
Subtype Differences in the Interaction of HIV-1 Matrix with Calmodulin: Implications for Biological Functions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Protein Expression and Purification
2.2. SPR Direct Interaction Analysis
2.3. In Silico Homology Modeling, Optimization, and Conservation Analysis
3. Results
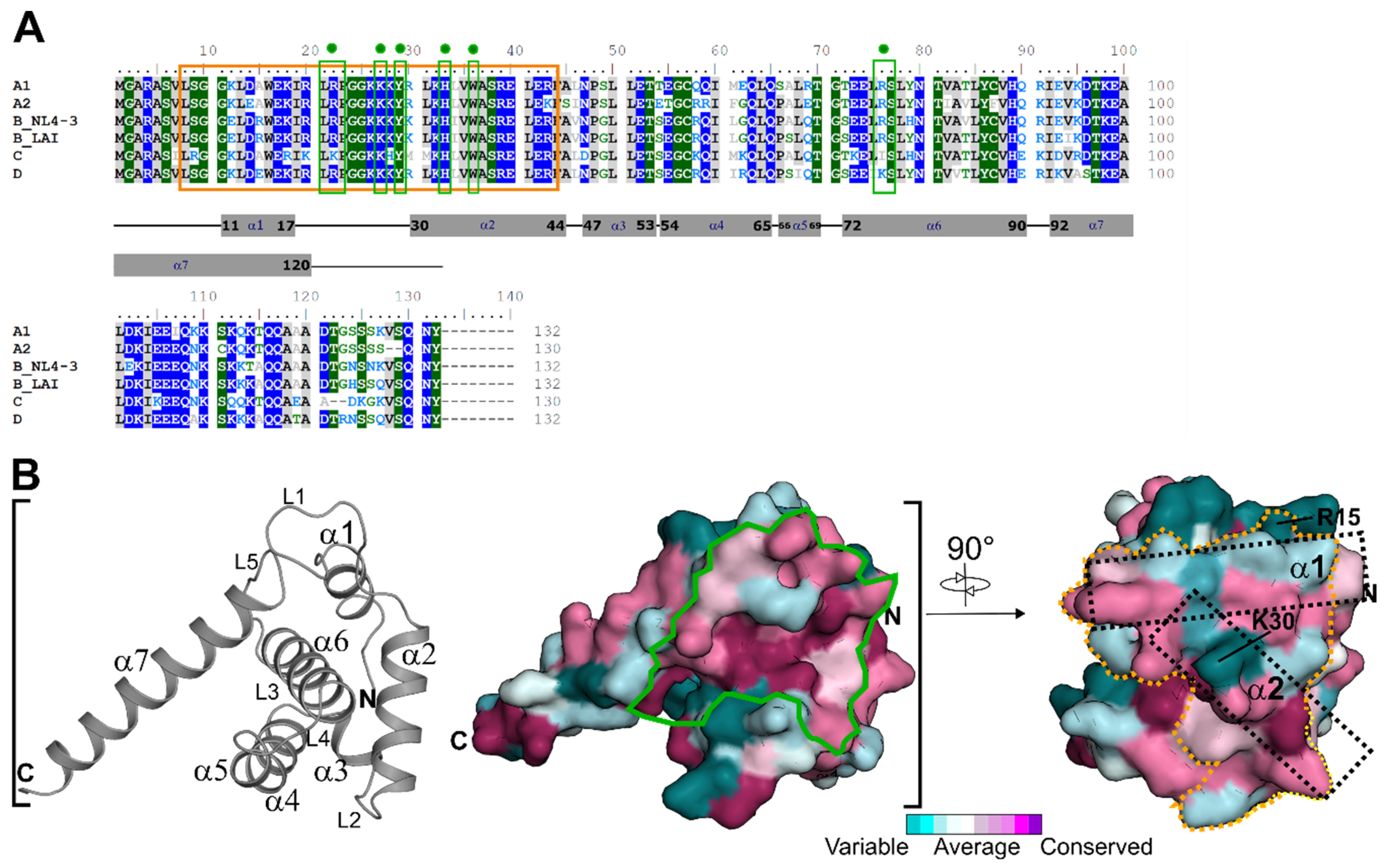
3.1. HIV-1 MA Conservation within the CaM Binding Motif
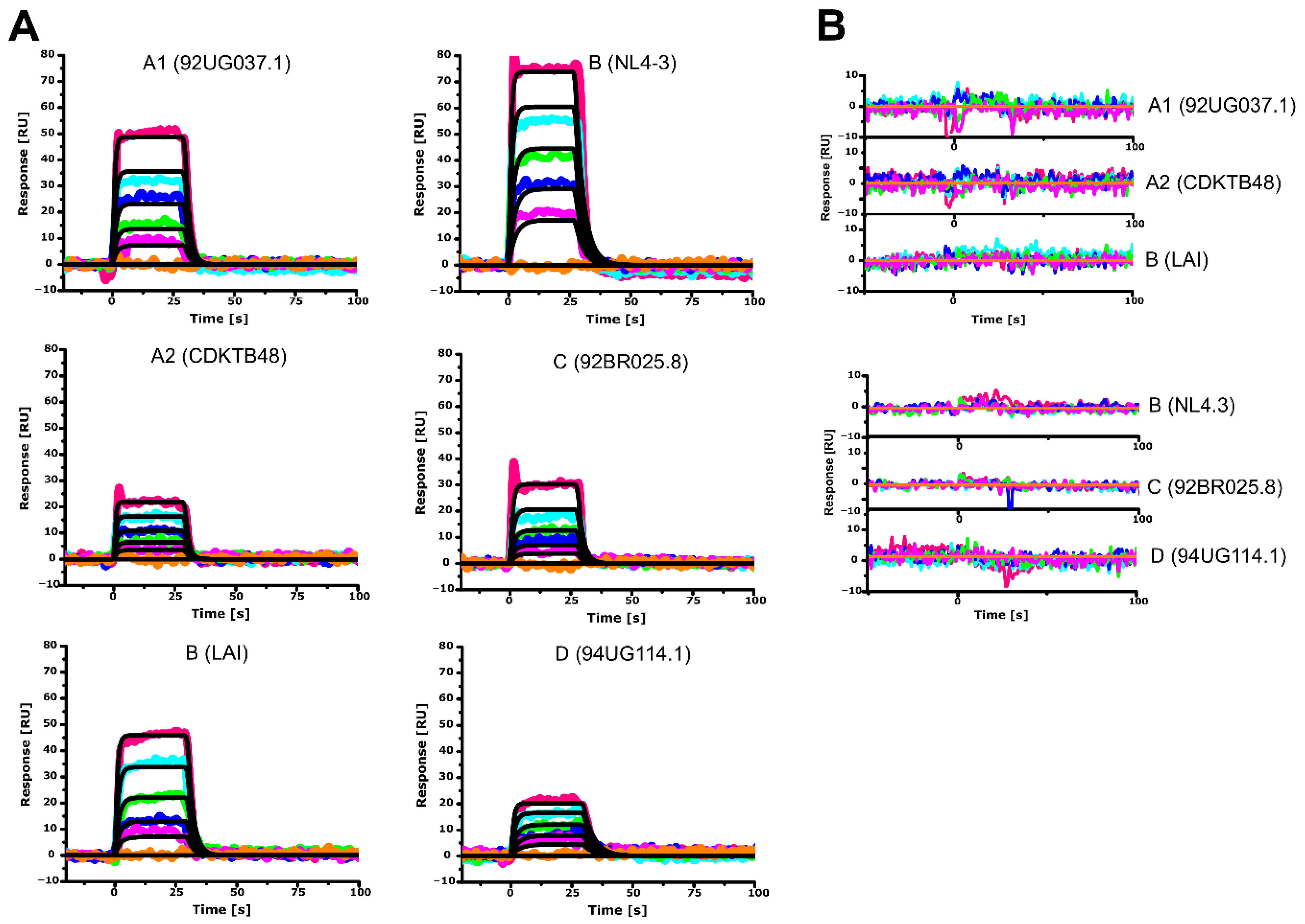
