
Inhibition of Autophagy Promotes the Elimination of Liver Cancer Stem Cells by CD133 Aptamer-Targeted Delivery of Doxorubicin

 , , ,
, , ,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Generation and Characterization of the CD133 Aptamer-Doxorubicin Conjugates (CD133 Aptamer-DOX)
2.3. Isothermal Titration Calorimetry (ITC)
2.4. Determination of Binding Affinity
2.5. Annexin V Assay
2.6. In Vitro Tumorsphere Formation Assay
2.7. Statistical Analysis
3. Results
3.1. Development and Characterization of a CD133 Aptamer-DOX Conjugate
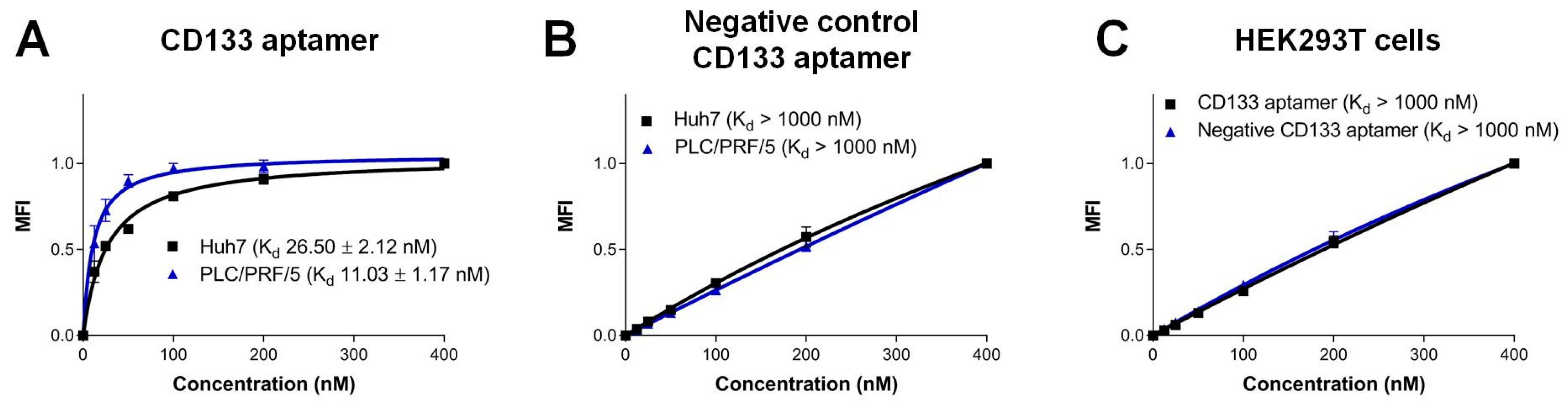
3.2. The CD133 Aptamer Specifically Binds to CD133-Positive Human Liver Cancer Cells
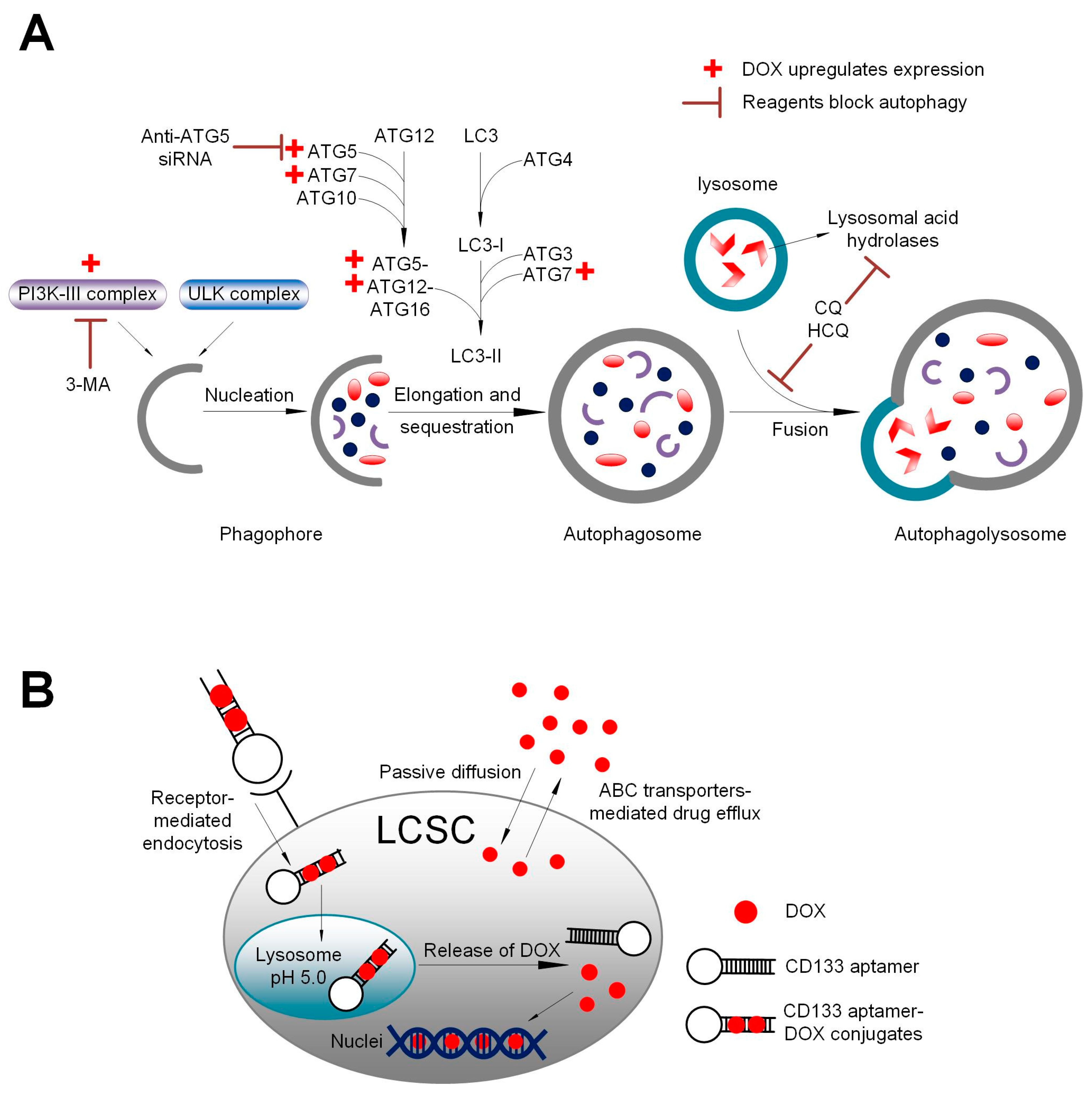
3.3. Enhanced Delivery of DOX into Liver Cancer Stem Cells via CD133 Aptamer
3.4. Autophagy Inhibition Augmented Apoptosis Elicited by DOX
3.5. Enhanced Elimination of Liver Cancer Stem Cells via CD133-Targeted Delivery of DOX and Inhibition of Autophagy
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Nio, K.; Yamashita, T.; Kaneko, S. The evolving concept of liver cancer stem cells. Mol. Cancer 2017, 16, 4. [Google Scholar] [CrossRef] [PubMed]
- Alisi, A.; Cho, W.C.; Locatelli, F.; Fruci, D. Multidrug resistance and cancer stem cells in neuroblastoma and hepatoblastoma. Int. J. Mol. Sci. 2013, 14, 24706–24725. [Google Scholar] [CrossRef]
- Li, L.; Xu, S.; Yan, H.; Li, X.; Yazd, H.S.; Li, X.; Huang, T.; Cui, C.; Jiang, J.; Tan, W. Nucleic Acid Aptamers for Molecular Diagnostics and Therapeutics: Advances and Perspectives. Angew. Chem. Int. Ed. Engl. 2021, 60, 2221–2231. [Google Scholar] [CrossRef]
- Shigdar, S.; Qiao, L.; Zhou, S.F.; Xiang, D.; Wang, T.; Li, Y.; Lim, L.Y.; Kong, L.; Li, L.; Duan, W. RNA aptamers targeting cancer stem cell marker CD133. Cancer Lett. 2013, 330, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.J.; Zhang, S.S.; Guo, X.L.; Sun, K.; Han, Z.P.; Li, R.; Zhao, Q.D.; Deng, W.J.; Xie, X.Q.; Zhang, J.W.; et al. Autophagy contributes to the survival of CD133+ liver cancer stem cells in the hypoxic and nutrient-deprived tumor microenvironment. Cancer Lett. 2013, 339, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Xiang, D.; Shigdar, S.; Bean, A.G.; Bruce, M.; Yang, W.; Mathesh, M.; Wang, T.; Yin, W.; Tran, P.H.; Al Shamaileh, H.; et al. Transforming doxorubicin into a cancer stem cell killer via EpCAM aptamer-mediated delivery. Theranostics 2017, 7, 4071–4086. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.R.; Weber, J.K.; Yin, W.; Huynh, T.; Duan, W.; Zhou, R. In silico design and validation of high-affinity RNA aptamers targeting epithelial cellular adhesion molecule dimers. Proc. Natl. Acad. Sci. USA 2020, 117, 8486–8493. [Google Scholar] [CrossRef]
