
Structural Consequences of Deproteinating the 50S Ribosome


Abstract
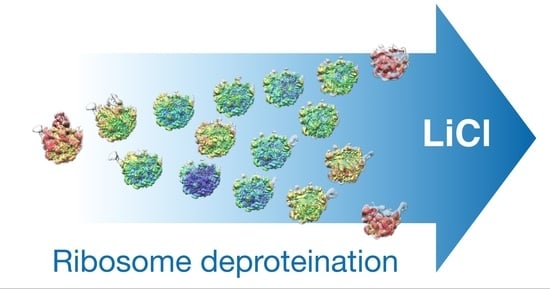
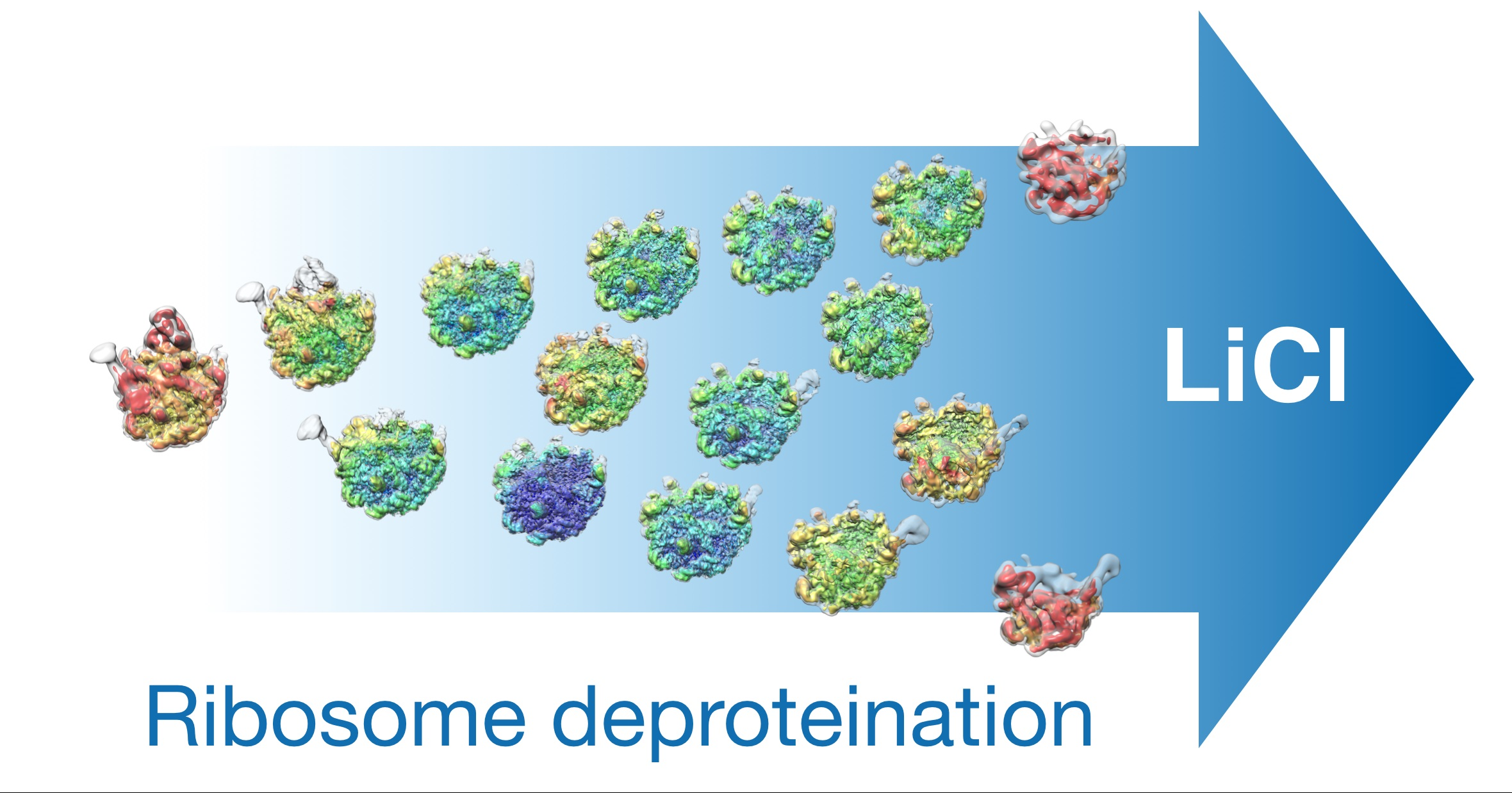{kind=link}
{kind=link}
{kind=link}
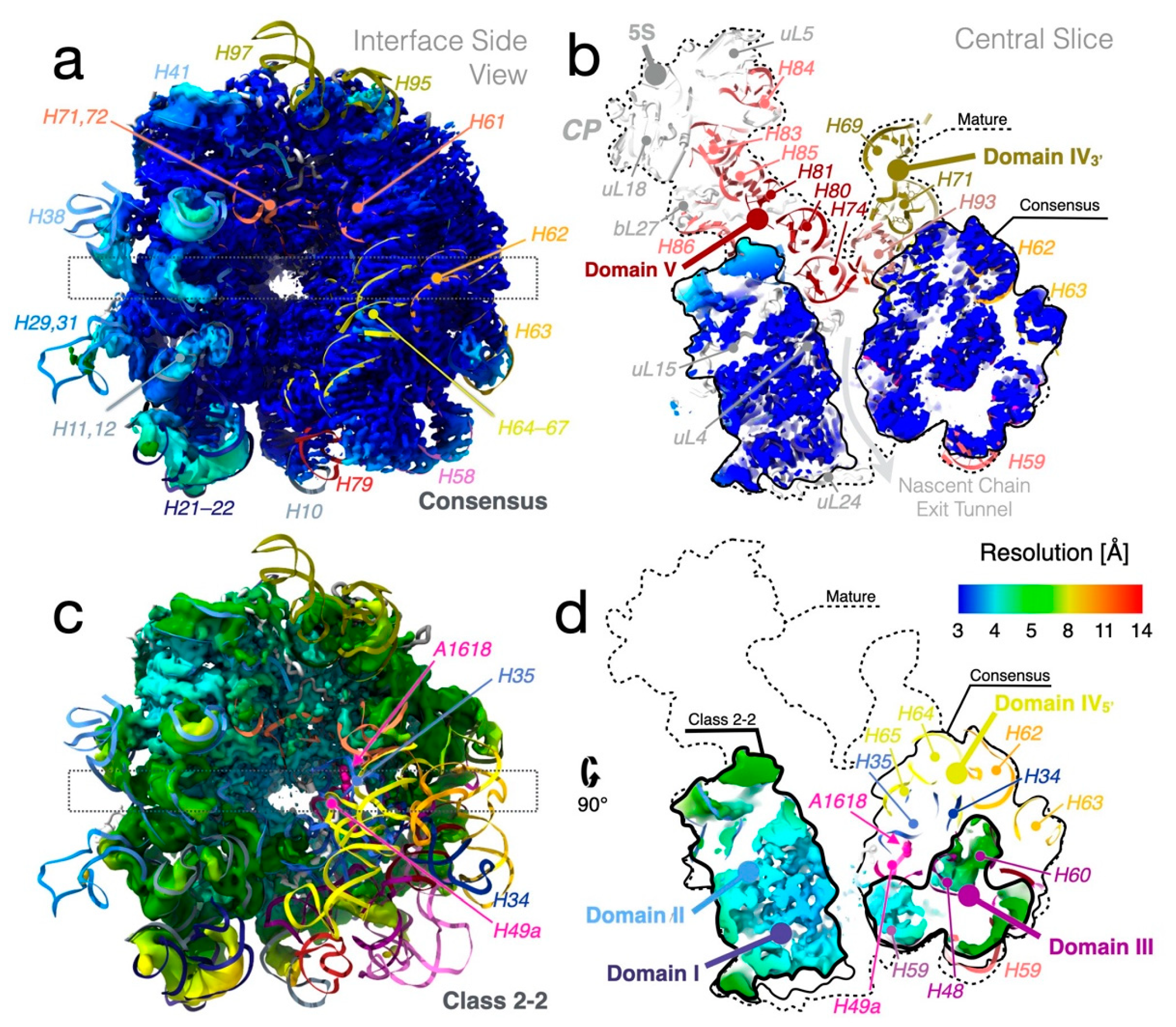{kind=link}
{kind=link}
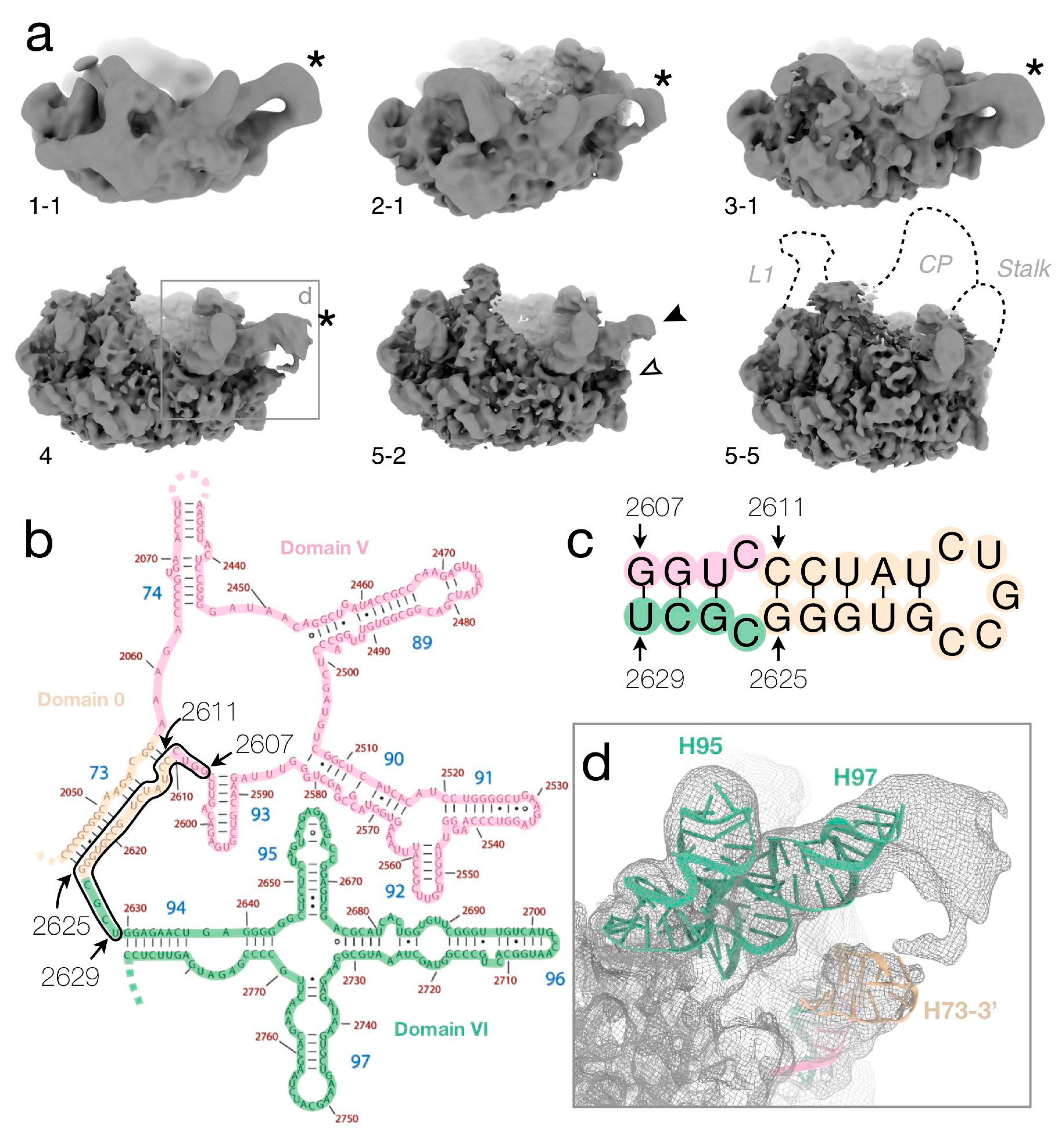{kind=link}
{kind=link}
{kind=link}
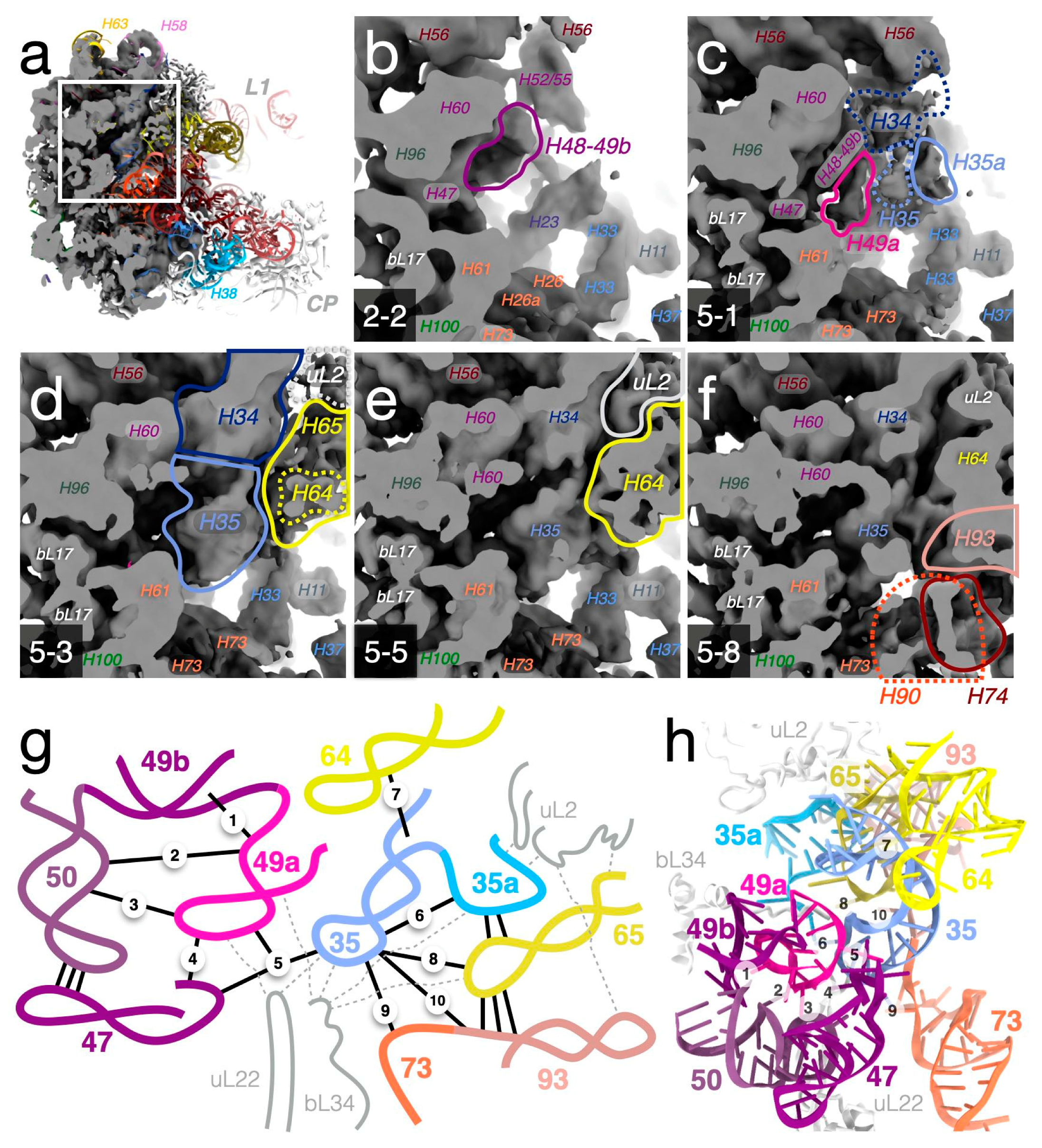{kind=link}
1. Introduction
2. Materials and Methods
2.1. Preparation of LiCl Core Particles
2.2. In Vitro Methylation Assay
2.3. Cryo-EM Sample Preparation
2.4. Cryo-EM Grid Freezing and Data Collection
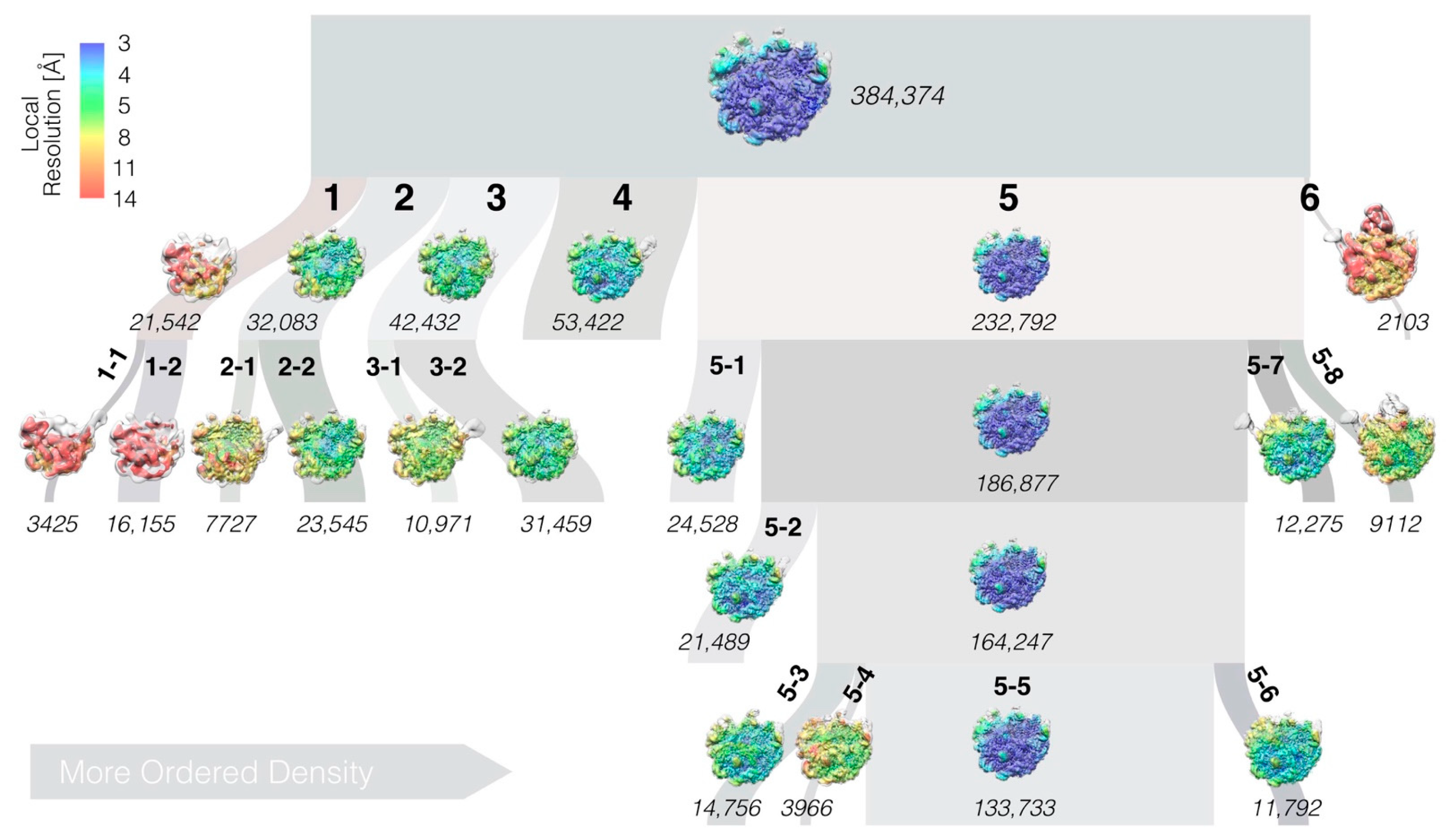
2.5. Cryo-EM Data Processing
2.6. Model Refinement in Cryo-EM Maps
2.7. Occupancy of Structural Elements and Hierarchical Clustering
3. Results
3.1. Sample Preparation
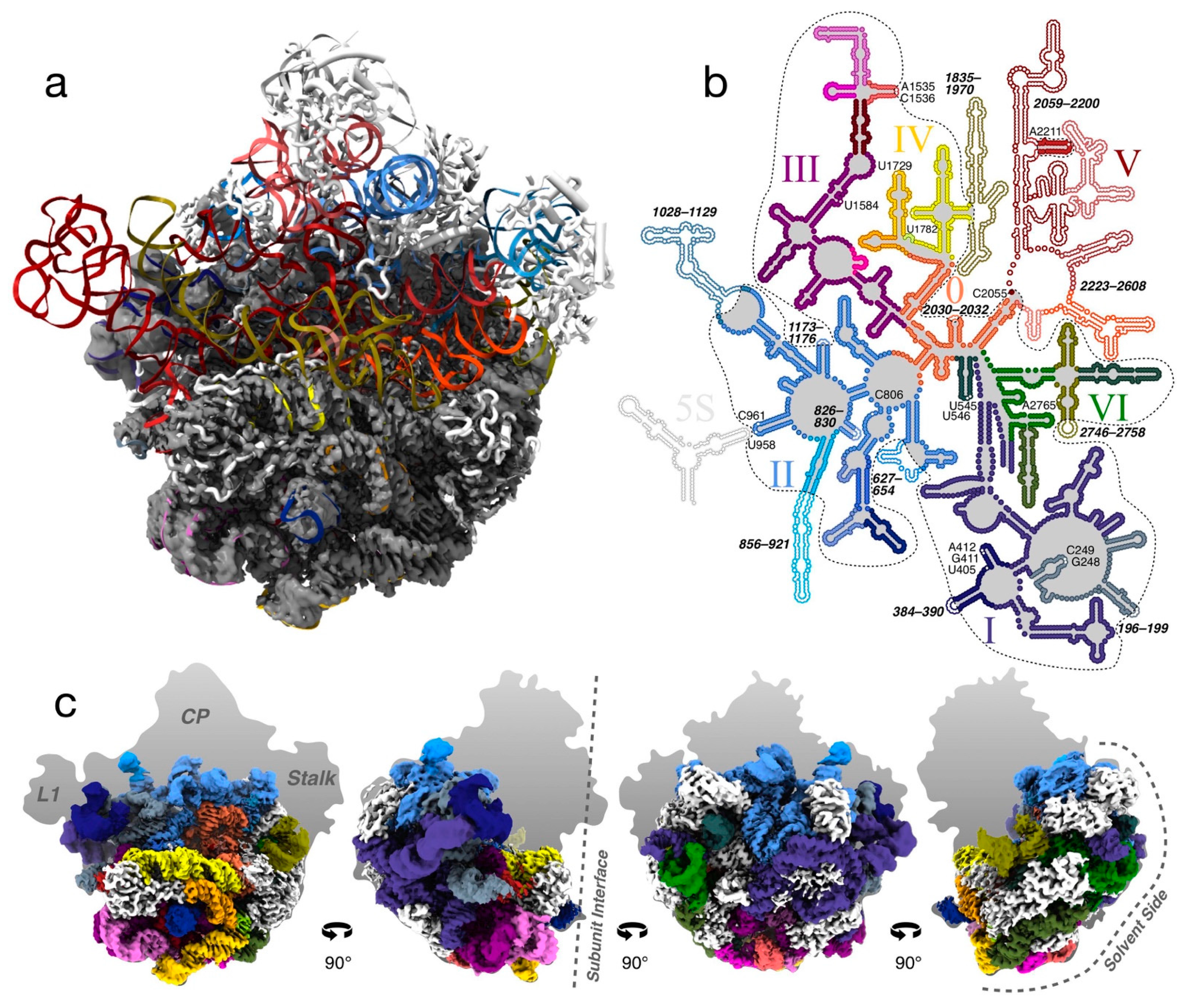
3.2. High-Resolution Cryo-EM Reconstruction of the 50S LiCl Core Particle
3.3. LiCl Washing of the LSU Produces a Range of Sub-Particles
3.4. The LSU Is the Most Stable at the Solvent-Side
3.5. The LiCl Core Particle Classes Are Analogous to In Vivo and In Vitro Assembly Intermediates
3.6. The Misfolded 3′ Strand of Helix H73
3.7. R-Proteins Leave the 50S According to In Vivo Assembly Groups
3.8. Comparison of Particle Classes Show Dependencies between Different Structural Elements
3.8.1. Sub-Domain IIItail Can Stabilize Sub-Domain IV5′ in Absence of uL2
3.8.2. R-Protein uL3 Is Important for the Stability of Domains 0 and VI
3.8.3. R-Protein uL2 Stabilizes Sub-Domain IV5′
3.8.4. Helices H79 and H88 Anchor the Core of Domain V
3.8.5. Stability of Sub-Domain IV3′ Depends on Domain V
