplification of uPAR gene is an adverse prognostic parameter compared with cases with no detectable amplifications. ↑ uPA associated with ↑ proliferation and ↓ apoptosis. | [35] |
| uPA | Tumour | 21 | RT-qPCR, IHC | ↑ uPA in 71% of PDAC samples, ↑ 9-fold relative to benign tumours (p = 0.002). All PDAC sections showed grade 2–3 immunostaining for uPA antibody vs. no staining in negative control sections or normal pancreas. ↑ uPA associated with degraded ECM and poor tissue morphology. ↑ uPA associated with ↑ tumour stage (↑ 75-fold in stage III PDAC relative to normal pancreatic tissue). | [123] |
| uPA, uPAR, PAI-1, PAI-2 | Tumour | 46 | RT-qPCR, IHC | ↑ uPA (p = 0.004) and uPAR (p = 0.025) in PC tissue relative to adjacent uninvolved pancreatic tissue. ↑ uPA, uPAR, and PAI-1 in PC tissue independently correlates to a ↑ UICC stage (p < 0.001). ↑ uPAR correlates to ↓ survival following surgery (p = 0.03). ↑ PAI-2 (46%) associated with ↑ survival (p < 0.007) and ↓ tumour size (p = 0.008) | [112] |
| uPAR, suPAR | Serum and Tumour | 127 | ELISA | ↑ uPAR in tumour tissue and ↑ circulating levels of suPAR in PDAC patients relative to healthy controls. ↑ suPAR levels associated with ↑ risk for acute kidney injury and surgical complications post-resection. ↑ pre-operative suPAR serum levels >5.956 × 10−6 g/L associated with ↓ patient OS of 231 days following resection vs. 756 days for patients with suPAR serum levels <5.956 × 10−6 g/L (p = 0.001). Postoperative suPAR serum levels are unsuitable for the prediction of OS. | [124] |
| uPA, uPAR, PAI-1, PAI-2 | Tumour | 46 | RT-qPCR | uPA ↑ 7.6-fold, uPAR ↑ 9.6-fold and PAI-1 ↑ 3.3-fold in PDAC tissue relative to adjacent uninvolved pancreatic tissue. From 15 genes from 3 gene families, PAI-2 was an independent prognostic marker for improved survival for patients with PC (p = 0.006), more significant than UICC stage (p = 0.012) | [125] |
| uPA | Serum | 40 | ELISA | uPA ↑ 3-fold in PDAC patients compared to control group (p < 0.01). Positive correlation between uPA serum level and CA19-9 (p < 0.05). ↑ uPA serum concentration associated with ↓ survival time (p < 0.05) | [32] |
| uPA, MMP-1, uPAR | Tumour | 25 | Gene ontology, RT-qPCR | ↑ uPA, ↑ MMP-1 and ↑ IL1-R1 in human pancreatic tumours. ↑ MMP-1 expression associated with ↑ PDAC tumour stage. | [134] |
| suPAR | Serum | 25 | ELISA | ↑ plasma suPAR in PC patients (median 3.7 × 10−6 g/L) relative to CP patients (2.6 × 10−6 g/L) (p = 0.014). Plasma suPAR cut-off value of 2.8 × 10−6 g/L (p = 0.009) determined for differentiation between PC and CP with a sensitivity and a specificity of 88% and 70%, respectively. | [136] |
| tPA | Tumour | 35 | ELISA | ↑ tPA in PDAC tumour homogenates relative to both CP and benign pancreatic tumour homogenates; tissue homogenate tPA levels ↑ 7.45 ng/mL indicative of PDAC | [137] |
| suPAR | Urine | 94 | ELISA | ↑ suPAR/creatinine in PDAC patients (median 9.8 ng/mg) relative to patients with CP (median 2.7 ng/mg) and healthy controls (median 0 ng/mg). ↑ suPAR positively associated with tumour stage (stage III p = 0.0017; stage IV p < 0.0001) and ↓ survival among all PDAC patients (p = 0.0023). | [138] |
| uPA, uPAR | Tumour | 101 | IHC | ↑ uPAR and ↑ uPA in PDAC tumours, with co-localization present in most tissues. | [34] |
| PAI-1 | Tumour | 93 | IHC | ↑ PAI-1 in tumour tissue relative to healthy tissue. ↑ PAI-1 positively associated with tumour stage and poor prognosis. | [139] |
| uPA, uPAR, MMP-2, -9 | Tumour | 20 | IHC | ↑ uPA in 85% of PC tissues. ↑ uPA, ↑ fibroblastic uPAR expression associated with liver metastases (p = 0.001). ↑ MMP-2 expression in all PC tissue. | [140] |
| uPA, uPAR | Tumour | 70 | IHC | ↑ uPA, ↑ uPAR in primary pancreatic tumour specimens from patients with lymph node and/or distant metastases relative to patients without metastases (p < 0.05). | [141] |
| uPA, uPAR, plasmin(ogen) | Tumour | 37 | IHC, ELISA | ↑ uPA, ↑ uPAR and ↑ plasmin(ogen) expression in malignant PC tissue versus non-malignant tissue. ↑ uPAR and ↑ plasmin(ogen) at the invasive front of PC tissue relative to the centre of the same PC tissue. | [142] |
| uPA | Tumour | 30 | IHC | ↑ uPAR found in 87% (n = 30) PC tissues and 100% (n = 6) of matched lymph node metastases, nil immunostaining in normal PC tissue. | [143] |
4. Evidence of the Upregulation of Plasminogen Receptors in Pancreatic Cancer
4.1. Alpha-Enolase
4.2. S100A10/Annexin A2 Complex
4.3. Cytokeratin 8
4.4. Plasminogen Receptors and Immune Function
5. The uPAS as a Target for Pancreatic Cancer Therapy
6. The uPAS as a Targeted Imaging Biomarker for the Detection and Monitoring of Pancreatic Cancer
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Orth, M.; Metzger, P.; Gerum, S.; Mayerle, J.; Schneider, G.; Belka, C.; Schnurr, M.; Lauber, K. Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat. Oncol. 2019, 14, 141. [Google Scholar] [CrossRef]
- Halbrook, C.J.; Lyssiotis, C.A. Employing Metabolism to Improve the Diagnosis and Treatment of Pancreatic Cancer. Cancer Cell 2017, 31, 5–19. [Google Scholar] [CrossRef]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef]
- Huang, L.; Jansen, L.; Balavarca, Y.; Babaei, M.; Van Der Geest, L.; Lemmens, V.; Van Eycken, L.; De Schutter, H.; Johannesen, T.B.; Primic-Žakelj, M.; et al. Stratified survival of resected and overall pancreatic cancer patients in Europe and the USA in the early twenty-first century: A large, international population-based study. BMC Med. 2018, 16, 1–15. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Kleeff, J.; Michl, P.; Costello, E.; Greenhalf, W.; Palmer, D.H. Therapeutic developments in pancreatic cancer: Current and future perspectives. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 333–348. [Google Scholar] [CrossRef]
- Breidert, M.; Keck, T.; Makowiec, F.; Lohrmann, C.; Harder, J.; Fischer, R. Early recurrence of pancreatic cancer after resection and during adjuvant chemotherapy. Saudi J. Gastroenterol. 2012, 18, 118–121. [Google Scholar] [CrossRef]
- Sperti, C.; Pasquali, C.; Piccoli, A.; Pedrazzoli, S. Recurrence after resection for ductal adenocarcinoma of the pancreas. World J. Surg. 1997, 21, 195–200. [Google Scholar] [CrossRef]
- Van Roessel, S.; Kasumova, G.G.; Verheij, J.; Najarian, R.M.; Maggino, L.; De Pastena, M.; Malleo, G.; Marchegiani, G.; Salvia, R.; Ng, S.C.; et al. International Validation of the Eighth Edition of the American Joint Committee on Cancer (AJCC) TNM Staging System in Patients With Resected Pancreatic Cancer. JAMA Surg. 2018, 153, e183617. [Google Scholar] [CrossRef]
- Neesse, A.; Michl, P.; Frese, K.K.; Feig, C.; Cook, N.; Jacobetz, M.A.; Lolkema, M.P.; Buchholz, M.; Olive, K.P.; Gress, T.M.; et al. Stromal biology and therapy in pancreatic cancer. Gut 2011, 60, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, R.; Walsh, N.; Clynes, M.; O’Connor, R.; McDermott, R. Challenges of drug resistance in the management of pancreatic cancer. Expert Rev. Anticancer. Ther. 2010, 10, 1647–1661. [Google Scholar] [CrossRef] [PubMed]
- Adamska, A.; Elaskalani, O.; Emmanouilidi, A.; Kim, M.; Razak, N.B.A.; Metharom, P.; Falasca, M. Molecular and cellular mechanisms of chemoresistance in pancreatic cancer. Adv. Biol. Regul. 2018, 68, 77–87. [Google Scholar] [CrossRef]
- Amrutkar, M.; Gladhaug, I.P. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers 2017, 9, 157. [Google Scholar] [CrossRef] [PubMed]
- Oberstein, P.E.; Olive, K.P. Pancreatic cancer: Why is it so hard to treat? Therap. Adv. Gastroenterol. 2013, 6, 321–337. [Google Scholar] [CrossRef]
- Oettle, H.; Neuhaus, P.; Hochhaus, A.; Hartmann, J.T.; Gellert, K.; Ridwelski, K.; Niedergethmann, M.; Zülke, C.; Fahlke, J.; Arning, M.B. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: The CONKO-001 randomized trial. JAMA 2013, 310, 1473–1481. [Google Scholar] [CrossRef]
- Heestand, G.M.; Kurzrock, R. Molecular landscape of pancreatic cancer: Implications for current clinical trials. Oncotarget 2015, 6, 4553–4561. [Google Scholar] [CrossRef]
- Katayama, E.S.; Hue, J.J.; Bajor, D.L.; Ocuin, L.M.; Ammori, J.B.; Hardacre, J.M.; Winter, J.M. A comprehensive analysis of clinical trials in pancreatic cancer: What is coming down the pike? Oncotarget 2020, 11, 3489–3501. [Google Scholar] [CrossRef]
- Wong, H.H.; Lemoine, N.R. Pancreatic cancer: Molecular pathogenesis and new therapeutic targets. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 412–422. [Google Scholar] [CrossRef]
- Bengtsson, A.; Andersson, R.; Ansari, D. The actual 5-year survivors of pancreatic ductal adenocarcinoma based on real-world data. Sci. Rep. 2020, 10, 16425. [Google Scholar] [CrossRef]
