


Macrophages in Skin Wounds: Functions and Therapeutic Potential


Abstract
1. Introduction
2. Macrophage Subsets and Their Plasticity in Healthy Skin
2.1. Murine Dermal Tissue-Resident Macrophages
2.1.1. Perineural Macrophages
2.1.2. Perivascular Macrophages
2.1.3. Perifollicular Macrophages
2.2. Human Dermal-Resident Macrophages
2.3. Monocyte-Derived Macrophages
3. Macrophages’ Role in Various Stages of Wound Healing
3.1. Haemostasis Phase (Day 0)
3.2. Macrophages in the Inflammatory Phase (Day 1–3/4)
3.2.1. Macrophage Regulation of Neutrophil Recruitment and Pro-Inflammatory Macrophage Polarisation
3.2.2. Macrophage Regulation of Neutrophils and Anti-Inflammatory Macrophage Polarisation
3.3. Macrophages in the Wound Proliferation Phase (Day 3/4–7)
3.3.1. Macrophage Regulation of Wound Angiogenesis
3.3.2. Macrophage Regulation of Fibroblasts and Myofibroblasts
3.3.3. Macrophage Regulation of Keratinocyte Re-Epithelisation
3.4. Macrophage Regulation in Remodelling Phase (Day 7–Months)
4. Macrophage Dysfunction in Chronic Wound
5. Strategies to Modulate Wound Inflammation and Macrophage Activity
5.1. NF-κB Signalling–A Friend or Foe of Wound Healing
5.1.1. Toll-like Receptors (TLRs) and MyD88 Adaptor Protein
5.1.2. IL-1β/IL-1R1 Signalling
5.1.3. IL-1β and IL-17A Signalling Crosstalk
5.1.4. TNF/TNFR1 Signalling
5.1.5. Microbiome
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Krzyszczyk, P.; Schloss, R.; Palmer, A.; Berthiaume, F. The Role of Macrophages in Acute and Chronic Wound Healing and Interventions to Promote Pro-wound Healing Phenotypes. Front. Physiol. 2018, 9, 419. [Google Scholar] [CrossRef] [PubMed]
- Lucas, T.; Waisman, A.; Ranjan, R.; Roes, J.; Krieg, T.; Müller, W.; Roers, A.; Eming, S.A. Differential Roles of Macrophages in Diverse Phases of Skin Repair. J. Immunol. 2010, 184, 3964–3977. [Google Scholar] [CrossRef] [PubMed]
- Sindrilaru, A.; Peters, T.; Wieschalka, S.; Baican, C.; Baican, A.; Peter, H.; Hainzl, A.; Schatz, S.; Qi, Y.; Schlecht, A.; et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J. Clin. Investig. 2011, 121, 985–997. [Google Scholar] [CrossRef]
- Ramasamy, R.; Yan, S.F.; Schmidt, A.M. Receptor for AGE (RAGE): Signaling mechanisms in the pathogenesis of diabetes and its complications. Ann. N. Y. Acad. Sci. 2011, 1243, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Abtin, A.; Jain, R.; Mitchell, A.J.; Roediger, B.; Brzoska, A.J.; Tikoo, S.; Cheng, Q.; Ng, L.G.; Cavanagh, L.L.; von Andrian, U.H.; et al. Perivascular macrophages mediate neutrophil recruitment during bacterial skin infection. Nat. Immunol. 2014, 15, 45–53. [Google Scholar] [CrossRef]
- Barreiro, O.; Cibrian, D.; Clemente, C.; Alvarez, D.; Moreno, V.; Valiente, Í.; Bernad, A.; Vestweber, D.; Arroyo, A.G.; Martín, P.A.-O.; et al. Pivotal role for skin transendothelial radio-resistant anti-inflammatory macrophages in tissue repair. eLife 2016, 5, e15251. [Google Scholar] [CrossRef]
- Chakarov, S.; Lim, H.Y.; Tan, L.; Lim, S.Y.; See, P.; Lum, J.; Zhang, X.-M.; Foo, S.; Nakamizo, S.; Duan, K.; et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science 2019, 363, eaau0964. [Google Scholar] [CrossRef]
- Kolter, J.; Feuerstein, R.; Zeis, P.; Hagemeyer, N.; Paterson, N.; d’Errico, P.; Baasch, S.; Amann, L.; Masuda, T.; Lösslein, A.; et al. A Subset of Skin Macrophages Contributes to the Surveillance and Regeneration of Local Nerves. Immunity 2019, 50, 1482–1497.e1487. [Google Scholar] [CrossRef]
- Mass, E.; Ballesteros, I.; Farlik, M.; Halbritter, F.; Günther, P.; Crozet, L.; Jacome-Galarza, C.E.; Händler, K.; Klughammer, J.; Kobayashi, Y.; et al. Specification of tissue-resident macrophages during organogenesis. Science 2016, 353, aaf4238. [Google Scholar] [CrossRef]
- West, H.C.; Bennett, C.L. Redefining the Role of Langerhans Cells As Immune Regulators within the Skin. Front. Immunol. 2018, 8, 1941. [Google Scholar] [CrossRef]
- Hoeffel, G.; Wang, Y.; Greter, M.; See, P.; Teo, P.; Malleret, B.; Leboeuf, M.; Low, D.; Oller, G.; Almeida, F.; et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac–derived macrophages. J. Exp. Med. 2012, 209, 1167–1181. [Google Scholar] [CrossRef] [PubMed]
- Ghigo, C.; Mondor, I.; Jorquera, A.; Nowak, J.; Wienert, S.; Zahner, S.P.; Clausen, B.E.; Luche, H.; Malissen, B.; Klauschen, F.; et al. Multicolor fate mapping of Langerhans cell homeostasis. J. Exp. Med. 2013, 210, 1657–1664. [Google Scholar] [CrossRef] [PubMed]
- Seré, K.; Baek, J.-H.; Ober-Blöbaum, J.; Müller-Newen, G.; Tacke, F.; Yokota, Y.; Zenke, M.; Hieronymus, T. Two Distinct Types of Langerhans Cells Populate the Skin during Steady State and Inflammation. Immunity 2012, 37, 905–916. [Google Scholar] [CrossRef]
- Merad, M.; Manz, M.G.; Karsunky, H.; Wagers, A.; Peters, W.; Charo, I.; Weissman, I.L.; Cyster, J.G.; Engleman, E.G. Langerhans cells renew in the skin throughout life under steady-state conditions. Nat. Immunol. 2002, 3, 1135–1141. [Google Scholar] [CrossRef] [PubMed]
- Olingy, C.E.; San Emeterio, C.L.; Ogle, M.E.; Krieger, J.R.; Bruce, A.C.; Pfau, D.D.; Jordan, B.T.; Peirce, S.M.; Botchwey, E.A. Non-classical monocytes are biased progenitors of wound healing macrophages during soft tissue injury. Sci. Rep. 2017, 7, 447. [Google Scholar] [CrossRef] [PubMed]
- Tamoutounour, S.; Guilliams, M.; Montanana Sanchis, F.; Liu, H.; Terhorst, D.; Malosse, C.; Pollet, E.; Ardouin, L.; Luche, H.; Sanchez, C.; et al. Origins and Functional Specialization of Macrophages and of Conventional and Monocyte-Derived Dendritic Cells in Mouse Skin. Immunity 2013, 39, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Lavin, Y.; Winter, D.; Blecher-Gonen, R.; David, E.; Keren-Shaul, H.; Merad, M.; Jung, S.; Amit, I. Tissue-Resident Macrophage Enhancer Landscapes Are Shaped by the Local Microenvironment. Cell 2014, 159, 1312–1326. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.C.E.; Dai, Z.; Ferrante, A.W.; Drake, C.G.; Christiano, A.M. A Subset of TREM2+ Dermal Macrophages Secretes Oncostatin M to Maintain Hair Follicle Stem Cell Quiescence and Inhibit Hair Growth. Cell Stem Cell 2019, 24, 654–669.e6. [Google Scholar] [CrossRef]
- Castellana, D.; Paus, R.; Perez-Moreno, M. Macrophages Contribute to the Cyclic Activation of Adult Hair Follicle Stem Cells. PLoS Biol. 2014, 12, e1002002. [Google Scholar] [CrossRef]
- Joost, S.; Jacob, T.; Sun, X.; Annusver, K.; La Manno, G.; Sur, I.; Kasper, M. Single-Cell Transcriptomics of Traced Epidermal and Hair Follicle Stem Cells Reveals Rapid Adaptations during Wound Healing. Cell Rep. 2018, 25, 585–597.e7. [Google Scholar] [CrossRef]