3.8.6. The CP Is Severely Destabilized in the LiCl Core Particles
3.9. Folding of H49a and H35 Demonstrate Analogies between Disassembly and Assembly
3.9.1. Folding and Tertiary Interactions of H49a
3.9.2. Folding and Tertiary Interactions of H35
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nierhaus, K.H.; Dohme, F. Total Reconstitution of Functionally Active 50S Ribosomal Subunits from Escherichia coli. Proc. Natl. Acad. Sci. USA 1974, 71, 4713–4717. [Google Scholar] [CrossRef] [PubMed]
- Homann, H.E.; Nierhaus, K.H. Ribosomal Proteins. Protein Compositions of Biosynthetic Precursors and Artifical Subparticles from Ribosomal Subunits in Escherichia coli K 12. JBIC J. Biol. Inorg. Chem. 1971, 20, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C.A.; Bradbury, E.; Littlechild, J.; Dijk, J. Proton magnetic resonance studies to compare Escherichia coli ribosomal proteins prepared by two different methods. FEBS Lett. 1977, 83, 348–352. [Google Scholar] [CrossRef]
- Dijk, J.; Littlechild, J. Purification of ribosomal proteins from Escherichia coli under nondenaturing conditions. Methods Enzym. 1979, 59, 481–502. [Google Scholar] [CrossRef]
- Chen, S.S.; Williamson, J.R. Characterization of the Ribosome Biogenesis Landscape in E. coli Using Quantitative Mass Spectrometry. J. Mol. Biol. 2012, 425, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Spillmann, S.; Dohme, F.; Nierhaus, K.H. Assembly in Vitro of the 50 S subunit from Escherichia coli ribosomes: Proteins essential for the first heat-dependent conformational change. J. Mol. Biol. 1977, 115, 513–523. [Google Scholar] [CrossRef]