- Xiang, D.; Zheng, C.; Zhou, S.F.; Qiao, S.; Tran, P.H.; Pu, C.; Li, Y.; Kong, L.; Kouzani, A.Z.; Lin, J.; et al. Superior Performance of Aptamer in Tumor Penetration over Antibody: Implication of Aptamer-Based Theranostics in Solid Tumors. Theranostics 2015, 5, 1083–1097. [Google Scholar] [CrossRef]
- Phillips, D.R.; White, R.J.; Cullinane, C. DNA sequence-specific adducts of adriamycin and mitomycin C. FEBS Lett. 1989, 246, 233–240. [Google Scholar] [CrossRef]
- Chushak, Y.; Stone, M.O. In silico selection of RNA aptamers. Nucleic Acids Res. 2009, 37, e87. [Google Scholar] [CrossRef]
- Turnbull, W.B.; Daranas, A.H. On the value of c: Can low affinity systems be studied by isothermal titration calorimetry? J. Am. Chem. Soc. 2003, 125, 14859–14866. [Google Scholar] [CrossRef] [PubMed]
- Erijman, A.; Rosenthal, E.; Shifman, J.M. How structure defines affinity in protein-protein interactions. PLoS ONE 2014, 9, e110085. [Google Scholar] [CrossRef] [PubMed]
- Pineiro, A.; Munoz, E.; Sabin, J.; Costas, M.; Bastos, M.; Velazquez-Campoy, A.; Garrido, P.F.; Dumas, P.; Ennifar, E.; Garcia-Rio, L.; et al. AFFINImeter: A software to analyze molecular recognition processes from experimental data. Anal. Biochem. 2019, 577, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Hirota, M.; Waugh, S.M.; Murakami, I.; Suzuki, T.; Muraguchi, M.; Shibamori, M.; Ishikawa, Y.; Jarvis, T.C.; Carter, J.D.; et al. Chemically modified DNA aptamers bind interleukin-6 with high affinity and inhibit signaling by blocking its interaction with interleukin-6 receptor. J. Biol. Chem. 2014, 289, 8706–8719. [Google Scholar] [CrossRef]
- Kaur, G.; Dufour, J.M. Cell lines: Valuable tools or useless artifacts. Spermatogenesis 2012, 2, 1–5. [Google Scholar] [CrossRef]
- Yin, W.; Xiang, D.; Wang, T.; Zhang, Y.; Pham, C.V.; Zhou, S.; Jiang, G.; Hou, Y.; Zhu, Y.; Han, Y.; et al. The inhibition of ABCB1/MDR1 or ABCG2/BCRP enables doxorubicin to eliminate liver cancer stem cells. Sci. Rep. 2021, 11, 10791. [Google Scholar] [CrossRef]