- Daszczuk, P.; Mazurek, P.; Pieczonka, T.D.; Olczak, A.; Boryń, Ł.M.; Kobielak, K. An Intrinsic Oscillation of Gene Networks Inside Hair Follicle Stem Cells: An Additional Layer That Can Modulate Hair Stem Cell Activities. Front. Cell Dev. Biol. 2020, 8, 1511. [Google Scholar] [CrossRef] [PubMed]
- Hardman, J.A.; Muneeb, F.; Pople, J.; Bhogal, R.; Shahmalak, A.; Paus, R. Human Perifollicular Macrophages Undergo Apoptosis, Express Wnt Ligands, and Switch their Polarization during Catagen. J. Investig. Dermatol. 2019, 139, 2543–2546.e9. [Google Scholar] [CrossRef] [PubMed]
- Bigley, V.; Haniffa, M.; Doulatov, S.; Wang, X.-N.; Dickinson, R.; McGovern, N.; Jardine, L.; Pagan, S.; Dimmick, I.; Chua, I.; et al. The human syndrome of dendritic cell, monocyte, B and NK lymphoid deficiency. J. Exp. Med. 2011, 208, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, G.; Vegh, P.; Fletcher, J.; Poyner Elizabeth, F.M.; Stephenson, E.; Goh, I.; Botting Rachel, A.; Huang, N.; Olabi, B.; Dubois, A.; et al. Developmental cell programs are co-opted in inflammatory skin disease. Science 2021, 371, eaba6500. [Google Scholar] [CrossRef]
- Xue, D.; Tabib, T.; Morse, C.; Lafyatis, R. Transcriptome landscape of myeloid cells in human skin reveals diversity, rare populations and putative DC progenitors. J. Dermatol. Sci. 2020, 97, 41–49. [Google Scholar] [CrossRef]
- He, H.; Suryawanshi, H.; Morozov, P.; Gay-Mimbrera, J.; Del Duca, E.; Kim, H.J.; Kameyama, N.; Estrada, Y.; Der, E.; Krueger, J.G.; et al. Single-cell transcriptome analysis of human skin identifies novel fibroblast subpopulation and enrichment of immune subsets in atopic dermatitis. J. Allergy Clin. Immunol. 2020, 145, 1615–1628. [Google Scholar] [CrossRef]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Rahmani, W.; Liu, Y.; Rosin, N.L.; Kline, A.; Raharjo, E.; Yoon, J.; Stratton, J.A.; Sinha, S.; Biernaskie, J. Macrophages Promote Wound-Induced Hair Follicle Regeneration in a CX3CR1 and TGF-β1 Dependent Manner. J. Investig. Dermatol. 2018, 138, 2111–2122. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef]
- Rodrigues, M.; Kosaric, N.; Bonham, C.A.; Gurtner, G.C. Wound Healing: A Cellular Perspective. Physiol. Rev. 2019, 99, 665–706. [Google Scholar] [CrossRef]
- Golebiewska, E.M.; Poole, A.W. Platelet secretion: From haemostasis to wound healing and beyond. Blood Rev. 2015, 29, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Nishihira, K.; Yamashita, A.; Tanaka, N.; Moriguchi-Goto, S.; Imamura, T.; Ishida, T.; Kawashima, S.; Yamamoto, R.; Kitamura, K.; Asada, Y. Serotonin induces vasoconstriction of smooth muscle cell-rich neointima through 5-hydroxytryptamine2A receptor in rabbit femoral arteries. J. Thromb. Haemost. 2008, 6, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Cediel, E.; Vázquez-Cruz, B.; Navarro-Cid, J.; De Las Heras, N.; Sanz-Rosa, D.; Cachofeiro, V.; Lahera, V. Role of endothelin-1 and thromboxane A2 in renal vasoconstriction induced by angiotensin II in diabetes and hypertension. Kidney Int. 2002, 62, S2–S7. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; DiPietro, L.A. Toll-Like Receptor Function in Acute Wounds. Adv. Wound Care 2017, 6, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Minutti, C.M.; Knipper, J.A.; Allen, J.E.; Zaiss, D.M.W. Tissue-specific contribution of macrophages to wound healing. Semin. Cell Dev. Biol. 2017, 61, 3–11. [Google Scholar]
- Rodero, M.P.; Licata, F.; Poupel, L.; Hamon, P.; Khosrotehrani, K.; Combadiere, C.; Boissonnas, A. In Vivo Imaging Reveals a Pioneer Wave of Monocyte Recruitment into Mouse Skin Wounds. PLoS ONE 2014, 9, e108212. [Google Scholar] [CrossRef]
- de Oliveira, S.; López-Muñoz, A.; Candel, S.; Pelegrín, P.; Calado, Â.; Mulero, V. ATP Modulates Acute Inflammation In Vivo through Dual Oxidase 1–Derived H2O2 Production and NF-κB Activation. J. Immunol. 2014, 192, 5710–5719. [Google Scholar] [CrossRef]
- Sipka, T.; Peroceschi, R.; Hassan-Abdi, R.; Groß, M.; Ellett, F.; Begon-Pescia, C.; Gonzalez, C.; Lutfalla, G.; Nguyen-Chi, M. Damage-Induced Calcium Signaling and Reactive Oxygen Species Mediate Macrophage Activation in Zebrafish. Front. Immunol. 2021, 12, 636585. [Google Scholar] [CrossRef]
- Jang, H.-S.; Rabb, H.; Padanilam, B.J. CD169+ Macrophages: Regulators of Neutrophil Trafficking to Injured Kidneys. J. Am. Soc. Nephrol. 2015, 26, 769–771. [Google Scholar] [CrossRef]
- Nourshargh, S.; Alon, R. Leukocyte Migration into Inflamed Tissues. Immunity 2014, 41, 694–707. [Google Scholar] [CrossRef]
- Wilgus, T.A.; Roy, S.; McDaniel, J.C. Neutrophils and Wound Repair: Positive Actions and Negative Reactions. Adv. Wound Care 2013, 2, 379–388. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss David, S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, H.N.; Hardman, M.J. Wound healing: Cellular mechanisms and pathological outcomes. Open Biol. 2020, 10, 200223. [Google Scholar] [CrossRef] [PubMed]
- Sharif, O.; Bolshakov, V.N.; Raines, S.; Newham, P.; Perkins, N.D. Transcriptional profiling of the LPS induced NF-κB response in macrophages. BMC Immunol. 2007, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hossain, M.; Thanabalasuriar, A.; Gunzer, M.; Meininger, C.; Kubes, P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science 2017, 358, 111–116. [Google Scholar] [CrossRef]
- Tauzin, S.; Starnes, T.W.; Becker, F.B.; Lam, P.-Y.; Huttenlocher, A. Redox and Src family kinase signaling control leukocyte wound attraction and neutrophil reverse migration. J. Cell Biol. 2014, 207, 589–598. [Google Scholar] [CrossRef]
- Marwick, J.A.; Mills, R.; Kay, O.; Michail, K.; Stephen, J.; Rossi, A.G.; Dransfield, I.; Hirani, N. Neutrophils induce macrophage anti-inflammatory reprogramming by suppressing NF-κB activation. Cell Death Dis. 2018, 9, 665. [Google Scholar] [CrossRef]
- Elliott, M.R.; Koster, K.M.; Murphy, P.S. Efferocytosis Signaling in the Regulation of Macrophage Inflammatory Responses. J. Immunol. 2017, 198, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Srivastava, S.; Singh, M.R.; Singh, D. Mechanistic insight into diabetic wounds: Pathogenesis, molecular targets and treatment strategies to pace wound healing. Biomed. Pharmacother. 2019, 112, 108615. [Google Scholar] [CrossRef]
- Ayala, T.S.; Tessaro, F.H.G.; Jannuzzi, G.P.; Bella, L.M.; Ferreira, K.S.; Martins, J.O. High Glucose Environments Interfere with Bone Marrow-Derived Macrophage Inflammatory Mediator Release, the TLR4 Pathway and Glucose Metabolism. Sci. Rep. 2019, 9, 11447. [Google Scholar] [CrossRef]