- Pulk, A.; Liiv, A.; Peil, L.; Maiväli, U.; Nierhaus, K.; Remme, J. Ribosome reactivation by replacement of damaged proteins. Mol. Microbiol. 2010, 75, 801–814. [Google Scholar] [CrossRef]
- Shajani, Z.; Sykes, M.T.; Williamson, J.R. Assembly of Bacterial Ribosomes. Annu. Rev. Biochem. 2011, 80, 501–526. [Google Scholar] [CrossRef]
- Rodgers, M.L.; Woodson, S.A. A roadmap for rRNA folding and assembly during transcription. Trends Biochem. Sci. 2021, 46, 889–901. [Google Scholar] [CrossRef]
- Chen, S.S.; Sperling, E.; Silverman, J.M.; Davis, J.H.; Williamson, J.R. Measuring the dynamics of E. coli ribosome biogenesis using pulse-labeling and quantitative mass spectrometry. Mol. BioSyst. 2012, 8, 3325–3334. [Google Scholar] [CrossRef]
- Dagley, S.; Sykes, J. Effect of Drugs upon Components of Bacterial Cytoplasm. Nature 1959, 183, 1608–1609. [Google Scholar] [CrossRef]
- Siibak, T.; Peil, L.; Dönhöfer, A.; Tats, A.; Remm, M.; Wilson, D.N.; Tenson, T.; Remme, J. Antibiotic-induced ribosomal assembly defects result from changes in the synthesis of ribosomal proteins. Mol. Microbiol. 2011, 80, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.H.; Tan, Y.Z.; Carragher, B.; Potter, C.S.; Lyumkis, D.; Williamson, J.R. Modular Assembly of the Bacterial Large Ribosomal Subunit. Cell 2016, 167, 1610–1622.e15. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, W.; Ge, X.; Yan, K.; Mandava, C.S.; Sanyal, S.; Gao, N. Loss of a single methylation in 23S rRNA delays 50S assembly at multiple late stages and impairs translation initiation and elongation. Proc. Natl. Acad. Sci. USA 2020, 117, 15609–15619. [Google Scholar] [CrossRef] [PubMed]