- Kaur, S.P.; Talat, A.; Karimi-Sari, H.; Grees, A.; Chen, H.W.; Lau, D.T.Y.; Catana, A.M. Hepatocellular Carcinoma in Hepatitis B Virus-Infected Patients and the Role of Hepatitis B Surface Antigen (HBsAg). J. Clin. Med. 2022, 11, 1126. [Google Scholar] [CrossRef]
- Liu, D.X.; Li, P.P.; Guo, J.P.; Li, L.L.; Guo, B.; Jiao, H.B.; Wu, J.H.; Chen, J.M. Exosomes derived from HBV-associated liver cancer promote chemoresistance by upregulating chaperone-mediated autophagy. Oncol. Lett. 2019, 17, 323–331. [Google Scholar] [CrossRef]
- Wang, C.; Wang, M.D.; Cheng, P.; Huang, H.; Dong, W.; Zhang, W.W.; Li, P.P.; Lin, C.; Pan, Z.Y.; Wu, M.C.; et al. Hepatitis B virus X protein promotes the stem-like properties of OV6+ cancer cells in hepatocellular carcinoma. Cell Death Dis. 2017, 8, e2560. [Google Scholar] [CrossRef]
- Zhao, X.; Guo, X.; Xing, L.; Yue, W.; Yin, H.; He, M.; Wang, J.; Yang, J.; Chen, J. HBV infection potentiates resistance to S-phase arrest-inducing chemotherapeutics by inhibiting CHK2 pathway in diffuse large B-cell lymphoma. Cell Death Dis. 2018, 9, 61. [Google Scholar] [CrossRef]
- MacNab, G.M.; Alexander, J.J.; Lecatsas, G.; Bey, E.M.; Urbanowicz, J.M. Hepatitis B surface antigen produced by a human hepatoma cell line. Br. J. Cancer 1976, 34, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, H.; Taketa, K.; Yamane, T.; Miyazaki, M.; Miyano, K.; Sato, J. Phenotypical stability of a human hepatoma cell line, HuH-7, in long-term culture with chemically defined medium. Gan 1984, 75, 151–158. [Google Scholar] [PubMed]
- Xue, X.; Liao, W.; Xing, Y. Comparison of clinical features and outcomes between HBV-related and non-B non-C hepatocellular carcinoma. Infect. Agents Cancer 2020, 15, 11. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yu, D.; Zhang, H.; He, H.; Zhang, C.; Zhao, W.; Shao, R.G. CD133+EpCAM+ phenotype possesses more characteristics of tumor initiating cells in hepatocellular carcinoma Huh7 cells. Int. J. Biol. Sci. 2012, 8, 992–1004. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Zhou, Y.; Zhai, B.; Liao, J.; Xu, W.; Zhang, R.; Li, J.; Zhang, Y.; Chen, L.; Qian, H.; et al. Sphere-forming cell subpopulations with cancer stem cell properties in human hepatoma cell lines. BMC Gastroenterol. 2011, 11, 71. [Google Scholar] [CrossRef]
- Lupertz, R.; Watjen, W.; Kahl, R.; Chovolou, Y. Dose- and time-dependent effects of doxorubicin on cytotoxicity, cell cycle and apoptotic cell death in human colon cancer cells. Toxicology 2010, 271, 115–121. [Google Scholar] [CrossRef]