- Winter, J.M.; Brennan, M.; Tang, L.H.; D’Angelica, M.I.; DeMatteo, R.P.; Fong, Y.; Klimstra, D.S.; Jarnagin, W.; Allen, P.J. Survival after Resection of Pancreatic Adenocarcinoma: Results from a Single Institution over Three Decades. Ann. Surg. Oncol. 2011, 19, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J. Proteases as prognostic markers in cancer. Clin. Cancer Res. 1996, 2, 613–618. [Google Scholar] [PubMed]
- Koblinski, J.E.; Ahram, M.; Sloane, B.F. Unraveling the role of proteases in cancer. Clin. Chim. Acta 2000, 291, 113–135. [Google Scholar] [CrossRef]
- Martin, C.E.; List, K. Cell surface–anchored serine proteases in cancer progression and metastasis. Cancer Metastasis Rev. 2019, 38, 357–387. [Google Scholar] [CrossRef]
- Brassart-Pasco, S.; Brézillon, S.; Brassart, B.; Ramont, L.; Oudart, J.B.; Monboisse, J.C. Tumor Microenvironment: Extracellular Matrix Alterations Influence Tumor Progression. Front. Oncol. 2020, 10, 397. [Google Scholar] [CrossRef]
- Procacci, P.; Moscheni, C.; Sartori, P.; Sommariva, M.; Gagliano, N. Tumor–Stroma Cross-Talk in Human Pancreatic Ductal Adenocarcinoma: A Focus on the Effect of the Extracellular Matrix on Tumor Cell Phenotype and Invasive Potential. Cells 2018, 7, 158. [Google Scholar] [CrossRef] [PubMed]
- Vassalli, J.D.; Sappino, P.; Belin, D. The plasminogen activator/plasmin system. J. Clin. Investig. 1991, 88, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Grøndahl-Hansen, J.; Peters, H.A.; Van Putten, W.L.; Look, M.P.; Pappot, H.; Rønne, E.; Dano, K.; Klijn, J.G.; Brünner, N.; Foekens, A.J. Prognostic significance of the receptor for urokinase plasminogen activator in breast cancer. Clin. Cancer Res. 1995, 1, 1079–1087. [Google Scholar] [PubMed]
- Look, M.P.; Van Putten, W.L.J.; Duffy, M.J.; Harbeck, N.; Christensen, I.J.; Thomssen, C.; Kates, R.; Spyratos, F.; Fernö, M.; Eppenberger-Castori, S.; et al. Pooled Analysis of Prognostic Impact of Urokinase-Type Plasminogen Activator and Its Inhibitor PAI-1 in 8377 Breast Cancer Patients. J. Natl. Cancer Inst. 2002, 94, 116–128. [Google Scholar] [CrossRef]
- Nielsen, T.O.; Andrews, H.N.; Cheang, M.; Kucab, J.E.; Hsu, F.D.; Ragaz, J.; Gilks, C.B.; Makretsov, N.; Bajdik, C.D.; Brookes, C.; et al. Expression of the insulin-like growth factor I receptor and urokinase plasminogen activator in breast cancer is associated with poor survival: Potential for intervention with 17-allylamino geldanamycin. Cancer Res. 2004, 64, 286–291. [Google Scholar] [CrossRef]
- Winter, K.; Szcześniak, P.; Bulska, M.; Kumor-Kisielewska, A.; Durko, L.; Gąsiorowska, A.; Małecka-Panas, E. Serum level of Urokinase Plasminogen Activator (uPA) Correlates with the Survival of Patients with Pancreatic Ductal Adenocarcinoma (PDAC). Pancreat Disord Ther. 2015, 5, 1–6. [Google Scholar]
- Cantero, D.; Friess, H.; Deflorin, J.; Zimmermann, A.; Bründler, M.A.; Riesle, E.; Korc, M.; Büchler, M.W. Enhanced expression of urokinase plasminogen activator and its receptor in pancreatic carcinoma. Br. J. Cancer 1997, 75, 388–395. [Google Scholar] [CrossRef]
- Gorantla, B.; Asuthkar, S.; Rao, J.S.; Patel, J.; Gondi, C.S. Suppression of the uPAR–uPA System Retards Angiogenesis, Invasion, andIn VivoTumor Development in Pancreatic Cancer Cells. Mol. Cancer Res. 2011, 9, 377–389. [Google Scholar] [CrossRef]
- Hildenbrand, R.; Niedergethmann, M.; Marx, A.; Belharazem, D.; Allgayer, H.; Schleger, C.; Ströbel, P. Amplification of the Urokinase-Type Plasminogen Activator Receptor (uPAR) Gene in Ductal Pancreatic Carcinomas Identifies a Clinically High-Risk Group. Am. J. Pathol. 2009, 174, 2246–2253. [Google Scholar] [CrossRef]
- Halamkova, J.; Kiss, I.; Pavlovsky, Z.; Jarkovsky, J.; Tomasek, J.; Tucek, S.; Hanakova, L.; Moulis, M.; Cech, Z.; Zavrelova, J.; et al. Clinical relevance of uPA, uPAR, PAI 1 and PAI 2 tissue expression and plasma PAI 1 level in colorectal carcinoma patients. Hepatogastroenterology 2012, 58, 1918–1925. [Google Scholar] [CrossRef]
- Märkl, B.; Renk, I.; Oruzio, D.; Jähnig, H.; Schenkirsch, G.; Schöler, C.; Ehret, W.; Arnholdt, H.; Anthuber, M.; Spatz, H. Tumour budding, uPA and PAI-1 are associated with aggressive behaviour in colon cancer. J. Surg. Oncol. 2010, 102, 235–241. [Google Scholar] [CrossRef]
- Brungs, D.; Chen, J.Y.J.; Aghmesheh, M.; Vine, K.; Becker, T.; Carolan, M.G.; Ranson, M. The urokinase plasminogen activation system in gastroesophageal cancer: A systematic review and meta-analysis. Oncotarget 2017, 8, 23099–23109. [Google Scholar] [CrossRef]
- Kaneko, T.; Konno, H.; Baba, M.; Tanaka, T.; Nakamura, S. Urokinase-type plasminogen activator expression correlates with tumor angiogenesis and poor outcome in gastric cancer. Cancer Sci. 2003, 94, 43–49. [Google Scholar] [CrossRef]
- Rømer, J.; Pyke, C.; Lund, L.R.; Danø, K.; Ralfkiær, E. Cancer Cell Expression of Urokinase-Type Plasminogen Activator Receptor mRNA in Squamous Cell Carcinomas of the Skin. J. Investig. Dermatol. 2001, 116, 353–358. [Google Scholar] [CrossRef]
- Santibanez, J.F. Transforming Growth Factor-Beta and Urokinase-Type Plasminogen Activator: Dangerous Partners in Tumorigenesis—Implications in Skin Cancer. ISRN Dermatol. 2013, 2013, 1–26. [Google Scholar] [CrossRef]
- Borgfeldt, C.; Hansson, S.R.; Gustavsson, B. Dedifferentiation of serous ovarian cancer from cystic to solid tumors is associated with increased expression of mRNA for urokinase plasminogen activator (uPA), its receptor (uPAR) and its inhibitor (PAI-1). Int. J. Cancer 2001, 92, 497–502. [Google Scholar] [CrossRef]
- Konecny, G.; Untch, M.; Pihan, A.; Kimmig, R.; Gropp, M.; Stieber, P.; Hepp, H.; Slamon, D.; Pegram, M. Association of urokinase-type plasminogen activator and its inhibitor with disease progression and prognosis in ovarian cancer. Clin. Cancer Res. 2001, 7, 1743–1749. [Google Scholar]
- Sier, C.; Stephens, R.W.; Bizik, J.; Mariani, A.; Bassan, M.; Pedersen, N.; Frigerio, L.; Ferrari, A.; Danø, K.; Brünner, N.; et al. The level of urokinase-type plasminogen activator receptor is increased in serum of ovarian cancer patients. Cancer Res. 1998, 58, 1843–1849. [Google Scholar]
- Duffy, M.J.; McGowan, P.M.; Harbeck, N.; Thomssen, C.; Schmitt, M. uPA and PAI-1 as biomarkers in breast cancer: Validated for clinical use in level-of-evidence-1 studies. Breast Cancer Res. 2014, 16, 1–10. [Google Scholar] [CrossRef]
- Alpízar-Alpízar, W.; Christensen, I.J.; Santoni-Rugiu, E.; Skarstein, A.; Ovrebo, K.; Illemann, M.; Laerum, O.D. Urokinase plasminogen activator receptor on invasive cancer cells: A prognostic factor in distal gastric adenocarcinoma. Int. J. Cancer 2012, 131, E329–E336. [Google Scholar] [CrossRef]
- Buø, L.; Meling, G.I.; Karlsrud, T.S.; Johansen, H.T.; Aasen, A.O. Antigen levels of urokinase plasminogen activator and its receptor at the tumor-host interface of colorectal adenocarcinomas are related to tumor aggressiveness. Hum. Pathol. 1995, 26, 1133–1138. [Google Scholar] [CrossRef]
- Kwaan, H.C.; Keer, H.N.; Radosevich, J.A.; Cajot, J.-F.; Ernst, R. Components of the Plasminogen-Plasmin System in Human Tumor Cell Lines. Semin. Thromb. Hemost. 1991, 17, 175–182. [Google Scholar] [CrossRef]
- Magnussen, S.N.; Hadler-Olsen, E.S.; Costea, D.E.; Berg, E.; Jacobsen, C.D.A.C.; Mortensen, B.; Salo, T.; Martinez-Zubiaurre, I.; Winberg, J.-O.; Uhlin-Hansen, L.; et al. Cleavage of the urokinase receptor (uPAR) on oral cancer cells: Regulation by transforming growth factor—β1 (TGF-β1) and potential effects on migration and invasion. BMC Cancer 2017, 17, 1–16. [Google Scholar] [CrossRef]
- Zlobec, I.; Lugli, A. Invasive front of colorectal cancer: Dynamic interface of pro-/anti-tumor factors. World J. Gastroenterol. 2009, 15, 5898–5906. [Google Scholar] [CrossRef]
- Gaylis, F.D.; Keer, H.N.; Wilson, M.J.; Kwaan, H.C.; Sinha, A.A.; Kozlowski, J.M. Plasminogen Activators in Human Prostate Cancer Cell Lines and Tumors: Correlation with the Aggressive Phenotype. J. Urol. 1989, 142, 193–198. [Google Scholar] [CrossRef]
- Kohga, S.; Harvey, S.R.; Weaver, R.M.; Markus, G. Localization of plasminogen activators in human colon cancer by immunoperoxidase staining. Cancer Res. 1985, 45, 1787–1796. [Google Scholar]
- Novokhatny, V.; Medved, L.; Mazar, A.; Marcotte, P.; Henkin, J.; Ingham, K. Domain structure and interactions of recombinant urokinase-type plasminogen activator. J. Biol. Chem. 1992, 267, 3878–3885. [Google Scholar] [CrossRef]
