

Linking the Amyloid, Tau, and Mitochondrial Hypotheses of Alzheimer’s Disease and Identifying Promising Drug Targets

Abstract

1. Introduction
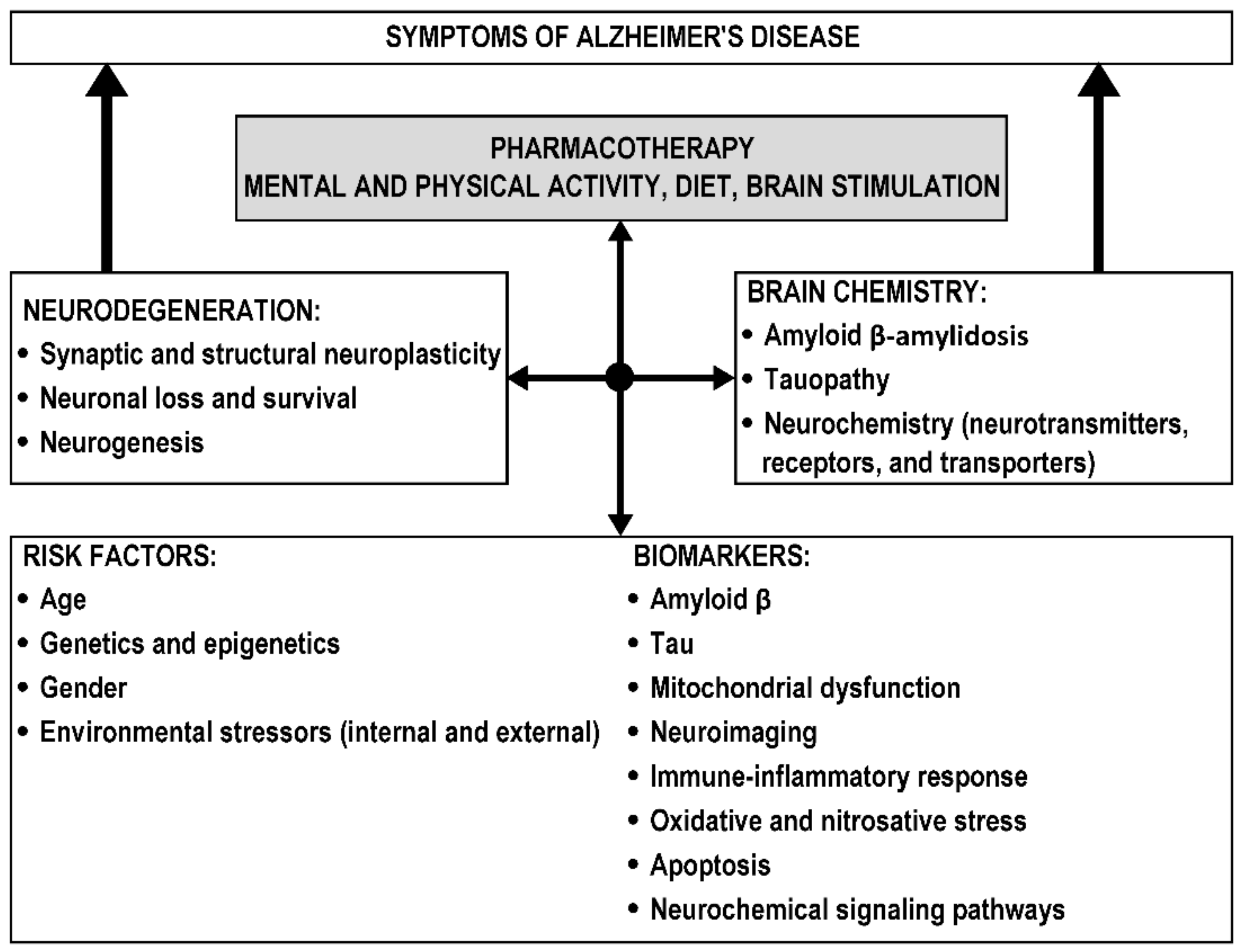
2. Risk Factors and Biomarkers of Alzheimer’s Disease
2.1. Risk Factors
2.1.1. Environmental
2.1.2. Genetic
2.2. Biomarkers
2.2.1. CSF and Neuroimaging
2.2.2. Blood-Based
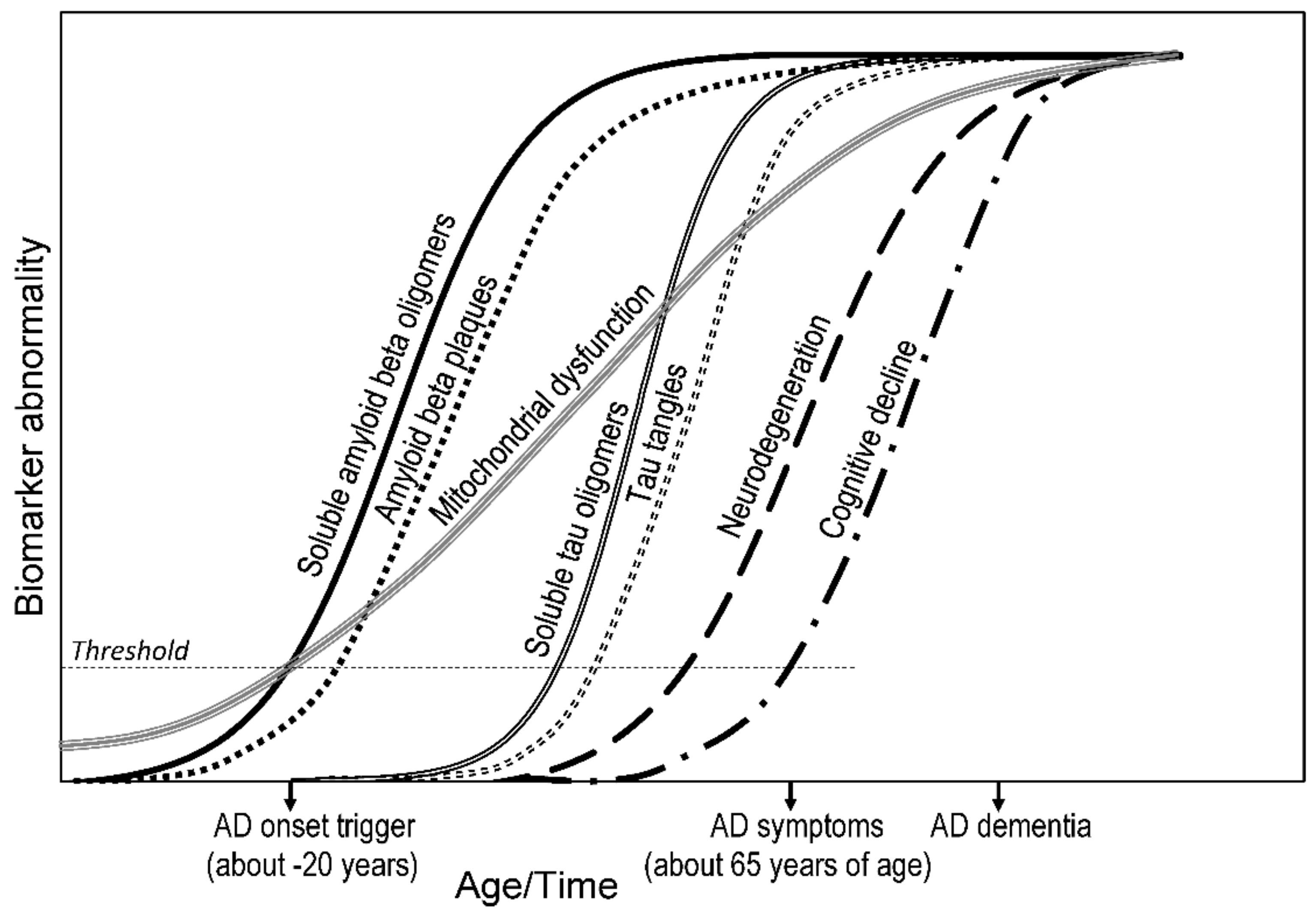
2.3. Time Course of Biomarkers

2.3.1. Clinical View
2.3.2. Research View
3. Hypotheses of Alzheimer’s Disease
3.1. Amyloid Hypothesis
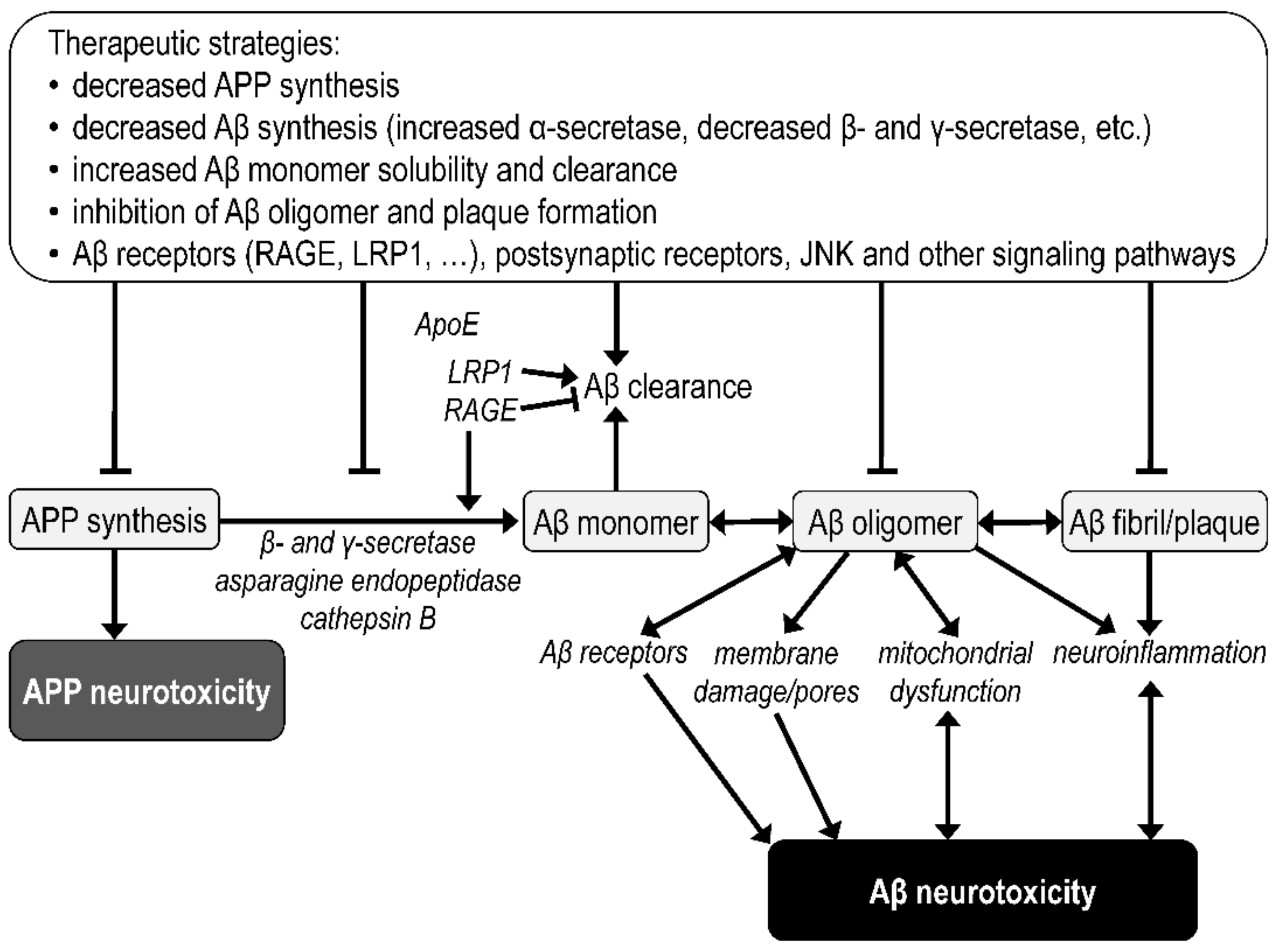
3.2. Amyloid Beta Pathology
3.3. Tau Hypothesis
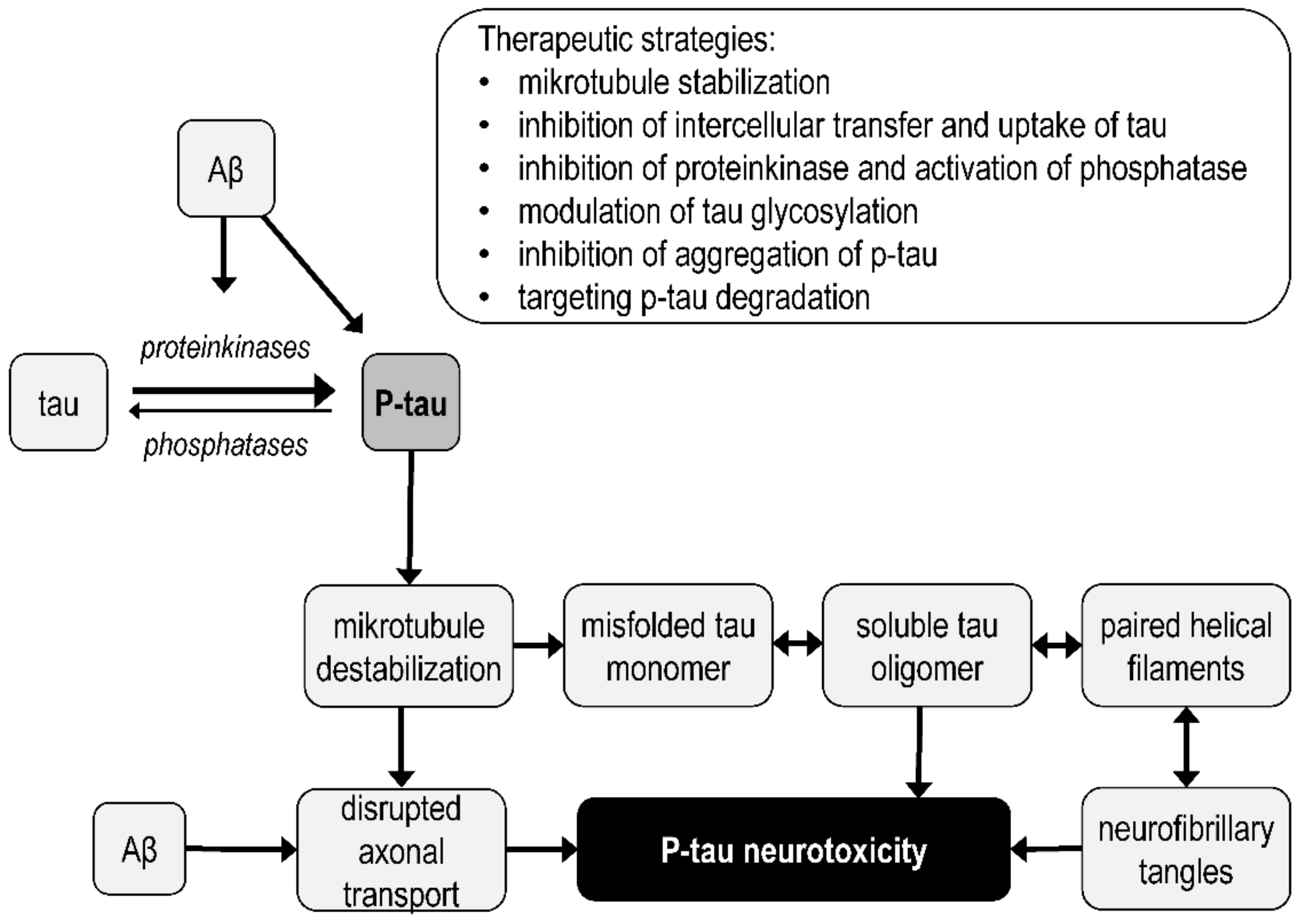
3.4. Tau Protein Pathology
3.5. Mitochondrial Hypothesis
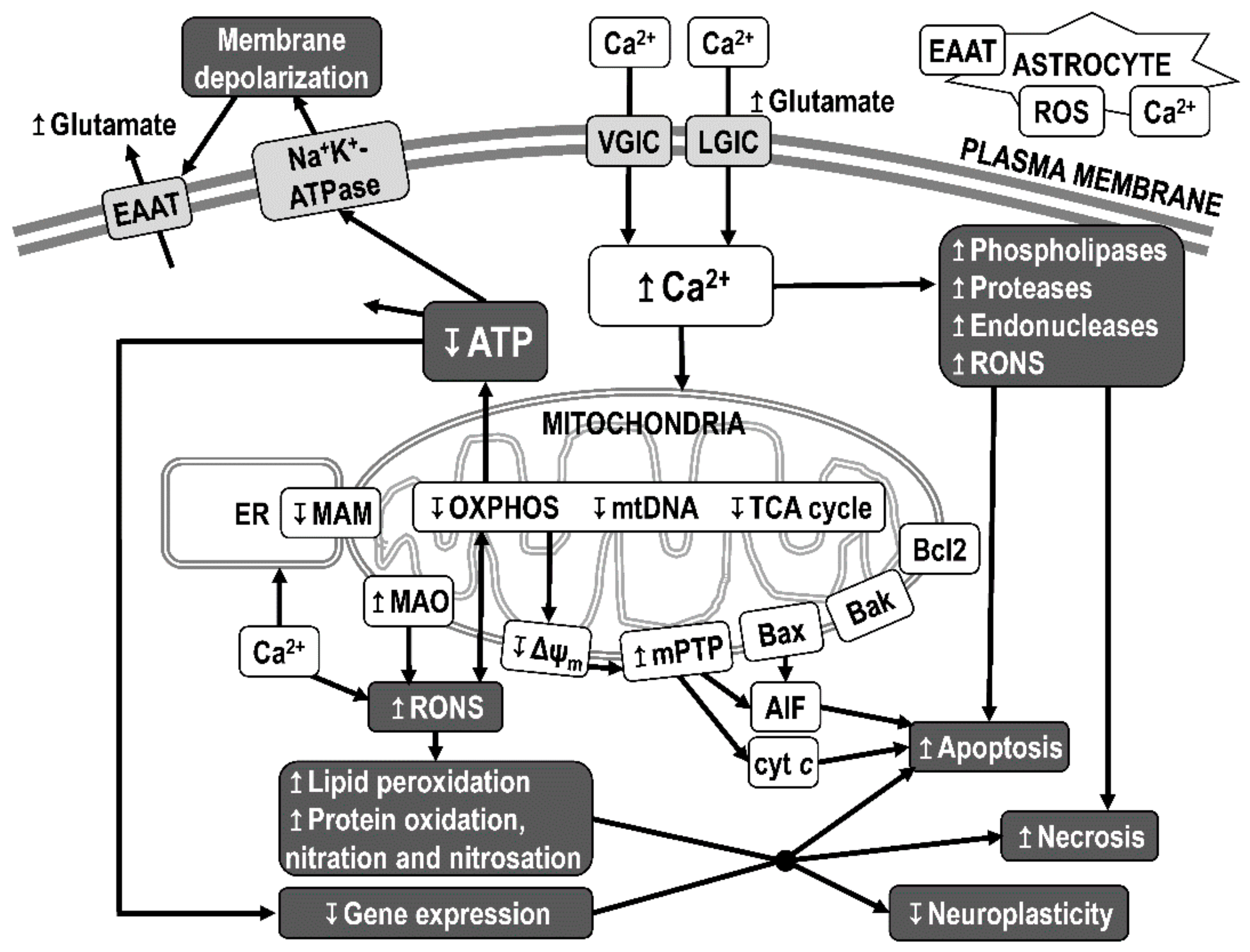
3.6. Mitochondrial Dysfunction
3.7. Synaptoplasticity Hypothesis
3.8. Neuroinflammation
3.9. Metabolic Dysregulation
3.10. Protein Degradation Deficiency
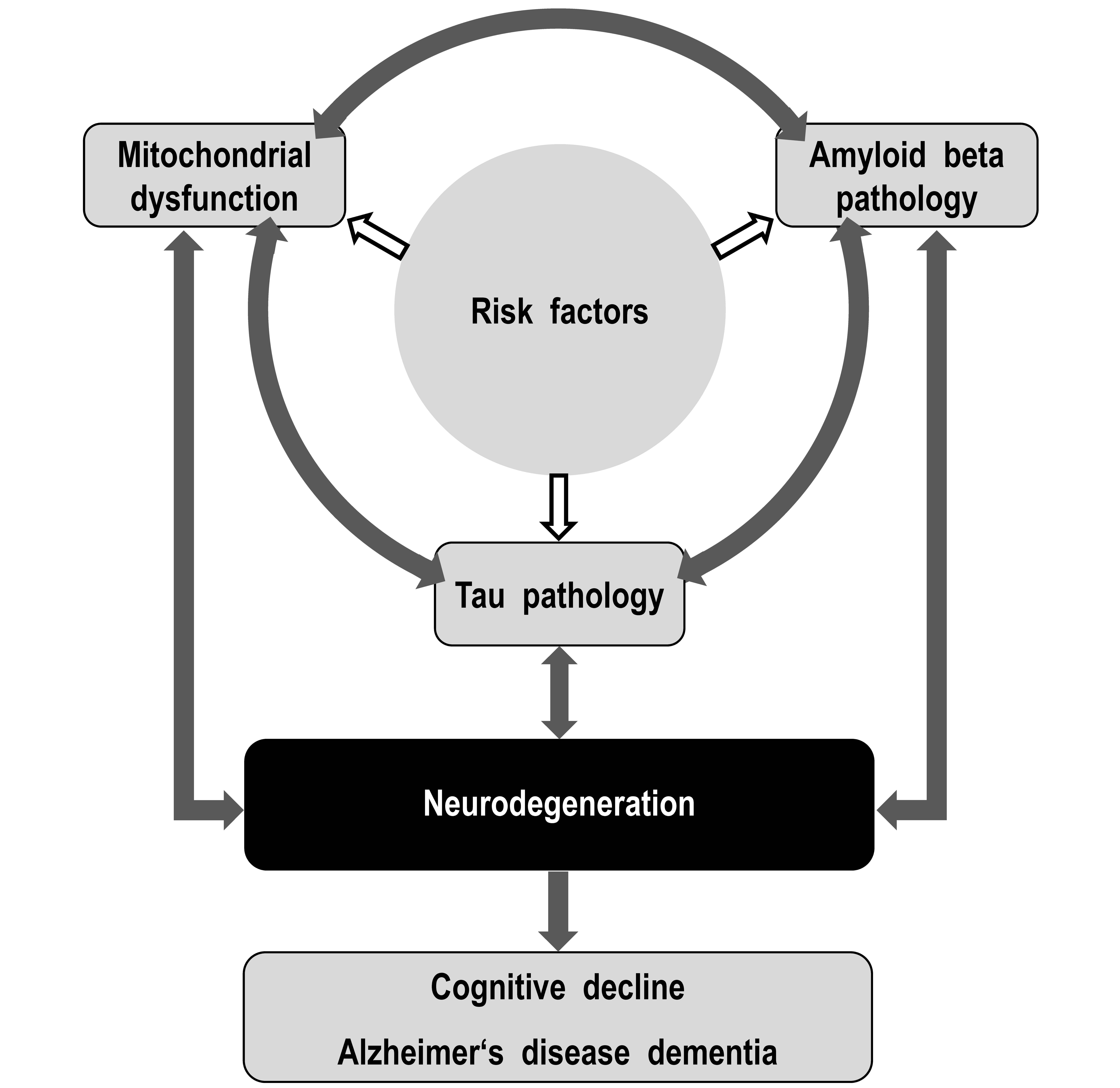
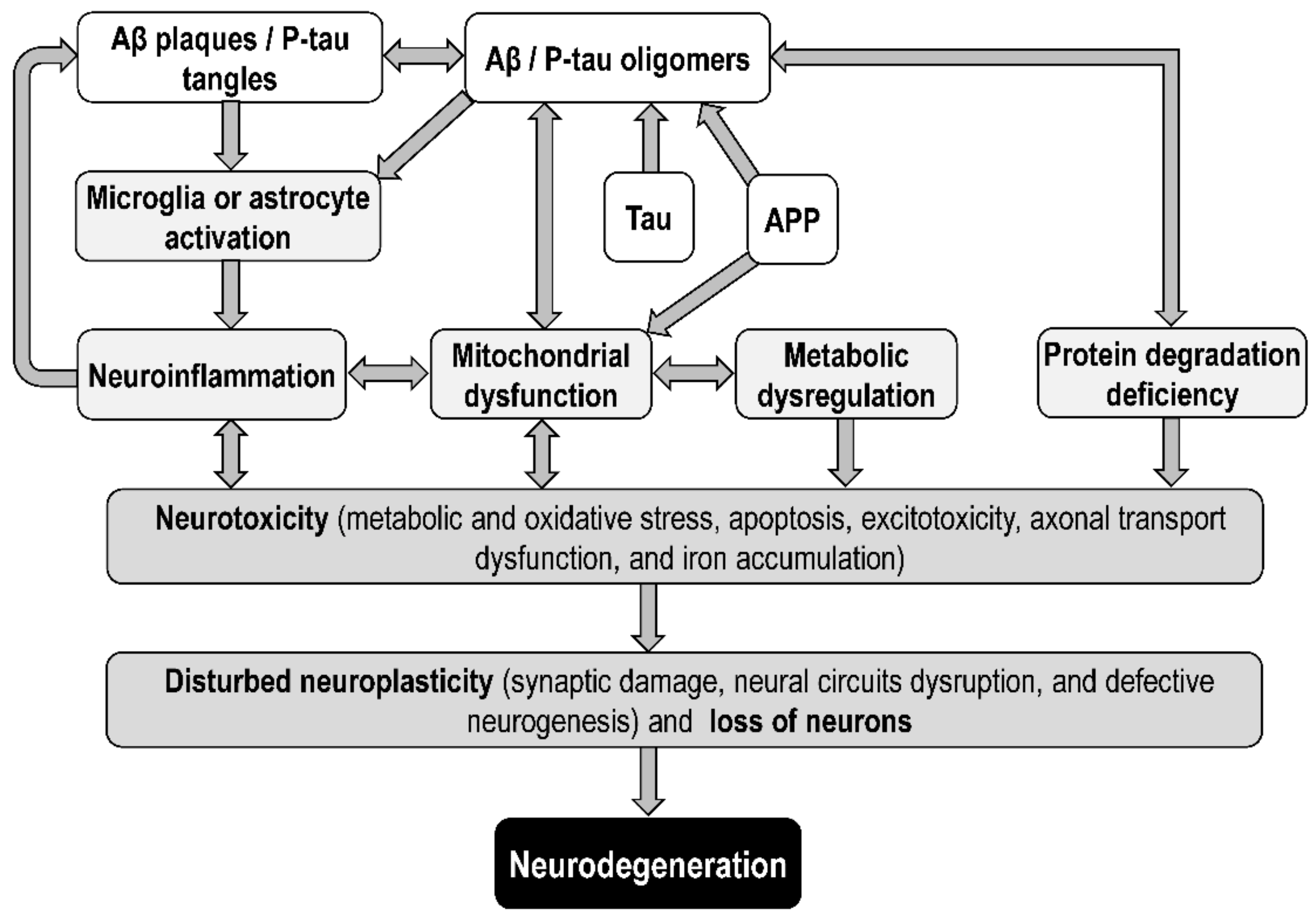
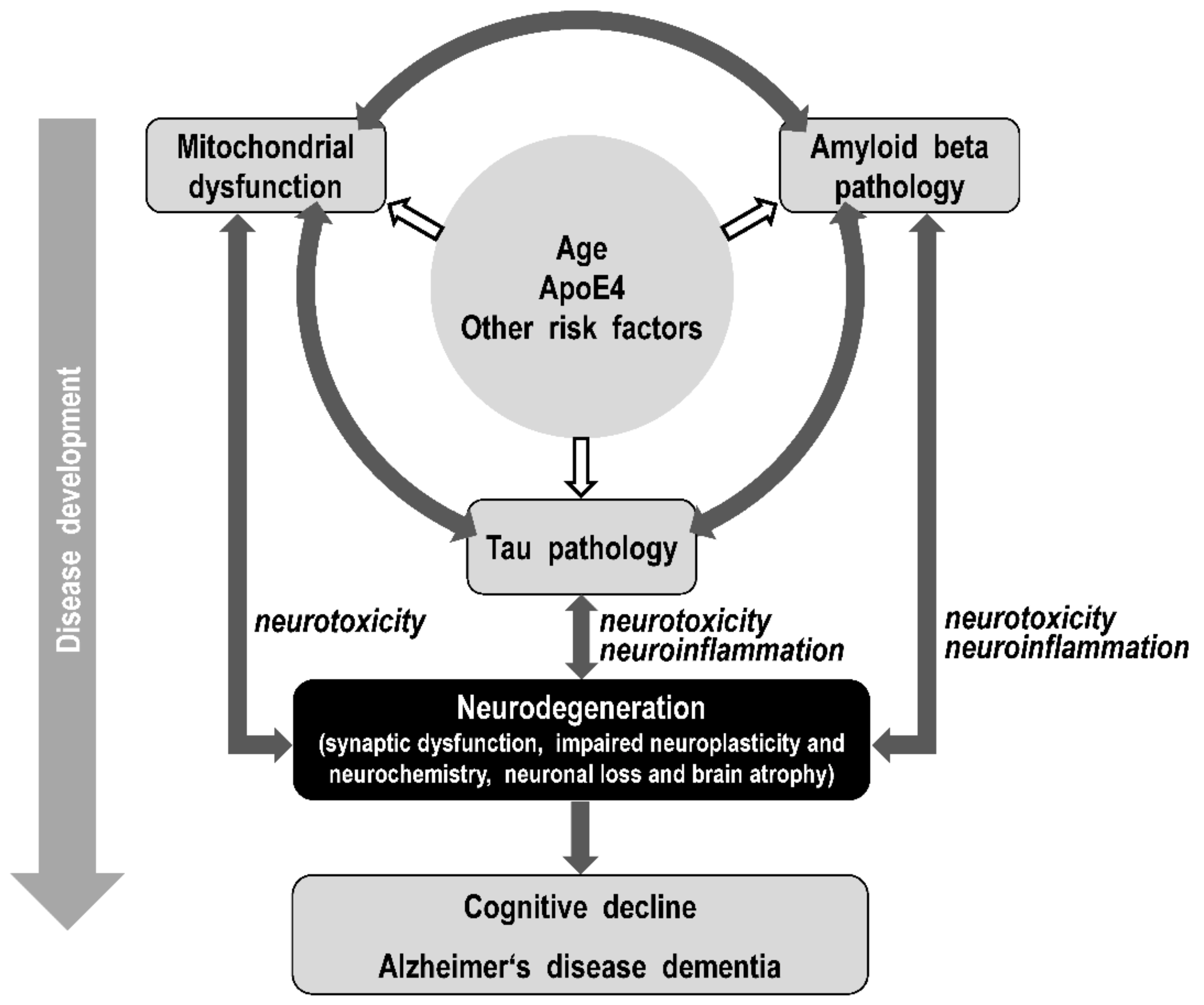
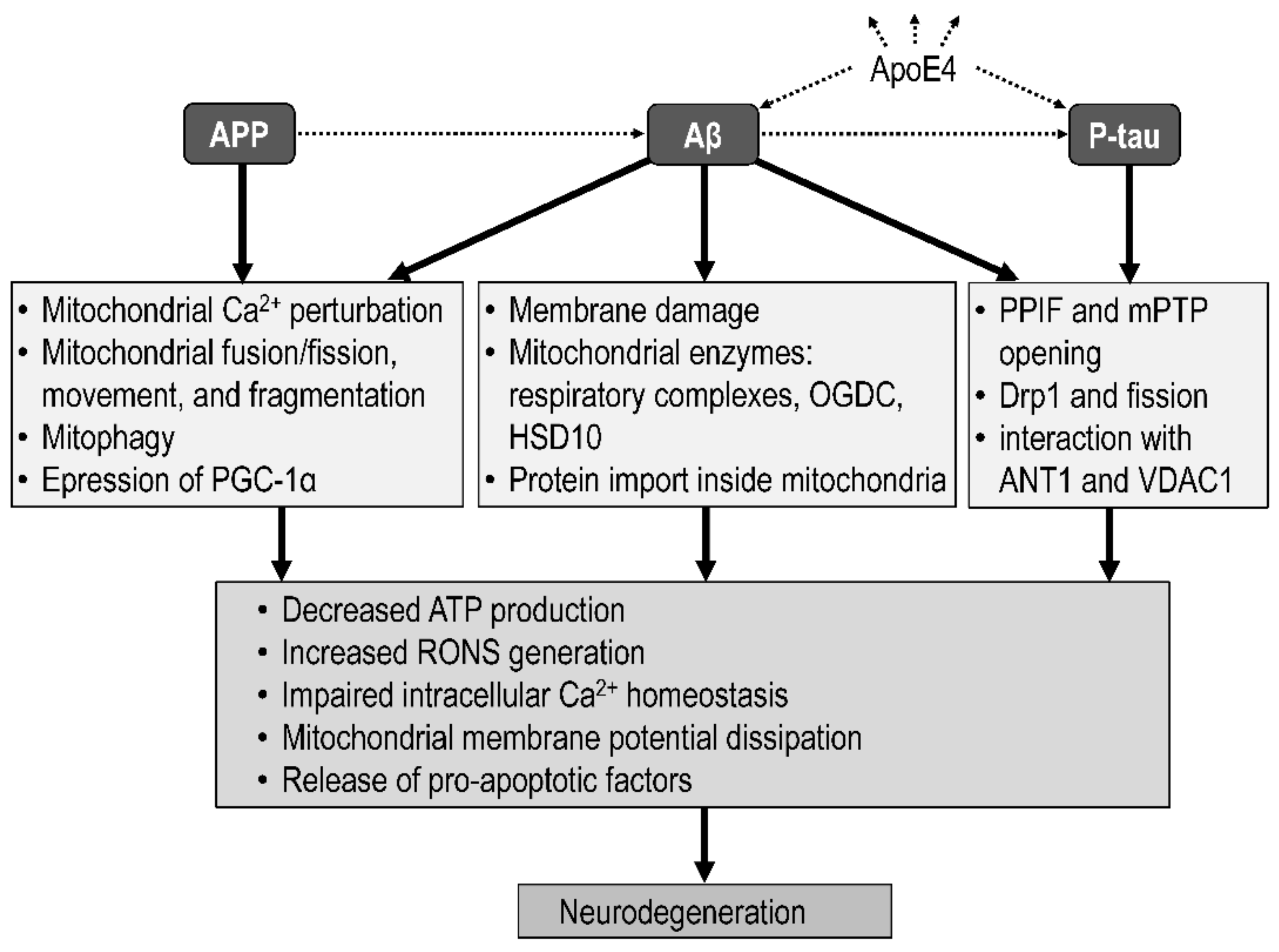
4. Linking the Amyloid, Tau, and the Mitochondrial Hypotheses of Alzheimer’s Disease
4.1. Integrative Models
4.2. Mitochondrial Targets of Amyloid Beta
4.3. Mitochondrial Targets of Tau
5. Alzheimer’s Drugs
5.1. Cellular Drug Targets
5.2. Approved Drugs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Primary Action Included | Reference |
|---|---|---|
| Rivastigmine | Cholinesterase inhibition | [351] |
| Galantamine | [352] | |
| Donepezil | [352] | |
| Memantine | Blockade of NMDA receptor | [353] |
| Aducanumab * | Monoclonal antibody directed at brain Aβ plaques and oligomers | [349] |
5.3. Drug Candidates
| Drug | Primary Brain Action Included | Clinical Trials | Reference |
|---|---|---|---|
| Disease-modifying molecules | |||
| Metformin | Improving of glucose metabolism; mitochondrial complex I inhibition | Phase 3 | [354,355] |
| Semaglutide | Glucagon-like peptide-1 agonism; improving of glycemic control; anti-inflammation | Phase 3 | [356] |
| Tricaprylin | Ketosis and improving of mitochondrial function | Phase 3 | [357,358] |
| Omega-3 PUFA | Anti-inflammation; antioxidant; synaptic plasticity; cerebrovascular function; blood flow | Phase 3 | [359] |
| Icosapent ethyl | Phase 3 | [360] | |
| Blarcamesine | σ1 receptor agonism; reduction of Aβ and NFTs; anti-inflammation; amelioration of mitochondrial dysfunction and oxidative stress; antiapoptotic; induction of neurogenesis | Phase 3 | [361,362] |
| Atuzaginstat | Inhibition of gingipains; reduction of neurodegeneration and neuroinflammation | Phase 3 | [363] |
| AGB101 | Inhibition of SV2A; reduction of Aβ pathology | Phase 3 | [364,365] |
| Simufilam | Reduction of P-tau and Aβ aggregates; reduction of α7 nicotinic acetylcholine, NMDA, and insulin receptor dysfunction | Phase 3 | [366] |
| Homotaurine (ALZ-801) | Inhibition of Aβ aggregation; GABAA receptor agonism | Phase 3 | [367,368] |
| NE3107 | Anti-inflammation | Phase 3 | [369] |
| Curcumin | Anti-inflammation; antioxidant; dual inhibition of Aβ and tau aggregation | Phase 2 | [337,371,372,373] |
| Gantenerumab | Monoclonal antibody directed at brain Aβ | Phase 3 | [374,375] |
| Lecanemab | Phase 3 | [375] | |
| Donanemab | Phase 3 | [376] | |
| Solanezumab | Phase 3 | [374,377] | |
| Neuropsychiatric drugs | |||
| Escitalopram | Selective serotonin reuptake inhibition | Phase 3 | [385] |
| Brexpiprazole | D2 receptor antagonism | Phase 3 | [386] |
| Dextromethorphan/bupropion | NMDA receptor agonism | Phase 3 | [387] |
| Deudextromethorphan/quinidine | Agonism σ1 and antagonism NMDA receptor; serotonin–norepinephrine reuptake inhibition | Phase 3 | [388] |
| Nabilone | Cannabinoid receptors agonism | Phase 3 | [389] |
| Dronabinol | Phase 2 | [389] | |
| Cannabidiol | Phase 2 | [389,390] | |
| Cognitive enhancers | |||
| Caffeine | Antagonism of adenosine A2A receptor; mitochondrial function | Phase 3 | [378,379] |
| Guanfacine | α2A-adrenergic receptor agonism | Phase 3 | [380] |
| Octohydroaminoacridine succinate | Acetylcholinesterase inhibition | Phase 3 | [381] |
| Nicotine | Nicotinic acetylcholine receptor agonism | Phase 2 | [382,383] |
| AD-35 | Acetylcholinesterase inhibition; disassembly of Aβ aggregates | Phase 2 | [384] |
5.4. Supplements
6. Conclusions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Pereira, C.F.; Santos, A.E.; Moreira, P.I.; Pereira, A.C.; Sousa, F.J.; Cardoso, S.M.; Cruz, M.T. Is Alzheimer’s disease an inflammasomopathy? Ageing Res. Rev. 2019, 56, 100966. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gu, B.J.; Masters, C.L.; Wang, Y.J. A systemic view of Alzheimer disease—Insights from amyloid-β metabolism beyond the brain. Nat. Rev. Neurol. 2017, 13, 703. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Klug, A.; Crowther, R.A. Tau protein, the paired helical filament and Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta 2010, 1802, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Plascencia-Villa, G.; Perry, G. Neuropathologic Changes Provide Insights into Key Mechanisms Related to Alzheimer Disease and Related Dementia. Am. J. Pathol. 2022, 192, 1340–1346. [Google Scholar] [CrossRef]
- Hashimoto, M.; Rockenstein, E.; Crews, L.; Masliah, E. Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer’s and Parkinson’s diseases. Neuromol. Med. 2003, 4, 21–36. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Guerrero-Munoz, M.J.; Kiritoshi, T.; Neugebauer, V.; Jackson, G.R.; Kayed, R. Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci. Rep. 2012, 2, 700. [Google Scholar] [CrossRef]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef]
- Balducci, C.; Beeg, M.; Stravalaci, M.; Bastone, A.; Sclip, A.; Biasini, E.; Tapella, L.; Colombo, L.; Manzoni, C.; Borsello, T.; et al. Synthetic amyloid-β oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. USA 2010, 107, 2295–2300. [Google Scholar] [CrossRef]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimer’s Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef]
- Hartley, D.M.; Walsh, D.M.; Ye, C.P.; Diehl, T.; Vasquez, S.; Vassilev, P.M.; Teplow, D.B.; Selkoe, D.J. Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J. Neurosci. 1999, 19, 8876–8884. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Griffiths, J.; Grant, S.G.N. Synapse Pathology in Alzheimer’s Disease. Semin. Cell Dev. Biol. 2022; in press. [Google Scholar] [CrossRef]
- Forloni, G.; Artuso, V.; La Vitola, P.; Balducci, C. Oligomeropathies and pathogenesis of Alzheimer and Parkinson’s diseases. Mov. Disord. 2016, 31, 771–781. [Google Scholar] [CrossRef]
- Tasaki, M.; Ueda, M.; Ochiai, S.; Tanabe, Y.; Murata, S.; Misumi, Y.; Su, Y.; Sun, X.; Shinriki, S.; Jono, H.; et al. Transmission of circulating cell-free AA amyloid oligomers in exosomes vectors via a prion-like mechanism. Biochem. Biophys. Res. Commun. 2010, 400, 559–562. [Google Scholar] [CrossRef]
- Bloom, G.S. Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef]
- Swardfager, W.; Lanctot, K.; Rothenburg, L.; Wong, A.; Cappell, J.; Herrmann, N. A meta-analysis of cytokines in Alzheimer’s disease. Biol. Psychiatry 2010, 68, 930–941. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxid. Med. Cell. Longev. 2017, 2017, 2525967. [Google Scholar] [CrossRef]
- Bhatia, V.; Sharma, S. Role of mitochondrial dysfunction, oxidative stress and autophagy in progression of Alzheimer’s disease. J. Neurol. Sci. 2021, 421, 117253. [Google Scholar] [CrossRef]
- Demetrius, L.A.; Driver, J. Alzheimer’s as a metabolic disease. Biogerontology 2013, 14, 641–649. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M.; Tong, M. Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Bonda, D.J.; Wang, X.; Lee, H.G.; Smith, M.A.; Perry, G.; Zhu, X. Neuronal failure in Alzheimer’s disease: A view through the oxidative stress looking-glass. Neurosci. Bull. 2014, 30, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, T.; Li, P.; Wei, N.; Zhao, Z.; Liang, H.; Ji, X.; Chen, W.; Xue, M.; Wei, J. The Ambiguous Relationship of Oxidative Stress, Tau Hyperphosphorylation, and Autophagy Dysfunction in Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2015, 2015, 352723. [Google Scholar] [CrossRef]
- Eckert, A.; Schmitt, K.; Gotz, J. Mitochondrial dysfunction—The beginning of the end in Alzheimer’s disease? Separate and synergistic modes of tau and amyloid-β toxicity. Alzheimer’s Res. Ther. 2011, 3, 15. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef]
- Bateman, R.J.; Aisen, P.S.; De Strooper, B.; Fox, N.C.; Lemere, C.A.; Ringman, J.M.; Salloway, S.; Sperling, R.A.; Windisch, M.; Xiong, C. Autosomal-dominant Alzheimer’s disease: A review and proposal for the prevention of Alzheimer’s disease. Alzheimer’s Res. Ther. 2011, 3, 1. [Google Scholar] [CrossRef]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef]
- Hroudova, J.; Singh, N.; Fisar, Z.; Ghosh, K.K. Progress in drug development for Alzheimer’s disease: An overview in relation to mitochondrial energy metabolism. Eur. J. Med. Chem. 2016, 121, 774–784. [Google Scholar] [CrossRef]
- Atkinson, A.J.; Colburn, W.A.; DeGruttola, V.G.; DeMets, D.L.; Downing, G.J.; Hoth, D.F.; Oates, J.A.; Peck, C.C.; Schooley, R.T.; Spilker, B.A.; et al. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar] [CrossRef]
- Yu, L.; Boyle, P.A.; Segawa, E.; Leurgans, S.; Schneider, J.A.; Wilson, R.S.; Bennett, D.A. Residual decline in cognition after adjustment for common neuropathologic conditions. Neuropsychology 2015, 29, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Bennett, D.A.; Buchman, A.S.; Boyle, P.A.; Barnes, L.L.; Wilson, R.S.; Schneider, J.A. Religious Orders Study and Rush Memory and Aging Project. J. Alzheimer’s Dis. 2018, 64, S161–S189. [Google Scholar] [CrossRef] [PubMed]
- Van der Flier, W.M.; Scheltens, P. Epidemiology and risk factors of dementia. J. Neurol. Neurosurg. Psychiatry 2005, 76 (Suppl. S5), v2–v7. [Google Scholar] [CrossRef] [PubMed]
- Brookmeyer, R.; Gray, S.; Kawas, C. Projections of Alzheimer’s disease in the United States and the public health impact of delaying disease onset. Am. J. Public Health 1998, 88, 1337–1342. [Google Scholar] [CrossRef]
- Andrews, S.J.; Fulton-Howard, B.; Goate, A. Interpretation of risk loci from genome-wide association studies of Alzheimer’s disease. Lancet Neurol. 2020, 19, 326–335. [Google Scholar] [CrossRef]
- Morris, J.C.; Roe, C.M.; Xiong, C.; Fagan, A.M.; Goate, A.M.; Holtzman, D.M.; Mintun, M.A. APOE predicts amyloid-β but not tau Alzheimer pathology in cognitively normal aging. Ann. Neurol. 2010, 67, 122–131. [Google Scholar] [CrossRef]
- Troutwine, B.R.; Hamid, L.; Lysaker, C.R.; Strope, T.A.; Wilkins, H.M. Apolipoprotein E and Alzheimer’s disease. Acta Pharm. Sin. B 2022, 12, 496–510. [Google Scholar] [CrossRef]
- Ramos-Cejudo, J.; Wisniewski, T.; Marmar, C.; Zetterberg, H.; Blennow, K.; de Leon, M.J.; Fossati, S. Traumatic Brain Injury and Alzheimer’s Disease: The Cerebrovascular Link. EBioMedicine 2018, 28, 21–30. [Google Scholar] [CrossRef]
- Bellou, V.; Belbasis, L.; Tzoulaki, I.; Middleton, L.T.; Ioannidis, J.P.A.; Evangelou, E. Systematic evaluation of the associations between environmental risk factors and dementia: An umbrella review of systematic reviews and meta-analyses. Alzheimer’s Dement. 2017, 13, 406–418. [Google Scholar] [CrossRef]