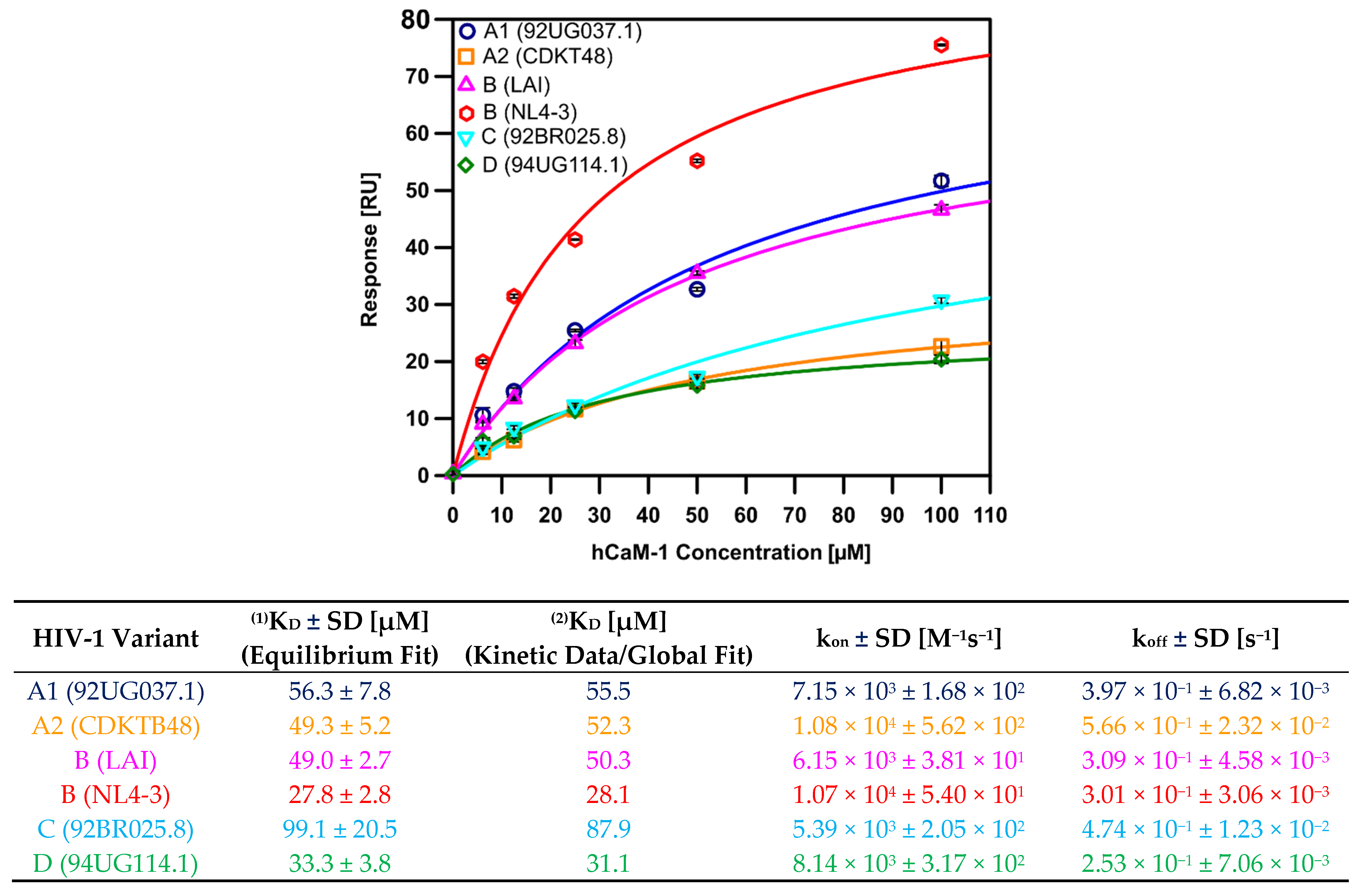
3.2. Differential Recognition of Human CaM-1 by HIV-1 MA Proteins
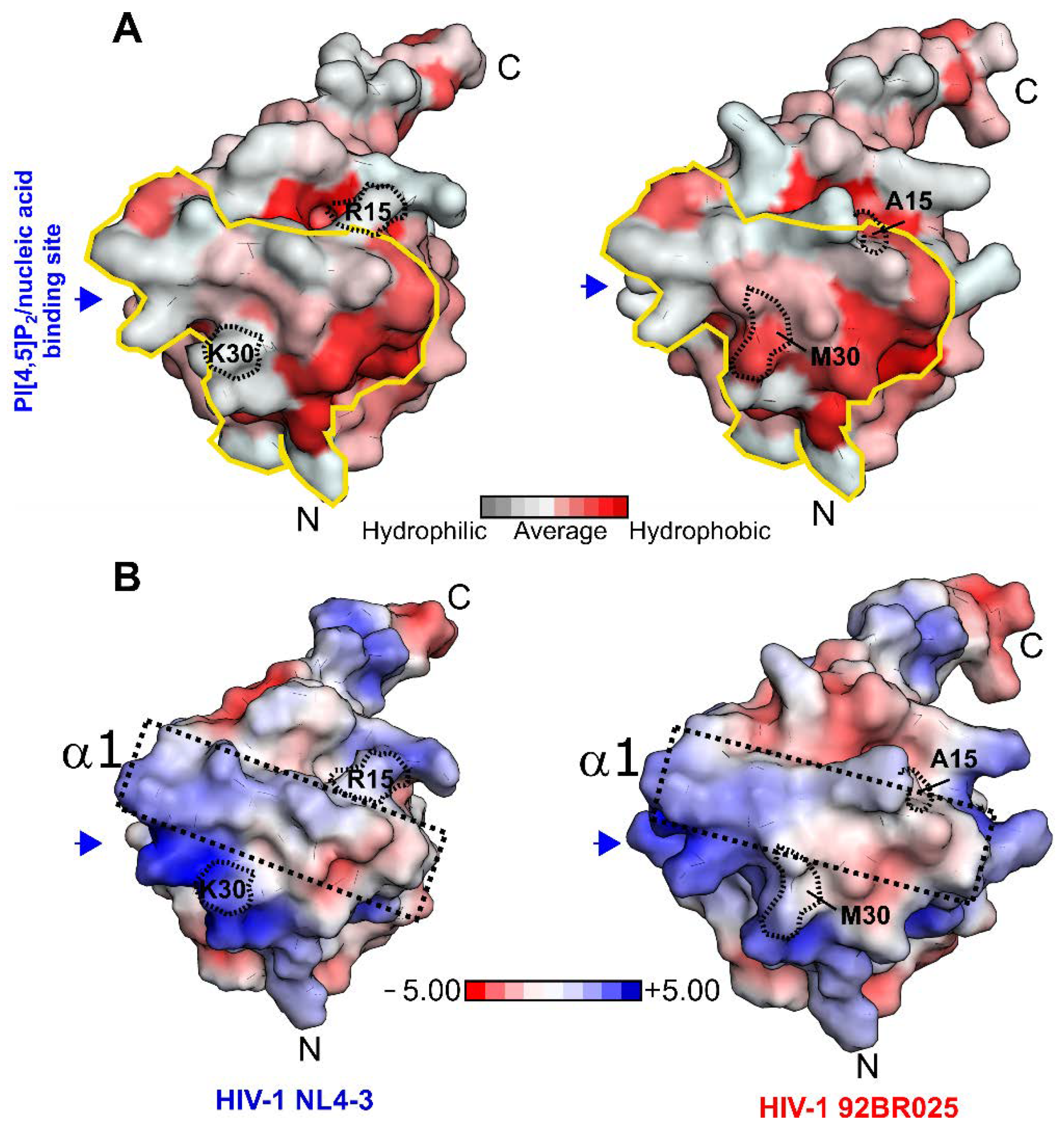
3.3. Structural and Electrostatic Alterations between HIV-1 Subtypes