- Kobayashi, H.; Kanayama, N.; Schmitt, M.; Goretzki, L.; Chucholowski, N.; Calvete, J.; Kramer, M.; Günzler, W.A.; Jänicke, F.; Terao, T.; et al. Cathepsin B Efficiently Activets the Soluble and the Tumor Cell Receptor-Bound Form of the Proenzyme Urokinase-Type Plasminogen Activator (Pro-Upa). Hemost. Circ. 1992, 266, 115–120. [Google Scholar] [CrossRef]
- Goretzki, L.; Schmitt, M.; Mann, K.; Calvete, J.; Chucholowski, N.; Kramer, M.; Günzler, W.A.; Jänicke, F.; Graeff, H. Effective activation of the proenzyme form of the urokinase-type plasminogen activator (pro-uPA) by the cysteine protease cathepsin L. FEBS Lett. 1992, 297, 112–118. [Google Scholar] [CrossRef]
- Ichinose, A.; Fujikawa, K.; Suyama, T. The activation of pro-urokinase by plasma kallikrein and its inactivation by thrombin. J. Biol. Chem. 1986, 261, 3486–3489. [Google Scholar] [CrossRef]
- Marcotte, P.A.; Henkin, J. Characterization of the activation of pro-urokinase by thermolysin. Biochim. et Biophys. Acta (BBA)—Protein Struct. Mol. Enzym. 1993, 1161, 105–112. [Google Scholar] [CrossRef]
- Stack, M.S.; Johnson, D.A. Human mast cell tryptase activates single-chain urinary-type plasminogen activator (pro-urokinase). J. Biol. Chem. 1994, 269, 9416–9419. [Google Scholar] [CrossRef]
- Petersen, L.C.; Lund, L.R.; Nielsen, L.S.; Danø, K.; Skriver, L. One-chain urokinase-type plasminogen activator from human sarcoma cells is a proenzyme with little or no intrinsic activity. J. Biol. Chem. 1988, 263, 11189–11195. [Google Scholar] [CrossRef]
- Ellis, V.; Behrendt, N.; Danø, K. Plasminogen activation by receptor-bound urokinase. A kinetic study with both cell-associated and isolated receptor. J. Biol. Chem. 1991, 266, 12752–12758. [Google Scholar] [CrossRef]
- Appella, E.; Robinson, E.A.; Ullrich, S.J.; Stoppelli, M.P.; Corti, A.; Cassani, G.; Blasi, F. The receptor-binding sequence of urokinase. A biological function for the growth-factor module of proteases. J. Biol. Chem. 1987, 262, 4437–4440. [Google Scholar] [CrossRef]
- Shetty, S.; Idell, S. FIBRINOLYSIS|Plasminogen Activator and Plasmin. In Encyclopedia of Respiratory Medicine; Academic Press: Oxford, UK, 2006; pp. 205–210. [Google Scholar]
- Kjaergaard, M.; Hansen, L.V.; Jacobsen, B.; Gardsvoll, H.; Ploug, M. Structure and ligand interactions of the urokinase receptor (uPAR). Front. Biosci. 2008, 5441–5461. [Google Scholar] [CrossRef]
- Gårdsvoll, H.; Ploug, M. Mapping of the Vitronectin-binding Site on the Urokinase Receptor: Involvement of a coherent receptor interface consisting of residues from both domain I and the flanking interdomain linker region. J. Biol. Chem. 2007, 282, 13561–13572. [Google Scholar] [CrossRef]
- Huai, Q.; Zhou, A.; Lin, L.; Mazar, A.P.; Parry, G.C.; Callahan, J.; Shaw, D.E.; Furie, B.; Furie, B.C.; Huang, M. Crystal structures of two human vitronectin, urokinase and urokinase receptor complexes. Nat. Struct. Mol. Biol. 2008, 15, 422–423. [Google Scholar] [CrossRef]
- Smith, H.W.; Marshall, C.J. Regulation of cell signalling by uPAR. Nat. Rev. Mol. Cell Biol. 2010, 11, 23–36. [Google Scholar] [CrossRef]
- Wei, Y.; Waltz, D.A.; Rao, N.; Drummond, R.J.; Rosenberg, S.; Chapman, H.A. Identification of the urokinase receptor as an adhesion receptor for vitronectin. J. Biol. Chem. 1994, 269, 32380–32388. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A.; Estrada, Y.; Liu, D.; Ossowski, L. ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK). Cancer Res. 2003, 63, 1684–1695. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A.; Liu, D.; Mignatti, A.; Kovalski, K.; Ossowski, L. Urokinase Receptor and Fibronectin Regulate the ERKMAPK to p38MAPK Activity Ratios That Determine Carcinoma Cell Proliferation or Dormancy In Vivo. Mol. Biol. Cell 2001, 12, 863–879. [Google Scholar] [CrossRef]
- Madsen, C.D.; Ferraris, G.M.; Andolfo, A.; Cunningham, O.; Sidenius, N. uPAR-induced cell adhesion and migration: Vitronectin provides the key. J. Cell Biol. 2007, 177, 927–939. [Google Scholar] [CrossRef]
- Chen, L.; Mayer, J.A.; Krisko, T.I.; Speers, C.W.; Wang, T.; Hilsenbeck, S.G.; Brown, P.H. Inhibition of the p38 Kinase Suppresses the Proliferation of Human ER-Negative Breast Cancer Cells. Cancer Res. 2009, 69, 8853–8861. [Google Scholar] [CrossRef]
- Parmer, R.J.; Miles, L.A. Plasminogen Receptors: The First Quarter Century. Semin. Thromb. Hemost. 2013, 39, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Alemany, R.; Mirshahi, S.; Faure, J.; Pouliquen, Y.; Burtin, P.; Mirshahi, M. Binding of plasminogen to corneal fibroblasts and their extracellular matrix. Evidence for a receptor in cell membranes. Fibrinolysis 1995, 9, 223–229. [Google Scholar] [CrossRef]
- Hajjar, K.A.; Harpel, P.C.; Jaffe, E.A.; Nachman, R.L. Binding of plasminogen to cultured human endothelial cells. J. Biol. Chem. 1986, 261, 11656–11662. [Google Scholar] [CrossRef]
- Reinartz, J. Binding and Activation of Plasminogen at the Surface of Human Keratinocytes. Exp. Cell Res. 1993, 208, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Miles, L.A.; Plow, E.F. Receptor mediated binding of the fibrinolytic components, plasminogen and urokinase, to peripheral blood cells. Thromb. Haemost. 1987, 58, 936–942. [Google Scholar] [CrossRef]
- Silverstein, R.L.; Friedlander, R.J.; Nicholas, R.L.; Nachman, R.L. Binding of Lys-plasminogen to monocytes/macrophages. J. Clin. Investig. 1988, 82, 1948–1955. [Google Scholar] [CrossRef]
- Miles, L.A.; Plow, E.F. Binding and activation of plasminogen on the platelet surface. J. Biol. Chem. 1985, 260, 4303–4311. [Google Scholar] [CrossRef]
- Ranson, M.; Andronicos, N.M. Plasminogen binding and cancer: Promises and pitfalls. Front. Biosci. 2003, 8, s294–s304. [Google Scholar] [CrossRef]
- Ponting, C.; Marshall, J.M.; A Cederholm-Williams, S. Plasminogen: A structural review. Blood Coagul. Fibrinolysis 1992, 3, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Law, R.H.; Caradoc-Davies, T.; Cowieson, N.; Horvath, A.J.; Quek, A.J.; Encarnacao, J.A.; Steer, D.; Cowan, A.; Zhang, Q.; Lu, B.G.; et al. The X-ray Crystal Structure of Full-length Human Plasminogen. Cell Rep. 2012, 1, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Miles, L.A.; Dahlberg, C.M.; Plow, E.F. The cell-binding domains of plasminogen and their function in plasma. J. Biol. Chem. 1988, 263, 11928–11934. [Google Scholar] [CrossRef]
- Marti, D.N.; Schaller, J.; Ochensberger, B.; Rickli, E.E. Expression, purification and characterization of the recombinant kringle 2 and kringle 3 domains of human plasminogen and analysis of their binding affinity for omega-aminocarboxylic acids. JBIC J. Biol. Inorg. Chem. 1994, 219, 455–462. [Google Scholar] [CrossRef]
- Pluskota, E.; Soloviev, D.A.; Bdeir, K.; Cines, D.B.; Plow, E.F. Integrin αMβ2 Orchestrates and Accelerates Plasminogen Activation and Fibrinolysis by Neutrophils. J. Biol. Chem. 2004, 279, 18063–18072. [Google Scholar] [CrossRef]
- Gonzalez-Gronow, M.; Stack, S.; Pizzo, S.V. Plasmin binding to the plasminogen receptor enhances catalytic efficiency and activates the receptor for subsequent ligand binding. Arch. Biochem. Biophys. 1991, 286, 625–628. [Google Scholar] [CrossRef]
- Stillfried, G.E.; Saunders, D.N.; Ranson, M. Plasminogen binding and activation at the breast cancer cell surface: The integral role of urokinase activity. Breast Cancer Res. 2007, 9, R14. [Google Scholar] [CrossRef]
- Hall, S.W.; Humphries, J.E.; Gonias, S.L. Inhibition of cell surface receptor-bound plasmin by alpha 2-antiplasmin and alpha 2-macroglobulin. J. Biol. Chem. 1991, 266, 12329–12336. [Google Scholar] [CrossRef]
- Didiasova, M.; Wujak, L.; Wygrecka, M.; Zakrzewicz, D. From Plasminogen to Plasmin: Role of Plasminogen Receptors in Human Cancer. Int. J. Mol. Sci. 2014, 15, 21229–21252. [Google Scholar] [CrossRef]
- Ranson, M.; Andronicos, N.; O’Mullane, M.; Baker, M.; Baker, M. Increased plasminogen binding is associated with metastatic breast cancer cells: Differential expression of plasminogen binding proteins. Br. J. Cancer 1998, 77, 1586–1597. [Google Scholar] [CrossRef]
- Andreasen, P.A.; Egelund, R.; Petersen, H.H. The plasminogen activation system in tumor growth, invasion, and metastasis. Cell. Mol. Life Sci. 2000, 57, 25–40. [Google Scholar] [CrossRef]
- Balduyck, M.; Zerimech, F.; Gouyer, V.; Lemaire, R.; Hemon, B.; Grard, G.; Thiebaut, C.; Lemaire, V.; Dacquembronne, E.; Duhem, T.; et al. Specific expression of matrix metalloproteinases 1, 3, 9 and 13 associated with invasiveness of breast cancer cells in vitro. Clin. Exp. Metastasis 2000, 18, 171–178. [Google Scholar] [CrossRef]
- Deryugina, E.I.; Quigley, J.P. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006, 25, 9–34. [Google Scholar] [CrossRef]