- Seffouh, A.; Jain, N.; Jahagirdar, D.; Basu, K.; Razi, A.; Ni, X.; Guarné, A.; Britton, R.A.; Ortega, J. Structural consequences of the interaction of RbgA with a 50S ribosomal subunit assembly intermediate. Nucleic Acids Res. 2019, 47, 10414–10425. [Google Scholar] [CrossRef]
- Zhou, D.; Zhu, X.; Zheng, S.; Tan, D.; Dong, M.-Q.; Ye, K. Cryo-EM structure of an early precursor of large ribosomal subunit reveals a half-assembled intermediate. Protein Cell 2018, 10, 120–130. [Google Scholar] [CrossRef]
- Nikolay, R.; Hilal, T.; Schmidt, S.; Qin, B.; Schwefel, D.; Vieira-Vieira, C.H.; Mielke, T.; Bürger, J.; Loerke, J.; Amikura, K.; et al. Snapshots of native pre-50S ribosomes reveal a biogenesis factor network and evolutionary specialization. Mol. Cell 2021, 81, 1200–1215.e9. [Google Scholar] [CrossRef]
- Nikolay, R.; Hilal, T.; Qin, B.; Mielke, T.; Bürger, J.; Loerke, J.; Textoris-Taube, K.; Nierhaus, K.H.; Spahn, C.M. Structural Visualization of the Formation and Activation of the 50S Ribosomal Subunit during In Vitro Reconstitution. Mol. Cell 2018, 70, 881–893.e3. [Google Scholar] [CrossRef] [PubMed]
- Sergiev, P.V.; Serebryakova, M.V.; Bogdanov, A.A.; Dontsova, O.A. The ybiN Gene of Escherichia coli Encodes Adenine-N6 Methyltransferase Specific for Modification of A1618 of 23 S Ribosomal RNA, a Methylated Residue Located Close to the Ribosomal Exit Tunnel. J. Mol. Biol. 2008, 375, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Boublik, M.; Spiess, E.; Roth, H.E.; Hellmann, W.; Jenkins, F. Structure of LiCl core particles of 50 S ribosomal subunits from Escherichia coli by electron microscopy. Cytobiologie 1978, 18, 309–319. [Google Scholar]