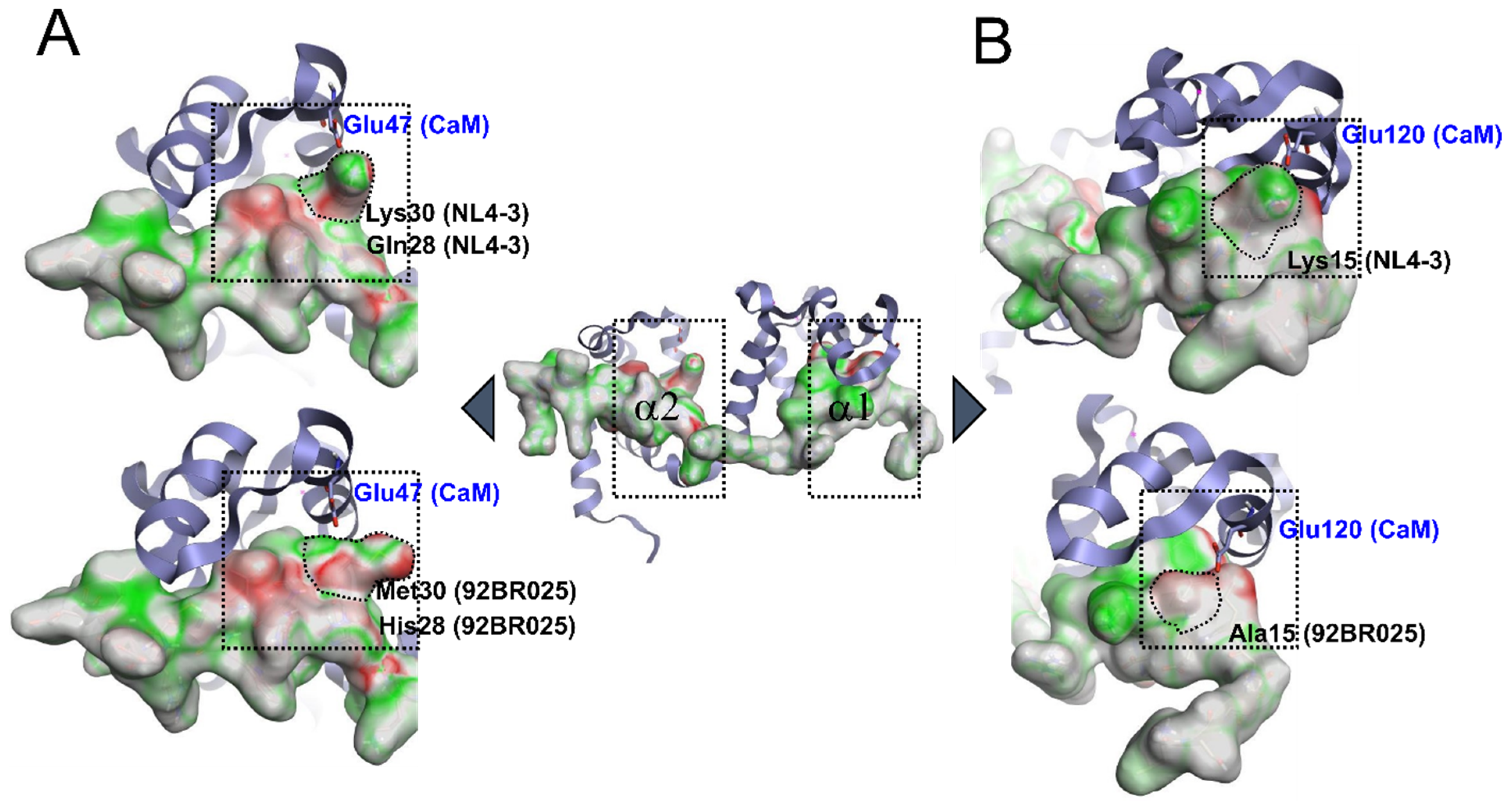
3.4. Electrostatic Complementarity of the HIV-1 MA/rnCaM Complex
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sharp, P.M.; Hahn, B.H. Origins of HIV and the AIDS pandemic. Cold Spring Harb. Perspect. Med. 2011, 1, a006841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UNAIDS. Global HIV & AIDS Statistics—2019 Fact Sheet. Available online: https://www.unaids.org/en/resources/fact-sheet (accessed on 6 May 2021).