- Pepper, M.S. Role of the Matrix Metalloproteinase and Plasminogen Activator–Plasmin Systems in Angiogenesis. Arter. Thromb. Vasc. Biol. 2001, 21, 1104–1117. [Google Scholar] [CrossRef]
- Schmalfeldt, B.; Prechtel, D.; Härting, K.; Späthe, K.; Rutke, S.; Konik, E.; Fridman, R.; Berger, U.; Schmitt, M.; Kuhn, W.; et al. Increased expression of matrix metalloproteinases (MMP)-2, MMP-9, and the urokinase-type plasminogen activator is associated with progression from benign to advanced ovarian cancer. Clin. Cancer Res. 2001, 7, 2396–2404. [Google Scholar]
- Zhang, G.; Miyake, M.; Lawton, A.; Goodison, S.; Rosser, C.J. Matrix metalloproteinase-10 promotes tumor progression through regulation of angiogenic and apoptotic pathways in cervical tumors. BMC Cancer 2014, 14, 310. [Google Scholar] [CrossRef]
- Dunn, S.; Torres, J.; Nihei, N.; Barrett, J. The insulin-like growth factor-1 elevates urokinase-type plasminogen activator-1 in human breast cancer cells: A new avenue for breast cancer therapy. Mol. Carcinog. 2000, 27, 10–17. [Google Scholar] [CrossRef]
- Matsuoka, H.; Sisson, T.H.; Nishiuma, T.; Simon, R.H. Plasminogen-Mediated Activation and Release of Hepatocyte Growth Factor from Extracellular Matrix. Am. J. Respir. Cell Mol. Biol. 2006, 35, 705–713. [Google Scholar] [CrossRef]
- Hannocks, M.J.; Oliver, L.; Gabrilove, J.L.; Wilson, E.L. Regulation of Proteolytic Activity in Human Bone Marrow Stromal Cells by Basic Fibroblast Growth Factor, Interleukin-1, and Transforming Growth Factor β. Blood 1992, 79, 1178–1184. [Google Scholar] [CrossRef]
- Breuss, J.M.; Uhrin, P. VEGF-initiated angiogenesis and the uPA/uPAR system. Cell Adhes. Migr. 2012, 6, 535–540. [Google Scholar] [CrossRef]
- McColl, B.K.; Baldwin, M.E.; Roufail, S.; Freeman, C.; Moritz, R.L.; Simpson, R.; Alitalo, K.; Stacker, S.; Achen, M.G. Plasmin Activates the Lymphangiogenic Growth Factors VEGF-C and VEGF-D. J. Exp. Med. 2003, 198, 863–868. [Google Scholar] [CrossRef]
- Jaiswal, R.K.; Varshney, A.K.; Yadava, P.K. Diversity and functional evolution of the plasminogen activator system. Biomed. Pharmacother. 2018, 98, 886–898. [Google Scholar] [CrossRef]
- Oh, J.; Kucab, J.; Bushel, P.; Martin, K.; Bennett, L.; Collins, J.; DiAugustine, R.; Barrett, J.; Afshari, C.; Dunn, S. Insulin-Like Growth Factor-1 Inscribes a Gene Expression Profile for Angiogenic Factors and Cancer Progression in Breast Epithelial Cells. Neoplasia 2002, 4, 204–217. [Google Scholar] [CrossRef]
- Law, R.H.P.; Zhang, Q.; McGowan, S.; Buckle, A.M.; Silverman, G.A.; Wong, W.; Rosado, C.J.; Langendorf, C.G.; Pike, R.N.; Bird, P.I.; et al. An overview of the serpin superfamily. Genome Biol. 2006, 7, 216. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kubala, M.H.; Declerck, Y.A. The plasminogen activator inhibitor-1 paradox in cancer: A mechanistic understanding. Cancer Metastasis Rev. 2019, 38, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, T.; Hibi, K.; Koike, M.; Fujiwara, M.; Kodera, Y.; Ito, K.; Nakao, A. Plasminogen activator inhibitor-1 as a potential marker for the malignancy of colorectal cancer. Br. J. Cancer 2005, 93, 799–803. [Google Scholar] [CrossRef]
- Pedersen, H.; Brünner, N.; Francis, D.; Osterlind, K.; Rønne, E.; Hansen, H.H.; Danø, K.; Grøndahl-Hansen, J. Prognostic impact of urokinase, urokinase receptor, and type 1 plasminogen activator inhibitor in squamous and large cell lung cancer tissue. Cancer Res. 1994, 54, 4671–4675. [Google Scholar]
- van der Burg, M.E.; Henzen-Logmans, S.C.; Berns, E.M.; van Putten, W.L.; Klijn, J.G.; Foekens, J.A. Expression of urokinase-type plasminogen activator (uPA) and its inhibitor PAI-1 in benign, borderline, malignant primary and metastatic ovarian tumors. Int. J. Cancer 1996, 69, 475–479. [Google Scholar] [CrossRef]
- Chambers, S.K.; Ivins, C.M.; Carcangiu, M.L. Plasminogen activator inhibitor-1 is an independent poor prognostic factor for survival in advanced stage epithelial ovarian cancer patients. Int. J. Cancer 1998, 79, 449–454. [Google Scholar] [CrossRef]
- Duggan, C.; Kennedy, S.P.; Kramer, M.D.; Barnes, C.; Elvin, P.; McDermott, E.; O’Higgins, N.; Duffy, M.J. Plasminogen activator inhibitor type 2 in breast cancer. Br. J. Cancer 1997, 76, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Konno, H.; Tanaka, T.; Maruo, Y.; Nishino, N.; Aoki, K.; Baba, S.; Sakaguchi, S.; Takada, Y.; Takada, A. Possible role of plasminogen activator inhibitor 2 in the prevention of the metastasis of gastric cancer tissues. Thromb. Res. 1992, 65, 709–719. [Google Scholar] [CrossRef][Green Version]
- Robert, C.; Bolon, I.; Gazzeri, S.; Veyrenc, S.; Brambilla, C.; Brambilla, E. Expression of plasminogen activator inhibitors 1 and 2 in lung cancer and their role in tumor progression. Clin. Cancer Res. 1999, 5, 2094–2102. [Google Scholar]
- Smith, R.; Xue, A.; Gill, A.; Scarlett, C.; Saxby, A.; Clarkson, A.; Hugh, T. High Expression of Plasminogen Activator Inhibitor-2 (PAI-2) is a Predictor of Improved Survival in Patients with Pancreatic Adenocarcinoma. World J. Surg. 2007, 31, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, H.; Endo, Y.; Watanabe, Y.; Sasaki, T. Significance of plasminogen activator inhibitor 2 as a prognostic marker in primary lung cancer: Association of decreased plasminogen activator inhibitor 2 with lymph node metastasis. Br. J. Cancer 1998, 78, 833–839. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhao, E.; Han, D.; Yu, Z.; Fan, E.; Li, Y.; Zhou, Z. Prognostic value of the urokinase-type plasminogen activator and its inhibitors in squamous cell carcinoma of human larynx. Lin Chuang Er Bi Yan Hou Ke Za Zhi = J. Clin. Otorhinolaryngol. 2002, 16, 599–602. [Google Scholar]
- Nordengren, J.; Lidebring, M.F.; Bendahl, P.-O.; Brünner, N.; Fernö, M.; Högberg, T.; Stephens, R.W.; Willén, R.; Casslén, B. High tumor tissue concentration of plasminogen activator inhibitor 2 (PAI-2) is an independent marker for shorter progression-free survival in patients with early stage endometrial cancer. Int. J. Cancer 2001, 97, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, S.; Sier, C.; Griffioen, G.; Vloedgraven, H.J.; De Boer, A.; Welvaart, K.; Van De Velde, C.J.; Van Krieken, J.H.; Verheijen, J.H.; Lamers, C.B. Prognostic relevance of plasminogen activators and their inhibitors in colorectal cancer. Cancer Res. 1994, 54, 4065–4071. [Google Scholar] [PubMed]
- Croucher, D.R.; Saunders, D.N.; Lobov, S.; Ranson, M. Revisiting the biological roles of PAI2 (SERPINB2) in cancer. Nat. Cancer 2008, 8, 535–545. [Google Scholar] [CrossRef]
- Croucher, D.R.; Saunders, D.N.; Stillfried, G.E.; Ranson, M. A structural basis for differential cell signalling by PAI-1 and PAI-2 in breast cancer cells. Biochem. J. 2007, 408, 203–210. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Nakao, A.; Harada, A.; Nonami, T.; Fukatsu, T.; Takagi, H. Expression of plasminogen activators and their inhibitors in human pancreatic carcinoma: Immunohistochemical study. Am. J. Gastroenterol. 1993, 88. [Google Scholar]
- Harvey, S.R.; Hurd, T.C.; Markus, G.; Martinick, M.I.; Penetrante, R.M.; Tan, D.; Venkataraman, P.; DeSouza, N.; Sait, S.N.J.; Driscoll, D.L.; et al. Evaluation of urinary plasminogen activator, its receptor, matrix metalloproteinase-9, and von Willebrand factor in pancreatic cancer. Clin. Cancer Res. 2003, 9, 4935–4943. [Google Scholar]
- de Geus, S.W.; Baart, V.M.; Boonstra, M.C.; Kuppen, P.J.; Prevoo, H.A.; Mazar, A.P.; Bonsing, B.A.; Morreau, H.; van de Velde, C.J.; Vahrmeijer, A.L.; et al. Prognostic impact of urokinase plasminogen activator receptor expression in pancreatic cancer: Malignant versus stromal cells. Biomark. Insights 2017, 12, 1177271917715443. [Google Scholar] [CrossRef]
- Harris, N.L.E.; Vennin, C.; Conway, J.R.W.; Vine, K.L.; Pinese, M.; Cowley, M.J.; Shearer, R.F.; Lucas, M.C.; Herrmann, D.; Allam, A.H.; et al. SerpinB2 regulates stromal remodelling and local invasion in pancreatic cancer. Oncogene 2017, 36, 4288–4298. [Google Scholar] [CrossRef]
- Nielsen, A.; Scarlett, C.J.; Samra, J.S.; Gill, A.; Li, Y.; Allen, B.J.; Smith, R.C. Significant overexpression of urokinase-type plasminogen activator in pancreatic adenocarcinoma using real-time quantitative reverse transcription polymerase chain reaction. J. Gastroenterol. Hepatol. 2005, 20, 256–263. [Google Scholar] [CrossRef]