- Gaugler, J.; James, B.; Johnson, T.; Reimer, J.; Solis, M.; Weuve, J.; Buckley, R.F.; Hohman, T.J. Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2022, 18, 700–789. [Google Scholar] [CrossRef]
- Ott, A.; Breteler, M.M.; van Harskamp, F.; Claus, J.J.; van der Cammen, T.J.; Grobbee, D.E.; Hofman, A. Prevalence of Alzheimer’s disease and vascular dementia: Association with education. The Rotterdam study. BMJ 1995, 310, 970–973. [Google Scholar] [CrossRef]
- Evans, D.A.; Hebert, L.E.; Beckett, L.A.; Scherr, P.A.; Albert, M.S.; Chown, M.J.; Pilgrim, D.M.; Taylor, J.O. Education and other measures of socioeconomic status and risk of incident Alzheimer disease in a defined population of older persons. Arch. Neurol. 1997, 54, 1399–1405. [Google Scholar] [CrossRef]
- Flicker, L. Modifiable lifestyle risk factors for Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 20, 803–811. [Google Scholar] [CrossRef]
- Douros, A.; Santella, C.; Dell’Aniello, S.; Azoulay, L.; Renoux, C.; Suissa, S.; Brassard, P. Infectious Disease Burden and the Risk of Alzheimer’s Disease: A Population-Based Study. J. Alzheimer’s Dis. 2021, 81, 329–338. [Google Scholar] [CrossRef]
- Hofman, A.; Ott, A.; Breteler, M.M.; Bots, M.L.; Slooter, A.J.; van Harskamp, F.; van Duijn, C.N.; Van Broeckhoven, C.; Grobbee, D.E. Atherosclerosis, apolipoprotein E, and prevalence of dementia and Alzheimer’s disease in the Rotterdam Study. Lancet 1997, 349, 151–154. [Google Scholar] [CrossRef]
- Luchsinger, J.A.; Reitz, C.; Honig, L.S.; Tang, M.X.; Shea, S.; Mayeux, R. Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology 2005, 65, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, C.M. Type 2 diabetes mellitus, dyslipidemia, and Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 20, 711–722. [Google Scholar] [CrossRef]
- Patel, V.N.; Chorawala, M.R.; Shah, M.B.; Shah, K.C.; Dave, B.P.; Shah, M.P.; Patel, T.M. Emerging Pathophysiological Mechanisms Linking Diabetes Mellitus and Alzheimer’s Disease: An Old Wine in a New Bottle. J. Alzheimer’s Dis. Rep. 2022, 6, 349–357. [Google Scholar] [CrossRef]
- Yan, X.; Hu, Y.; Wang, B.; Wang, S.; Zhang, X. Metabolic Dysregulation Contributes to the Progression of Alzheimer’s Disease. Front. Neurosci. 2020, 14, 530219. [Google Scholar] [CrossRef]
- Nelson, P.T.; Head, E.; Schmitt, F.A.; Davis, P.R.; Neltner, J.H.; Jicha, G.A.; Abner, E.L.; Smith, C.D.; Van Eldik, L.J.; Kryscio, R.J.; et al. Alzheimer’s disease is not “brain aging”: Neuropathological, genetic, and epidemiological human studies. Acta Neuropathol. 2011, 121, 571–587. [Google Scholar] [CrossRef] [PubMed]
- Corral-Debrinski, M.; Horton, T.; Lott, M.T.; Shoffner, J.M.; Beal, M.F.; Wallace, D.C. Mitochondrial DNA deletions in human brain: Regional variability and increase with advanced age. Nat. Genet. 1992, 2, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Ojaimi, J.; Masters, C.L.; McLean, C.; Opeskin, K.; McKelvie, P.; Byrne, E. Irregular distribution of cytochrome c oxidase protein subunits in aging and Alzheimer’s disease. Ann. Neurol. 1999, 46, 656–660. [Google Scholar] [CrossRef]
- Weidling, I.; Swerdlow, R.H. Mitochondrial Dysfunction and Stress Responses in Alzheimer’s Disease. Biology 2019, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Weidling, I.W.; Swerdlow, R.H. Mitochondria in Alzheimer’s disease and their potential role in Alzheimer’s proteostasis. Exp. Neurol. 2020, 330, 113321. [Google Scholar] [CrossRef]
- Mielke, M.M.; Vemuri, P.; Rocca, W.A. Clinical epidemiology of Alzheimer’s disease: Assessing sex and gender differences. Clin. Epidemiol. 2014, 6, 37–48. [Google Scholar] [CrossRef]
- Snyder, H.M.; Asthana, S.; Bain, L.; Brinton, R.; Craft, S.; Dubal, D.B.; Espeland, M.A.; Gatz, M.; Mielke, M.M.; Raber, J.; et al. Sex biology contributions to vulnerability to Alzheimer’s disease: A think tank convened by the Women’s Alzheimer’s Research Initiative. Alzheimer’s Dement. 2016, 12, 1186–1196. [Google Scholar] [CrossRef]
- Demetrius, L.A.; Eckert, A.; Grimm, A. Sex differences in Alzheimer’s disease: Metabolic reprogramming and therapeutic intervention. Trends Endocrinol. Metab. 2021, 32, 963–979. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Samieri, C.; Perier, M.C.; Gaye, B.; Proust-Lima, C.; Helmer, C.; Dartigues, J.F.; Berr, C.; Tzourio, C.; Empana, J.P. Association of Cardiovascular Health Level in Older Age with Cognitive Decline and Incident Dementia. JAMA 2018, 320, 657–664. [Google Scholar] [CrossRef]
- Ogino, E.; Manly, J.J.; Schupf, N.; Mayeux, R.; Gu, Y. Current and past leisure time physical activity in relation to risk of Alzheimer’s disease in older adults. Alzheimer’s Dement. 2019, 15, 1603–1611. [Google Scholar] [CrossRef]
- Morris, M.C.; Tangney, C.C.; Wang, Y.; Sacks, F.M.; Bennett, D.A.; Aggarwal, N.T. MIND diet associated with reduced incidence of Alzheimer’s disease. Alzheimer’s Dement. 2015, 11, 1007–1014. [Google Scholar] [CrossRef]
- Sando, S.B.; Melquist, S.; Cannon, A.; Hutton, M.; Sletvold, O.; Saltvedt, I.; White, L.R.; Lydersen, S.; Aasly, J. Risk-reducing effect of education in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2008, 23, 1156–1162. [Google Scholar] [CrossRef]
- Fann, J.R.; Ribe, A.R.; Pedersen, H.S.; Fenger-Gron, M.; Christensen, J.; Benros, M.E.; Vestergaard, M. Long-term risk of dementia among people with traumatic brain injury in Denmark: A population-based observational cohort study. Lancet Psychiatry 2018, 5, 424–431. [Google Scholar] [CrossRef]
- Plassman, B.L.; Havlik, R.J.; Steffens, D.C.; Helms, M.J.; Newman, T.N.; Drosdick, D.; Phillips, C.; Gau, B.A.; Welsh-Bohmer, K.A.; Burke, J.R.; et al. Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology 2000, 55, 1158–1166. [Google Scholar] [CrossRef]
- Cherbuin, N.; Kim, S.; Anstey, K.J. Dementia risk estimates associated with measures of depression: A systematic review and meta-analysis. BMJ Open 2015, 5, e008853. [Google Scholar] [CrossRef]
- Terracciano, A.; Sutin, A.R.; An, Y.; O’Brien, R.J.; Ferrucci, L.; Zonderman, A.B.; Resnick, S.M. Personality and risk of Alzheimer’s disease: New data and meta-analysis. Alzheimer’s Dement. 2014, 10, 179–186. [Google Scholar] [CrossRef]
- Avramopoulos, D. Genetics of Alzheimer’s disease: Recent advances. Genome Med. 2009, 1, 34. [Google Scholar] [CrossRef]
- Loy, C.T.; Schofield, P.R.; Turner, A.M.; Kwok, J.B. Genetics of dementia. Lancet 2014, 383, 828–840. [Google Scholar] [CrossRef]
- Ramos, C.; Aguillon, D.; Cordano, C.; Lopera, F. Genetics of dementia: Insights from Latin America. Dement. Neuropsychol. 2020, 14, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chen, F.; Jiang, Y.; Zhang, L.; Hu, G.; Sun, F.; Zhang, M.; Ji, Y.; Chen, Y.; Che, G.; et al. A Review of ApoE4 Interference Targeting Mitophagy Molecular Pathways for Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 881239. [Google Scholar] [CrossRef]
- Zhao, N.; Liu, C.C.; Van Ingelgom, A.J.; Martens, Y.A.; Linares, C.; Knight, J.A.; Painter, M.M.; Sullivan, P.M.; Bu, G. Apolipoprotein E4 Impairs Neuronal Insulin Signaling by Trapping Insulin Receptor in the Endosomes. Neuron 2017, 96, 115–129.e5. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hagg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef]
- Marioni, R.E.; Harris, S.E.; Zhang, Q.; McRae, A.F.; Hagenaars, S.P.; Hill, W.D.; Davies, G.; Ritchie, C.W.; Gale, C.R.; Starr, J.M.; et al. GWAS on family history of Alzheimer’s disease. Transl. Psychiatry 2018, 8, 99. [Google Scholar] [CrossRef]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef]
- Murray, M.E.; Lowe, V.J.; Graff-Radford, N.R.; Liesinger, A.M.; Cannon, A.; Przybelski, S.A.; Rawal, B.; Parisi, J.E.; Petersen, R.C.; Kantarci, K.; et al. Clinicopathologic and 11C-Pittsburgh compound B implications of Thal amyloid phase across the Alzheimer’s disease spectrum. Brain 2015, 138, 1370–1381. [Google Scholar] [CrossRef]
- Bellenguez, C.; Charbonnier, C.; Grenier-Boley, B.; Quenez, O.; Le Guennec, K.; Nicolas, G.; Chauhan, G.; Wallon, D.; Rousseau, S.; Richard, A.C.; et al. Contribution to Alzheimer’s disease risk of rare variants in TREM2, SORL1, and ABCA7 in 1779 cases and 1273 controls. Neurobiol. Aging 2017, 59, 220.e1–220.e9. [Google Scholar] [CrossRef]
- Fu, W.Y.; Ip, N.Y. The role of genetic risk factors of Alzheimer’s disease in synaptic dysfunction. Semin. Cell Dev. Biol. 2022, in press. [Google Scholar] [CrossRef]
- Nikolac Perkovic, M.; Videtic Paska, A.; Konjevod, M.; Kouter, K.; Svob Strac, D.; Nedic Erjavec, G.; Pivac, N. Epigenetics of Alzheimer’s Disease. Biomolecules 2021, 11, 195. [Google Scholar] [CrossRef]
- Villa, C.; Stoccoro, A. Epigenetic Peripheral Biomarkers for Early Diagnosis of Alzheimer’s Disease. Genes 2022, 13, 1308. [Google Scholar] [CrossRef]
- Gao, X.; Chen, Q.; Yao, H.; Tan, J.; Liu, Z.; Zhou, Y.; Zou, Z. Epigenetics in Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 911635. [Google Scholar] [CrossRef]
- Maity, S.; Farrell, K.; Navabpour, S.; Narayanan, S.N.; Jarome, T.J. Epigenetic Mechanisms in Memory and Cognitive Decline Associated with Aging and Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 12280. [Google Scholar] [CrossRef]
- Pellegrini, C.; Pirazzini, C.; Sala, C.; Sambati, L.; Yusipov, I.; Kalyakulina, A.; Ravaioli, F.; Kwiatkowska, K.M.; Durso, D.F.; Ivanchenko, M.; et al. A Meta-Analysis of Brain DNA Methylation across Sex, Age, and Alzheimer’s Disease Points for Accelerated Epigenetic Aging in Neurodegeneration. Front. Aging Neurosci. 2021, 13, 639428. [Google Scholar] [CrossRef]
- Zhao, Y.; Jaber, V.; Alexandrov, P.N.; Vergallo, A.; Lista, S.; Hampel, H.; Lukiw, W.J. microRNA-Based Biomarkers in Alzheimer’s Disease (AD). Front. Neurosci. 2020, 14, 585432. [Google Scholar] [CrossRef]
- Arora, T.; Prashar, V.; Singh, R.; Barwal, T.S.; Changotra, H.; Sharma, A.; Parkash, J. Dysregulated miRNAs in Progression and Pathogenesis of Alzheimer’s Disease. Mol. Neurobiol. 2022, 59, 6107–6124. [Google Scholar] [CrossRef]
- Zetterberg, H. Blood-based biomarkers for Alzheimer’s disease—An update. J. Neurosci. Methods 2019, 319, 2–6. [Google Scholar] [CrossRef]
- Marquez, F.; Yassa, M.A. Neuroimaging Biomarkers for Alzheimer’s Disease. Mol. Neurodegener. 2019, 14, 21. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Feldman, H.H.; Frisoni, G.B.; Hampel, H.; Jagust, W.J.; Johnson, K.A.; Knopman, D.S.; et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016, 87, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Cummings, J.; Blennow, K.; Gao, P.; Jack, C.R., Jr.; Vergallo, A. Developing the ATX(N) classification for use across the Alzheimer disease continuum. Nat. Rev. Neurol. 2021, 17, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Baldeiras, I.; Santana, I.; Leitao, M.J.; Vieira, D.; Duro, D.; Mroczko, B.; Kornhuber, J.; Lewczuk, P. Erlangen Score as a tool to predict progression from mild cognitive impairment to dementia in Alzheimer’s disease. Alzheimer’s Res. Ther. 2019, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Leuzy, A.; Chiotis, K.; Lemoine, L.; Gillberg, P.G.; Almkvist, O.; Rodriguez-Vieitez, E.; Nordberg, A. Tau PET imaging in neurodegenerative tauopathies-still a challenge. Mol. Psychiatry 2019, 24, 1112–1134. [Google Scholar] [CrossRef]
- Vlassenko, A.G.; Benzinger, T.L.; Morris, J.C. PET amyloid-β imaging in preclinical Alzheimer’s disease. Biochim. Biophys. Acta 2012, 1822, 370–379. [Google Scholar] [CrossRef]