- Dick, A.; Cocklin, S. Recent Advances in HIV-1 Gag Inhibitor Design and Development. Molecules 2020, 25, 1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, R.E.; Saad, J.S. The Interplay between HIV-1 Gag Binding to the Plasma Membrane and Env Incorporation. Viruses 2020, 12, 548. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.D.; Musier-Forsyth, K. Retroviral Gag protein-RNA interactions: Implications for specific genomic RNA packaging and virion assembly. Semin. Cell Dev. Biol. 2019, 86, 129–139. [Google Scholar] [CrossRef]
- Ganser-Pornillos, B.K.; Yeager, M.; Sundquist, W.I. The structural biology of HIV assembly. Curr. Opin. Struct. Biol. 2008, 18, 203–217. [Google Scholar] [CrossRef] [Green Version]
- Jouvenet, N.; Neil, S.J.; Bess, C.; Johnson, M.C.; Virgen, C.A.; Simon, S.M.; Bieniasz, P.D. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 2006, 4, e435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saad, J.S.; Miller, J.; Tai, J.; Kim, A.; Ghanam, R.H.; Summers, M.F. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc. Natl. Acad. Sci. USA 2006, 103, 11364–11369. [Google Scholar] [CrossRef] [Green Version]
- Vlach, J.; Saad, J.S. Trio engagement via plasma membrane phospholipids and the myristoyl moiety governs HIV-1 matrix binding to bilayers. Proc. Natl. Acad. Sci. USA 2013, 110, 3525–3530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chukkapalli, V.; Ono, A. Molecular determinants that regulate plasma membrane association of HIV-1 Gag. J. Mol. Biol. 2011, 410, 512–524. [Google Scholar] [CrossRef] [Green Version]
- Chukkapalli, V.; Hogue, I.B.; Boyko, V.; Hu, W.S.; Ono, A. Interaction between the human immunodeficiency virus type 1 Gag matrix domain and phosphatidylinositol-(4,5)-bisphosphate is essential for efficient gag membrane binding. J. Virol. 2008, 82, 2405–2417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chukkapalli, V.; Oh, S.J.; Ono, A. Opposing mechanisms involving RNA and lipids regulate HIV-1 Gag membrane binding through the highly basic region of the matrix domain. Proc. Natl. Acad. Sci. USA 2010, 107, 1600–1605. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.; Garg, H.; Nagashima, K.; Bonifacino, J.S.; Freed, E.O. GGA and Arf proteins modulate retrovirus assembly and release. Mol. Cell 2008, 30, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Batonick, M.; Favre, M.; Boge, M.; Spearman, P.; Honing, S.; Thali, M. Interaction of HIV-1 Gag with the clathrin-associated adaptor AP-2. Virology 2005, 342, 190–200. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Li, H.; Derdowski, A.; Ding, L.; Burnett, A.; Chen, X.; Peters, T.R.; Dermody, T.S.; Woodruff, E.; Wang, J.J.; et al. AP-3 directs the intracellular trafficking of HIV-1 Gag and plays a key role in particle assembly. Cell 2005, 120, 663–674. [Google Scholar] [CrossRef] [Green Version]
- Camus, G.; Segura-Morales, C.; Molle, D.; Lopez-Verges, S.; Begon-Pescia, C.; Cazevieille, C.; Schu, P.; Bertrand, E.; Berlioz-Torrent, C.; Basyuk, E. The clathrin adaptor complex AP-1 binds HIV-1 and MLV Gag and facilitates their budding. Mol. Biol. Cell 2007, 18, 3193–3203. [Google Scholar] [CrossRef]
- Lopez-Verges, S.; Camus, G.; Blot, G.; Beauvoir, R.; Benarous, R.; Berlioz-Torrent, C. Tail-interacting protein TIP47 is a connector between Gag and Env and is required for Env incorporation into HIV-1 virions. Proc. Natl. Acad. Sci. USA 2006, 103, 14947–14952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryo, A.; Tsurutani, N.; Ohba, K.; Kimura, R.; Komano, J.; Nishi, M.; Soeda, H.; Hattori, S.; Perrem, K.; Yamamoto, M.; et al. SOCS1 is an inducible host factor during HIV-1 infection and regulates the intracellular trafficking and stability of HIV-1 Gag. Proc. Natl. Acad. Sci. USA 2008, 105, 294–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strasner, A.B.; Natarajan, M.; Doman, T.; Key, D.; August, A.; Henderson, A.J. The Src kinase Lck facilitates assembly of HIV-1 at the plasma membrane. J. Immunol. 