- Pavlou, S.; Lindsay, J.; Ingram, R.; Xu, H.; Chen, M. Sustained high glucose exposure sensitizes macrophage responses to cytokine stimuli but reduces their phagocytic activity. BMC Immunol. 2018, 19, 24. [Google Scholar] [CrossRef] [PubMed]
- Boniakowski, A.E.; Kimball, A.S.; Joshi, A.; Schaller, M.; Davis, F.M.; denDekker, A.; Obi, A.T.; Moore, B.B.; Kunkel, S.L.; Gallagher, K.A. Murine macrophage chemokine receptor CCR2 plays a crucial role in macrophage recruitment and regulated inflammation in wound healing. Eur. J. Immunol. 2018, 48, 1445–1455. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Kuninaka, Y.; Nosaka, M.; Furuta, M.; Kimura, A.; Taruya, A.; Yamamoto, H.; Shimada, E.; Akiyama, M.; Mukaida, N.; et al. CCL2-Mediated Reversal of Impaired Skin Wound Healing in Diabetic Mice by Normalization of Neovascularization and Collagen Accumulation. J. Investig. Dermatol. 2019, 139, 2517–2527.e5. [Google Scholar] [PubMed]
- Wood, S.; Jayaraman, V.; Huelsmann, E.J.; Bonish, B.; Burgad, D.; Sivaramakrishnan, G.; Qin, S.; DiPietro, L.A.; Zloza, A.; Zhang, C.; et al. Pro-inflammatory chemokine CCL2 (MCP-1) promotes healing in diabetic wounds by restoring the macrophage response. PLoS ONE 2014, 9, e91574. [Google Scholar] [CrossRef]
- Jetten, N.; Roumans, N.; Gijbels, M.J.; Romano, A.; Post, M.J.; de Winther, M.P.J.; van der Hulst, R.R.W.J.; Xanthoulea, S. Wound administration of M2-polarized macrophages does not improve murine cutaneous healing responses. PLoS ONE 2014, 9, e102994. [Google Scholar] [CrossRef]
- Shook, B.; Xiao, E.; Kumamoto, Y.; Iwasaki, A.; Horsley, V. CD301b+ Macrophages Are Essential for Effective Skin Wound Healing. J. Investig. Dermatol. 2016, 136, 1885–1891. [Google Scholar] [CrossRef]
- Ishida, Y.; Gao, J.-L.; Murphy, P.M. Chemokine Receptor CX3CR1 Mediates Skin Wound Healing by Promoting Macrophage and Fibroblast Accumulation and Function. J. Immunol. 2008, 180, 569. [Google Scholar] [CrossRef]
- Daley, J.M.; Brancato, S.K.; Thomay, A.A.; Reichner, J.S.; Albina, J.E. The phenotype of murine wound macrophages. J. Leukoc. Biol. 2010, 87, 59–67. [Google Scholar] [CrossRef]
- Saraiva, M.; Vieira, P.; O’Garra, A. Biology and therapeutic potential of interleukin-10. J. Exp. Med. 2019, 217, e20190418. [Google Scholar] [CrossRef]
- Kimball, A.; Schaller, M.; Joshi, A.; Davis, F.M.; denDekker, A.; Boniakowski, A.; Bermick, J.; Obi, A.; Moore, B.; Henke, P.K.; et al. Ly6CHi Blood Monocyte/Macrophage Drive Chronic Inflammation and Impair Wound Healing in Diabetes Mellitus. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1102–1114. [Google Scholar] [CrossRef]
- Crane, M.J.; Daley, J.M.; van Houtte, O.; Brancato, S.K.; Henry, W.L., Jr.; Albina, J.E. The Monocyte to Macrophage Transition in the Murine Sterile Wound. PLoS ONE 2014, 9, e86660. [Google Scholar] [CrossRef]
- Hanna, R.N.; Carlin, L.M.; Hubbeling, H.G.; Nackiewicz, D.; Green, A.M.; Punt, J.A.; Geissmann, F.; Hedrick, C.C. The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C− monocytes. Nat. Immunol. 2011, 12, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Landsman, L.; Bar-On, L.; Zernecke, A.; Kim, K.-W.; Krauthgamer, R.; Shagdarsuren, E.; Lira, S.A.; Weissman, I.L.; Weber, C.; Jung, S. CX3CR1 is required for monocyte homeostasis and atherogenesis by promoting cell survival. Blood 2009, 113, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Rodero, M.P.; Khosrotehrani, K. Skin wound healing modulation by macrophages. Int. J. Clin. Exp. Pathol. 2010, 3, 643–653. [Google Scholar] [PubMed]
- Gurevich, D.B.; Severn, C.E.; Twomey, C.; Greenhough, A.; Cash, J.; Toye, A.M.; Mellor, H.; Martin, P. Live imaging of wound angiogenesis reveals macrophage orchestrated vessel sprouting and regression. EMBO J. 2018, 37, e97786. [Google Scholar]
- Blanco, R.; Gerhardt, H. VEGF and Notch in tip and stalk cell selection. Cold Spring Harb. Perspect. Med. 2013, 3, a006569. [Google Scholar] [CrossRef] [PubMed]
- Fantin, A.; Vieira, J.M.; Gestri, G.; Denti, L.; Schwarz, Q.; Prykhozhij, S.; Peri, F.; Wilson, S.W.; Ruhrberg, C. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood 2010, 116, 829–840. [Google Scholar] [CrossRef]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Patel, J.; Seppanen, E.J.; Rodero, M.P.; Wong, H.Y.; Donovan, P.; Neufeld, Z.; Fisk, N.M.; Francois, M.; Khosrotehrani, K. Functional Definition of Progenitors Versus Mature Endothelial Cells Reveals Key SoxF-Dependent Differentiation Process. Circulation 2017, 135, 786–805. [Google Scholar] [CrossRef]
- Denton, C.P.; Khan, K.; Hoyles, R.K.; Shiwen, X.; Leoni, P.; Chen, Y.; Eastwood, M.; Abraham, D.J. Inducible Lineage-Specific Deletion of TβRII in Fibroblasts Defines a Pivotal Regulatory Role during Adult Skin Wound Healing. J. Investig. Dermatol. 2009, 129, 194–204. [Google Scholar] [CrossRef]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Rodero, M.P.; Legrand, J.M.D.; Bou-Gharios, G.; Khosrotehrani, K. Wound-associated macrophages control collagen 1α2 transcription during the early stages of skin wound healing. Exp. Dermatol. 2013, 22, 143–145. [Google Scholar] [CrossRef] [PubMed]
- Shook, B.A.; Wasko, R.R.; Rivera-Gonzalez, G.C.; Salazar-Gatzimas, E.; López-Giráldez, F.; Dash, B.C.; Muñoz-Rojas, A.R.; Aultman, K.D.; Zwick, R.K.; Lei, V.; et al. Myofibroblast proliferation and heterogeneity are supported by macrophages during skin repair. Science 2018, 362, eaar2971. [Google Scholar] [CrossRef] [PubMed]
- Campbell Jean, S.; Hughes Steven, D.; Gilbertson Debra, G.; Palmer Thomas, E.; Holdren Matthew, S.; Haran Aaron, C.; Odell Melissa, M.; Bauer Renay, L.; Ren, H.-P.; Haugen Harald, S.; et al. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2005, 102, 3389–3394. [Google Scholar] [CrossRef] [PubMed]
- van Roeyen, C.R.C.; Martin, I.V.; Drescher, A.; Schuett, K.A.; Hermert, D.; Raffetseder, U.; Otten, S.; Buhl, E.M.; Braun, G.S.; Kuppe, C.; et al. Identification of platelet-derived growth factor C as a mediator of both renal fibrosis and hypertension. Kidney Int. 2019, 95, 1103–1119. [Google Scholar] [CrossRef] [PubMed]
- Pontén, A.; Li, X.; Thorén, P.; Aase, K.; Sjöblom, T.; Östman, A.; Eriksson, U. Transgenic Overexpression of Platelet-Derived Growth Factor-C in the Mouse Heart Induces Cardiac Fibrosis, Hypertrophy, and Dilated Cardiomyopathy. Am. J. Pathol. 2003, 163, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Sommerfeld Sven, D.; Cherry, C.; Schwab Remi, M.; Chung, L.; Maestas David, R.; Laffont, P.; Stein Julie, E.; Tam, A.; Ganguly, S.; Housseau, F.; et al. Interleukin-36γ–producing macrophages drive IL-17–mediated fibrosis. Sci. Immunol. 2019, 4, eaax4783. [Google Scholar] [CrossRef]