- Loosen, S.H.; Tacke, F.; Püthe, N.; Binneboesel, M.; Wiltberger, G.; Alizai, P.H.; Kather, J.N.; Paffenholz, P.; Ritz, T.; Koch, A.; et al. High baseline soluble urokinase plasminogen activator receptor (suPAR) serum levels indicate adverse outcome after resection of pancreatic adenocarcinoma. Carcinogenesis 2019, 40, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Xue, A.; Scarlett, C.J.; Jackson, C.; Allen, B.J.; Smith, R.C. Prognostic Significance of Growth Factors and the Urokinase-Type Plasminogen Activator System in Pancreatic Ductal Adenocarcinoma. Pancreas 2008, 36, 160–167. [Google Scholar] [CrossRef]
- Albert, M.B.; Steinberg, W.M.; Henry, J.P. Elevated serum levels of tumor marker CA19-9 in acute cholangitis. Dig. Dis. Sci. 1988, 33, 1223–1225. [Google Scholar] [CrossRef] [PubMed]
- Ballehaninna, U.K.; Chamberlain, R.S. The clinical utility of serum CA 19-9 in the diagnosis, prognosis and management of pancreatic adenocarcinoma: An evidence based appraisal. J. Gastrointest. Oncol. 2012, 3, 105–119. [Google Scholar] [CrossRef]
- Salleh, S.; Thyagarajan, A.; Sahu, R.P. Exploiting the relevance of CA 19-9 in pancreatic cancer. J. Cancer Metastasis Treat. 2020, 2020, 31. [Google Scholar] [CrossRef]
- Kim, S.; Park, B.K.; Seo, J.H.; Choi, J.; Choi, J.W.; Lee, C.K.; Chung, J.B.; Park, Y.; Kim, D.W. Carbohydrate antigen 19-9 elevation without evidence of malignant or pancreatobiliary diseases. Sci. Rep. 2020, 10, 8820. [Google Scholar] [CrossRef] [PubMed]
- Azizian, A.; Rühlmann, F.; Krause, T.; Bernhardt, M.; Jo, P.; König, A.; Kleiß, M.; Leha, A.; Ghadimi, M.; Gaedcke, J. CA19-9 for detecting recurrence of pancreatic cancer. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kang, K.N.; Shin, Y.S.; Byun, Y.; Han, Y.; Kwon, W.; Kim, C.W.; Jang, J.Y. Biomarker Panel for the Diagnosis of Pancreatic Ductal Adenocarcinoma. Cancers 2020, 12, 1443. [Google Scholar] [CrossRef]
- Rieser, C.J.; Zenati, M.; Hamad, A.; Al Abbas, A.; Bahary, N.; Zureikat, A.; Zeh, H.J.; Hogg, M.E. CA19-9 on Postoperative Surveillance in Pancreatic Ductal Adenocarcinoma: Predicting Recurrence and Changing Prognosis over Time. Ann. Surg. Oncol. 2018, 25, 3483–3491. [Google Scholar] [CrossRef]
- Herszényi, L.; Farinati, F.; Cardin, R.; István, G.; Molnár, L.D.; Hritz, I.; De Paoli, M.; Plebani, M.; Tulassay, Z. Tumor marker utility and prognostic relevance of cathepsin B, cathepsin L, urokinase-type plasminogen activator, plasminogen activator inhibitor type-1, CEA and CA 19-9 in colorectal cancer. BMC Cancer 2008, 8, 194. [Google Scholar] [CrossRef]
- Rogers, A.; Smith, M.J.; Doolan, P.; Clarke, C.; Clynes, M.; Murphy, J.F.; McDermott, A.; Swan, N.; Crotty, P.; Ridgway, P.F.; et al. Invasive markers identified by gene expression profiling in pancreatic cancer. Pancreatology 2012, 12, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zheng, B.; Robbins, D.H.; Lewin, D.N.; Mikhitarian, K.; Graham, A.; Rumpp, L.; Glenn, T.; Gillanders, W.E.; Cole, D.J.; et al. Accurate discrimination of pancreatic ductal adenocarcinoma and chronic pancreatitis using multimarker expression data and samples obtained by minimally invasive fine needle aspiration. Int. J. Cancer 2007, 120, 1511–1517. [Google Scholar] [CrossRef] [PubMed]
- Aronen, A.; Aittoniemi, J.; Huttunen, R.; Nikkola, A.; Rinta-Kiikka, I.; Nikkola, J.; Limnell, O.; Nordback, I.; Sand, J.; Laukkarinen, J. Plasma suPAR may help to distinguish between chronic pancreatitis and pancreatic cancer. Scand. J. Gastroenterol. 2021, 56, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Baluka, D.; Urbanek, T.; Lekstan, A.; Swietochowska, E.; Wiaderkiewicz, R.; Kajor, M.; Jedrzejewska-Szypulka, H.; Kusnierz, K.; Lampe, P. The role of the tissue plasminogen activator as a prognostic and differentiation factor in patients with pancreatic cancer and chronic pancreatitis. J. Physiol. Pharmacol. 2016, 67, 93–101. [Google Scholar]
- Sorio, C.; Mafficini, A.; Furlan, F.; Barbi, S.; Bonora, A.; Brocco, G.; Blasi, F.; Talamini, G.; Bassi, C.; Scarpa, A. Elevated urinary levels of urokinase-type plasminogen activator receptor (uPAR) in pancreatic ductal adenocarcinoma identify a clinically high-risk group. BMC Cancer 2011, 11, 448. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-J.; Zhou, L.; Liang, Z.-Y.; Zhou, W.-X.; You, L.; Zhang, T.-P.; Zhao, Y.P. Plasminogen Activator Inhibitor 1 as a Poor Prognostic Indicator in Resectable Pancreatic Ductal Adenocarcinoma. Chin. Med. J. 2018, 131, 2947–2952. [Google Scholar] [CrossRef]
- He, Y.; Liu, X.D.; Chen, Z.Y.; Zhu, J.; Xiong, Y.; Li, K.; Dong, J.H.; Li, X. Interaction between cancer cells and stromal fibroblasts is required for activation of the uPAR-uPA-MMP-2 cascade in pancreatic cancer metastasis. Clin. Cancer Res. 2007, 13, 3115–3124. [Google Scholar] [CrossRef]
- Huang, C.; Xie, D.; Cui, J.; Li, Q.; Gao, Y.; Xie, K. FOXM1c Promotes Pancreatic Cancer Epithelial-to-Mesenchymal Transition and Metastasis via Upregulation of Expression of the Urokinase Plasminogen Activator System. Clin. Cancer Res. 2014, 20, 1477–1488. [Google Scholar] [CrossRef]
- Tan, X.; Egami, H.; Nozawa, F.; Abe, M.; Baba, H. Analysis of the invasion-metastasis mechanism in pancreatic cancer: Involvement of plasmin(ogen) cascade proteins in the invasion of pancreatic cancer cells. Int. J. Oncol. 2006, 28, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.J.; Rizvi, S.M.A.; Qu, C.F.; Smith, R.C. Targeted Alpha Therapy Approach to the Management of Pancreatic Cancer. Cancers 2011, 3, 1821–1843. [Google Scholar] [CrossRef]
- Miles, L.A.; Dahlberg, C.M.; Plescia, J.; Felez, J.; Kato, K.; Plow, E.F. Role of cell-surface lysines in plasminogen binding to cells: Identification of .alpha.-enolase as a candidate plasminogen receptor. Biochemistry 1991, 30, 1682–1691. [Google Scholar] [CrossRef] [PubMed]
- Redlitz, A.; Fowler, B.J.; Plow, E.F.; Miles, L.A. The Role of an Enolase-Related Molecule in Plasminogen Binding to Cells. JBIC J. Biol. Inorg. Chem. 1995, 227, 407–415. [Google Scholar] [CrossRef]
- Almaguel, F.A.; Sanchez, T.W.; Ortiz-Hernandez, G.L.; Casiano, C.A. Alpha-Enolase: Emerging Tumor-Associated Antigen, Cancer Biomarker, and Oncotherapeutic Target. Front. Genet. 2021, 11. [Google Scholar] [CrossRef]
- Capello, M.; Borgogno, S.F.; Riganti, C.; Chattaragada, M.S.; Principe, M.; Roux, C.; Zhou, W.; Petricoin, E.F.; Cappello, P.; Novelli, F. Targeting the Warburg effect in cancer cells through ENO1 knockdown rescues oxidative phosphorylation and induces growth arrest. Oncotarget 2015, 7, 5598–5612. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Principe, M.; Ceruti, P.; Shih, N.-Y.; Chattaragada, M.S.; Rolla, S.; Conti, L.; Bestagno, M.; Zentilin, L.; Yang, S.H.; Migliorini, P.; et al. Targeting of surface alpha-enolase inhibits the invasiveness of pancreatic cancer cells. Oncotarget 2015, 6, 11098–11113. [Google Scholar] [CrossRef]
- Sun, L.; Guo, C.; Cao, J.; Burnett, J.; Yang, Z.; Ran, Y.; Sun, D. Over-Expression of Alpha-Enolase as a Prognostic Biomarker in Patients with Pancreatic Cancer. Int. J. Med. Sci. 2017, 14, 655–661. [Google Scholar] [CrossRef]
- Principe, M.; Borgoni, S.; Cascione, M.; Chattaragada, M.S.; Ferri-Borgogno, S.; Capello, M.; Bulfamante, S.; Chapelle, J.; Di Modugno, F.; Defilippi, P.; et al. Alpha-enolase (ENO1) controls alpha v/beta 3 integrin expression and regulates pancreatic cancer adhesion, invasion, and metastasis. J. Hematol. Oncol. 2017, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hosotani, R.; Kawaguchi, M.; Masui, T.; Koshiba, T.; Ida, J.; Fujimoto, K.; Wada, M.; Doi, R.; Imamura, M. Expression of Integrin αVβ3 in Pancreatic Carcinoma: Relation to MMP-2 Activation and Lymph Node Metastasis. Pancreas 2002, 25, e30–e35. [Google Scholar] [CrossRef]
- Leavesley, D.; Ferguson, G.; Wayner, E.; Cheresh, D. Requirement of the integrin beta 3 subunit for carcinoma cell spreading or migration on vitronectin and fibrinogen. J. Cell Biol. 1992, 117, 1101–1107. [Google Scholar] [CrossRef]
- Takayama, S.; Ishii, S.; Ikeda, T.; Masamura, S.; Doi, M.; Kitajima, M. The relationship between bone metastasis from human breast cancer and integrin αvβ3 expression. Anticancer Res. 2005, 25, 79–83. [Google Scholar] [PubMed]
- Cagnol, S.; Chambard, J.C. ERK and cell death: Mechanisms of ERK-induced cell death—Apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef]
- Yin, H.; Wang, L.; Liu, H.-L. ENO1 Overexpression in Pancreatic Cancer Patients and Its Clinical and Diagnostic Significance. Gastroenterol. Res. Pract. 2018, 2018, 3842198. [Google Scholar] [CrossRef] [PubMed]
- Rokhgireh, S.; Kashi, A.M.; Chaichian, S.; Delbandi, A.A.; Allahqoli, L.; Ahmadi-Pishkuhi, M.; Khodaverdi, S.; Alkatout, I. The Diagnostic Accuracy of Combined Enolase/Cr, CA125, and CA19-9 in the Detection of Endometriosis. BioMed Res. Int. 2020, 2020, 5208279. [Google Scholar] [CrossRef]