- Korkaya, H.; Paulson, A.; Charafe-Jauffret, E.; Ginestier, C.; Brown, M.; Dutcher, J.; Clouthier, S.G.; Wicha, M.S. Regulation of mammary stem/progenitor cells by PTEN/Akt/beta-catenin signaling. PLoS Biol. 2009, 7, e1000121. [Google Scholar] [CrossRef]
- Xue, F.; Hu, L.; Ge, R.; Yang, L.; Liu, K.; Li, Y.; Sun, Y.; Wang, K. Autophagy-deficiency in hepatic progenitor cells leads to the defects of stemness and enhances susceptibility to neoplastic transformation. Cancer Lett. 2016, 371, 38–47. [Google Scholar] [CrossRef]
- Chen, Y.L.; Lin, P.Y.; Ming, Y.Z.; Huang, W.C.; Chen, R.F.; Chen, P.M.; Chu, P.Y. The effects of the location of cancer stem cell marker CD133 on the prognosis of hepatocellular carcinoma patients. BMC Cancer 2017, 17, 474. [Google Scholar] [CrossRef]
- Chen, W.; Li, F.; Xue, Z.M.; Wu, H.R. Anti-human CD133 monoclonal antibody that could inhibit the proliferation of colorectal cancer cells. Hybridoma 2010, 29, 305–310. [Google Scholar] [CrossRef]
- Dai, X.; Guo, Y.; Hu, Y.; Bao, X.; Zhu, X.; Fu, Q.; Zhang, H.; Tong, Z.; Liu, L.; Zheng, Y.; et al. Immunotherapy for targeting cancer stem cells in hepatocellular carcinoma. Theranostics 2021, 11, 3489–3501. [Google Scholar] [CrossRef] [PubMed]
- Glumac, P.M.; LeBeau, A.M. The role of CD133 in cancer: A concise review. Clin. Transl. Med. 2018, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Kemper, K.; Sprick, M.R.; de Bree, M.; Scopelliti, A.; Vermeulen, L.; Hoek, M.; Zeilstra, J.; Pals, S.T.; Mehmet, H.; Stassi, G.; et al. The AC133 epitope, but not the CD133 protein, is lost upon cancer stem cell differentiation. Cancer Res. 2010, 70, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Piao, L.S.; Hur, W.; Kim, T.K.; Hong, S.W.; Kim, S.W.; Choi, J.E.; Sung, P.S.; Song, M.J.; Lee, B.C.; Hwang, D.; et al. CD133+ liver cancer stem cells modulate radioresistance in human hepatocellular carcinoma. Cancer Lett. 2012, 315, 129–137. [Google Scholar] [CrossRef]
- Wu, Y.T.; Tan, H.L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.N.; Codogno, P.; Shen, H.M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef]
- Wu, D.H.; Jia, C.C.; Chen, J.; Lin, Z.X.; Ruan, D.Y.; Li, X.; Lin, Q.; Min, D.; Ma, X.K.; Wan, X.B.; et al. Autophagic LC3B overexpression correlates with malignant progression and predicts a poor prognosis in hepatocellular carcinoma. Tumor Biol. 2014, 35, 12225–12233. [Google Scholar] [CrossRef]