- Campese, N.; Palermo, G.; Del Gamba, C.; Beatino, M.F.; Galgani, A.; Belli, E.; Del Prete, E.; Della Vecchia, A.; Vergallo, A.; Siciliano, G.; et al. Progress regarding the context-of-use of tau as biomarker of Alzheimer’s disease and other neurodegenerative diseases. Expert Rev. Proteomics 2021, 18, 27–48. [Google Scholar] [CrossRef]
- Hampel, H.; Burger, K.; Teipel, S.J.; Bokde, A.L.; Zetterberg, H.; Blennow, K. Core candidate neurochemical and imaging biomarkers of Alzheimer’s disease. Alzheimer’s Dement. 2008, 4, 38–48. [Google Scholar] [CrossRef]
- Zetterberg, H. Applying fluid biomarkers to Alzheimer’s disease. Am. J. Physiol. Cell Physiol. 2017, 313, C3–C10. [Google Scholar] [CrossRef]
- Gordon, B.A.; Blazey, T.M.; Su, Y.; Hari-Raj, A.; Dincer, A.; Flores, S.; Christensen, J.; McDade, E.; Wang, G.; Xiong, C.; et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer’s disease: A longitudinal study. Lancet Neurol. 2018, 17, 241–250. [Google Scholar] [CrossRef]
- Murray, M.E.; Kouri, N.; Lin, W.L.; Jack, C.R., Jr.; Dickson, D.W.; Vemuri, P. Clinicopathologic assessment and imaging of tauopathies in neurodegenerative dementias. Alzheimer’s Res. Ther. 2014, 6, 1. [Google Scholar] [CrossRef]
- Nakamura, A.; Kaneko, N.; Villemagne, V.L.; Kato, T.; Doecke, J.; Dore, V.; Fowler, C.; Li, Q.X.; Martins, R.; Rowe, C.; et al. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature 2018, 554, 249–254. [Google Scholar] [CrossRef]
- Li, Y.; Schindler, S.E.; Bollinger, J.G.; Ovod, V.; Mawuenyega, K.G.; Weiner, M.W.; Shaw, L.M.; Masters, C.L.; Fowler, C.J.; Trojanowski, J.Q.; et al. Validation of Plasma Amyloid-β 42/40 for Detecting Alzheimer Disease Amyloid Plaques. Neurology 2022, 98, e688–e699. [Google Scholar] [CrossRef]
- Leuzy, A.; Mattsson-Carlgren, N.; Palmqvist, S.; Janelidze, S.; Dage, J.L.; Hansson, O. Blood-based biomarkers for Alzheimer’s disease. EMBO Mol. Med. 2022, 14, e14408. [Google Scholar] [CrossRef]
- Cummings, J.; Kinney, J. Biomarkers for Alzheimer’s Disease: Context of Use, Qualification, and Roadmap for Clinical Implementation. Medicina 2022, 58, 952. [Google Scholar] [CrossRef]
- Saint-Pol, J.; Gosselet, F.; Duban-Deweer, S.; Pottiez, G.; Karamanos, Y. Targeting and Crossing the Blood-Brain Barrier with Extracellular Vesicles. Cells 2020, 9, 851. [Google Scholar] [CrossRef]
- Park, S.A.; Jang, Y.J.; Kim, M.K.; Lee, S.M.; Moon, S.Y. Promising Blood Biomarkers for Clinical Use in Alzheimer’s Disease: A Focused Update. J. Clin. Neurol. 2022, 18, 401–409. [Google Scholar] [CrossRef]
- Carmona, P.; Molina, M.; Lopez-Tobar, E.; Toledano, A. Vibrational spectroscopic analysis of peripheral blood plasma of patients with Alzheimer’s disease. Anal. Bioanal. Chem. 2015, 407, 7747–7756. [Google Scholar] [CrossRef]
- O’Bryant, S.E.; Edwards, M.; Johnson, L.; Hall, J.; Villarreal, A.E.; Britton, G.B.; Quiceno, M.; Cullum, C.M.; Graff-Radford, N.R. A blood screening test for Alzheimer’s disease. Alzheimer’s Dement. 2016, 3, 83–90. [Google Scholar] [CrossRef]
- Wang, G.; Zhou, Y.; Huang, F.J.; Tang, H.D.; Xu, X.H.; Liu, J.J.; Wang, Y.; Deng, Y.L.; Ren, R.J.; Xu, W.; et al. Plasma metabolite profiles of Alzheimer’s disease and mild cognitive impairment. J. Proteome Res. 2014, 13, 2649–2658. [Google Scholar] [CrossRef]
- Platenik, J.; Fisar, Z.; Buchal, R.; Jirak, R.; Kitzlerova, E.; Zverova, M.; Raboch, J. GSK3β, CREB, and BDNF in peripheral blood of patients with Alzheimer’s disease and depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 50, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Nistico, R.; Seyfried, N.T.; Levey, A.I.; Modeste, E.; Lemercier, P.; Baldacci, F.; Toschi, N.; Garaci, F.; Perry, G.; et al. Omics sciences for systems biology in Alzheimer’s disease: State-of-the-art of the evidence. Ageing Res. Rev. 2021, 69, 101346. [Google Scholar] [CrossRef] [PubMed]
- Habartova, L.; Hrubesova, K.; Syslova, K.; Vondrousova, J.; Fisar, Z.; Jirak, R.; Raboch, J.; Setnicka, V. Blood-based molecular signature of Alzheimer’s disease via spectroscopy and metabolomics. Clin. Biochem. 2019, 72, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Reed, T.; Newman, S.F.; Sultana, R. Roles of amyloid β-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radic. Biol. Med. 2007, 43, 658–677. [Google Scholar] [CrossRef] [PubMed]
- Fišar, Z.; Hroudová, J.; Hansiková, H.; Spáčilová, J.; Lelková, P.; Wenchich, L.; Jirák, R.; Zeman, J.; Martásek, P.; Raboch, J. Mitochondrial respiration in the platelets of patients with Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 930–941. [Google Scholar] [CrossRef]
- Fisar, Z.; Hansikova, H.; Krizova, J.; Jirak, R.; Kitzlerova, E.; Zverova, M.; Hroudova, J.; Wenchich, L.; Zeman, J.; Raboch, J. Activities of mitochondrial respiratory chain complexes in platelets of patients with Alzheimer’s disease and depressive disorder. Mitochondrion 2019, 48, 67–77. [Google Scholar] [CrossRef]
- Zheng, C.; Zhou, X.W.; Wang, J.Z. The dual roles of cytokines in Alzheimer’s disease: Update on interleukins, TNF-α, TGF-β and IFN-γ. Transl. Neurodegener. 2016, 5, 7. [Google Scholar] [CrossRef]
- Mazzucchi, S.; Palermo, G.; Campese, N.; Galgani, A.; Della Vecchia, A.; Vergallo, A.; Siciliano, G.; Ceravolo, R.; Hampel, H.; Baldacci, F. The role of synaptic biomarkers in the spectrum of neurodegenerative diseases. Expert Rev. Proteom. 2020, 17, 543–559. [Google Scholar] [CrossRef]
- Alawode, D.O.T.; Fox, N.C.; Zetterberg, H.; Heslegrave, A.J. Alzheimer’s Disease Biomarkers Revisited from the Amyloid Cascade Hypothesis Standpoint. Front. Neurosci. 2022, 16, 837390. [Google Scholar] [CrossRef]
- Chatterjee, P.; Pedrini, S.; Stoops, E.; Goozee, K.; Villemagne, V.L.; Asih, P.R.; Verberk, I.M.W.; Dave, P.; Taddei, K.; Sohrabi, H.R.; et al. Plasma glial fibrillary acidic protein is elevated in cognitively normal older adults at risk of Alzheimer’s disease. Transl. Psychiatry 2021, 11, 27. [Google Scholar] [CrossRef]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef]
- Wolfe, C.M.; Fitz, N.F.; Nam, K.N.; Lefterov, I.; Koldamova, R. The Role of APOE and TREM2 in Alzheimer’s Disease-Current Understanding and Perspectives. Int. J. Mol. Sci. 2018, 20, 81. [Google Scholar] [CrossRef]
- Lewczuk, P.; Riederer, P.; O’Bryant, S.E.; Verbeek, M.M.; Dubois, B.; Visser, P.J.; Jellinger, K.A.; Engelborghs, S.; Ramirez, A.; Parnetti, L.; et al. Cerebrospinal fluid and blood biomarkers for neurodegenerative dementias: An update of the Consensus of the Task Force on Biological Markers in Psychiatry of the World Federation of Societies of Biological Psychiatry. World J. Biol. Psychiatry 2018, 19, 244–328. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Holtzman, D.M. Biomarker modeling of Alzheimer’s disease. Neuron 2013, 80, 1347–1358. [Google Scholar] [CrossRef]
- Leuzy, A.; Ashton, N.J.; Mattsson-Carlgren, N.; Dodich, A.; Boccardi, M.; Corre, J.; Drzezga, A.; Nordberg, A.; Ossenkoppele, R.; Zetterberg, H.; et al. 2020 update on the clinical validity of cerebrospinal fluid amyloid, tau, and phospho-tau as biomarkers for Alzheimer’s disease in the context of a structured 5-phase development framework. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2121–2139. [Google Scholar] [CrossRef]
- Li, R.X.; Ma, Y.H.; Tan, L.; Yu, J.T. Prospective biomarkers of Alzheimer’s disease: A systematic review and meta-analysis. Ageing Res. Rev. 2022, 81, 101699. [Google Scholar] [CrossRef]
- Cullen, N.C.; Leuzy, A.; Janelidze, S.; Palmqvist, S.; Svenningsson, A.L.; Stomrud, E.; Dage, J.L.; Mattsson-Carlgren, N.; Hansson, O. Plasma biomarkers of Alzheimer’s disease improve prediction of cognitive decline in cognitively unimpaired elderly populations. Nat. Commun. 2021, 12, 3555. [Google Scholar] [CrossRef]
- Fisar, Z.; Jirak, R.; Zverova, M.; Setnicka, V.; Habartova, L.; Hroudova, J.; Vanickova, Z.; Raboch, J. Plasma amyloid β levels and platelet mitochondrial respiration in patients with Alzheimer’s disease. Clin. Biochem. 2019, 72, 71–80. [Google Scholar] [CrossRef]
- Kitzlerova, E.; Fisar, Z.; Jirak, R.; Zverova, M.; Hroudova, J.; Benakova, H.; Raboch, J. Plasma homocysteine in Alzheimer’s disease with or without co-morbid depressive symptoms. Neuro Endocrinol. Lett. 2014, 35, 42–49. [Google Scholar] [PubMed]
- Zverova, M.; Fisar, Z.; Jirak, R.; Kitzlerova, E.; Hroudova, J.; Raboch, J. Plasma cortisol in Alzheimer’s disease with or without depressive symptoms. Med. Sci. Monit. 2013, 19, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Zverova, M.; Kitzlerova, E.; Fisar, Z.; Jirak, R.; Hroudova, J.; Benakova, H.; Lelkova, P.; Martasek, P.; Raboch, J. Interplay between the APOE Genotype and Possible Plasma Biomarkers in Alzheimer’s Disease. Curr. Alzheimer Res. 2018, 15, 938–950. [Google Scholar] [CrossRef] [PubMed]
- Hansson, O.; Edelmayer, R.M.; Boxer, A.L.; Carrillo, M.C.; Mielke, M.M.; Rabinovici, G.D.; Salloway, S.; Sperling, R.; Zetterberg, H.; Teunissen, C.E. The Alzheimer’s Association appropriate use recommendations for blood biomarkers in Alzheimer’s disease. Alzheimer’s Dement. 2022, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef]
- Villemagne, V.L.; Burnham, S.; Bourgeat, P.; Brown, B.; Ellis, K.A.; Salvado, O.; Szoeke, C.; Macaulay, S.L.; Martins, R.; Maruff, P.; et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. Lancet Neurol. 2013, 12, 357–367. [Google Scholar] [CrossRef]
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s Dement. 2012, 8, 1–13. [Google Scholar] [CrossRef]
- Thal, D.R.; Rub, U.; Orantes, M.; Braak, H. Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef]
- Mirra, S.S.; Heyman, A.; McKeel, D.; Sumi, S.M.; Crain, B.J.; Brownlee, L.M.; Vogel, F.S.; Hughes, J.P.; van Belle, G.; Berg, L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991, 41, 479–486. [Google Scholar] [CrossRef]
- Alafuzoff, I.; Arzberger, T.; Al-Sarraj, S.; Bodi, I.; Bogdanovic, N.; Braak, H.; Bugiani, O.; Del-Tredici, K.; Ferrer, I.; Gelpi, E.; et al. Staging of neurofibrillary pathology in Alzheimer’s disease: A study of the BrainNet Europe Consortium. Brain Pathol. 2008, 18, 484–496. [Google Scholar] [CrossRef]
- Tissot, C.; Therriault, J.; Kunach, P.; Benedet, A.L.; Pascoal, T.A.; Ashton, N.J.; Karikari, T.K.; Servaes, S.; Lussier, F.Z.; Chamoun, M.; et al. Comparing tau status determined via plasma pTau181, pTau231 and [(18)F]MK6240 tau-PET. EBioMedicine 2022, 76, 103837. [Google Scholar] [CrossRef]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R., Jr.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Lowe, V.J.; Lundt, E.S.; Albertson, S.M.; Przybelski, S.A.; Senjem, M.L.; Parisi, J.E.; Kantarci, K.; Boeve, B.; Jones, D.T.; Knopman, D.; et al. Neuroimaging correlates with neuropathologic schemes in neurodegenerative disease. Alzheimer’s Dement. 2019, 15, 927–939. [Google Scholar] [CrossRef]
- Sato, C.; Barthelemy, N.R.; Mawuenyega, K.G.; Patterson, B.W.; Gordon, B.A.; Jockel-Balsarotti, J.; Sullivan, M.; Crisp, M.J.; Kasten, T.; Kirmess, K.M.; et al. Tau Kinetics in Neurons and the Human Central Nervous System. Neuron 2018, 97, 1284–1298.e7. [Google Scholar] [CrossRef]
- Di, J.; Cohen, L.S.; Corbo, C.P.; Phillips, G.R.; El Idrissi, A.; Alonso, A.D. Abnormal tau induces cognitive impairment through two different mechanisms: Synaptic dysfunction and neuronal loss. Sci. Rep. 2016, 6, 20833. [Google Scholar] [CrossRef]
- Barthelemy, N.R.; Li, Y.; Joseph-Mathurin, N.; Gordon, B.A.; Hassenstab, J.; Benzinger, T.L.S.; Buckles, V.; Fagan, A.M.; Perrin, R.J.; Goate, A.M.; et al. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer’s disease. Nat. Med. 2020, 26, 398–407. [Google Scholar] [CrossRef]