2008, 181, 3706–3713. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.; Garg, H.; Ablan, S.D.; Freed, E.O. Evidence of a role for soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) machinery in HIV-1 assembly and release. J. Biol. Chem. 2011, 286, 29861–29871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, J.; Liu, L.; Woodruff, E.A.; Taylor, H.E.; Goodwin, J.S.; D’Aquila, R.T.; Spearman, P.; Hildreth, J.E.; Dong, X. Filamin A protein interacts with human immunodeficiency virus type 1 Gag protein and contributes to productive particle assembly. J. Biol. Chem. 2011, 286, 28498–28510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomita, Y.; Noda, T.; Fujii, K.; Watanabe, T.; Morikawa, Y.; Kawaoka, Y. The cellular factors Vps18 and Mon2 are required for efficient production of infectious HIV-1 particles. J. Virol. 2011, 85, 5618–5627. [Google Scholar] [CrossRef] [Green Version]
- Engeland, C.E.; Oberwinkler, H.; Schumann, M.; Krause, E.; Muller, G.A.; Krausslich, H.G. The cellular protein lyric interacts with HIV-1 Gag. J. Virol. 2011, 85, 13322–13332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meusser, B.; Purfuerst, B.; Luft, F.C. HIV-1 Gag release from yeast reveals ESCRT interaction with the Gag N-terminal protein region. J. Biol. Chem. 2020, 295, 17950–17972. [Google Scholar] [CrossRef] [PubMed]
- Ono, A.; Ablan, S.D.; Lockett, S.J.; Nagashima, K.; Freed, E.O. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc. Natl. Acad. Sci. USA 2004, 101, 14889–14894. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.S.; Sobieszczyk, M.E.; McCutchan, F.E.; Hammer, S.M. The challenge of HIV-1 subtype diversity. N. Engl. J. Med. 2008, 358, 1590–1602. [Google Scholar] [CrossRef] [Green Version]
- Stefic, K.; Bouvin-Pley, M.; Braibant, M.; Barin, F. Impact of HIV-1 Diversity on Its Sensitivity to Neutralization. Vaccines 2019, 7, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, A.; Hogan, J.W.; Istrail, S.; Delong, A.; Katzenstein, D.A.; Kantor, R. Global analysis of sequence diversity within HIV-1 subtypes across geographic regions. Future Virol. 2012, 7, 505–517. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Piampongsant, S.; Faria, N.R.; Voet, A.; Pineda-Pena, A.C.; Khouri, R.; Lemey, P.; Vandamme, A.M.; Theys, K. An integrated map of HIV genome-wide variation from a population perspective. Retrovirology 2015, 12, 18. [Google Scholar] [CrossRef] [Green Version]
- Weeks, S.D.; Drinker, M.; Loll, P.J. Ligation independent cloning vectors for expression of SUMO fusions. Protein Expr. Purif. 2007, 53, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Studier, F.W. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef]
- Bravman, T.; Bronner, V.; Lavie, K.; Notcovich, A.; Papalia, G.A.; Myszka, D.G. Exploring “one-shot” kinetics and small molecule analysis using the ProteOn XPR36 array biosensor. Anal. Biochem. 2006, 358, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77 (Suppl. S9), 114–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [Green Version]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [Green Version]
- Vlach, J.; Saad, J.S. Structural and molecular determinants of HIV-1 Gag binding to the plasma membrane. Front. Microbiol. 2015, 6, 232. [Google Scholar] [CrossRef] [Green Version]