- Koss, C.K.; Wohnhaas, C.T.; Baker, J.R.; Tilp, C.; Przibilla, M.; Lerner, C.; Frey, S.; Keck, M.; Williams, C.M.M.; Peter, D.; et al. IL36 is a critical upstream amplifier of neutrophilic lung inflammation in mice. Commun. Biol. 2021, 4, 172. [Google Scholar] [CrossRef]
- Zhang, J.; Qiao, Q.; Liu, M.; He, T.; Shi, J.; Bai, X.; Zhang, Y.; Li, Y.; Cai, W.; Han, S.; et al. IL-17 Promotes Scar Formation by Inducing Macrophage Infiltration. Am. J. Pathol. 2018, 188, 1693–1702. [Google Scholar] [CrossRef]
- Park, M.-J.; Moon, S.-J.; Lee, E.-J.; Jung, K.-A.; Kim, E.-K.; Kim, D.-S.; Lee, J.-H.; Kwok, S.-K.; Min, J.-K.; Park, S.-H.; et al. IL-1-IL-17 Signaling Axis Contributes to Fibrosis and Inflammation in Two Different Murine Models of Systemic Sclerosis. Front. Immunol. 2018, 9, 1611. [Google Scholar]
- Patel, J.; Baz, B.; Wong, H.Y.; Lee, J.S.; Khosrotehrani, K. Accelerated Endothelial to Mesenchymal Transition Increased Fibrosis via Deleting Notch Signaling in Wound Vasculature. J. Investig. Dermatol. 2018, 138, 1166–1175. [Google Scholar] [PubMed]
- Zhao, J.; Patel, J.; Kaur, S.; Sim, S.-L.; Wong, H.Y.; Styke, C.; Hogan, I.; Kahler, S.; Hamilton, H.; Wadlow, R.; et al. Sox9 and Rbpj differentially regulate endothelial to mesenchymal transition and wound scarring in murine endovascular progenitors. Nat. Commun. 2021, 12, 2564. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Chaboissier, M.-C.; Behringer Richard, R.; Rowitch David, H.; Schedl, A.; Epstein Jonathan, A.; de Crombrugghe, B. Essential role of Sox9 in the pathway that controls formation of cardiac valves and septa. Proc. Natl. Acad. Sci. USA 2004, 101, 6502–6507. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, J.; Kist, R.; Scherer, G.; Yutzey, K.E. Sox9 is required for precursor cell expansion and extracellular matrix organization during mouse heart valve development. Dev. Biol. 2007, 305, 120–132. [Google Scholar] [CrossRef]
- Alonso-Herranz, L.; Sahún-Español, Á.; Paredes, A.; Gonzalo, P.; Gkontra, P.; Núñez, V.; Clemente, C.; Cedenilla, M.; Villalba-Orero, M.; Inserte, J.; et al. Macrophages promote endothelial-to-mesenchymal transition via MT1-MMP/TGFβ1 after myocardial infarction. eLife 2020, 9, e57920. [Google Scholar] [CrossRef]
- Mascré, G.; Dekoninck, S.; Drogat, B.; Youssef, K.K.; Brohée, S.; Sotiropoulou, P.A.; Simons, B.D.; Blanpain, C. Distinct contribution of stem and progenitor cells to epidermal maintenance. Nature 2012, 489, 257–262. [Google Scholar] [CrossRef]
- Aragona, M.; Dekoninck, S.; Rulands, S.; Lenglez, S.; Mascré, G.; Simons, B.D.; Blanpain, C. Defining stem cell dynamics and migration during wound healing in mouse skin epidermis. Nat. Commun. 2017, 8, 14684. [Google Scholar] [CrossRef]
- Ito, M.; Liu, Y.; Yang, Z.; Nguyen, J.; Liang, F.; Morris, R.J.; Cotsarelis, G. Stem cells in the hair follicle bulge contribute to wound repair but not to homeostasis of the epidermis. Nat. Med. 2005, 11, 1351–1354. [Google Scholar] [CrossRef]
- Huang, S.; Kuri, P.; Aubert, Y.; Brewster, M.; Li, N.; Farrelly, O.; Rice, G.; Bae, H.; Prouty, S.; Dentchev, T.; et al. Lgr6 marks epidermal stem cells with a nerve-dependent role in wound re-epithelialization. Cell Stem Cell 2021, 28, 1582–1596.e6. [Google Scholar] [CrossRef]
- Villarreal-Ponce, A.; Tiruneh, M.W.; Lee, J.; Guerrero-Juarez, C.F.; Kuhn, J.; David, J.A.; Dammeyer, K.; Mc Kell, R.; Kwong, J.; Rabbani, P.S.; et al. Keratinocyte-macrophage crosstalk by the Nrf2/Ccl2/EGF signaling axis orchestrates tissue repair. Cell Rep. 2020, 33, 108417. [Google Scholar] [CrossRef]
- Zhou, X.; Brown, B.A.; Siegel, A.P.; El Masry, M.S.; Zeng, X.; Song, W.; Das, A.; Khandelwal, P.; Clark, A.; Singh, K.; et al. Exosome-Mediated Crosstalk between Keratinocytes and Macrophages in Cutaneous Wound Healing. ACS Nano 2020, 14, 12732–12748. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Dong, Z.; Cao, Y.; Wang, H.; Liu, S.; Liao, L.; Jin, Y.; Yuan, L.; Li, B. MSC-Derived Exosome Promotes M2 Polarization and Enhances Cutaneous Wound Healing. Stem Cells Int. 2019, 2019, 7132708. [Google Scholar] [CrossRef] [PubMed]
- Kreuger, J.; Phillipson, M. Targeting vascular and leukocyte communication in angiogenesis, inflammation and fibrosis. Nat. Rev. Drug Discov. 2016, 15, 125–142. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Guo, Y.; Lin, C.; Xu, P.; Wu, S.; Fu, X.; Xia, W.; Yao, M. AGEs Induced Autophagy Impairs Cutaneous Wound Healing via Stimulating Macrophage Polarization to M1 in Diabetes. Sci. Rep. 2016, 6, 36416. [Google Scholar] [CrossRef]
- Jin, X.; Yao, T.; Zhou, Z.E.; Zhu, J.; Zhang, S.; Hu, W.; Shen, C. Advanced Glycation End Products Enhance Macrophages Polarization into M1 Phenotype through Activating RAGE/NF-κB Pathway. BioMed Res. Int. 2015, 2015, 732450. [Google Scholar] [CrossRef]
- Hoyer, F.F.; Zhang, X.; Coppin, E.; Vasamsetti, S.B.; Modugu, G.; Schloss, M.J.; Rohde, D.; McAlpine, C.S.; Iwamoto, Y.; Libby, P.; et al. Bone Marrow Endothelial Cells Regulate Myelopoiesis in Diabetes Mellitus. Circulation 2020, 142, 244–258. [Google Scholar] [CrossRef]
- Barman, P.K.; Urao, N.; Koh, T.J. Diabetes induces myeloid bias in bone marrow progenitors associated with enhanced wound macrophage accumulation and impaired healing. J. Pathol. 2019, 249, 435–446. [Google Scholar]
- Bannon, P.; Wood, S.; Restivo, T.; Campbell, L.; Hardman, M.J.; Mace, K.A. Diabetes induces stable intrinsic changes to myeloid cells that contribute to chronic inflammation during wound healing in mice. Dis. Model. Mech. 2013, 6, 1434–1447. [Google Scholar] [CrossRef]
- Lecube, A.; Pachón, G.; Petriz, J.; Hernández, C.; Simó, R. Phagocytic activity is impaired in type 2 diabetes mellitus and increases after metabolic improvement. PLoS ONE 2011, 6, e23366. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhu, G.; Cao, X.; Dong, J.; Song, F.; Niu, Y. Blocking AGE-RAGE Signaling Improved Functional Disorders of Macrophages in Diabetic Wound. J. Diabetes Res. 2017, 2017, 1428537. [Google Scholar] [CrossRef] [PubMed]