- Koss, D.J.; Jones, G.; Cranston, A.; Gardner, H.; Kanaan, N.M.; Platt, B. Soluble pre-fibrillar tau and β-amyloid species emerge in early human Alzheimer’s disease and track disease progression and cognitive decline. Acta Neuropathol. 2016, 132, 875–895. [Google Scholar] [CrossRef]
- Pimplikar, S.W.; Nixon, R.A.; Robakis, N.K.; Shen, J.; Tsai, L.H. Amyloid-independent mechanisms in Alzheimer’s disease pathogenesis. J. Neurosci. 2010, 30, 14946–14954. [Google Scholar] [CrossRef]
- Herrup, K. Reimagining Alzheimer’s disease—An age-based hypothesis. J. Neurosci. 2010, 30, 16755–16762. [Google Scholar] [CrossRef]
- Wareham, L.K.; Liddelow, S.A.; Temple, S.; Benowitz, L.I.; Di Polo, A.; Wellington, C.; Goldberg, J.L.; He, Z.; Duan, X.; Bu, G.; et al. Solving neurodegeneration: Common mechanisms and strategies for new treatments. Mol. Neurodegener. 2022, 17, 23. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.P.; Xie, Y.; Meng, X.Y.; Kang, J.S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Karran, E.; De Strooper, B. The amyloid cascade hypothesis: Are we poised for success or failure? J. Neurochem. 2016, 139 (Suppl. S2), 237–252. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Walsh, D.M.; Selkoe, D.J. Aβ oligomers—A decade of discovery. J. Neurochem. 2007, 101, 1172–1184. [Google Scholar] [CrossRef]
- Yang, T.; Li, S.; Xu, H.; Walsh, D.M.; Selkoe, D.J. Large Soluble Oligomers of Amyloid β-Protein from Alzheimer Brain Are Far Less Neuroactive Than the Smaller Oligomers to Which They Dissociate. J. Neurosci. 2017, 37, 152–163. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Jellinger, K.A. Basic mechanisms of neurodegeneration: A critical update. J. Cell. Mol. Med. 2010, 14, 457–487. [Google Scholar] [CrossRef]
- Harris, L.D.; Jasem, S.; Licchesi, J.D.F. The Ubiquitin System in Alzheimer’s Disease. Adv. Exp. Med. Biol. 2020, 1233, 195–221. [Google Scholar] [CrossRef]
- Richard, R.; Mousa, S. Necroptosis in Alzheimer’s disease: Potential therapeutic target. Biomed. Pharmacother. 2022, 152, 113203. [Google Scholar] [CrossRef]
- Mangalmurti, A.; Lukens, J.R. How neurons die in Alzheimer’s disease: Implications for neuroinflammation. Curr. Opin. Neurobiol. 2022, 75, 102575. [Google Scholar] [CrossRef]
- Musiek, E.S.; Holtzman, D.M. Three dimensions of the amyloid hypothesis: Time, space and ‘wingmen’. Nat. Neurosci. 2015, 18, 800–806. [Google Scholar] [CrossRef]
- Folch, J.; Ettcheto, M.; Petrov, D.; Abad, S.; Pedros, I.; Marin, M.; Olloquequi, J.; Camins, A. Review of the advances in treatment for Alzheimer disease: Strategies for combating β-amyloid protein. Neurol. Engl. Ed. 2018, 33, 47–58. [Google Scholar] [CrossRef]
- De Strooper, B.; Karran, E. The Cellular Phase of Alzheimer’s Disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef]
- Delport, A.; Hewer, R. The amyloid precursor protein: A converging point in Alzheimer’s disease. Mol. Neurobiol. 2022, 59, 4501–4516. [Google Scholar] [CrossRef]
- Li, X.; Uemura, K.; Hashimoto, T.; Nasser-Ghodsi, N.; Arimon, M.; Lill, C.M.; Palazzolo, I.; Krainc, D.; Hyman, B.T.; Berezovska, O. Neuronal activity and secreted amyloid β lead to altered amyloid β precursor protein and presenilin 1 interactions. Neurobiol. Dis. 2013, 50, 127–134. [Google Scholar] [CrossRef]
- Chang, Y.J.; Chen, Y.R. The coexistence of an equal amount of Alzheimer’s amyloid-β 40 and 42 forms structurally stable and toxic oligomers through a distinct pathway. FEBS J. 2014, 281, 2674–2687. [Google Scholar] [CrossRef]
- Verma, M.; Vats, A.; Taneja, V. Toxic species in amyloid disorders: Oligomers or mature fibrils. Ann. Indian Acad. Neurol. 2015, 18, 138–145. [Google Scholar] [CrossRef]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid β: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- Jarosz-Griffiths, H.H.; Noble, E.; Rushworth, J.V.; Hooper, N.M. Amyloid-β Receptors: The Good, the Bad, and the Prion Protein. J. Biol. Chem. 2016, 291, 3174–3183. [Google Scholar] [CrossRef] [PubMed]
- Canale, C.; Seghezza, S.; Vilasi, S.; Carrotta, R.; Bulone, D.; Diaspro, A.; San Biagio, P.L.; Dante, S. Different effects of Alzheimer’s peptide Aβ(1–40) oligomers and fibrils on supported lipid membranes. Biophys. Chem. 2013, 182, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, E.; Cascella, R.; Becatti, M.; Marrazza, G.; Dobson, C.M.; Chiti, F.; Stefani, M.; Cecchi, C. Binding affinity of amyloid oligomers to cellular membranes is a generic indicator of cellular dysfunction in protein misfolding diseases. Sci. Rep. 2016, 6, 32721. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Lasagna-Reeves, C.A. Molecular mechanisms of amyloid oligomers toxicity. J. Alzheimer’s Dis. 2013, 33 (Suppl. S1), S67–S78. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef]
- Muller-Schiffmann, A.; Herring, A.; Abdel-Hafiz, L.; Chepkova, A.N.; Schable, S.; Wedel, D.; Horn, A.H.; Sticht, H.; de Souza Silva, M.A.; Gottmann, K.; et al. Amyloid-β dimers in the absence of plaque pathology impair learning and synaptic plasticity. Brain 2016, 139, 509–525. [Google Scholar] [CrossRef]
- Narayan, P.; Meehan, S.; Carver, J.A.; Wilson, M.R.; Dobson, C.M.; Klenerman, D. Amyloid-β oligomers are sequestered by both intracellular and extracellular chaperones. Biochemistry 2012, 51, 9270–9276. [Google Scholar] [CrossRef]
- Overk, C.R.; Masliah, E. Toward a unified therapeutics approach targeting putative amyloid-β oligomer receptors. Proc. Natl. Acad. Sci. USA 2014, 111, 13680–13681. [Google Scholar] [CrossRef]
- Takuma, K.; Fang, F.; Zhang, W.; Yan, S.; Fukuzaki, E.; Du, H.; Sosunov, A.; McKhann, G.; Funatsu, Y.; Nakamichi, N.; et al. RAGE-mediated signaling contributes to intraneuronal transport of amyloid-β and neuronal dysfunction. Proc. Natl. Acad. Sci. USA 2009, 106, 20021–20026. [Google Scholar] [CrossRef]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef]
- Li, X.H.; Du, L.L.; Cheng, X.S.; Jiang, X.; Zhang, Y.; Lv, B.L.; Liu, R.; Wang, J.Z.; Zhou, X.W. Glycation exacerbates the neuronal toxicity of β-amyloid. Cell Death Dis. 2013, 4, e673. [Google Scholar] [CrossRef]
- Batkulwar, K.; Godbole, R.; Banarjee, R.; Kassaar, O.; Williams, R.J.; Kulkarni, M.J. Advanced Glycation End Products Modulate Amyloidogenic APP Processing and Tau Phosphorylation: A Mechanistic Link between Glycation and the Development of Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 988–1000. [Google Scholar] [CrossRef]
- Deane, R.; Singh, I.; Sagare, A.P.; Bell, R.D.; Ross, N.T.; LaRue, B.; Love, R.; Perry, S.; Paquette, N.; Deane, R.J.; et al. A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Investig. 2012, 122, 1377–1392. [Google Scholar] [CrossRef]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef]
- Zerbinatti, C.V.; Bu, G. LRP and Alzheimer’s disease. Rev. Neurosci. 2005, 16, 123–135. [Google Scholar] [CrossRef]
- Shinohara, M.; Tachibana, M.; Kanekiyo, T.; Bu, G. Role of LRP1 in the pathogenesis of Alzheimer’s disease: Evidence from clinical and preclinical studies. J. Lipid Res. 2017, 58, 1267–1281. [Google Scholar] [CrossRef]
- Shibata, M.; Yamada, S.; Kumar, S.R.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.M.; Miller, C.A.; Strickland, D.K.; Ghiso, J.; et al. Clearance of Alzheimer’s amyloid-β(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Investig. 2000, 106, 1489–1499. [Google Scholar] [CrossRef]
- Viola, K.L.; Klein, W.L. Amyloid β oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 2015, 129, 183–206. [Google Scholar] [CrossRef]
- Kosik, K.S.; Joachim, C.L.; Selkoe, D.J. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1986, 83, 4044–4048. [Google Scholar] [CrossRef]
- Martin, L.; Latypova, X.; Terro, F. Post-translational modifications of tau protein: Implications for Alzheimer’s disease. Neurochem. Int. 2011, 58, 458–471. [Google Scholar] [CrossRef]
- Arnsten, A.F.T.; Datta, D.; Del Tredici, K.; Braak, H. Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer’s disease. Alzheimer’s Dement. 2021, 17, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Ward, S.M.; Himmelstein, D.S.; Lancia, J.K.; Binder, L.I. Tau oligomers and tau toxicity in neurodegenerative disease. Biochem. Soc. Trans. 2012, 40, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Cardenas-Aguayo Mdel, C.; Gomez-Virgilio, L.; DeRosa, S.; Meraz-Rios, M.A. The role of tau oligomers in the onset of Alzheimer’s disease neuropathology. ACS Chem. Neurosci. 2014, 5, 1178–1191. [Google Scholar] [CrossRef]
- Berger, Z.; Roder, H.; Hanna, A.; Carlson, A.; Rangachari, V.; Yue, M.; Wszolek, Z.; Ashe, K.; Knight, J.; Dickson, D.; et al. Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J. Neurosci. 2007, 27, 3650–3662. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Clos, A.L.; Jackson, G.R.; Kayed, R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener. 2011, 6, 39. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.A.; Sengupta, U.; Castillo-Carranza, D.; Gerson, J.E.; Guerrero-Munoz, M.; Troncoso, J.C.; Jackson, G.R.; Kayed, R. The formation of tau pore-like structures is prevalent and cell specific: Possible implications for the disease phenotypes. Acta Neuropathol. Commun. 2014, 2, 56. [Google Scholar] [CrossRef]
- Gerson, J.E.; Sengupta, U.; Lasagna-Reeves, C.A.; Guerrero-Munoz, M.J.; Troncoso, J.; Kayed, R. Characterization of tau oligomeric seeds in progressive supranuclear palsy. Acta Neuropathol. Commun. 2014, 2, 73. [Google Scholar] [CrossRef]
- Hernandez, F.; Lucas, J.J.; Avila, J. GSK3 and tau: Two convergence points in Alzheimer’s disease. J. Alzheimer’s Dis. 2013, 33 (Suppl. S1), S141–S144. [Google Scholar] [CrossRef]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef]
- Muralidar, S.; Ambi, S.V.; Sekaran, S.; Thirumalai, D.; Palaniappan, B. Role of tau protein in Alzheimer’s disease: The prime pathological player. Int. J. Biol. Macromol. 2020, 163, 1599–1617. [Google Scholar] [CrossRef]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar] [CrossRef]
- Wang, J.Z.; Xia, Y.Y.; Grundke-Iqbal, I.; Iqbal, K. Abnormal hyperphosphorylation of tau: Sites, regulation, and molecular mechanism of neurofibrillary degeneration. J. Alzheimer’s Dis. 2013, 33 (Suppl. S1), S123–S139. [Google Scholar] [CrossRef]
- Soeda, Y.; Takashima, A. New Insights Into Drug Discovery Targeting Tau Protein. Front. Mol. Neurosci. 2020, 13, 590896. [Google Scholar] [CrossRef]
- Zhang, H.; Cao, Y.; Ma, L.; Wei, Y.; Li, H. Possible Mechanisms of Tau Spread and Toxicity in Alzheimer’s Disease. Front. Cell Dev. Biol. 2021, 9, 707268. [Google Scholar] [CrossRef]
- Blurton-Jones, M.; Laferla, F.M. Pathways by which Aβ facilitates tau pathology. Curr. Alzheimer Res. 2006, 3, 437–448. [Google Scholar] [CrossRef]
- Pascoal, T.A.; Mathotaarachchi, S.; Shin, M.; Benedet, A.L.; Mohades, S.; Wang, S.; Beaudry, T.; Kang, M.S.; Soucy, J.P.; Labbe, A.; et al. Synergistic interaction between amyloid and tau predicts the progression to dementia. Alzheimer’s Dement. 2017, 13, 644–653. [Google Scholar] [CrossRef]
- Um, J.W.; Kaufman, A.C.; Kostylev, M.; Heiss, J.K.; Stagi, M.; Takahashi, H.; Kerrisk, M.E.; Vortmeyer, A.; Wisniewski, T.; Koleske, A.J.; et al. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer aβ oligomer bound to cellular prion protein. Neuron 2013, 79, 887–902. [Google Scholar] [CrossRef]