- Barros, M.; Heinrich, F.; Datta, S.A.K.; Rein, A.; Karageorgos, I.; Nanda, H.; Losche, M. Membrane Binding of HIV-1 Matrix Protein: Dependence on Bilayer Composition and Protein Lipidation. J. Virol. 2016, 90, 4544–4555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercredi, P.Y.; Bucca, N.; Loeliger, B.; Gaines, C.R.; Mehta, M.; Bhargava, P.; Tedbury, P.R.; Charlier, L.; Floquet, N.; Muriaux, D.; et al. Structural and Molecular Determinants of Membrane Binding by the HIV-1 Matrix Protein. J. Mol. Biol. 2016, 428, 1637–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monje-Galvan, V.; Voth, G.A. Binding mechanism of the matrix domain of HIV-1 gag on lipid membranes. Elife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Vlach, J.; Samal, A.B.; Saad, J.S. Solution structure of calmodulin bound to the binding domain of the HIV-1 matrix protein. J. Biol. Chem. 2014, 289, 8697–8705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghanam, R.H.; Fernandez, T.F.; Fledderman, E.L.; Saad, J.S. Binding of calmodulin to the HIV-1 matrix protein triggers myristate exposure. J. Biol. Chem. 2010, 285, 41911–41920. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.E.; Chow, J.Y.; Jeffries, C.M.; Kwan, A.H.; Duff, A.P.; Hamilton, W.A.; Trewhella, J. Calmodulin binds a highly extended HIV-1 MA protein that refolds upon its release. Biophys. J. 2012, 103, 541–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samal, A.B.; Ghanam, R.H.; Fernandez, T.F.; Monroe, E.B.; Saad, J.S. NMR, biophysical, and biochemical studies reveal the minimal Calmodulin binding domain of the HIV-1 matrix protein. J. Biol. Chem. 2011, 286, 33533–33543. [Google Scholar] [CrossRef] [Green Version]
- Chin, D.; Means, A.R. Calmodulin: A prototypical calcium sensor. Trends Cell Biol. 2000, 10, 322–328. [Google Scholar] [CrossRef]
- Eisenberg, D.; Schwarz, E.; Komaromy, M.; Wall, R. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J. Mol. Biol. 1984, 179, 125–142. [Google Scholar] [CrossRef]
- Tang, C.; Loeliger, E.; Luncsford, P.; Kinde, I.; Beckett, D.; Summers, M.F. Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc. Natl. Acad. Sci. USA 2004, 101, 517–522. [Google Scholar] [CrossRef] [Green Version]
- Saad, J.S.; Loeliger, E.; Luncsford, P.; Liriano, M.; Tai, J.; Kim, A.; Miller, J.; Joshi, A.; Freed, E.O.; Summers, M.F. Point mutations in the HIV-1 matrix protein turn off the myristyl switch. J. Mol. Biol. 2007, 366, 574–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakiuchi, S.; Yasuda, S.; Yamazaki, R.; Teshima, Y.; Kanda, K.; Kakiuchi, R.; Sobue, K. Quantitative determinations of calmodulin in the supernatant and particulate fractions of mammalian tissues. J. Biochem. 1982, 92, 1041–1048. [Google Scholar] [CrossRef]
- Black, D.J.; Tran, Q.K.; Persechini, A. Monitoring the total available calmodulin concentration in intact cells over the physiological range in free Ca2+. Cell Calcium 2004, 35, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Chukkapalli, V.; Inlora, J.; Todd, G.C.; Ono, A. Evidence in support of RNA-mediated inhibition of phosphatidylserine-dependent HIV-1 Gag membrane binding in cells. J. Virol. 2013, 87, 7155–7159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfadhli, A.; McNett, H.; Tsagli, S.; Bachinger, H.P.; Peyton, D.H.; Barklis, E. HIV-1 matrix protein binding to RNA. J. Mol. Biol. 2011, 410, 653–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dick, A.; Cocklin, S. Subtype Differences in the Interaction of HIV-1 Matrix with Calmodulin: Implications for Biological Functions. Biomolecules 2021, 11, 1294. https://doi.org/10.3390/biom11091294
Dick A, Cocklin S. Subtype Differences in the Interaction of HIV-1 Matrix with Calmodulin: Implications for Biological Functions. Biomolecules. 2021; 11(9):1294. https://doi.org/10.3390/biom11091294
Chicago/Turabian StyleDick, Alexej, and Simon Cocklin. 2021. "Subtype Differences in the Interaction of HIV-1 Matrix with Calmodulin: Implications for Biological Functions" Biomolecules 11, no. 9: 1294. https://doi.org/10.3390/biom11091294
APA StyleDick, A., & Cocklin, S. (2021). Subtype Differences in the Interaction of HIV-1 Matrix with Calmodulin: Implications for Biological Functions. Biomolecules, 11(9), 1294. https://doi.org/10.3390/biom11091294