- Baba, T.; Ara, T.; Hasegawa, M.; Takai, Y.; Okumura, Y.; Baba, M.; Datsenko, K.A.; Tomita, M.; Wanner, B.L.; Mori, H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol. Syst. Biol. 2006, 2, 2006.0008. [Google Scholar] [CrossRef]
- Zivanov, J.; Nakane, T.; Forsberg, B.O.; Kimanius, D.; Hagen, W.J.; Lindahl, E.; Scheres, S.H. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 2018, 7, e42166. [Google Scholar] [CrossRef]
- Scheres, S. Processing of Structurally Heterogeneous Cryo-EM Data in RELION. Methods Enzymol. 2016, 579, 125–157. [Google Scholar] [CrossRef]
- Zheng, S.Q.; Palovcak, E.; Armache, J.-P.; Verba, K.A.; Cheng, Y.; Agard, D.A. MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 2017, 14, 331–332. [Google Scholar] [CrossRef]
- Zhang, K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 2016, 193, 1–12. [Google Scholar] [CrossRef]
- Zivanov, J.; Nakane, T.; Scheres, S.H.W. Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION-3.1. IUCrJ 2020, 7, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Naydenova, K.; Russo, C.J. Measuring the effects of particle orientation to improve the efficiency of electron cryomicroscopy. Nat. Commun. 2017, 8, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Noeske, J.; Wasserman, M.R.; Terry, D.S.; Altman, R.; Blanchard, S.C.; Cate, J.H.D. High-resolution structure of the Escherichia coli ribosome. Nat. Struct. Mol. Biol. 2015, 22, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkóczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.-W.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. Sect. D Struct. Biol. 2019, 75, 861–877. [Google Scholar] [CrossRef] [PubMed]