- Zhao, Y.; Kuca, K.; Wu, W.; Wang, X.; Nepovimova, E.; Musilek, K.; Wu, Q. Hypothesis: JNK signaling is a therapeutic target of neurodegenerative diseases. Alzheimer’s Dement. 2022, 18, 152–158. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. The mitochondrial hypothesis: Dysfunction, bioenergetic defects, and the metabolic link to Alzheimer’s disease. Int. Rev. Neurobiol. 2020, 154, 207–233. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.K.; Jara, C.; Park-Kang, H.S.; Polanco, C.M.; Tapia, D.; Alarcon, F.; de la Pena, A.; Llanquinao, J.; Vargas-Mardones, G.; Indo, J.A.; et al. Synaptic Mitochondria: An Early Target of Amyloid-β and Tau in Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 84, 1391–1414. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. MAM (mitochondria-associated membranes) in mammalian cells: Lipids and beyond. Biochim. Biophys. Acta 2014, 1841, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Giacomello, M.; Pellegrini, L. The coming of age of the mitochondria-ER contact: A matter of thickness. Cell Death Differ. 2016, 23, 1417–1427. [Google Scholar] [CrossRef]
- Area-Gomez, E.; de Groof, A.J.; Boldogh, I.; Bird, T.D.; Gibson, G.E.; Koehler, C.M.; Yu, W.H.; Duff, K.E.; Yaffe, M.P.; Pon, L.A.; et al. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am. J. Pathol. 2009, 175, 1810–1816. [Google Scholar] [CrossRef]
- Del Prete, D.; Suski, J.M.; Oules, B.; Debayle, D.; Gay, A.S.; Lacas-Gervais, S.; Bussiere, R.; Bauer, C.; Pinton, P.; Paterlini-Brechot, P.; et al. Localization and Processing of the Amyloid-β Protein Precursor in Mitochondria-Associated Membranes. J. Alzheimer’s Dis. 2017, 55, 1549–1570. [Google Scholar] [CrossRef]
- Schon, E.A.; Area-Gomez, E. Mitochondria-associated ER membranes in Alzheimer disease. Mol. Cell. Neurosci. 2013, 55, 26–36. [Google Scholar] [CrossRef]
- Eysert, F.; Kinoshita, P.F.; Mary, A.; Vaillant-Beuchot, L.; Checler, F.; Chami, M. Molecular Dysfunctions of Mitochondria-Associated Membranes (MAMs) in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 9521. [Google Scholar] [CrossRef]
- Bishop, N.A.; Lu, T.; Yankner, B.A. Neural mechanisms of ageing and cognitive decline. Nature 2010, 464, 529–535. [Google Scholar] [CrossRef]
- Payne, B.A.; Chinnery, P.F. Mitochondrial dysfunction in aging: Much progress but many unresolved questions. Biochim. Biophys. Acta 2015, 1847, 1347–1353. [Google Scholar] [CrossRef]
- Cenini, G.; Voos, W. Mitochondria as Potential Targets in Alzheimer Disease Therapy: An Update. Front. Pharmacol. 2019, 10, 902. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Koppel, S.; Weidling, I.; Hayley, C.; Ji, Y.; Wilkins, H.M. Mitochondria, Cybrids, Aging, and Alzheimer’s Disease. Prog. Mol. Biol. Transl. Sci. 2017, 146, 259–302. [Google Scholar] [CrossRef]
- Jia, K.; Du, H. Mitochondrial Permeability Transition: A Pore Intertwines Brain Aging and Alzheimer’s Disease. Cells 2021, 10, 649. [Google Scholar] [CrossRef]
- Hroudova, J.; Singh, N.; Fisar, Z. Mitochondrial dysfunctions in neurodegenerative diseases: Relevance to Alzheimer’s disease. BioMed Res. Int. 2014, 2014, 175062. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Changes in the mitochondrial permeability transition pore in aging and age-associated diseases. Mech. Ageing Dev. 2013, 134, 1–9. [Google Scholar] [CrossRef]
- Reiss, A.B.; Ahmed, S.; Dayaramani, C.; Glass, A.D.; Gomolin, I.H.; Pinkhasov, A.; Stecker, M.M.; Wisniewski, T.; De Leon, J. The role of mitochondrial dysfunction in Alzheimer’s disease: A potential pathway to treatment. Exp. Gerontol. 2022, 164, 111828. [Google Scholar] [CrossRef]
- Blagov, A.V.; Grechko, A.V.; Nikiforov, N.G.; Borisov, E.E.; Sadykhov, N.K.; Orekhov, A.N. Role of Impaired Mitochondrial Dynamics Processes in the Pathogenesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 6954. [Google Scholar] [CrossRef]
- Hroudova, J.; Fisar, Z. Connectivity between mitochondrial functions and psychiatric disorders. Psychiatry Clin. Neurosci. 2011, 65, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P. Why F-ATP Synthase Remains a Strong Candidate as the Mitochondrial Permeability Transition Pore. Front. Physiol. 2018, 9, 1543. [Google Scholar] [CrossRef]
- Amadoro, G.; Corsetti, V.; Atlante, A.; Florenzano, F.; Capsoni, S.; Bussani, R.; Mercanti, D.; Calissano, P. Interaction between NH2-tau fragment and Aβ in Alzheimer’s disease mitochondria contributes to the synaptic deterioration. Neurobiol. Aging 2012, 33, 833.e1–833.e25. [Google Scholar] [CrossRef] [PubMed]
- John, A.; Reddy, P.H. Synaptic basis of Alzheimer’s disease: Focus on synaptic amyloid β, P-tau and mitochondria. Ageing Res. Rev. 2021, 65, 101208. [Google Scholar] [CrossRef] [PubMed]
- Kocahan, S.; Dogan, Z. Mechanisms of Alzheimer’s Disease Pathogenesis and Prevention: The Brain, Neural Pathology, N-methyl-D-aspartate Receptors, Tau Protein and Other Risk Factors. Clin. Psychopharmacol. Neurosci. 2017, 15, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Neill, D. Alzheimer’s disease: Maladaptive synaptoplasticity hypothesis. Neurodegeneration 1995, 4, 217–232. [Google Scholar] [CrossRef] [PubMed]
- Mesulam, M.M. Neuroplasticity failure in Alzheimer’s disease: Bridging the gap between plaques and tangles. Neuron 1999, 24, 521–529. [Google Scholar] [CrossRef]
- Citri, A.; Malenka, R.C. Synaptic plasticity: Multiple forms, functions, and mechanisms. Neuropsychopharmacology 2008, 33, 18–41. [Google Scholar] [CrossRef]
- Bello-Medina, P.C.; Gonzalez-Franco, D.A.; Vargas-Rodriguez, I.; Diaz-Cintra, S. Oxidative stress, the immune response, synaptic plasticity, and cognition in transgenic models of Alzheimer disease. Neurol. Engl. Ed. 2022, 37, 682–690. [Google Scholar] [CrossRef]
- Merceron-Martinez, D.; Ibaceta-Gonzalez, C.; Salazar, C.; Almaguer-Melian, W.; Bergado-Rosado, J.A.; Palacios, A.G. Alzheimer’s Disease, Neural Plasticity, and Functional Recovery. J. Alzheimer’s Dis. 2021, 82, S37–S50. [Google Scholar] [CrossRef]
- Teter, B.; Ashford, J.W. Neuroplasticity in Alzheimer’s disease. J. Neurosci. Res. 2002, 70, 402–437. [Google Scholar] [CrossRef]
- Bailey, C.H.; Giustetto, M.; Huang, Y.Y.; Hawkins, R.D.; Kandel, E.R. Is heterosynaptic modulation essential for stabilizing Hebbian plasticity and memory? Nat. Rev. Neurosci. 2000, 1, 11–20. [Google Scholar] [CrossRef]
- Abraham, W.C. Metaplasticity: Tuning synapses and networks for plasticity. Nat. Rev. Neurosci. 2008, 9, 387. [Google Scholar] [CrossRef]
- Jang, S.S.; Chung, H.J. Emerging Link between Alzheimer’s Disease and Homeostatic Synaptic Plasticity. Neural Plast. 2016, 2016, 7969272. [Google Scholar] [CrossRef]
- Li, S.; Selkoe, D.J. A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Aβ oligomers from Alzheimer’s brain. J. Neurochem. 2020, 154, 583–597. [Google Scholar] [CrossRef]
- Walsh, C.; Drinkenburg, W.H.; Ahnaou, A. Neurophysiological assessment of neural network plasticity and connectivity: Progress towards early functional biomarkers for disease interception therapies in Alzheimer’s disease. Neurosci. Biobehav. Rev. 2017, 73, 340–358. [Google Scholar] [CrossRef]
- Long, H.Z.; Zhou, Z.W.; Cheng, Y.; Luo, H.Y.; Li, F.J.; Xu, S.G.; Gao, L.C. The Role of Microglia in Alzheimer’s Disease from the Perspective of Immune Inflammation and Iron Metabolism. Front. Aging Neurosci. 2022, 14, 888989. [Google Scholar] [CrossRef]
- Vainchtein, I.D.; Molofsky, A.V. Astrocytes and Microglia: In Sickness and in Health. Trends Neurosci. 2020, 43, 144–154. [Google Scholar] [CrossRef]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10248. [Google Scholar] [CrossRef]
- Hylén, U.; Eklund, D.; Humble, M.; Bartoszek, J.; Sarndahl, E.; Bejerot, S. Increased inflammasome activity in markedly ill psychiatric patients: An explorative study. J. Neuroimmunol. 2020, 339, 577119. [Google Scholar] [CrossRef]
- Weng, S.; Lai, Q.L.; Wang, J.; Zhuang, L.; Cheng, L.; Mo, Y.; Liu, L.; Zhao, Z.; Zhang, Y.; Qiao, S. The Role of Exosomes as Mediators of Neuroinflammation in the Pathogenesis and Treatment of Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 899944. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, B.; Wang, J.; Xu, L.; Yu, S.; Fu, J.; Yan, X.; Su, J. Aβ and Tau Regulate Microglia Metabolism via Exosomes in Alzheimer’s Disease. Biomedicines 2022, 10, 1800. [Google Scholar] [CrossRef]
- Noonin, C.; Thongboonkerd, V. Exosome-inflammasome crosstalk and their roles in inflammatory responses. Theranostics 2021, 11, 4436–4451. [Google Scholar] [CrossRef] [PubMed]
- Efthymiou, A.G.; Goate, A.M. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 2017, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Forloni, G.; Balducci, C. Alzheimer’s Disease, Oligomers, and Inflammation. J. Alzheimer’s Dis. 2018, 62, 1261–1276. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.S.; Welsh, C.A.; Barres, B.A.; Stevens, B. Do glia drive synaptic and cognitive impairment in disease? Nat. Neurosci. 2015, 18, 1539–1545. [Google Scholar] [CrossRef]
- Meraz-Rios, M.A.; Toral-Rios, D.; Franco-Bocanegra, D.; Villeda-Hernandez, J.; Campos-Pena, V. Inflammatory process in Alzheimer’s Disease. Front. Integr. Neurosci. 2013, 7, 59. [Google Scholar] [CrossRef]
- Ghosh, S.; Wu, M.D.; Shaftel, S.S.; Kyrkanides, S.; LaFerla, F.M.; Olschowka, J.A.; O’Banion, M.K. Sustained interleukin-1β overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J. Neurosci. 2013, 33, 5053–5064. [Google Scholar] [CrossRef]
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 2011, 187, 6539–6549. [Google Scholar] [CrossRef]
- Rivers-Auty, J.; Mather, A.E.; Peters, R.; Lawrence, C.B.; Brough, D. Anti-inflammatories in Alzheimer’s disease-potential therapy or spurious correlate? Brain Commun. 2020, 2, fcaa109. [Google Scholar] [CrossRef]
- Wei, Z.; Koya, J.; Reznik, S.E. Insulin Resistance Exacerbates Alzheimer Disease via Multiple Mechanisms. Front. Neurosci. 2021, 15, 687157. [Google Scholar] [CrossRef]
- Murrow, B.A.; Hoehn, K.L. Mitochondrial regulation of insulin action. Int. J. Biochem. Cell Biol. 2010, 42, 1936–1939. [Google Scholar] [CrossRef]
- Clark, I.A.; Vissel, B. Therapeutic implications of how TNF links apolipoprotein E, phosphorylated tau, alpha-synuclein, amyloid-β and insulin resistance in neurodegenerative diseases. Br. J. Pharmacol. 2018, 175, 3859–3875. [Google Scholar] [CrossRef]
- Munoz, S.S.; Garner, B.; Ooi, L. Understanding the Role of ApoE Fragments in Alzheimer’s Disease. Neurochem. Res. 2019, 44, 1297–1305. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Mattson, M.P. Apolipoprotein E and oxidative stress in brain with relevance to Alzheimer’s disease. Neurobiol. Dis. 2020, 138, 104795. [Google Scholar] [CrossRef]
- Harris, F.M.; Brecht, W.J.; Xu, Q.; Tesseur, I.; Kekonius, L.; Wyss-Coray, T.; Fish, J.D.; Masliah, E.; Hopkins, P.C.; Scearce-Levie, K.; et al. Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer’s disease-like neurodegeneration and behavioral deficits in transgenic mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10966–10971. [Google Scholar] [CrossRef]
- Chang, S.; ran Ma, T.; Miranda, R.D.; Balestra, M.E.; Mahley, R.W.; Huang, Y. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc. Natl. Acad. Sci. USA 2005, 102, 18694–18699. [Google Scholar] [CrossRef]
- Dafnis, I.; Tzinia, A.K.; Tsilibary, E.C.; Zannis, V.I.; Chroni, A. An apolipoprotein E4 fragment affects matrix metalloproteinase 9, tissue inhibitor of metalloproteinase 1 and cytokine levels in brain cell lines. Neuroscience 2012, 210, 21–32. [Google Scholar] [CrossRef]