- Macedo, L.; Pinhal-Enfield, G.; Alshits, V.; Elson, G.; Cronstein, B.N.; Leibovich, S.J. Wound Healing Is Impaired in MyD88-Deficient Mice: A Role for MyD88 in the Regulation of Wound Healing by Adenosine A2A Receptors. Am. J. Pathol. 2007, 171, 1774–1788. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Sweren, E.; Liu, H.; Wier, E.; Alphonse, M.P.; Chen, R.; Islam, N.; Li, A.; Xue, Y.; Chen, J.; et al. Bacteria induce skin regeneration via IL-1β signaling. Cell Host Microbe 2021, 29, 777–791.e6. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Fang, D.; Fang, J.; Ren, X.; Yang, X.; Wen, F.; Su, S.B. Impaired Wound Healing with Defective Expression of Chemokines and Recruitment of Myeloid Cells in TLR3-Deficient Mice. J. Immunol. 2011, 186, 3710. [Google Scholar] [CrossRef]
- Chen, L.; Guo, S.; Ranzer, M.J.; DiPietro, L.A. Toll-Like Receptor 4 Has an Essential Role in Early Skin Wound Healing. J. Investig. Dermatol. 2013, 133, 258–267. [Google Scholar] [CrossRef]
- Thomay, A.A.; Daley, J.M.; Sabo, E.; Worth, P.J.; Shelton, L.J.; Harty, M.W.; Reichner, J.S.; Albina, J.E. Disruption of Interleukin-1 Signaling Improves the Quality of Wound Healing. Am. J. Pathol. 2009, 174, 2129–2136. [Google Scholar] [CrossRef]
- Takagi, N.; Kawakami, K.; Kanno, E.; Tanno, H.; Takeda, A.; Ishii, K.; Imai, Y.; Iwakura, Y.; Tachi, M. IL-17A promotes neutrophilic inflammation and disturbs acute wound healing in skin. Exp. Dermatol. 2017, 26, 137–144. [Google Scholar] [CrossRef]
- MacLeod, A.S.; Hemmers, S.; Garijo, O.; Chabod, M.; Mowen, K.; Witherden, D.A.; Havran, W.L. Dendritic epidermal T cells regulate skin antimicrobial barrier function. J. Clin. Investig. 2013, 123, 4364–4374. [Google Scholar] [CrossRef]
- Rodero, M.P.; Hodgson, S.S.; Hollier, B.; Combadiere, C.; Khosrotehrani, K. Reduced Il17a Expression Distinguishes a Ly6cloMHCIIhi Macrophage Population Promoting Wound Healing. J. Investig. Dermatol. 2013, 133, 783–792. [Google Scholar] [CrossRef]
- Lee, J.; Rodero, M.P.; Patel, J.; Moi, D.; Mazzieri, R.; Khosrotehrani, K. Interleukin-23 regulates interleukin-17 expression in wounds, and its inhibition accelerates diabetic wound healing through the alteration of macrophage polarization. FASEB J. 2018, 32, 2086–2094. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Y.; Zhou, L.; Liu, M.; Liang, G.; Yan, R.; Jiang, Y.; Hao, J.; Zhang, X.; Hu, X.; et al. Vγ4 T Cells Inhibit the Pro-healing Functions of Dendritic Epidermal T Cells to Delay Skin Wound Closure Through IL-17A. Front. Immunol. 2018, 9, 240. [Google Scholar] [CrossRef] [PubMed]
- Mori, R.; Kondo, T.; Ohshima, T.; Ishida, Y.; Mukaida, N. Accelerated wound healing in tumor necrosis factor receptor p55-deficient mice with reduced leukocyte infiltration. FASEB J. 2002, 16, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cai, G.; Liu, C.; Zhao, J.; Gu, C.; Wu, L.; Hamilton, T.A.; Zhang, C.-J.; Ko, J.; Zhu, L.; et al. IL-17R–EGFR axis links wound healing to tumorigenesis in Lrig1+ stem cells. J. Exp. Med. 2018, 216, 195–214. [Google Scholar] [CrossRef] [PubMed]
- Konieczny, P.; Xing, Y.; Sidhu, I.; Subudhi, I.; Mansfield Kody, P.; Hsieh, B.; Biancur Douglas, E.; Larsen Samantha, B.; Cammer, M.; Li, D.; et al. Interleukin-17 governs hypoxic adaptation of injured epithelium. Science 2022, 377, eabg9302. [Google Scholar] [CrossRef]
- Tan, J.L.; Lash, B.; Karami, R.; Nayer, B.; Lu, Y.-Z.; Piotto, C.; Julier, Z.; Martino, M.M. Restoration of the healing microenvironment in diabetic wounds with matrix-binding IL-1 receptor antagonist. Commun. Biol. 2021, 4, 422. [Google Scholar] [CrossRef]
- Mirza, R.E.; Fang, M.M.; Ennis, W.J.; Koh, T.J. Blocking Interleukin-1β Induces a Healing-Associated Wound Macrophage Phenotype and Improves Healing in Type 2 Diabetes. Diabetes 2013, 62, 2579–2587. [Google Scholar] [CrossRef]
- Kondylis, V.; Kumari, S.; Vlantis, K.; Pasparakis, M. The interplay of IKK, NF-κB and RIPK1 signaling in the regulation of cell death, tissue homeostasis and inflammation. Immunol. Rev. 2017, 277, 113–127. [Google Scholar]
- Ding, Y.; Ding, X.; Zhang, H.; Li, S.; Yang, P.; Tan, Q. Relevance of NLRP3 Inflammasome-Related Pathways in the Pathology of Diabetic Wound Healing and Possible Therapeutic Targets. Oxid. Med. Cell. Longev. 2022, 2022, 9687925. [Google Scholar] [CrossRef]
- Ishida, Y.; Kondo, T.; Kimura, A.; Matsushima, K.; Mukaida, N. Absence of IL-1 Receptor Antagonist Impaired Wound Healing along with Aberrant NF-κB Activation and a Reciprocal Suppression of TGF-β Signal Pathway. J. Immunol. 2006, 176, 5598. [Google Scholar] [CrossRef]
- Huang, Y.Y.; Lin, C.W.; Cheng, N.C.; Cazzell, S.M.; Chen, H.H.; Huang, K.F.; Tung, K.Y.; Huang, H.L.; Lin, P.Y.; Perng, C.K.; et al. Effect of a Novel Macrophage-Regulating Drug on Wound Healing in Patients with Diabetic Foot Ulcers: A Randomized Clinical Trial. JAMA Netw. Open 2021, 4, e2122607. [Google Scholar] [PubMed]
- Leu, W.J.; Chen, J.C.; Guh, J.H. Extract From Plectranthus amboinicus Inhibit Maturation and Release of Interleukin 1β through Inhibition of NF-κB Nuclear Translocation and NLRP3 Inflammasome Activation. Front. Pharmacol. 2019, 10, 573. [Google Scholar] [CrossRef] [PubMed]
- Bylka, W.; Znajdek-Awiżeń, P.; Studzińska-Sroka, E.; Dańczak-Pazdrowska, A.; Brzezińska, M. Centella asiatica in Dermatology: An Overview. Phytother. Res. 2014, 28, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.M.; Lovato, P.; MacLeod, A.S.; Witherden, D.A.; Skov, L.; Dyring-Andersen, B.; Dabelsteen, S.; Woetmann, A.; Ødum, N.; Havran, W.L.; et al. IL-1β–Dependent Activation of Dendritic Epidermal T Cells in Contact Hypersensitivity. J. Immunol. 2014, 192, 2975. [Google Scholar] [CrossRef] [PubMed]
- Yoshiki, R.; Kabashima, K.; Honda, T.; Nakamizo, S.; Sawada, Y.; Sugita, K.; Yoshioka, H.; Ohmori, S.; Malissen, B.; Tokura, Y.; et al. IL-23 from Langerhans Cells Is Required for the Development of Imiquimod-Induced Psoriasis-Like Dermatitis by Induction of IL-17A-Producing γδ T Cells. J. Investig. Dermatol. 2014, 134, 1912–1921. [Google Scholar]
- Wu, L.; Chen, X.; Zhao, J.; Martin, B.; Zepp, J.A.; Ko, J.S.; Gu, C.; Cai, G.; Ouyang, W.; Sen, G.; et al. A novel IL-17 signaling pathway controlling keratinocyte proliferation and tumorigenesis via the TRAF4–ERK5 axis. J. Exp. Med. 2015, 212, 1571–1587. [Google Scholar] [CrossRef]
- Sharp, L.L.; Jameson, J.M.; Cauvi, G.; Havran, W.L. Dendritic epidermal T cells regulate skin homeostasis through local production of insulin-like growth factor 1. Nat. Immunol. 2005, 6, 73–79. [Google Scholar] [CrossRef]
- Vallerand, I.A.; Hardin, J. Ustekinumab for the treatment of recalcitrant pyoderma gangrenosum: A case report. SAGE Open Med. Case Rep. 2019, 7, 2050313X19845206. [Google Scholar] [CrossRef]