- Luo, X.; Wei, Y.Q.; Hai, L.; Hu, Y.C.; Zhao, Z.J.; Ma, W.L.; Ma, L.N.; Liu, X.Y.; Ding, X.C. A preliminary study of serum marker alpha-enolase in the diagnosis of hepatocellular carcinoma. Zhonghua Gan Zang Bing Za Zhi 2019, 27, 505–510. [Google Scholar]
- Zhu, W.; Li, H.; Yu, Y.; Chen, J.; Chen, X.; Ren, F.; Ren, Z.; Cui, G. Enolase-1 serves as a biomarker of diagnosis and prognosis in hepatocellular carcinoma patients. Cancer Manag. Res. 2018, 10, 5735–5745. [Google Scholar] [CrossRef]
- Cappello, P.; Tomaino, B.; Chiarle, R.; Ceruti, P.; Novarino, A.; Castagnoli, C.; Migliorini, P.; Perconti, G.; Giallongo, A.; Milella, M.; et al. An integrated humoral and cellular response is elicited in pancreatic cancer by α-enolase, a novel pancreatic ductal adenocarcinoma-associated antigen. Int. J. Cancer 2009, 125, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Cappello, P.; Rolla, S.; Chiarle, R.; Principe, M.; Cavallo, F.; Perconti, G.; Feo, S.; Giovarelli, M.; Novelli, F. Vaccination with ENO1 DNA Prolongs Survival of Genetically Engineered Mice with Pancreatic Cancer. Gastroenterology 2013, 144, 1098–1106. [Google Scholar] [CrossRef]
- Tomaino, B.; Cappello, P.; Capello, M.; Fredolini, C.; Sperduti, I.; Migliorini, P.; Salacone, P.; Novarino, A.; Giacobino, A.; Ciuffreda, L.; et al. Circulating Autoantibodies to Phosphorylated α-Enolase are a Hallmark of Pancreatic Cancer. J. Proteome Res. 2011, 10, 105–112. [Google Scholar] [CrossRef]
- Capello, M.; Ferri-Borgogno, S.; Cappello, P.; Novelli, F. α-enolase: A promising therapeutic and diagnostic tumor target. FEBS J. 2011, 278, 1064–1074. [Google Scholar] [CrossRef]
- Christensen, M.V.; Høgdall, C.K.; Jochumsen, K.M.; Høgdall, E.V.S. Annexin A2 and cancer: A systematic review. Int. J. Oncol. 2017, 52, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Lokman, N.A.; Ween, M.P.; Oehler, M.K.; Ricciardelli, C. The Role of Annexin A2 in Tumorigenesis and Cancer Progression. Cancer Microenviron. 2011, 4, 199–208. [Google Scholar] [CrossRef]
- Sharma, M.C.; Sharma, M. The role of annexin II in angiogenesis and tumor progression: A potential therapeutic target. Curr. Pharm. Des. 2007, 13, 3568–3575. [Google Scholar] [CrossRef] [PubMed]
- Madureira, P.A.; Surette, A.P.; Phipps, K.D.; Taboski, M.A.S.; Miller, V.A.; Waisman, D.M. The role of the annexin A2 heterotetramer in vascular fibrinolysis. Blood 2011, 118, 4789–4797. [Google Scholar] [CrossRef]
- Kassam, G.; Le, B.-H.; Choi, K.-S.; Kang, H.-M.; Fitzpatrick, S.L.; Louie, P.; Waisman, D. The p11 Subunit of the Annexin II Tetramer Plays a Key Role in the Stimulation of t-PA-Dependent Plasminogen Activation. Biochemistry 1998, 37, 16958–16966. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, A.; Bydoun, M.; Holloway, R.; Waisman, D. Annexin A2 Heterotetramer: Structure and Function. Int. J. Mol. Sci. 2013, 14, 6259–6305. [Google Scholar] [CrossRef] [PubMed]
- Kassam, G.; Choi, K.-S.; Ghuman, J.; Kang, H.-M.; Fitzpatrick, S.L.; Zackson, T.; Zackson, S.; Toba, M.; Shinomiya, A.; Waisman, D. The Role of Annexin II Tetramer in the Activation of Plasminogen. J. Biol. Chem. 1998, 273, 4790–4799. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Havens, A.M.; Jung, Y.; Ziegler, A.M.; Pedersen, E.A.; Wang, J.; Wang, J.; Lu, G.; Roodman, G.D.; Loberg, R.D.; et al. Annexin II/Annexin II receptor axis regulates adhesion, migration, homing, and growth of prostate cancer. J. Cell. Biochem. 2008, 105, 370–380. [Google Scholar] [CrossRef]
- Takano, S.; Togawa, A.; Yoshitomi, H.; Shida, T.; Kimura, F.; Shimizu, H.; Yoshidome, H.; Ohtsuka, M.; Kato, A.; Tomonaga, T.; et al. Annexin II Overexpression Predicts Rapid Recurrence after Surgery in Pancreatic Cancer Patients Undergoing Gemcitabine-Adjuvant Chemotherapy. Ann. Surg. Oncol. 2008, 15, 3157–3168. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.G.; Foley, K.; Rucki, A.A.; Xia, T.; Jaffee, E.; Huang, C.-Y.; Zheng, L. Stromal Annexin A2 expression is predictive of decreased survival in pancreatic cancer. Oncotarget 2017, 8, 106405–106414. [Google Scholar] [CrossRef]
- Vishwanatha, J.; Chiang, Y.; Kumble, K.D.; Hollingsworth, M.A.; Pour, P.M. Enhanced expression of annexin II in human pancreatic carcinoma cells and primary pancreatic cancers. Carcinogenesis 1993, 14, 2575–2579. [Google Scholar] [CrossRef]
- Díaz, V.M.; Hurtado, M.; Thomson, T.; Reventós, J.; Paciucci, R. Specific interaction of tissue-type plasminogen activator (t-PA) with annexin II on the membrane of pancreatic cancer cells activates plasminogen and promotes invasion in vitro. Gut 2004, 53, 993–1000. [Google Scholar] [CrossRef]
- Paciucci, R.; Torà, M.; Díaz, V.M.; Real, F.X. The plasminogen activator system in pancreas cancer: Role of t-PA in the invasive potential in vitro. Oncogene 1998, 16, 625–633. [Google Scholar] [CrossRef][Green Version]
- Zheng, L.; Foley, K.; Huang, L.; Leubner, A.; Mo, G.; Olino, K.; Edil, B.H.; Mizuma, M.; Sharma, R.; Le, D.T.; et al. Tyrosine 23 Phosphorylation-Dependent Cell-Surface Localization of Annexin A2 Is Required for Invasion and Metastases of Pancreatic Cancer. PLoS ONE 2011, 6, e19390. [Google Scholar] [CrossRef] [PubMed]
- Nedjadi, T.; Kitteringham, N.; Campbell, F.; Jenkins, R.E.; Park, B.K.; Navarro, P.; Ashcroft, F.; Tepikin, A.; Neoptolemos, J.P.; Costello, E. S100A6 binds to annexin 2 in pancreatic cancer cells and promotes pancreatic cancer cell motility. Br. J. Cancer 2009, 101, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Sitek, B.; Sipos, B.; Alkatout, I.; Poschmann, G.; Stephan, C.; Schulenborg, T.; Marcus, K.; Lüttges, J.; Dittert, D.-D.; Baretton, G.; et al. Analysis of the Pancreatic Tumor Progression by a Quantitative Proteomic Approach and Immunhistochemical Validation. J. Proteome Res. 2009, 8, 1647–1656. [Google Scholar] [CrossRef]
- Yu, D.-Y.; Yu, Y.-D.; Kim, W.-B.; Han, H.-J.; Choi, S.-B.; Kim, D.-S.; Choi, S.-Y.; Kim, J.-Y.; Chang, H.; Kim, B.-H. Clinical significance of pancreatic intraepithelial neoplasia in resectable pancreatic cancer on survivals. Ann. Surg. Treat. Res. 2018, 94, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Bydoun, M.; Sterea, A.; Liptay, H.; Uzans, A.; Huang, W.; Rodrigues, G.J.; Weaver, I.; Gu, H.; Waisman, D.M. S100A10, a novel biomarker in pancreatic ductal adenocarcinoma. Mol. Oncol. 2018, 12, 1895–1916. [Google Scholar] [CrossRef]
- Zhuang, H.; Chen, X.; Dong, F.; Zhang, Z.; Zhou, Z.; Ma, Z.; Huang, S.; Chen, B.; Zhang, C.; Hou, B. Prognostic values and immune suppression of the S100A family in pancreatic cancer. J. Cell. Mol. Med. 2021, 25, 3006–3018. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-B.; Wang, J.-L.; Jin, X.-D.; Zhao, L.; Ye, H.-L.; Kuang, Y.-B.; Ma, Y.; Jiang, X.-Y.; Yu, Z.-Y. Comprehensive analysis of the transcriptional expressions and prognostic value of S100A family in pancreatic ductal adenocarcinoma. BMC Cancer 2021, 21, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Nakamura, Y.; Morinaga, S.; Numata, K.; Sawazaki, S.; Watanabe, T.; Numata, M.; Tamagawa, H.; Godai, T.; Shiozawa, M.; et al. The clinical significance of S100A10 in pancreatic cancer. J. Clin. Oncol. 2013, 31, 194. [Google Scholar] [CrossRef]
- Hatzfeld, M.; Franke, W.W. Pair formation and promiscuity of cytokeratins: Formation in vitro of heterotypic complexes and intermediate-sized filaments by homologous and heterologous recombinations of purified polypeptides. J. Cell Biol. 1985, 101, 1826–1841. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Lee, H.S.; Kim, W.H.; Choi, Y.; Yang, M. Expression of Mucins and Cytokeratins in Primary Carcinomas of the Digestive System. Mod. Pathol. 2003, 16, 403–410. [Google Scholar] [CrossRef]
- Steven, L.G.; Gonias, S.L.; Hembrough, T.A.; Sankovic, M. Cytokeratin 8 functions as a major plasminogen receptor in select epithelial and carcinoma cells. Front. Biosci. 2001, 6, d1403–d1411. [Google Scholar] [CrossRef][Green Version]
- Kralovich, K.R.; Li, L.; Hembrough, T.A.; Webb, N.J.; Karns, L.R.; Gonias, S.L. Characterization of the binding sites for plasminogen and tissue-type plasminogen activator in cytokeratin 8 and cytokeratin 18. Protein J. 1998, 17, 845–854. [Google Scholar] [CrossRef]
- Gires, O.; Mack, B.; Rauch, J.; Matthias, C. CK8 correlates with malignancy in leukoplakia and carcinomas of the head and neck. Biochem. Biophys. Res. Commun. 2006, 343, 252–259. [Google Scholar] [CrossRef]
- Xu, X.C.; Lee, J.S.; Lippman, S.M.; Ro, J.Y.; Hong, W.K.; Lotan, R. Increased expression of cytokeratins CK8 and CK19 is associated with head and neck carcinogenesis. Cancer Epidemiol. Biomark. Prev. 1995, 4, 871–876. [Google Scholar]