- Hohn, A.; Tramutola, A.; Cascella, R. Proteostasis Failure in Neurodegenerative Diseases: Focus on Oxidative Stress. Oxid. Med. Cell. Longev. 2020, 2020, 5497046. [Google Scholar] [CrossRef]
- Tramutola, A.; Di Domenico, F.; Barone, E.; Perluigi, M.; Butterfield, D.A. It Is All about (U)biquitin: Role of Altered Ubiquitin-Proteasome System and UCHL1 in Alzheimer Disease. Oxid. Med. Cell. Longev. 2016, 2016, 2756068. [Google Scholar] [CrossRef]
- Hong, L.; Huang, H.C.; Jiang, Z.F. Relationship between amyloid-β and the ubiquitin-proteasome system in Alzheimer’s disease. Neurol. Res. 2014, 36, 276–282. [Google Scholar] [CrossRef]
- Reddy, P.H.; Oliver, D.M. Amyloid β and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A.; Ojala, J.; Haapasalo, A.; Soininen, H.; Hiltunen, M. Impaired autophagy and APP processing in Alzheimer’s disease: The potential role of Beclin 1 interactome. Prog. Neurobiol. 2013, 106–107, 33–54. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Lee, J.H.; Rubinsztein, D.C. Tau degradation: The ubiquitin-proteasome system versus the autophagy-lysosome system. Prog. Neurobiol. 2013, 105, 49–59. [Google Scholar] [CrossRef]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of Aβ accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef] [PubMed]
- Supnet, C.; Bezprozvanny, I. The dysregulation of intracellular calcium in Alzheimer disease. Cell Calcium 2010, 47, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef]
- Small, S.A.; Duff, K. Linking Aβ and tau in late-onset Alzheimer’s disease: A dual pathway hypothesis. Neuron 2008, 60, 534–542. [Google Scholar] [CrossRef]
- Han, P.; Shi, J. A Theoretical Analysis of the Synergy of Amyloid and Tau in Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 52, 1461–1470. [Google Scholar] [CrossRef]
- Reiman, E.M.; Chen, K.; Alexander, G.E.; Caselli, R.J.; Bandy, D.; Osborne, D.; Saunders, A.M.; Hardy, J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc. Natl. Acad. Sci. USA 2004, 101, 284–289. [Google Scholar] [CrossRef]
- Mosconi, L.; Brys, M.; Switalski, R.; Mistur, R.; Glodzik, L.; Pirraglia, E.; Tsui, W.; De Santi, S.; de Leon, M.J. Maternal family history of Alzheimer’s disease predisposes to reduced brain glucose metabolism. Proc. Natl. Acad. Sci. USA 2007, 104, 19067–19072. [Google Scholar] [CrossRef]
- Jagust, W.J.; Mormino, E.C. Lifespan brain activity, β-amyloid, and Alzheimer’s disease. Trends Cogn. Sci. 2011, 15, 520–526. [Google Scholar] [CrossRef]
- Kukreja, L.; Kujoth, G.C.; Prolla, T.A.; Van Leuven, F.; Vassar, R. Increased mtDNA mutations with aging promotes amyloid accumulation and brain atrophy in the APP/Ld transgenic mouse model of Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 16. [Google Scholar] [CrossRef]
- Goyal, S.; Chaturvedi, R.K. Mitochondrial Protein Import Dysfunction in Pathogenesis of Neurodegenerative Diseases. Mol. Neurobiol. 2021, 58, 1418–1437. [Google Scholar] [CrossRef]
- Fernandez-Vizarra, P.; Fernandez, A.P.; Castro-Blanco, S.; Serrano, J.; Bentura, M.L.; Martinez-Murillo, R.; Martinez, A.; Rodrigo, J. Intra- and extracellular Aβ and PHF in clinically evaluated cases of Alzheimer’s disease. Histol. Histopathol. 2004, 19, 823–844. [Google Scholar] [CrossRef]
- Hu, W.; Wang, Z.; Zheng, H. Mitochondrial accumulation of amyloid β (Aβ) peptides requires TOMM22 as a main Aβ receptor in yeast. J. Biol. Chem. 2018, 293, 12681–12689. [Google Scholar] [CrossRef]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid β-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef]
- Kawahara, M. Neurotoxicity of β-amyloid protein: Oligomerization, channel formation, and calcium dyshomeostasis. Curr. Pharm. Des. 2010, 16, 2779–2789. [Google Scholar] [CrossRef]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links Aβ to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef]
- Takuma, K.; Yao, J.; Huang, J.; Xu, H.; Chen, X.; Luddy, J.; Trillat, A.C.; Stern, D.M.; Arancio, O.; Yan, S.S. ABAD enhances Aβ-induced cell stress via mitochondrial dysfunction. FASEB J. 2005, 19, 597–598. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef]
- Bogorodskiy, A.; Okhrimenko, I.; Burkatovskii, D.; Jakobs, P.; Maslov, I.; Gordeliy, V.; Dencher, N.A.; Gensch, T.; Voos, W.; Altschmied, J.; et al. Role of Mitochondrial Protein Import in Age-Related Neurodegenerative and Cardiovascular Diseases. Cells 2021, 10, 3528. [Google Scholar] [CrossRef] [PubMed]
- Mary, A.; Eysert, F.; Checler, F.; Chami, M. Mitophagy in Alzheimer’s disease: Molecular defects and therapeutic approaches. Mol. Psychiatry 2022, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Yan, S.S. Mitochondrial permeability transition pore in Alzheimer’s disease: Cyclophilin D and amyloid β. Biochim. Biophys. Acta 2010, 1802, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, R.K.; Flint Beal, M. Mitochondrial diseases of the brain. Free Radic. Biol. Med. 2013, 63, 1–29. [Google Scholar] [CrossRef]
- Caspersen, C.; Wang, N.; Yao, J.; Sosunov, A.; Chen, X.; Lustbader, J.W.; Xu, H.W.; Stern, D.; McKhann, G.; Yan, S.D. Mitochondrial Aβ: A potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005, 19, 2040–2041. [Google Scholar] [CrossRef]
- Chen, J.X.; Yan, S.D. Amyloid-β-induced mitochondrial dysfunction. J. Alzheimer’s Dis. 2007, 12, 177–184. [Google Scholar] [CrossRef]
- Shearman, M.S.; Ragan, C.I.; Iversen, L.L. Inhibition of PC12 cell redox activity is a specific, early indicator of the mechanism of β-amyloid-mediated cell death. Proc. Natl. Acad. Sci. USA 1994, 91, 1470–1474. [Google Scholar] [CrossRef]
- Casley, C.S.; Canevari, L.; Land, J.M.; Clark, J.B.; Sharpe, M.A. Β-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J. Neurochem. 2002, 80, 91–100. [Google Scholar] [CrossRef]
- Moreira, P.I.; Santos, M.S.; Moreno, A.; Rego, A.C.; Oliveira, C. Effect of amyloid β-peptide on permeability transition pore: A comparative study. J. Neurosci. Res. 2002, 69, 257–267. [Google Scholar] [CrossRef]
- Tillement, L.; Lecanu, L.; Yao, W.G.; Greeson, J.; Papadopoulos, V. The spirostenol (22R, 25R)-20 alpha-spirost-5-en-3 β-yl hexanoate blocks mitochondrial uptake of Aβ in neuronal cells and prevents Aβ-induced impairment of mitochondrial function. Steroids 2006, 71, 725–735. [Google Scholar] [CrossRef]
- Falkevall, A.; Alikhani, N.; Bhushan, S.; Pavlov, P.F.; Busch, K.; Johnson, K.A.; Eneqvist, T.; Tjernberg, L.; Ankarcrona, M.; Glaser, E. Degradation of the amyloid β-protein by the novel mitochondrial peptidasome, PreP. J. Biol. Chem. 2006, 281, 29096–29104. [Google Scholar] [CrossRef]
- De Strooper, B. Proteases and proteolysis in Alzheimer disease: A multifactorial view on the disease process. Physiol. Rev. 2010, 90, 465–494. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal interaction of VDAC1 with amyloid β and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum. Mol. Genet. 2012, 21, 5131–5146. [Google Scholar] [CrossRef]
- Atlante, A.; Valenti, D.; Latina, V.; Amadoro, G. Dysfunction of Mitochondria in Alzheimer’s Disease: ANT and VDAC Interact with Toxic Proteins and Aid to Determine the Fate of Brain Cells. Int. J. Mol. Sci. 2022, 23, 7722. [Google Scholar] [CrossRef]
- Karikari, T.K.; Nagel, D.A.; Grainger, A.; Clarke-Bland, C.; Hill, E.J.; Moffat, K.G. Preparation of stable tau oligomers for cellular and biochemical studies. Anal. Biochem. 2019, 566, 67–74. [Google Scholar] [CrossRef]
- Cieri, D.; Vicario, M.; Vallese, F.; D’Orsi, B.; Berto, P.; Grinzato, A.; Catoni, C.; De Stefani, D.; Rizzuto, R.; Brini, M.; et al. Tau localises within mitochondrial sub-compartments and its caspase cleavage affects ER-mitochondria interactions and cellular Ca2+ handling. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3247–3256. [Google Scholar] [CrossRef]
- Shafiei, S.S.; Guerrero-Munoz, M.J.; Castillo-Carranza, D.L. Tau Oligomers: Cytotoxicity, Propagation, and Mitochondrial Damage. Front. Aging Neurosci. 2017, 9, 83. [Google Scholar] [CrossRef]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid β with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef]
- Camilleri, A.; Ghio, S.; Caruana, M.; Weckbecker, D.; Schmidt, F.; Kamp, F.; Leonov, A.; Ryazanov, S.; Griesinger, C.; Giese, A.; et al. Tau-induced mitochondrial membrane perturbation is dependent upon cardiolipin. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183064. [Google Scholar] [CrossRef]
- Li, X.C.; Hu, Y.; Wang, Z.H.; Luo, Y.; Zhang, Y.; Liu, X.P.; Feng, Q.; Wang, Q.; Ye, K.; Liu, G.P.; et al. Human wild-type full-length tau accumulation disrupts mitochondrial dynamics and the functions via increasing mitofusins. Sci. Rep. 2016, 6, 24756. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.J.; Jara, C.; Quintanilla, R.A. Contribution of Tau Pathology to Mitochondrial Impairment in Neurodegeneration. Front. Neurosci. 2018, 12, 441. [Google Scholar] [CrossRef] [PubMed]
- Aleardi, A.M.; Benard, G.; Augereau, O.; Malgat, M.; Talbot, J.C.; Mazat, J.P.; Letellier, T.; Dachary-Prigent, J.; Solaini, G.C.; Rossignol, R. Gradual alteration of mitochondrial structure and function by β-amyloids: Importance of membrane viscosity changes, energy deprivation, reactive oxygen species production, and cytochrome c release. J. Bioenerg. Biomembr. 2005, 37, 207–225. [Google Scholar] [CrossRef]
- Kaminsky, Y.G.; Tikhonova, L.A.; Kosenko, E.A. Critical analysis of Alzheimer’s amyloid-β toxicity to mitochondria. Front. Biosci. 2015, 20, 173–197. [Google Scholar] [CrossRef] [PubMed]
- Pagani, L.; Eckert, A. Amyloid-β interaction with mitochondria. Int. J. Alzheimer’s Dis. 2011, 2011, 925050. [Google Scholar] [CrossRef]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Nahed, P.; Kambar, M.; Zhong, K.; Fonseca, J.; Taghva, K. Alzheimer’s disease drug development pipeline: 2022. Alzheimer’s Dement. 2022, 8, e12295. [Google Scholar] [CrossRef]
- Athar, T.; Al Balushi, K.; Khan, S.A. Recent advances on drug development and emerging therapeutic agents for Alzheimer’s disease. Mol. Biol. Rep. 2021, 48, 5629–5645. [Google Scholar] [CrossRef]
- Pardo-Moreno, T.; Gonzalez-Acedo, A.; Rivas-Dominguez, A.; Garcia-Morales, V.; Garcia-Cozar, F.J.; Ramos-Rodriguez, J.J.; Melguizo-Rodriguez, L. Therapeutic Approach to Alzheimer’s Disease: Current Treatments and New Perspectives. Pharmaceutics 2022, 14, 1117. [Google Scholar] [CrossRef]
- Fisar, Z.; Musilek, K.; Benek, O.; Hroch, L.; Vinklarova, L.; Schmidt, M.; Hroudova, J.; Raboch, J. Effects of novel 17β-hydroxysteroid dehydrogenase type 10 inhibitors on mitochondrial respiration. Toxicol. Lett. 2021, 339, 12–19. [Google Scholar] [CrossRef]
- Pogacnik, L.; Ota, A.; Ulrih, N.P. An Overview of Crucial Dietary Substances and Their Modes of Action for Prevention of Neurodegenerative Diseases. Cells 2020, 9, 576. [Google Scholar] [CrossRef]
- Thu Thuy Nguyen, V.; Endres, K. Targeting gut microbiota to alleviate neuroinflammation in Alzheimer’s disease. Adv. Drug Deliv. Rev. 2022, 188, 114418. [Google Scholar] [CrossRef]
- Meltzer, C.C.; Smith, G.; DeKosky, S.T.; Pollock, B.G.; Mathis, C.A.; Moore, R.Y.; Kupfer, D.J.; Reynolds, C.F., 3rd. Serotonin in aging, late-life depression, and Alzheimer’s disease: The emerging role of functional imaging. Neuropsychopharmacology 1998, 18, 407–430. [Google Scholar] [CrossRef]