- Guenova, E.; Teske, A.; Fehrenbacher, B.; Hoerber, S.; Adamczyk, A.; Schaller, M.; Hoetzenecker, W.; Biedermann, T. Interleukin 23 Expression in Pyoderma Gangrenosum and Targeted Therapy with Ustekinumab. Arch. Dermatol. 2011, 147, 1203–1205. [Google Scholar] [CrossRef]
- McPhie, M.L.; Kirchhof, M.G. Pyoderma gangrenosum treated with secukinumab: A case report. SAGE Open Med. Case Rep. 2020, 8, 2050313X20940430. [Google Scholar] [CrossRef]
- Kao, A.S.; King, A.D.; Daveluy, S. Successful treatment of cabozantinib-induced pyoderma gangrenosum with ixekizumab therapy: A case report. Dermatol. Ther. 2022, 35, e15716. [Google Scholar] [CrossRef] [PubMed]
- Lowes, M.A.; Kikuchi, T.; Fuentes-Duculan, J.; Cardinale, I.; Zaba, L.C.; Haider, A.S.; Bowman, E.P.; Krueger, J.G. Psoriasis Vulgaris Lesions Contain Discrete Populations of Th1 and Th17 T Cells. J. Investig. Dermatol. 2008, 128, 1207–1211. [Google Scholar] [CrossRef]
- Nussbaum, L.; Chen, Y.L.; Ogg, G.S. Role of regulatory T cells in psoriasis pathogenesis and treatment. Br. J. Dermatol. 2021, 184, 14–24. [Google Scholar] [PubMed]
- Nosbaum, A.; Prevel, N.; Truong, H.-A.; Mehta, P.; Ettinger, M.; Scharschmidt, T.C.; Ali, N.H.; Pauli, M.L.; Abbas, A.K.; Rosenblum, M.D. Cutting Edge: Regulatory T Cells Facilitate Cutaneous Wound Healing. J. Immunol. 2016, 196, 2010. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, Y.; Lischke, T.; Dahler, A.C.; Mages, H.W.; Lam, K.-P.; Coyle, A.J.; Kroczek, R.A.; Hutloff, A. ICOS Controls the Pool Size of Effector-Memory and Regulatory T Cells. J. Immunol. 2008, 180, 774. [Google Scholar] [CrossRef] [PubMed]
- Busse, M.; Krech, M.; Meyer-Bahlburg, A.; Hennig, C.; Hansen, G. ICOS Mediates the Generation and Function of CD4+CD25+Foxp3+ Regulatory T Cells Conveying Respiratory Tolerance. J. Immunol. 2012, 189, 1975. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Fujimoto, M.; Matsushita, T.; Hamaguchi, Y.; Takehara, K.; Hasegawa, M. Inducible Costimulator (ICOS) and ICOS Ligand Signaling Has Pivotal Roles in Skin Wound Healing via Cytokine Production. Am. J. Pathol. 2011, 179, 2360–2369. [Google Scholar] [PubMed]
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Eto, K.; Yamashita, H.; Ohsugi, M.; Otsu, M.; Hara, K.; Ueki, K.; Sugiura, S.; et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 2009, 15, 914–920. [Google Scholar] [CrossRef]
- Stoppa, I.; Gigliotti, C.L.; Clemente, N.; Pantham, D.; Dianzani, C.; Monge, C.; Puricelli, C.; Rolla, R.; Sutti, S.; Renò, F.; et al. ICOSL Stimulation by ICOS-Fc Accelerates Cutaneous Wound Healing In Vivo. Int. J. Mol. Sci. 2022, 23, 7363. [Google Scholar] [CrossRef] [PubMed]
- Piao, W.; Li, L.; Saxena, V.; Iyyathurai, J.; Lakhan, R.; Zhang, Y.; Lape, I.T.; Paluskievicz, C.; Hippen, K.L.; Lee, Y.; et al. PD-L1 signaling selectively regulates T cell lymphatic transendothelial migration. Nat. Commun. 2022, 13, 2176. [Google Scholar] [CrossRef]
- Kuai, L.; Xiang, Y.-W.; Chen, Q.-L.; Ru, Y.; Yin, S.-Y.; Li, W.; Jiang, J.-S.; Luo, Y.; Song, J.-K.; Lu, B.; et al. PD-L1 Triggered by Binding eIF3I Contributes to the Amelioration of Diabetes-Associated Wound Healing Defects by Regulating IRS4. J. Investig. Dermatol. 2022, 142, 220–231.e8. [Google Scholar] [CrossRef] [PubMed]
- Strauss, L.; Mahmoud, M.A.A.; Weaver, J.D.; Tijaro-Ovalle, N.M.; Christofides, A.; Wang, Q.; Pal, R.; Yuan, M.; Asara, J.; Patsoukis, N.; et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci. Immunol. 2020, 5, eaay1863. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.D.; Vucic, D. The Balance of TNF Mediated Pathways Regulates Inflammatory Cell Death Signaling in Healthy and Diseased Tissues. Front. Cell Dev. Biol. 2020, 8, 365. [Google Scholar] [CrossRef]
- Dondelinger, Y.; Jouan-Lanhouet, S.; Divert, T.; Theatre, E.; Bertin, J.; Gough, P.J.; Giansanti, P.; Heck, A.J.R.; Dejardin, E.; Vandenabeele, P.; et al. NF-κB-Independent Role of IKKα/IKKβ in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death during TNF Signaling. Mol. Cell 2015, 60, 63–76. [Google Scholar]
- Kumari, S.; Redouane, Y.; Lopez-Mosqueda, J.; Shiraishi, R.; Romanowska, M.; Lutzmayer, S.; Kuiper, J.; Martinez, C.; Dikic, I.; Pasparakis, M.; et al. Sharpin prevents skin inflammation by inhibiting TNFR1-induced keratinocyte apoptosis. eLife 2014, 3, e03422. [Google Scholar] [CrossRef]
- Kumari, S.; Van, T.-M.; Preukschat, D.; Schuenke, H.; Basic, M.; Bleich, A.; Klein, U.; Pasparakis, M. NF-κB inhibition in keratinocytes causes RIPK1-mediated necroptosis and skin inflammation. Life Sci. Alliance 2021, 4, e202000956. [Google Scholar] [CrossRef]
- Heckmann, B.L.; Tummers, B.; Green, D.R. Crashing the computer: Apoptosis vs. necroptosis in neuroinflammation. Cell Death Differ. 2019, 26, 41–52. [Google Scholar] [CrossRef]
- Green, D.R.; Ferguson, T.; Zitvogel, L.; Kroemer, G. Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 2009, 9, 353–363. [Google Scholar] [CrossRef]
- Lowe, M.M.; Naik, H.B.; Clancy, S.; Pauli, M.; Smith, K.M.; Bi, Y.; Dunstan, R.; Gudjonsson, J.E.; Paul, M.; Harris, H.; et al. Immunopathogenesis of hidradenitis suppurativa and response to anti–TNF-α therapy. JCI Insight 2020, 5, e139932. [Google Scholar] [CrossRef]
- Griffiths, C.E.M.; Barker, J.N.W.N. Pathogenesis and clinical features of psoriasis. Lancet 2007, 370, 263–271. [Google Scholar]
- Baliwag, J.; Barnes, D.H.; Johnston, A. Cytokines in psoriasis. Cytokine 2015, 73, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Bonnet, M.C.; Ulvmar, M.H.; Wolk, K.; Karagianni, N.; Witte, E.; Uthoff-Hachenberg, C.; Renauld, J.-C.; Kollias, G.; Toftgard, R.; et al. Tumor Necrosis Factor Receptor Signaling in Keratinocytes Triggers Interleukin-24-Dependent Psoriasis-like Skin Inflammation in Mice. Immunity 2013, 39, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, M.; Courtois, G.; Hafner, M.; Schmidt-Supprian, M.; Nenci, A.; Toksoy, A.; Krampert, M.; Goebeler, M.; Gillitzer, R.; Israel, A.; et al. TNF-mediated inflammatory skin disease in mice with epidermis-specific deletion of IKK2. Nature 2002, 417, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Stratis, A.; Pasparakis, M.; Rupec, R.A.; Markur, D.; Hartmann, K.; Scharffetter-Kochanek, K.; Peters, T.; van Rooijen, N.; Krieg, T.; Haase, I. Pathogenic role for skin macrophages in a mouse model of keratinocyte-induced psoriasis-like skin inflammation. J. Clin. Investig. 2006, 116, 2094–2104. [Google Scholar] [CrossRef] [PubMed]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef]