- Watson, Z.L.; Ward, F.R.; Méheust, R.; Ad, O.; Schepartz, A.; Banfield, J.F.; Cate, J.H. Structure of the bacterial ribosome at 2 Å resolution. eLife 2020, 9, e60482. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2017, 27, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Petrov, A.S.; Bernier, C.; Hershkovits, E.; Xue, Y.; Waterbury, C.C.; Hsiao, C.; Stepanov, V.G.; Gaucher, E.A.; Grover, M.A.; Harvey, S.C.; et al. Secondary structure and domain architecture of the 23S and 5S rRNAs. Nucleic Acids Res. 2013, 41, 7522–7535. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, J.; Xiao, Y. 3dRNA: Building RNA 3D structure with improved template library. Comput. Struct. Biotechnol. J. 2020, 18, 2416–2423. [Google Scholar] [CrossRef] [PubMed]
- Lanier, K.A.; Athavale, S.S.; Petrov, A.S.; Wartell, R.; Williams, L.D. Imprint of Ancient Evolution on rRNA Folding. Biochemistry 2016, 55, 4603–4613. [Google Scholar] [CrossRef]
- Athavale, S.S.; Gossett, J.J.; Hsiao, C.; Bowman, J.C.; O’Neill, E.; Hershkovitz, E.; Preeprem, T.; Hud, N.V.; Wartell, R.M.; Harvey, S.C.; et al. Domain III of the T. thermophilus 23S rRNA folds independently to a near-native state. RNA 2012, 18, 752–758. [Google Scholar] [CrossRef][Green Version]
- Sergiev, P.V.; Golovina, A.Y.; Osterman, I.A.; Nesterchuk, M.V.; Sergeeva, O.V.; Chugunova, A.A.; Evfratov, S.A.; Andreianova, E.S.; Pletnev, P.I.; Laptev, I.G.; et al. N6-Methylated Adenosine in RNA: From Bacteria to Humans. J. Mol. Biol. 2016, 428, 2134–2145. [Google Scholar] [CrossRef]