- Makino, T.; Yamasaki, M.; Takeno, A.; Shirakawa, M.; Miyata, H.; Takiguchi, S.; Nakajima, K.; Fujiwara, Y.; Nishida, T.; Matsuura, N.; et al. Cytokeratins 18 and 8 are poor prognostic markers in patients with squamous cell carcinoma of the oesophagus. Br. J. Cancer 2009, 101, 1298–1306. [Google Scholar] [CrossRef]
- Fillies, T.; Werkmeister, R.; Packeisen, J.; Brandt, B.; Morin, P.; Weingart, D.; Joos, U.; Buerger, H. Cytokeratin 8/18 expression indicates a poor prognosis in squamous cell carcinomas of the oral cavity. BMC Cancer 2006, 6, 1–8. [Google Scholar] [CrossRef]
- Fukunaga, Y.; Bandoh, S.; Fujita, J.; Yang, Y.; Ueda, Y.; Hojo, S.; Dohmoto, K.; Tojo, Y.; Takahara, J.; Ishida, T. Expression of cytokeratin 8 in lung cancer cell lines and measurement of serum cytokeratin 8 in lung cancer patients. Lung Cancer 2002, 38, 31–38. [Google Scholar] [CrossRef]
- Ishii, T.; Bandoh, S.; Fujita, J.; Ohtsuki, Y.; Tojo, Y.; Kanaji, N.; Fukunaga, Y.; Ueda, Y.; Ishida, T.; Kubo, A. Full-Length Cytokeratin 8 Is Released and Circulates in Patients with Non-Small Cell Lung Cancer. Tumor Biol. 2008, 29, 57–62. [Google Scholar] [CrossRef]
- Hembrough, T.A.; Li, L.; Gonias, S.L. Cell-surface Cytokeratin 8 Is the Major Plasminogen Receptor on Breast Cancer Cells and Is Required for the Accelerated Activation of Cell-associated Plasminogen by Tissue-type Plasminogen Activator. J. Biol. Chem. 1996, 271, 25684–25691. [Google Scholar] [CrossRef] [PubMed]
- Hembrough, T.A.; Vasudevan, J.; Allietta, M.M.; Glass, W.F.; Gonias, S.L. A cytokeratin 8-like protein with plasminogen-binding activity is present on the external surfaces of hepatocytes, HepG2 cells and breast carcinoma cell lines. J. Cell Sci. 1995, 108, 1071–1082. [Google Scholar] [CrossRef]
- Bonin, S.; Brunetti, D.; Benedetti, E.; Dotti, I.; Gorji, N.; Stanta, G. Molecular characterisation of breast cancer patients at high and low recurrence risk. Virchows Arch. 2008, 452, 241–250. [Google Scholar] [CrossRef][Green Version]
- Iyer, S.V.; Dange, P.P.; Alam, H.; Sawant, S.S.; Ingle, A.D.; Borges, A.M.; Shirsat, N.V.; Dalal, S.N.; Vaidya, M.M. Understanding the Role of Keratins 8 and 18 in Neoplastic Potential of Breast Cancer Derived Cell Lines. PLoS ONE 2013, 8, e53532. [Google Scholar] [CrossRef] [PubMed]
- Casanova, M.L.; Bravo, A.; Ramírez, A.; de Escobar, G.M.; Were, F.; Merlino, G.; Vidal, M.; Jorcano, J.L. Exocrine pancreatic disorders in transsgenic mice expressing human keratin 8. J. Clin. Investig. 1999, 103, 1587–1595. [Google Scholar] [CrossRef]
- Treiber, M.; Schulz, H.-U.; Landt, O.; Drenth, J.P.; Castellani, C.; Real, F.X.; Akar, N.; Ammann, R.W.; Bargetzi, M.; Bhatia, E.; et al. Keratin 8 sequence variants in patients with pancreatitis and pancreatic cancer. J. Mol. Med. 2006, 84, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Nakatochi, M.; Hosono, Y.; Ito, H.; Kamatani, Y.; Inoko, A.; Sakamoto, H.; Kinoshita, F.; Kobayashi, Y.; Ishii, H.; et al. Genome-wide association meta-analysis identifies GP2 gene risk variants for pancreatic cancer. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.K.D.; Limaye, M.; Schaffert, S.; West, R.; Ozawa, M.G.; Chu, P.; Nair, V.S.; Koong, A.C.; Khatri, P. A multi-scale integrated analysis identifies KRT8 as a pan-cancer early biomarker. Biocomputing 2021, 2020, 297–308. [Google Scholar]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef]
- Feinberg, A.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Liu, Q.; Liao, Q. Tumor-Associated Macrophages in Pancreatic Ductal Adenocarcinoma: Origin, Polarization, Function, and Reprogramming. Front. Cell Dev. Biol. 2020, 8, 607209. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Kim, H.; Lee, N.; Won, C.; Kim, H.-R.; Hwang, Y.-I.; Song, Y.W.; Kang, J.S.; Lee, W.J. α-Enolase Expressed on the Surfaces of Monocytes and Macrophages Induces Robust Synovial Inflammation in Rheumatoid Arthritis. J. Immunol. 2012, 189, 365–372. [Google Scholar] [CrossRef]
- Ceruti, P.; Principe, M.; Capello, M.; Cappello, P.; Novelli, F. Three are better than one: Plasminogen receptors as cancer theranostic targets. Exp. Hematol. Oncol. 2013, 2, 12. [Google Scholar] [CrossRef]
- O’Connell, P.A.; Surette, A.P.; Liwski, R.S.; Svenningsson, P.; Waisman, D.M. S100A10 regulates plasminogen-dependent macrophage invasion. Blood 2010, 116, 1136–1146. [Google Scholar] [CrossRef]
- Xu, W.; Yang, W.; Wu, C.; Ma, X.; Li, H.; Zheng, J. Enolase 1 Correlated with Cancer Progression and Immune-Infiltrating in Multiple Cancer Types: A Pan-Cancer Analysis. Front. Oncol. 2021, 10, 593706. [Google Scholar] [CrossRef]
- Andronicos, N.M.; Chen, E.I.; Baik, N.; Bai, H.; Parmer, C.M.; Kiosses, W.B.; Kamps, M.P.; Yates, J.R.; Parmer, R.J.; Miles, L.A. Proteomics-based discovery of a novel, structurally unique, and developmentally regulated plasminogen receptor, Plg-RKT, a major regulator of cell surface plasminogen activation. Blood 2010, 115, 1319–1330. [Google Scholar] [CrossRef]
- Miles, L.A.; Baik, N.; Krajewski, S.; Parmer, R.J.; Mueller, B.M. The Novel Plasminogen Receptor, Plg-RKT, and Breast Cancer Progression. Blood 2011, 118, 853. [Google Scholar] [CrossRef]
- Herren, T.; Burke, T.A.; Das, R.; Plow, E.F. Identification of Histone H2B as a Regulated Plasminogen Receptor. Biochemistry 2006, 45, 9463–9474. [Google Scholar] [CrossRef]
- Das, R.; Burke, T.; Plow, E.F. Histone H2B as a functionally important plasminogen receptor on macrophages. Blood 2007, 110, 3763–3772. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Sampedro, A.; Gaggia, G.; Ney, A.; Mahamed, I.; Acedo, P. The State-of-the-Art of Phase II/III Clinical Trials for Targeted Pancreatic Cancer Therapies. J. Clin. Med. 2021, 10, 566. [Google Scholar] [CrossRef]
- Heinemann, V.; Ebert, M.P.; Pinter, T.; Bevan, P.; Neville, N.G.; Mala, C. Randomized phase II trial with an uPA inhibitor (WX-671) in patients with locally advanced nonmetastatic pancreatic cancer. J. Clin. Oncol. 2010, 28, 4060. [Google Scholar] [CrossRef]
- Rockway, T.W.; Giranda, V.L. Inhibitors of the Proteolytic Activity of Urokinase Type Plasminogen Activator. Curr. Pharm. Des. 2003, 9, 1483–1498. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Lin, L.; Huai, Q.; Huang, M. Challenges for drug discovery—A case study of urokinase receptor inhibition. Comb. Chem. High Throughput Screen. 2009, 12, 961–967. [Google Scholar] [CrossRef]
- Salamouni, N.S.E.; Buckley, B.J.; Ranson, M.; Kelso, M.J.; Yu, H. Urokinase plasminogen activator as an anti-metastasis target: Inhibitor design principles, recent amiloride derivatives, and issues with human/mouse species selectivity. Biophys. Rev. 2022, 1–25. [Google Scholar] [CrossRef]
- Rockway, T.W. Small molecule inhibitors of urokinase-type plasminogen activator. Expert Opin. Ther. Pat. 2003, 13, 773–786. [Google Scholar] [CrossRef]
- Spraggon, G.; Phillips, C.; Nowak, U.K.; Ponting, C.P.; Saunders, D.; Dobson, C.M.; Stuart, D.I.; Jones, E. The crystal structure of the catalytic domain of human urokinase-type plasminogen activator. Structure 1995, 3, 681–691. [Google Scholar] [CrossRef]
- Nienaber, V.L.; Davidson, D.; Edalji, R.; Giranda, V.L.; Klinghofer, V.; Henkin, J.; Magdalinos, P.; Mantei, R.; Merrick, S.; Severin, J.M.; et al. Structure-directed discovery of potent non-peptidic inhibitors of human urokinase that access a novel binding subsite. Structure 2000, 8, 553–563. [Google Scholar] [CrossRef][Green Version]
- Schweinitz, A.; Steinmetzer, T.; Banke, I.J.; Arlt, M.J.E.; Stürzebecher, A.; Schuster, O.; Geissler, A.; Giersiefen, H.; Żesławska, E.; Jacob, U.; et al. Design of Novel and Selective Inhibitors of Urokinase-type Plasminogen Activator with Improved Pharmacokinetic Properties for Use as Antimetastatic Agents. J. Biol. Chem. 2004, 279, 33613–33622. [Google Scholar] [CrossRef] [PubMed]
- Sulimov, V.B.; Katkova, E.V.; Oferkin, I.V.; Sulimov, A.V.; Romanov, A.N.; Roschin, A.I.; Beloglazova, I.B.; Plekhanova, O.S.; Tkachuk, V.A.; Sadovnichiy, V.A. Application of Molecular Modeling to Urokinase Inhibitors Development. BioMed Res. Int. 2014, 2014, 625176. [Google Scholar] [CrossRef] [PubMed]
- Buckley, B.; Aboelela, A.; Minaei, E.; Jiang, L.X.; Xu, Z.; Ali, U.; Fildes, K.; Cheung, C.-Y.; Cook, S.M.; Johnson, D.; et al. 6-Substituted Hexamethylene Amiloride (HMA) Derivatives as Potent and Selective Inhibitors of the Human Urokinase Plasminogen Activator for Use in Cancer. J. Med. Chem. 2018, 61, 8299–8320. [Google Scholar] [CrossRef]