- Ashcroft, G.S.; Jeong, M.J.; Ashworth, J.J.; Hardman, M.; Jin, W.; Moutsopoulos, N.; Wild, T.; McCartney-Francis, N.; Sim, D.; McGrady, G.; et al. Tumor necrosis factor-alpha (TNF-α) is a therapeutic target for impaired cutaneous wound healing. Wound Repair Regen. 2012, 20, 38–49. [Google Scholar] [CrossRef]
- Siqueira, M.F.; Li, J.; Chehab, L.; Desta, T.; Chino, T.; Krothpali, N.; Behl, Y.; Alikhani, M.; Yang, J.; Braasch, C.; et al. Impaired wound healing in mouse models of diabetes is mediated by TNF-alpha dysregulation and associated with enhanced activation of forkhead box O1 (FOXO1). Diabetologia 2010, 53, 378–388. [Google Scholar] [CrossRef]
- Rai, N.K.; Suryabhan; Ansari, M.; Kumar, M.; Shukla, V.K.; Tripathi, K. Effect of glycaemic control on apoptosis in diabetic wounds. J. Wound Care 2005, 14, 277–281. [Google Scholar] [CrossRef]
- Hasnan, J.; Yusoff, M.I.; Damitri, T.D.; Faridah, A.R.; Adenan, A.S.; Norbaini, T.H. Relationship between apoptotic markers (Bax and Bcl-2) and biochemical markers in type 2 diabetes mellitus. Singap. Med. J. 2010, 51, 50. [Google Scholar]
- Liu, R.; Bal, H.S.; Desta, T.; Behl, Y.; Graves, D.T. Tumor Necrosis Factor-α Mediates Diabetes-Enhanced Apoptosis of Matrix-Producing Cells and Impairs Diabetic Healing. Am. J. Pathol. 2006, 168, 757–764. [Google Scholar] [CrossRef]
- Banno, T.; Gazel, A.; Blumenberg, M. Effects of Tumor Necrosis Factor-α (TNFα) in Epidermal Keratinocytes Revealed Using Global Transcriptional Profiling. J. Biol. Chem. 2004, 279, 32633–32642. [Google Scholar] [PubMed]
- Alikhani, M.; Alikhani, Z.; Raptis, M.; Graves, D.T. TNF-α in vivo stimulates apoptosis in fibroblasts through caspase-8 activation and modulates the expression of pro-apoptotic genes. J. Cell. Physiol. 2004, 201, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, S.; Rizwani, W.; Joshi, B.; Kunigal, S.; Chellappan, S.P. TNF-α response of vascular endothelial and vascular smooth muscle cells involve differential utilization of ASK1 kinase and p73. Cell Death Differ. 2012, 19, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.D.; Baquerizo-Nole, K.L.; Keegan, B.R.; Macquhae, F.; Escandon, J.; Espinosa, A.; Perez, C.; Romanelli, P.; Kirsner, R.S. Adalimumab treatment leads to reduction of tissue tumor necrosis factor-alpha correlated with venous leg ulcer improvement: A pilot study. Int. Wound J. 2016, 13, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Streit, M.; Beleznay, Z.; Braathen, L.R. Topical application of the tumour necrosis factor-α antibody infliximab improves healing of chronic wounds. Int. Wound J. 2006, 3, 171–179. [Google Scholar] [CrossRef]
- Demidova-Rice, T.N.; Hamblin, M.R.; Herman, I.M. Acute and impaired wound healing: Pathophysiology and current methods for drug delivery, part 1: Normal and chronic wounds: Biology, causes, and approaches to care. Adv. Ski. Wound Care 2012, 25, 304–314. [Google Scholar] [CrossRef]
- Wolcott, R.D.; Hanson, J.D.; Rees, E.J.; Koenig, L.D.; Phillips, C.D.; Wolcott, R.A.; Cox, S.B.; White, J.S. Analysis of the chronic wound microbiota of 2,963 patients by 16S rDNA pyrosequencing. Wound Repair Regen. 2016, 24, 163–174. [Google Scholar] [CrossRef]
- Secor, P.R.; James, G.A.; Fleckman, P.; Olerud, J.E.; McInnerney, K.; Stewart, P.S. Staphylococcus aureus Biofilm and Planktonic cultures differentially impact gene expression, mapk phosphorylation, and cytokine production in human keratinocytes. BMC Microbiol. 2011, 11, 143. [Google Scholar] [CrossRef]
- Pastar, I.; Sawaya, A.P.; Marjanovic, J.; Burgess, J.L.; Strbo, N.; Rivas, K.E.; Wikramanayake, T.C.; Head, C.R.; Stone, R.C.; Jozic, I.; et al. Intracellular Staphylococcus aureus triggers pyroptosis and contributes to inhibition of healing due to perforin-2 suppression. J. Clin. Investig. 2021, 131, e133727. [Google Scholar] [CrossRef]
- Chambers, H.F.; DeLeo, F.R. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 2009, 7, 629–641. [Google Scholar] [CrossRef]
- Guo, Y.; Song, G.; Sun, M.; Wang, J.; Wang, Y. Prevalence and Therapies of Antibiotic-Resistance in Staphylococcus aureus. Front. Cell. Infect. Microbiol. 2020, 10, 107. [Google Scholar] [PubMed]
- Harris-Tryon, T.A.; Grice, E.A. Microbiota and maintenance of skin barrier function. Science 2022, 376, 940–945. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuji, T.; Chen, T.H.; Narala, S.; Chun, K.A.; Two, A.M.; Yun, T.; Shafiq, F.; Kotol, P.F.; Bouslimani, A.; Melnik, A.V.; et al. Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci. Transl. Med. 2017, 9, eaah4680. [Google Scholar] [CrossRef] [PubMed]
- Paharik, A.E.; Parlet, C.P.; Chung, N.; Todd, D.A.; Rodriguez, E.I.; Van Dyke, M.J.; Cech, N.B.; Horswill, A.R. Coagulase-Negative Staphylococcal Strain Prevents Staphylococcus aureus Colonization and Skin Infection by Blocking Quorum Sensing. Cell Host Microbe 2017, 22, 746–756.e5. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.R.; Costa, S.K.; Zaramela, L.S.; Khalil, S.; Todd, D.A.; Winter, H.L.; Sanford, J.A.; O’Neill, A.M.; Liggins, M.C.; Nakatsuji, T.; et al. Quorum sensing between bacterial species on the skin protects against epidermal injury in atopic dermatitis. Sci. Transl. Med. 2019, 11, eaat8329. [Google Scholar] [CrossRef]
- Bitschar, K.; Sauer, B.; Focken, J.; Dehmer, H.; Moos, S.; Konnerth, M.; Schilling, N.A.; Grond, S.; Kalbacher, H.; Kurschus, F.C.; et al. Lugdunin amplifies innate immune responses in the skin in synergy with host- and microbiota-derived factors. Nat. Commun. 2019, 10, 2730. [Google Scholar] [CrossRef]
- Stefia, L.V.; Lee, J.; Patel, J.; de Sousa, S.R.; Legrand, J.; Rodero, M.; Burman, S.; Linedale, R.; Morrison, M.; Khosrotehrani, K. Secretome Components from Faecalibacterium prausnitzii Strains A2-165 and AHMP21 Modulate Cutaneous Wound Inflammation. J. Investig. Dermatol. 2020, 140, 2312–2315.e6. [Google Scholar] [CrossRef]


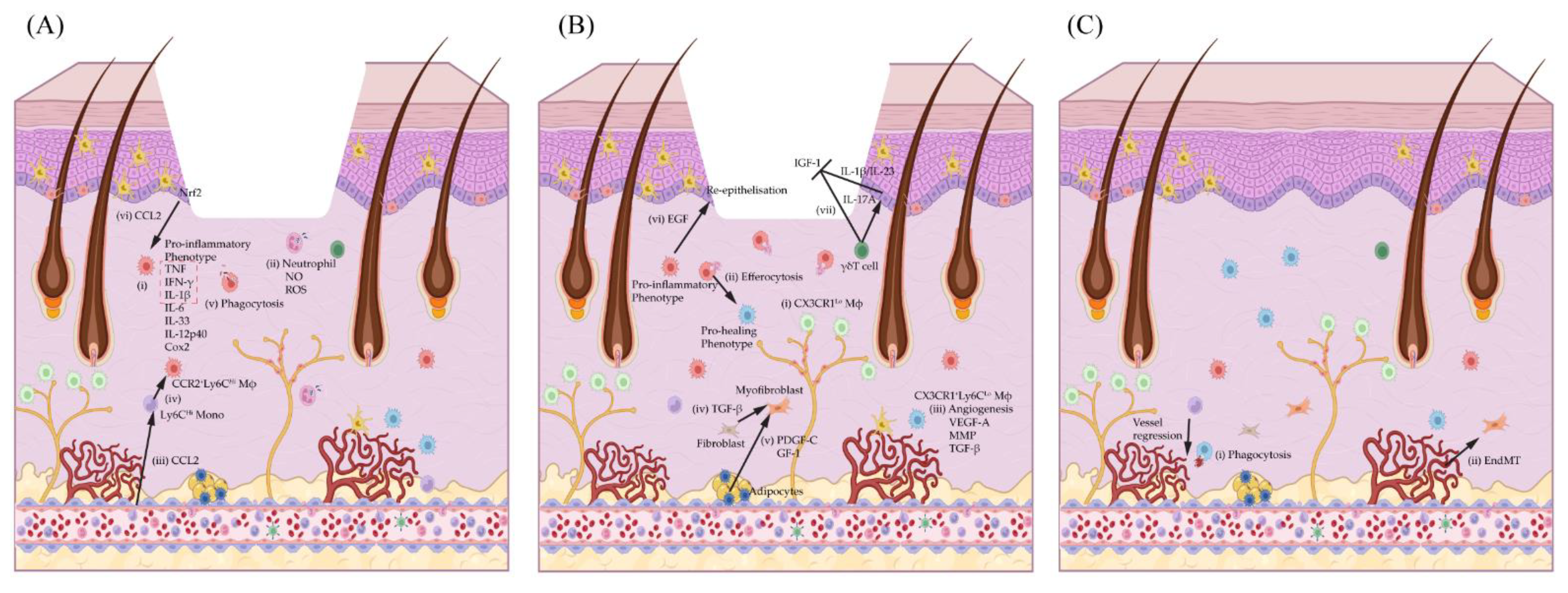{kind=link}
| Model | Wound Type | Strain | Age (Weeks) | Functions/Phenotype | Method | Reference |
|---|---|---|---|---|---|---|
| Ccr2 KO | Skin wound (4 mm) | C57BL/6 | 20–32 | CCR2 is required for CCR2+Ly6C+ Mϕ recruitment. Knockout (KO) model
Adoptive transfer
| CCR2 global knockout and adoptive transfer of WT CD11b+ cells into CCR2 KO wounded mice | [18] |
| Cx3cr1 KO | Skin wound (4 mm) | C57BL/6 | 8–10 | CX3CR1 is required for CX3CR1+Ly6Cneg Mϕ recruitment. Knockout model
BM transplant study
| Cx3cr1 global knockout and BM transplant of WT or Cx3cr1 KO BM cells into WT or Cx3cr1 KO wounded mice | [19] |
| Anti-CX3CR1 antibody | Skin wound (4 mm) | C57BL/6 | 8–10 | Neutralising CX3CR1 prohibit CX3CR1+Ly6Cneg Mϕ recruitment.