- Dringenberg, H.C. Alzheimer’s disease: More than a ‘cholinergic disorder’—Evidence that cholinergic-monoaminergic interactions contribute to EEG slowing and dementia. Behav. Brain Res. 2000, 115, 235–249. [Google Scholar] [CrossRef]
- Guzman-Ramos, K.; Osorio-Gomez, D.; Bermudez-Rattoni, F. Cognitive Impairment in Alzheimer’s and Metabolic Diseases: A Catecholaminergic Hypothesis. Neuroscience 2022, 497, 308–323. [Google Scholar] [CrossRef]
- Wang, W.; Karamanlidis, G.; Tian, R. Novel targets for mitochondrial medicine. Sci. Transl. Med. 2016, 8, 326rv323. [Google Scholar] [CrossRef]
- Fisar, Z.; Hroudova, J. Measurement of Mitochondrial Respiration in Platelets. Methods Mol. Biol. 2021, 2277, 269–276. [Google Scholar] [CrossRef]
- Grimm, A.; Schmitt, K.; Eckert, A. Advanced Mitochondrial Respiration Assay for Evaluation of Mitochondrial Dysfunction in Alzheimer’s Disease. Methods Mol. Biol. 2016, 1303, 171–183. [Google Scholar] [CrossRef]
- Hroudova, J.; Fisar, Z. Assessment of the Effects of Drugs on Mitochondrial Respiration. Methods Mol. Biol. 2021, 2277, 133–142. [Google Scholar] [CrossRef]
- Singh, N.; Hroudova, J.; Fisar, Z. In Vitro Effects of Cognitives and Nootropics on Mitochondrial Respiration and Monoamine Oxidase Activity. Mol. Neurobiol. 2017, 54, 5894–5904. [Google Scholar] [CrossRef]
- Hroudova, J.; Novakova, T.; Korabecny, J.; Malinak, D.; Gorecki, L.; Fisar, Z. Effects of Novel Tacrine Derivatives on Mitochondrial Energy Metabolism and Monoamine Oxidase Activity—In Vitro Study. Mol. Neurobiol. 2021, 58, 1102–1113. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, J.A.R.; Buckland, G.R.; Harrison, C.H.; Page, A.; Harris, S.; Love, S.; Neal, J.W.; Holmes, C.; Boche, D. Persistent neuropathological effects 14 years following amyloid-β immunization in Alzheimer’s disease. Brain 2019, 142, 2113–2126. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.P.; Clark, I.A.; Vissel, B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 2, 135. [Google Scholar] [CrossRef]
- Tolar, M.; Hey, J.; Power, A.; Abushakra, S. Neurotoxic Soluble Amyloid Oligomers Drive Alzheimer’s Pathogenesis and Represent a Clinically Validated Target for Slowing Disease Progression. Int. J. Mol. Sci. 2021, 22, 6355. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, D.; Privitera, L.; Fa, M.; Staniszewski, A.; Hashimoto, G.; Aziz, F.; Sakurai, M.; Ribe, E.M.; Troy, C.M.; Mercken, M.; et al. Endogenous amyloid-β is necessary for hippocampal synaptic plasticity and memory. Ann. Neurol. 2011, 69, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Ghazanfari, D.; Noori, M.S.; Bergmeier, S.C.; Hines, J.V.; McCall, K.D.; Goetz, D.J. A novel GSK-3 inhibitor binds to GSK-3β via a reversible, time and Cys-199-dependent mechanism. Bioorg. Med. Chem. 2021, 40, 116179. [Google Scholar] [CrossRef]
- Wang, L.; Bharti; Kumar, R.; Pavlov, P.F.; Winblad, B. Small molecule therapeutics for tauopathy in Alzheimer’s disease: Walking on the path of most resistance. Eur. J. Med. Chem. 2021, 209, 112915. [Google Scholar] [CrossRef]
- Medina, M. An Overview on the Clinical Development of Tau-Based Therapeutics. Int. J. Mol. Sci. 2018, 19, 1160. [Google Scholar] [CrossRef]
- Tolar, M.; Abushakra, S.; Sabbagh, M. The path forward in Alzheimer’s disease therapeutics: Reevaluating the amyloid cascade hypothesis. Alzheimer’s Dement 2020, 16, 1553–1560. [Google Scholar] [CrossRef]
- Sengupta, U.; Nilson, A.N.; Kayed, R. The Role of Amyloid-β Oligomers in Toxicity, Propagation, and Immunotherapy. EBioMedicine 2016, 6, 42–49. [Google Scholar] [CrossRef]
- Krafft, G.A.; Jerecic, J.; Siemers, E.; Cline, E.N. ACU193: An Immunotherapeutic Poised to Test the Amyloid β Oligomer Hypothesis of Alzheimer’s Disease. Front. Neurosci. 2022, 16, 848215. [Google Scholar] [CrossRef]
- Youdim, M.B.; Buccafusco, J.J. Multi-functional drugs for various CNS targets in the treatment of neurodegenerative disorders. Trends Pharmacol. Sci. 2005, 26, 27–35. [Google Scholar] [CrossRef]
- Hroch, L.; Guest, P.; Benek, O.; Soukup, O.; Janockova, J.; Dolezal, R.; Kuca, K.; Aitken, L.; Smith, T.K.; Gunn-Moore, F.; et al. Synthesis and evaluation of frentizole-based indolyl thiourea analogues as MAO/ABAD inhibitors for Alzheimer’s disease treatment. Bioorg. Med. Chem. 2017, 25, 1143–1152. [Google Scholar] [CrossRef]
- Benek, O.; Hroch, L.; Aitken, L.; Gunn-Moore, F.; Vinklarova, L.; Kuca, K.; Perez, D.I.; Perez, C.; Martinez, A.; Fisar, Z.; et al. 1-(Benzo[d]thiazol-2-yl)-3-phenylureas as dual inhibitors of casein kinase 1 and ABAD enzymes for treatment of neurodegenerative disorders. J. Enzyme Inhib. Med. Chem. 2018, 33, 665–670. [Google Scholar] [CrossRef]
- Moreira, N.; Lima, J.; Marchiori, M.F.; Carvalho, I.; Sakamoto-Hojo, E.T. Neuroprotective Effects of Cholinesterase Inhibitors: Current Scenario in Therapies for Alzheimer’s Disease and Future Perspectives. J. Alzheimer’s Dis. Rep. 2022, 6, 177–193. [Google Scholar] [CrossRef]
- Sang, Z.; Wang, K.; Dong, J.; Tang, L. Alzheimer’s disease: Updated multi-targets therapeutics are in clinical and in progress. Eur. J. Med. Chem. 2022, 238, 114464. [Google Scholar] [CrossRef]
- Weinreb, O.; Mandel, S.; Bar-Am, O.; Yogev-Falach, M.; Avramovich-Tirosh, Y.; Amit, T.; Youdim, M.B. Multifunctional neuroprotective derivatives of rasagiline as anti-Alzheimer’s disease drugs. Neurotherapeutics 2009, 6, 163–174. [Google Scholar] [CrossRef]
- Weinstock, M.; Luques, L.; Bejar, C.; Shoham, S. Ladostigil, a novel multifunctional drug for the treatment of dementia co-morbid with depression. J. Neural Transm. Suppl. 2006, 443–446. [Google Scholar] [CrossRef]
- Haddad, H.W.; Malone, G.W.; Comardelle, N.J.; Degueure, A.E.; Poliwoda, S.; Kaye, R.J.; Murnane, K.S.; Kaye, A.M.; Kaye, A.D. Aduhelm, a novel anti-amyloid monoclonal antibody, for the treatment of Alzheimer’s Disease: A comprehensive review. Health Psychol. Res. 2022, 10, 37023. [Google Scholar] [CrossRef]
- Vaz, M.; Silva, V.; Monteiro, C.; Silvestre, S. Role of Aducanumab in the Treatment of Alzheimer’s Disease: Challenges and Opportunities. Clin. Interv. Aging 2022, 17, 797–810. [Google Scholar] [CrossRef]
- Khoury, R.; Rajamanickam, J.; Grossberg, G.T. An update on the safety of current therapies for Alzheimer’s disease: Focus on rivastigmine. Ther. Adv. Drug Saf. 2018, 9, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Birks, J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst. Rev. 2006, 2006, CD005593. [Google Scholar] [CrossRef] [PubMed]
- McShane, R.; Westby, M.J.; Roberts, E.; Minakaran, N.; Schneider, L.; Farrimond, L.E.; Maayan, N.; Ware, J.; Debarros, J. Memantine for dementia. Cochrane Database Syst. Rev. 2019, 3, CD003154. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Ports, K.D.; Corrada, M.M.; Odegaard, A.O.; O’Connell, J.; Jiang, L. Metformin and Dementia Risk: A Systematic Review with Respect to Time Related Biases. J. Alzheimer’s Dis. Rep. 2022, 6, 443–459. [Google Scholar] [CrossRef] [PubMed]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, M.K.; Karuppasamy, M.; Sahoo, B.M. Therapeutic Potential of Semaglutide, a Newer GLP-1 Receptor Agonist, in Abating Obesity, Non-Alcoholic Steatohepatitis and Neurodegenerative diseases: A Narrative Review. Pharm. Res. 2022, 39, 1233–1248. [Google Scholar] [CrossRef]
- Henderson, S.T.; Vogel, J.L.; Barr, L.J.; Garvin, F.; Jones, J.J.; Costantini, L.C. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: A randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. 2009, 6, 31. [Google Scholar] [CrossRef]
- Croteau, E.; Castellano, C.A.; Richard, M.A.; Fortier, M.; Nugent, S.; Lepage, M.; Duchesne, S.; Whittingstall, K.; Turcotte, E.E.; Bocti, C.; et al. Ketogenic Medium Chain Triglycerides Increase Brain Energy Metabolism in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 64, 551–561. [Google Scholar] [CrossRef]
- Wood, A.H.R.; Chappell, H.F.; Zulyniak, M.A. Dietary and supplemental long-chain omega-3 fatty acids as moderators of cognitive impairment and Alzheimer’s disease. Eur. J. Nutr. 2022, 61, 589–604. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Hull, M.A.; Song, M.; Van Hulle, C.; Carlsson, C.; Chapman, M.J.; Toth, P.P. Beyond cardiovascular medicine: Potential future uses of icosapent ethyl. Eur. Heart J. Suppl. 2020, 22, J54–J64. [Google Scholar] [CrossRef]
- Hampel, H.; Williams, C.; Etcheto, A.; Goodsaid, F.; Parmentier, F.; Sallantin, J.; Kaufmann, W.E.; Missling, C.U.; Afshar, M. A precision medicine framework using artificial intelligence for the identification and confirmation of genomic biomarkers of response to an Alzheimer’s disease therapy: Analysis of the blarcamesine (ANAVEX2-73) Phase 2a clinical study. Alzheimer’s Dement. 2020, 6, e12013. [Google Scholar] [CrossRef]
- Sanati, M.; Aminyavari, S.; Afshari, A.R.; Sahebkar, A. Mechanistic insight into the role of metformin in Alzheimer’s disease. Life Sci. 2022, 291, 120299. [Google Scholar] [CrossRef]
- Sabbagh, M.N.; Decourt, B. COR388 (atuzaginstat): An investigational gingipain inhibitor for the treatment of Alzheimer disease. Expert Opin. Investig. Drugs 2022, 31, 987–993. [Google Scholar] [CrossRef]
- Piscopo, P.; Crestini, A.; Carbone, E.; Rivabene, R.; Ancidoni, A.; Lo Giudice, M.; Corbo, M.; Vanacore, N.; Lacorte, E. A systematic review on drugs for synaptic plasticity in the treatment of dementia. Ageing Res. Rev. 2022, 81, 101726. [Google Scholar] [CrossRef]
- Zheng, X.Y.; Zhang, H.C.; Lv, Y.D.; Jin, F.Y.; Wu, X.J.; Zhu, J.; Ruan, Y. Levetiracetam alleviates cognitive decline in Alzheimer’s disease animal model by ameliorating the dysfunction of the neuronal network. Front. Aging Neurosci. 2022, 14, 888784. [Google Scholar] [CrossRef]
- Wang, H.Y.; Bakshi, K.; Frankfurt, M.; Stucky, A.; Goberdhan, M.; Shah, S.M.; Burns, L.H. Reducing amyloid-related Alzheimer’s disease pathogenesis by a small molecule targeting filamin A. J. Neurosci. 2012, 32, 9773–9784. [Google Scholar] [CrossRef]
- Tolar, M.; Abushakra, S.; Hey, J.A.; Porsteinsson, A.; Sabbagh, M. Aducanumab, gantenerumab, BAN2401, and ALZ-801-the first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimer’s Res. Ther. 2020, 12, 95. [Google Scholar] [CrossRef]
- Abushakra, S.; Porsteinsson, A.; Scheltens, P.; Sadowsky, C.; Vellas, B.; Cummings, J.; Gauthier, S.; Hey, J.A.; Power, A.; Wang, P.; et al. Clinical Effects of Tramiprosate in APOE4/4 Homozygous Patients with Mild Alzheimer’s Disease Suggest Disease Modification Potential. J. Prev. Alzheimer’s Dis. 2017, 4, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Reading, C.L.; Ahlem, C.N.; Murphy, M.F. NM101 Phase III study of NE3107 in Alzheimer’s disease: Rationale, design and therapeutic modulation of neuroinflammation and insulin resistance. Neurodegener. Dis. Manag. 2021, 11, 289–298. [Google Scholar] [CrossRef]
- Hamaguchi, T.; Ono, K.; Yamada, M. REVIEW: Curcumin and Alzheimer’s disease. CNS Neurosci. Ther. 2010, 16, 285–297. [Google Scholar] [CrossRef]
- Malafaia, D.; Albuquerque, H.M.T.; Silva, A.M.S. Amyloid-β and tau aggregation dual-inhibitors: A synthetic and structure-activity relationship focused review. Eur. J. Med. Chem. 2021, 214, 113209. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar] [CrossRef]
- Jakubowski, J.M.; Orr, A.A.; Le, D.A.; Tamamis, P. Interactions between Curcumin Derivatives and Amyloid-β Fibrils: Insights from Molecular Dynamics Simulations. J. Chem. Inf. Model. 2020, 60, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Arndt, J.W.; Qian, F.; Smith, B.A.; Quan, C.; Kilambi, K.P.; Bush, M.W.; Walz, T.; Pepinsky, R.B.; Bussiere, T.; Hamann, S.; et al. Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid-β. Sci. Rep. 2018, 8, 6412. [Google Scholar] [CrossRef] [PubMed]