- Meselson, M.; Nomura, M.; Brenner, S.; Davern, C.; Schlessinger, D. Conservation of ribosomes during bacterial growth. J. Mol. Biol. 1964, 9, 696–711. [Google Scholar] [CrossRef]
- Yang, Z.; Guo, Q.; Goto, S.; Chen, Y.; Li, N.; Yan, K.; Zhang, Y.; Muto, A.; Deng, H.; Himeno, H.; et al. Structural insights into the assembly of the 30S ribosomal subunit in vivo: Functional role of S5 and location of the 17S rRNA precursor sequence. Protein Cell 2014, 5, 394–407. [Google Scholar] [CrossRef]
- Nierhaus, K. The assembly of prokaryotic ribosomes. Biochimie 1991, 73, 739–755. [Google Scholar] [CrossRef]
- Nierhaus, K. Reconstitution of Ribosomes. In Ribosomes and Protein Synthesis; Spedding, G., Ed.; Oxford University Press: Oxford, UK, 1990; pp. 161–189. [Google Scholar]
- Orelle, C.; Carlson, E.D.; Szal, T.; Florin, T.; Jewett, M.; Mankin, A.S. Protein synthesis by ribosomes with tethered subunits. Nature 2015, 524, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Siibak, T.; Remme, J. Subribosomal particle analysis reveals the stages of bacterial ribosome assembly at which rRNA nucleotides are modified. RNA 2010, 16, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.H.; Williamson, J.R. Structure and dynamics of bacterial ribosome biogenesis. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160181. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, M.L.; Woodson, S.A. Transcription Increases the Cooperativity of Ribonucleoprotein Assembly. Cell 2019, 179, 1370–1381.e12. [Google Scholar] [CrossRef]