| Neutralising antibody | [19] |
| LysMCre-DTR | Skin wound (5 mm) | C57BL/6 | 10–12 | Myeloid cell depletion caused impaired wound closure. Early stage (DT injection from Day 2 to Day 4)
| Mo/Mϕ depletion | [2] |
| Mgl2—DTR/GFP (CD301b) | Skin wound (4 mm) | C57BL/6 | 7–9 | CD301+ Mϕ regulates adipocyte-derived myofibroblasts proliferation.
| CD301b+ Mϕ depletion | [20] |
| CSF1R inhibitor (BLZ945) andClodronate liposomes | Ear punch (2 mm) | C57BL/6 | 6–20 | Mϕ depletion impaired nerve regeneration and sprouting. BLZ945
| Mϕ depletion | [8] |
| OVA-coated nanoparticles adsorbed with DT (DT-OVA-NP) | Skin wound (5 mm) | C57BL/6 | 8–12 | Selective depletion of perivascular Mϕ negatively affects wound healing.
| Perivascular Mϕ depletion | [6] |
| Model | Wound Type | Strain | Age (Weeks) | Functions/Phenotype | Method | Reference |
|---|---|---|---|---|---|---|
| MyD88 KO | Skin wound (10 mm) | C57BL/6 | 10 | Gene knockout of adaptor protein MyD88, which is required for NF-κB signalling, impairs wound healing and reduces growth factor expression.
| Global MyD88 KO | [91] |
| MyD88 KO | Skin wound (1.44 cm2) | C57BL/6 | 3 | Abrogation of NF-κB signalling impairs wound healing outcome and affects host microbiota diversity.
| Global MyD88 KO | [92] |
| Tlr3 KO | Skin wound (4 mm) | C57BL/6 | 8–12 | Gene knockout of Tlr3 affects chemokine expression and myeloid cell recruitment.
| Global Tlr3 KO | [93] |
| Tlr4 KO | Skin wound (3 mm) | C3H/HeJ | 6–8 | Gene knockout of Tlr4 affects pro-inflammatory cytokines production and reduces keratinocytes’ proliferation.
| Global Tlr4 KO | [94] |
| Il-1β KO | Skin wound (1.44 cm2) | C57BL/6 | 3 | IL-1β is required for wound healing and promotes wound-induced hair neogenesis (WIHN).
| Global IL-1β KO | [92] |
| Il-1r KO | Skin incisional, 6 mm excisional and PVA implant | C57BL/6 | 8–12 | IL-1 signaling regulates pro-inflammatory cytokine expression and plays different regulatory roles in various wound types. PVA implant
| Global Il-1r KO | [95] |
| Il-17a KO | Skin wound (3 mm) | C57BL/6 | - | IL-17A is required for neutrophil recruitment and modulates collagen production in the granulation. Knockout model
| Global Il-17a KO and administration of rIL17A on WT wound | [96] |
| Il-17a KO | Skin wound (6 mm) | C57BL/6 | 6–8 | IL-17A/IL-1β/IL-23 forms a positive feedback loop in the epidermis around the wound and negatively regulates IGF-1 production. Knockout model
| Global Il-17a KO | [97] |
| Il-17a KO | Skin wound (6 mm) | C57BL/6 | 8–12 | Inhibition of IL-17A improves wound healing with increased percentage of pro-healing Mϕ
| Global Il-17a KO | [98,99] |
| Il-23p19 KO | Skin wound (6 mm) | C57BL/6 | 24 | IL23 is required for IL-17 production and its abrogation altered myeloid cells’ infiltration
| Global Il-23p19 KO | [99] |
| Anti-Vγ4 antibody (Vγ4D) | Skin wound (6 mm) | C57BL/6 | 6–8 | IL-17A regulates epidermal IL-1β, IL-23 and IGF-1 expression in a dose-dependent manner. Dermal γδ T cells infiltrate the epidermis and supply IL-17A to induce keratinocyte IL-1β production. Keratinocytes IL-1β negatively regulate DETC IGF-1 production. γδ T cells depletion model
| Neutralising Vγ4 T cells | [100] |
| TNF-Rp55 KO | Skin wound (4 mm) | BALB/c | 8–12 | TNF has negative effect on skin wound healing. Abrogating TNF signaling improves wound closure
| Global TNF-Rp55 KO | [101] |
| K14CreERT x Nrf2 fl/fl | Skin wound (10 mm) | - | 6–8 | Nrf2 induces CCL2 production in keratinocytes and promotes Mϕ infiltration. Reciprocally, Mϕ produces EGF to induce keratinocytes’ proliferation. Knockout model
| Epidermis-specific Nrf2 KO | [90] |
| Lrig1-EGFP-ires-CreERT x Act fl/fl | Skin wound (5 mm) | C57BL/6 | 6–8 | Gene knockout of IL-17R complex adaptor Act1 in the Lrig+ HF progenitors reduces Lrig1+ cells’ migration and the proliferation of Lrig1+ progenies to the IFE.
| HF progenitor-specific Act KO | [102] |
| Lrig1-EGFP-ires-CreERT x EGFR fl/fl | Skin wound (5 mm) | C57BL/6 | 6–8 | IL-17RA interacts with EGFR to promote keratinocytes’ proliferation and migration via p-ERK5.
| HF progenitor-specific EGFR KO | [102] |
| TcrdCreER x Rorc fl/fl and Rorgt-EGFP | Splinted skin wound (4 mm) | C57BL/6 | 7–8 | RORγt+γδT cells regulate wound epithelial tongue migration via IL-17A/IL-17RC signalling Control wound
| Mice lacking γδT cells | [103] |
| K14Cre x Il17rc fl/fl | Splinted skin wound (4 mm) | C57BL/6 | 7–8 | Gene knockout of Il17rc in the Krt14+ keratinocytes reduces wound epithelial tongue migration
| Epidermis-specific Il17rc KO | [103] |
| K14Cre x Hif1a fl/fl | IL-17A/IL17RC signalling is essential for HIF1α induction in the Krt14+ keratinocytes. HIF1α regulates keratinocytes’ glycolysis activity and helps with epithelial tongue migration. Knockout model
| Epidermis-specific Hif1a KO | [103] |
| Model | Wound Type | Strain | Age (Weeks) | Functions/Phenotype | Method | Reference |
|---|---|---|---|---|---|---|
| rCCL2 | Skin wound (4 mm) | Balb/c (STZ induced diabetes) | 8–10 | rCCL2 treatment rescues impaired wound healing by restoring Mϕ infiltration.
| Chemokine treatment | [55] |
| Il-1r1 KO x db/db | Skin wound (5 mm) | C57BLKS/J (db/db mice) | 12–14 | IL-1 receptor knockout attenuates IL-1β signaling in diabetic wound.
| Global Il-1r1 KO in obese mice | [119] |
| IL-1Ra/PIGF topical treatment | Skin wound (5 mm) | C57BLKS/J (db/db mice) | 12–14 | IL-1 receptor antagonist delivered with PIGF displays superior wound healing effect compared to IL-1 receptor antagonist alone in diabetic wound.
| Recombinant IL-1Ra/PIGF treatment on obese wound | [119] |
| Anti-IL-1β antibody | Skin wound (8 mm) | C57BL/6 (db/db mice) | 12–16 | Neutralising IL-1β promotes a pro-healing microenvironment by downregulating IL-1β activity.
| Neutralising antibody treatment | [120] |
| Anti-IL-17A or anti-IL23 antibody | Skin wound (6 mm) | C57BL/6 (ob/ob mice) | 24 | Neutralising IL-17A or -23 improved wound closure rate in obese mice and increased pro-healing Mϕ
| Neutralising antibody treatment | [98,99] |
| Il-17a KO x db/db | Skin wound (6 mm) | C57BL/6 (ob/ob mice) | 24 | Il-17 knockout in obese mice improved wound healing
| Global Il-17 KO in obese mice | [99] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sim, S.L.; Kumari, S.; Kaur, S.; Khosrotehrani, K. Macrophages in Skin Wounds: Functions and Therapeutic Potential. Biomolecules 2022, 12, 1659. https://doi.org/10.3390/biom12111659
Sim SL, Kumari S, Kaur S, Khosrotehrani K. Macrophages in Skin Wounds: Functions and Therapeutic Potential. Biomolecules. 2022; 12(11):1659. https://doi.org/10.3390/biom12111659
Chicago/Turabian StyleSim, Seen Ling, Snehlata Kumari, Simranpreet Kaur, and Kiarash Khosrotehrani. 2022. "Macrophages in Skin Wounds: Functions and Therapeutic Potential" Biomolecules 12, no. 11: 1659. https://doi.org/10.3390/biom12111659
APA StyleSim, S. L., Kumari, S., Kaur, S., & Khosrotehrani, K. (2022). Macrophages in Skin Wounds: Functions and Therapeutic Potential. Biomolecules, 12(11), 1659. https://doi.org/10.3390/biom12111659