- Soderberg, L.; Johannesson, M.; Nygren, P.; Laudon, H.; Eriksson, F.; Osswald, G.; Moller, C.; Lannfelt, L. Lecanemab, Aducanumab, and Gantenerumab—Binding Profiles to Different Forms of Amyloid-β Might Explain Efficacy and Side Effects in Clinical Trials for Alzheimer’s Disease. Neurotherapeutics 2022, 1–12. [Google Scholar] [CrossRef]
- Bouter, Y.; Liekefeld, H.; Pichlo, S.; Westhoff, A.C.; Fenn, L.; Bakrania, P.; Bayer, T.A. Donanemab detects a minor fraction of amyloid-β plaques in post-mortem brain tissue of patients with Alzheimer’s disease and Down syndrome. Acta Neuropathol. 2022, 143, 601–603. [Google Scholar] [CrossRef]
- Doody, R.S.; Thomas, R.G.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Raman, R.; Sun, X.; Aisen, P.S.; et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 311–321. [Google Scholar] [CrossRef]
- YM, M.Y.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. Neuroprotective Effect of Caffeine in Alzheimer’s Disease. Molecules 2022, 27, 3737. [Google Scholar] [CrossRef]
- Vaughan, R.A.; Garcia-Smith, R.; Bisoffi, M.; Trujillo, K.A.; Conn, C.A. Effects of caffeine on metabolism and mitochondria biogenesis in rhabdomyosarcoma cells compared with 2,4-dinitrophenol. Nutr. Metab. Insights 2012, 5, 59–70. [Google Scholar] [CrossRef]
- Hoang, K.; Watt, H.; Golemme, M.; Perry, R.J.; Ritchie, C.; Wilson, D.; Pickett, J.; Fox, C.; Howard, R.; Malhotra, P.A. Noradrenergic Add-on Therapy with Extended-Release Guanfacine in Alzheimer’s Disease (NorAD): Study protocol for a randomised clinical trial and COVID-19 amendments. Trials 2022, 23, 623. [Google Scholar] [CrossRef]
- Xiao, S.; Wang, T.; Ma, X.; Qin, Y.; Li, X.; Zhao, Z.; Liu, X.; Wang, X.; Xie, H.; Jiang, Q.; et al. Efficacy and safety of a novel acetylcholinesterase inhibitor octohydroaminoacridine in mild-to-moderate Alzheimer’s disease: A Phase II multicenter randomised controlled trial. Age Ageing 2017, 46, 767–773. [Google Scholar] [CrossRef]
- Rangani, R.J.; Upadhya, M.A.; Nakhate, K.T.; Kokare, D.M.; Subhedar, N.K. Nicotine evoked improvement in learning and memory is mediated through NPY Y1 receptors in rat model of Alzheimer’s disease. Peptides 2012, 33, 317–328. [Google Scholar] [CrossRef]
- Mihailescu, S.; Drucker-Colin, R. Nicotine, brain nicotinic receptors, and neuropsychiatric disorders. Arch. Med. Res. 2000, 31, 131–144. [Google Scholar] [CrossRef]
- Li, L.; Xu, S.; Liu, L.; Feng, R.; Gong, Y.; Zhao, X.; Li, J.; Cai, J.; Feng, N.; Wang, L.; et al. Multifunctional Compound AD-35 Improves Cognitive Impairment and Attenuates the Production of TNF-α and IL-1β in an Aβ25-35-induced Rat Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 56, 1403–1417. [Google Scholar] [CrossRef]
- Miziak, B.; Blaszczyk, B.; Czuczwar, S.J. Some Candidate Drugs for Pharmacotherapy of Alzheimer’s Disease. Pharmaceuticals 2021, 14, 458. [Google Scholar] [CrossRef]
- Grossberg, G.T.; Kohegyi, E.; Mergel, V.; Josiassen, M.K.; Meulien, D.; Hobart, M.; Slomkowski, M.; Baker, R.A.; McQuade, R.D.; Cummings, J.L. Efficacy and Safety of Brexpiprazole for the Treatment of Agitation in Alzheimer’s Dementia: Two 12-Week, Randomized, Double-Blind, Placebo-Controlled Trials. Am. J. Geriatr. Psychiatry 2020, 28, 383–400. [Google Scholar] [CrossRef]
- Ward, K.; Citrome, L. AXS-05: An investigational treatment for Alzheimer’s disease-associated agitation. Expert Opin. Investig. Drugs 2022, 31, 773–780. [Google Scholar] [CrossRef]
- Ahmed, M.; Malik, M.; Teselink, J.; Lanctot, K.L.; Herrmann, N. Current Agents in Development for Treating Behavioral and Psychological Symptoms Associated with Dementia. Drugs Aging 2019, 36, 589–605. [Google Scholar] [CrossRef]
- Bosnjak Kuharic, D.; Markovic, D.; Brkovic, T.; Jeric Kegalj, M.; Rubic, Z.; Vuica Vukasovic, A.; Jeroncic, A.; Puljak, L. Cannabinoids for the treatment of dementia. Cochrane Database Syst. Rev. 2021, 9, CD012820. [Google Scholar] [CrossRef]
- Ozarowski, M.; Karpinski, T.M.; Zielinska, A.; Souto, E.B.; Wielgus, K. Cannabidiol in Neurological and Neoplastic Diseases: Latest Developments on the Molecular Mechanism of Action. Int. J. Mol. Sci. 2021, 22, 4294. [Google Scholar] [CrossRef]
- Wang, J.H.; Wu, Y.J.; Tee, B.L.; Lo, R.Y. Medical Comorbidity in Alzheimer’s Disease: A Nested Case-Control Study. J. Alzheimer’s Dis. 2018, 63, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Eshetie, T.C.; Nguyen, T.A.; Gillam, M.H.; Kalisch Ellett, L.M. Medication Use for Comorbidities in People with Alzheimer’s Disease: An Australian Population-Based Study. Pharmacotherapy 2019, 39, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Majidazar, R.; Rezazadeh-Gavgani, E.; Sadigh-Eteghad, S.; Naseri, A. Pharmacotherapy of Alzheimer’s disease: An overview of systematic reviews. Eur. J. Clin. Pharmacol. 2022, 78, 1567–1587. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.; Nelson, V.A.; Davila, H.; Ratner, E.; Fink, H.A.; Hemmy, L.S.; McCarten, J.R.; Barclay, T.R.; Brasure, M.; Kane, R.L. Over-the-Counter Supplement Interventions to Prevent Cognitive Decline, Mild Cognitive Impairment, and Clinical Alzheimer-Type Dementia: A Systematic Review. Ann. Intern. Med. 2018, 168, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Park, H.H.; Koh, S.H.; Choi, N.Y.; Yu, H.J.; Park, J.; Lee, Y.J.; Lee, K.Y. Coenzyme Q10 protects against amyloid β-induced neuronal cell death by inhibiting oxidative stress and activating the P13K pathway. Neurotoxicology 2012, 33, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Mitochondrial bioenergetics decay in aging: Beneficial effect of melatonin. Cell. Mol. Life Sci. 2017, 74, 3897–3911. [Google Scholar] [CrossRef]
- De Castro, A.A.; Soares, F.V.; Pereira, A.F.; Polisel, D.A.; Caetano, M.S.; Leal, D.H.S.; da Cunha, E.F.F.; Nepovimova, E.; Kuca, K.; Ramalho, T.C. Non-conventional compounds with potential therapeutic effects against Alzheimer’s disease. Expert Rev. Neurother. 2019, 19, 375–395. [Google Scholar] [CrossRef]
- Lu, P.R.; Wong, S.Y.; Wu, L.; Lin, D.B. Carotenoid metabolism in mitochondrial function. Food Qual. Saf. 2020, 4, 115–122. [Google Scholar] [CrossRef]
- Shin, S.J.; Jeon, S.G.; Kim, J.I.; Jeong, Y.O.; Kim, S.; Park, Y.H.; Lee, S.K.; Park, H.H.; Hong, S.B.; Oh, S.; et al. Red Ginseng Attenuates Aβ-Induced Mitochondrial Dysfunction and Aβ-mediated Pathology in an Animal Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 3030. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Bioenergetics and metabolism: A bench to bedside perspective. J. Neurochem. 2016, 139 (Suppl. S2), 126–135. [Google Scholar] [CrossRef][Green Version]
- Gauthier, S.; Feldman, H.H.; Schneider, L.S.; Wilcock, G.K.; Frisoni, G.B.; Hardlund, J.H.; Moebius, H.J.; Bentham, P.; Kook, K.A.; Wischik, D.J.; et al. Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: A randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet 2016, 388, 2873–2884. [Google Scholar] [CrossRef]
- Shin, S.J.; Park, Y.H.; Jeon, S.G.; Kim, S.; Nam, Y.; Oh, S.M.; Lee, Y.Y.; Moon, M. Red Ginseng Inhibits Tau Aggregation and Promotes Tau Dissociation In Vitro. Oxid. Med. Cell. Longev. 2020, 2020, 7829842. [Google Scholar] [CrossRef]
- Chalatsa, I.; Arvanitis, D.A.; Koulakiotis, N.S.; Giagini, A.; Skaltsounis, A.L.; Papadopoulou-Daifoti, Z.; Tsarbopoulos, A.; Sanoudou, D. The Crocus sativus Compounds trans-Crocin 4 and trans-Crocetin Modulate the Amyloidogenic Pathway and Tau Misprocessing in Alzheimer Disease Neuronal Cell Culture Models. Front. Neurosci. 2019, 13, 249. [Google Scholar] [CrossRef]
- George, R.C.; Lew, J.; Graves, D.J. Interaction of cinnamaldehyde and epicatechin with tau: Implications of beneficial effects in modulating Alzheimer’s disease pathogenesis. J. Alzheimer’s Dis. 2013, 36, 21–40. [Google Scholar] [CrossRef]
- Viswanathan, G.K.; Shwartz, D.; Losev, Y.; Arad, E.; Shemesh, C.; Pichinuk, E.; Engel, H.; Raveh, A.; Jelinek, R.; Cooper, I.; et al. Purpurin modulates Tau-derived VQIVYK fibrillization and ameliorates Alzheimer’s disease-like symptoms in animal model. Cell. Mol. Life Sci. 2020, 77, 2795–2813. [Google Scholar] [CrossRef]
- Ghasemzadeh, S.; Riazi, G.H. Inhibition of Tau amyloid fibril formation by folic acid: In-vitro and theoretical studies. Int. J. Biol. Macromol. 2020, 154, 1505–1516. [Google Scholar] [CrossRef]
- Jia, Y.; Wang, N.; Liu, X. Resveratrol and Amyloid-β: Mechanistic Insights. Nutrients 2017, 9, 1122. [Google Scholar] [CrossRef]
- Yang, G.; Wang, Y.; Tian, J.; Liu, J.P. Huperzine A for Alzheimer’s disease: A systematic review and meta-analysis of randomized clinical trials. PLoS ONE 2013, 8, e74916. [Google Scholar] [CrossRef]
- Xiao, X.Q.; Wang, R.; Tang, X.C. Huperzine A and tacrine attenuate β-amyloid peptide-induced oxidative injury. J. Neurosci. Res. 2000, 61, 564–569. [Google Scholar] [CrossRef]
- Azizi, Z.; Majlessi, N.; Choopani, S.; Naghdi, N. Neuroprotective effects of carvacrol against Alzheimer’s disease and other neurodegenerative diseases: A review. Avicenna J. Phytomed. 2022, 12, 371–387. [Google Scholar] [CrossRef]
- Celik Topkara, K.; Kilinc, E.; Cetinkaya, A.; Saylan, A.; Demir, S. Therapeutic effects of carvacrol on β-amyloid-induced impairments in in vitro and in vivo models of Alzheimer’s disease. Eur. J. Neurosci. 2021, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Rubin, L.; Silva, M.; Li, S.; Yang, C.; Lazarovici, P.; Zheng, W. Current Progress on Neuroprotection Induced by Artemisia, Ginseng, Astragalus, and Ginkgo Traditional Chinese Medicines for the Therapy of Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2022, 2022, 3777021. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.Y.; Kang, E.S.; Park, J.H.; Cho, B.O.; Jang, S.I. Anti-inflammatory effect of red ginseng marc, Artemisia scoparia, Paeonia japonica and Angelica gigas extract mixture in LPS-stimulated RAW 264.7 cells. Biomed. Rep. 2022, 17, 63. [Google Scholar] [CrossRef]
- Chakraborty, B.; Mukerjee, N.; Maitra, S.; Zehravi, M.; Mukherjee, D.; Ghosh, A.; Massoud, E.E.S.; Rahman, M.H. Therapeutic Potential of Different Natural Products for the Treatment of Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2022, 2022, 6873874. [Google Scholar] [CrossRef] [PubMed]
- Aillaud, I.; Funke, S.A. Tau Aggregation Inhibiting Peptides as Potential Therapeutics for Alzheimer Disease. Cell. Mol. Neurobiol. 2022, 1–11. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fišar, Z. Linking the Amyloid, Tau, and Mitochondrial Hypotheses of Alzheimer’s Disease and Identifying Promising Drug Targets. Biomolecules 2022, 12, 1676. https://doi.org/10.3390/biom12111676
Fišar Z. Linking the Amyloid, Tau, and Mitochondrial Hypotheses of Alzheimer’s Disease and Identifying Promising Drug Targets. Biomolecules. 2022; 12(11):1676. https://doi.org/10.3390/biom12111676
Chicago/Turabian StyleFišar, Zdeněk. 2022. "Linking the Amyloid, Tau, and Mitochondrial Hypotheses of Alzheimer’s Disease and Identifying Promising Drug Targets" Biomolecules 12, no. 11: 1676. https://doi.org/10.3390/biom12111676
APA StyleFišar, Z. (2022). Linking the Amyloid, Tau, and Mitochondrial Hypotheses of Alzheimer’s Disease and Identifying Promising Drug Targets. Biomolecules, 12(11), 1676. https://doi.org/10.3390/biom12111676