- Lawrence, M.G.; Shamsuzzaman, M.; Kondopaka, M.; Pascual, C.; Zengel, J.M.; Lindahl, L. The extended loops of ribosomal proteins uL4 and uL22 of Escherichia coli contribute to ribosome assembly and protein translation. Nucleic Acids Res. 2016, 44, 5798–5810. [Google Scholar] [CrossRef]
- Talkington, M.W.T.; Siuzdak, G.; Williamson, J. An assembly landscape for the 30S ribosomal subunit. Nature 2005, 438, 628–632. [Google Scholar] [CrossRef]
- Zhong, E.D.; Bepler, T.; Berger, B.; Davis, J.H. CryoDRGN: Reconstruction of heterogeneous cryo-EM structures using neural networks. Nat. Methods 2021, 18, 176–185. [Google Scholar] [CrossRef]
- Rabuck-Gibbons, J.N.; Lyumkis, D.; Williamson, J.R. Quantitative mining of compositional heterogeneity in cryo-EM datasets of ribosome assembly intermediates. Structure 2022, 30, 498–509.e4. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larsson, D.S.D.; Kanchugal P, S.; Selmer, M. Structural Consequences of Deproteinating the 50S Ribosome. Biomolecules 2022, 12, 1605. https://doi.org/10.3390/biom12111605
Larsson DSD, Kanchugal P S, Selmer M. Structural Consequences of Deproteinating the 50S Ribosome. Biomolecules. 2022; 12(11):1605. https://doi.org/10.3390/biom12111605
Chicago/Turabian StyleLarsson, Daniel S. D., Sandesh Kanchugal P, and Maria Selmer. 2022. "Structural Consequences of Deproteinating the 50S Ribosome" Biomolecules 12, no. 11: 1605. https://doi.org/10.3390/biom12111605
APA StyleLarsson, D. S. D., Kanchugal P, S., & Selmer, M. (2022). Structural Consequences of Deproteinating the 50S Ribosome. Biomolecules, 12(11), 1605. https://doi.org/10.3390/biom12111605