- Buckley, B.; Majed, H.; Aboelela, A.; Minaei, E.; Jiang, L.; Fildes, K.; Cheung, C.-Y.; Johnson, D.; Bachovchin, D.; Cook, G.M.; et al. 6-Substituted amiloride derivatives as inhibitors of the urokinase-type plasminogen activator for use in metastatic disease. Bioorg. Med. Chem. Lett. 2019, 29, 126753. [Google Scholar] [CrossRef] [PubMed]
- Buckley, B.J.; Kumar, A.; Aboelela, A.; Bujaroski, R.S.; Li, X.; Majed, H.; Fliegel, L.; Ranson, M.; Kelso, M.J. Screening of 5- and 6-Substituted Amiloride Libraries Identifies Dual-uPA/NHE1 Active and Single Target-Selective Inhibitors. Int. J. Mol. Sci. 2021, 22, 2999. [Google Scholar] [CrossRef]
- Buckley, B.J.; Aboelela, A.; Majed, H.; Bujaroski, R.S.; White, K.L.; Powell, A.K.; Wang, W.; Katneni, K.; Saunders, J.; Shackleford, D.M.; et al. Systematic evaluation of structure–property relationships and pharmacokinetics in 6-(hetero)aryl-substituted matched pair analogs of amiloride and 5-(N,N-hexamethylene)amiloride. Bioorg. Med. Chem. 2021, 37, 116116. [Google Scholar] [CrossRef]
- National Lung Screening Trial Research Team. Reduced Lung-Cancer Mortality with Low-Dose Computed Tomographic Screening. N. Engl. J. Med. 2011, 365, 395–409. [Google Scholar]
- Corley, D.A.; Jensen, C.D.; Marks, A.; Zhao, W.K.; Lee, J.K.; Doubeni, C.; Zauber, A.G.; De Boer, J.; Fireman, B.H.; Schottinger, J.E.; et al. Adenoma Detection Rate and Risk of Colorectal Cancer and Death. N. Engl. J. Med. 2014, 370, 1298–1306. [Google Scholar] [CrossRef]
- Cuzick, J.; Thorat, M.; Andriole, G.; Brawley, O.W.; Brown, P.H.; Culig, Z.; Eeles, R.; Ford, L.G.; Hamdy, F.C.; Holmberg, L.; et al. Prevention and early detection of prostate cancer. Lancet Oncol. 2014, 15, e484–e492. [Google Scholar] [CrossRef]
- de Koning, H.J.; van der Aalst, C.M.; de Jong, P.A.; Scholten, E.T.; Nackaerts, K.; Heuvelmans, M.A.; Lammers, J.J.; Weenink, C.; Yousaf-Khan, U.; Horeweg, N.; et al. Reduced Lung-Cancer Mortality with Volume CT Screening in a Randomized Trial. N. Engl. J. Med. 2020, 382, 503–513. [Google Scholar] [CrossRef]
- Poruk, K.E.; Firpo, M.A.; Adler, D.G.; Mulvihill, S.J. Screening for pancreatic cancer: Why, how, and who? Ann. Surg. 2013, 257, 17–26. [Google Scholar] [CrossRef]
- Schröder, F.H.; Hugosson, J.; Carlsson, S.; Tammela, T.; Määttänen, L.; Auvinen, A.; Kwiatkowski, M.; Recker, F.; Roobol, M.J. Screening for Prostate Cancer Decreases the Risk of Developing Metastatic Disease: Findings from the European Randomized Study of Screening for Prostate Cancer (ERSPC). Eur. Urol. 2012, 62, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Tabár, L.; Vitak, B.; Chen, T.H.-H.; Yen, A.M.-F.; Cohen, A.; Tot, T.; Chiu, S.Y.-H.; Chen, S.L.-S.; Fann, J.C.-Y.; Rosell, J.; et al. Swedish Two-County Trial: Impact of Mammographic Screening on Breast Cancer Mortality during 3 Decades. Radiology 2011, 260, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.S.; Lee, J.M. Imaging diagnosis of pancreatic cancer: A state-of-the-art review. World J. Gastroenterol. 2014, 20, 7864–7877. [Google Scholar] [CrossRef]
- Harinck, F.; Konings, I.C.A.W.; Kluijt, I.; Poley, J.W.; Van Hooft, J.E.; Van Dullemen, H.M.; Nio, C.Y.; Krak, N.C.; Hermans, J.J.; Aalfs, C.M.; et al. A multicentre comparative prospective blinded analysis of EUS and MRI for screening of pancreatic cancer in high-risk individuals. Gut 2016, 65, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Singhi, A.D.; Koay, E.J.; Chari, S.T.; Maitra, A. Early Detection of Pancreatic Cancer: Opportunities and Challenges. Gastroenterology 2019, 156, 2024–2040. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y. Introduction; Value of Endoscopic Ultrasound-Guided Fine Needle Aspiration. Clin. Endosc. 2012, 45, 115–116. [Google Scholar] [CrossRef]
- Narkhede, R.A.; Desai, G.S.; Prasad, P.P.; Wagle, P.K. Diagnosis and Management of Pancreatic Adenocarcinoma in the Background of Chronic Pancreatitis: Core Issues. Dig. Dis. 2019, 37, 315–324. [Google Scholar] [CrossRef]
- Tummers, W.S.; Willmann, J.K.; Bonsing, B.A.; Vahrmeijer, A.L.; Gambhir, S.S.; Swijnenburg, R.-J. Advances in Diagnostic and Intraoperative Molecular Imaging of Pancreatic Cancer. Pancreas 2018, 47, 675–689. [Google Scholar] [CrossRef]
- Yang, D.; Severin, G.W.; Dougherty, C.A.; Lombardi, R.; Chen, D.; Van Dort, M.E.; Barnhart, T.E.; Ross, B.D.; Mazar, A.P.; Hong, H. Antibody-based PET of uPA/uPAR signaling with broad applicability for cancer imaging. Oncotarget 2016, 7, 73912–73924. [Google Scholar] [CrossRef]
- Kryza, T.; Khan, T.; Puttick, S.; Li, C.; Sokolowski, K.A.; Tse, B.W.; Cuda, T.; Lyons, N.; Gough, M.; Yin, J.; et al. Effective targeting of intact and proteolysed CDCP1 for imaging and treatment of pancreatic ductal adenocarcinoma. Theranostics 2020, 10, 4116–4133. [Google Scholar] [CrossRef]
- Moroz, A.; Wang, Y.-H.; Sharib, J.M.; Wei, J.; Zhao, N.; Huang, Y.; Chen, Z.; Martinko, A.J.; Zhuo, J.; Lim, S.A.; et al. Theranostic Targeting of CUB Domain Containing Protein 1 (CDCP1) in Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 3608–3615. [Google Scholar] [CrossRef]
- Miyazawa, Y.; Uekita, T.; Hiraoka, N.; Fujii, S.; Kosuge, T.; Kanai, Y.; Nojima, Y.; Sakai, R. CUB Domain–Containing Protein 1, a Prognostic Factor for Human Pancreatic Cancers, Promotes Cell Migration and Extracellular Matrix Degradation. Cancer Res. 2010, 70, 5136–5146. [Google Scholar] [CrossRef]
- Khan, T.; Kryza, T.; Lyons, N.J.; He, Y.; Hooper, J.D. The CDCP1 Signaling Hub: A Target for Cancer Detection and Therapeutic Intervention. Cancer Res. 2021, 81, 2259–2269. [Google Scholar] [CrossRef]
- Kryza, T.; Khan, T.; Lovell, S.; Harrington, B.S.; Yin, J.; Porazinski, S.; Pajic, M.; Koistinen, H.; Rantala, J.K.; Dreyer, T.; et al. Substrate-biased activity-based probes identify proteases that cleave receptor CDCP1. Nat. Chem. Biol. 2021, 17, 776–783. [Google Scholar] [CrossRef]
- Persson, M.; Madsen, J.; Østergaard, S.; Jensen, M.M.; Jørgensen, J.T.; Juhl, K.; Lehmann, C.; Ploug, M.; Kjaer, A. Quantitative PET of Human Urokinase-Type Plasminogen Activator Receptor with 64Cu-DOTA-AE105: Implications for Visualizing Cancer Invasion. J. Nucl. Med. 2012, 53, 138–145. [Google Scholar] [CrossRef]
- Persson, M.; Skovgaard, D.; Brandt-Larsen, M.; Christensen, C.; Madsen, J.; Nielsen, C.H.; Thurison, T.; Klausen, T.L.; Holm, S.; Loft, A.; et al. First-in-human uPAR PET: Imaging of Cancer Aggressiveness. Theranostics 2015, 5, 1303–1316. [Google Scholar] [CrossRef]
- Persson, M.; Kjaer, A. Urokinase-type plasminogen activator receptor (uPAR) as a promising new imaging target: Potential clinical applications. Clin. Physiol. Funct. Imaging 2013, 33, 329–337. [Google Scholar] [CrossRef]
- Persson, M.; Liu, H.; Madsen, J.; Cheng, Z.; Kjaer, A. First 18F-labeled ligand for PET imaging of uPAR: In vivo studies in human prostate cancer xenografts. Nucl. Med. Biol. 2013, 40, 618–624. [Google Scholar] [CrossRef]
- Skovgaard, D.; Persson, M.; Brandt-Larsen, M.; Christensen, C.; Madsen, J.; Klausen, T.L.; Holm, S.; Andersen, F.L.; Loft, A.; Berthelsen, A.K.; et al. Safety, Dosimetry, and Tumor Detection Ability of 68 Ga-NOTA-AE105: First-in-Human Study of a Novel Radioligand for uPAR PET Imaging. J. Nucl. Med. 2017, 58, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Hong, G.; Antaris, A.L.; Dai, H. Near-infrared fluorophores for biomedical imaging. Nat. Biomed. Eng. 2017, 1, 1–22. [Google Scholar] [CrossRef]
- Juhl, K.; Christensen, A.; Rubek, N.; Karnov, K.K.S.; Von Buchwald, C.; Kjaer, A. Improved surgical resection of metastatic pancreatic cancer using uPAR targeted in vivo fluorescent guidance: Comparison with traditional white light surgery. Oncotarget 2019, 10, 6308–6316. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, A.A.; Buckley, B.J.; Ranson, M. The Urokinase Plasminogen Activation System in Pancreatic Cancer: Prospective Diagnostic and Therapeutic Targets. Biomolecules 2022, 12, 152. https://doi.org/10.3390/biom12020152
Kumar AA, Buckley BJ, Ranson M. The Urokinase Plasminogen Activation System in Pancreatic Cancer: Prospective Diagnostic and Therapeutic Targets. Biomolecules. 2022; 12(2):152. https://doi.org/10.3390/biom12020152
Chicago/Turabian StyleKumar, Ashna A., Benjamin J. Buckley, and Marie Ranson. 2022. "The Urokinase Plasminogen Activation System in Pancreatic Cancer: Prospective Diagnostic and Therapeutic Targets" Biomolecules 12, no. 2: 152. https://doi.org/10.3390/biom12020152
APA StyleKumar, A. A., Buckley, B. J., & Ranson, M. (2022). The Urokinase Plasminogen Activation System in Pancreatic Cancer: Prospective Diagnostic and Therapeutic Targets. Biomolecules, 12(2), 152. https://doi.org/10.3390/biom12020152