- Haj, H.T.; Salerno, M.; Priebe, W.; Kozlowski, H.; Garnier-Suillerot, A. New findings in the study on the intercalation of bisdaunorubicin and its monomeric analogues with naked and nucleus DNA. Chem.-Biol. Interact. 2003, 145, 349–358. [Google Scholar] [CrossRef]
- Li, N.; Ebright, J.N.; Stovall, G.M.; Chen, X.; Nguyen, H.H.; Singh, A.; Syrett, A.; Ellington, A.D. Technical and biological issues relevant to cell typing with aptamers. J. Proteome Res. 2009, 8, 2438–2448. [Google Scholar] [CrossRef]
- Park, J.M.; Tougeron, D.; Huang, S.; Okamoto, K.; Sinicrope, F.A. Beclin 1 and UVRAG confer protection from radiation-induced DNA damage and maintain centrosome stability in colorectal cancer cells. PLoS ONE 2014, 9, e100819. [Google Scholar]
- Kim, D.H.; Behlke, M.A.; Rose, S.D.; Chang, M.S.; Choi, S.; Rossi, J.J. Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat. Biotechnol. 2005, 23, 222–226. [Google Scholar] [CrossRef]
- Heatwole, V.M. TUNEL assay for apoptotic cells. Methods Mol. Biol. 1999, 115, 141–148. [Google Scholar] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, W.; Pham, C.V.; Wang, T.; Al Shamaileh, H.; Chowdhury, R.; Patel, S.; Li, Y.; Kong, L.; Hou, Y.; Zhu, Y.; et al. Inhibition of Autophagy Promotes the Elimination of Liver Cancer Stem Cells by CD133 Aptamer-Targeted Delivery of Doxorubicin. Biomolecules 2022, 12, 1623. https://doi.org/10.3390/biom12111623
Yin W, Pham CV, Wang T, Al Shamaileh H, Chowdhury R, Patel S, Li Y, Kong L, Hou Y, Zhu Y, et al. Inhibition of Autophagy Promotes the Elimination of Liver Cancer Stem Cells by CD133 Aptamer-Targeted Delivery of Doxorubicin. Biomolecules. 2022; 12(11):1623. https://doi.org/10.3390/biom12111623
Chicago/Turabian StyleYin, Wang, Cuong V. Pham, Tao Wang, Hadi Al Shamaileh, Rocky Chowdhury, Shweta Patel, Yong Li, Lingxue Kong, Yingchu Hou, Yimin Zhu, and et al. 2022. "Inhibition of Autophagy Promotes the Elimination of Liver Cancer Stem Cells by CD133 Aptamer-Targeted Delivery of Doxorubicin" Biomolecules 12, no. 11: 1623. https://doi.org/10.3390/biom12111623
APA StyleYin, W., Pham, C. V., Wang, T., Al Shamaileh, H., Chowdhury, R., Patel, S., Li, Y., Kong, L., Hou, Y., Zhu, Y., Chen, S., Xu, H., Jia, L., Duan, W., & Xiang, D. (2022). Inhibition of Autophagy Promotes the Elimination of Liver Cancer Stem Cells by CD133 Aptamer-Targeted Delivery of Doxorubicin. Biomolecules, 12(11), 1623. https://doi.org/10.3390/biom12111623