
The Mechanism of CD8+ T Cells for Reducing Myofibroblasts Accumulation during Renal Fibrosis

{kind=link}
{kind=link}
Abstract
:1. Introduction
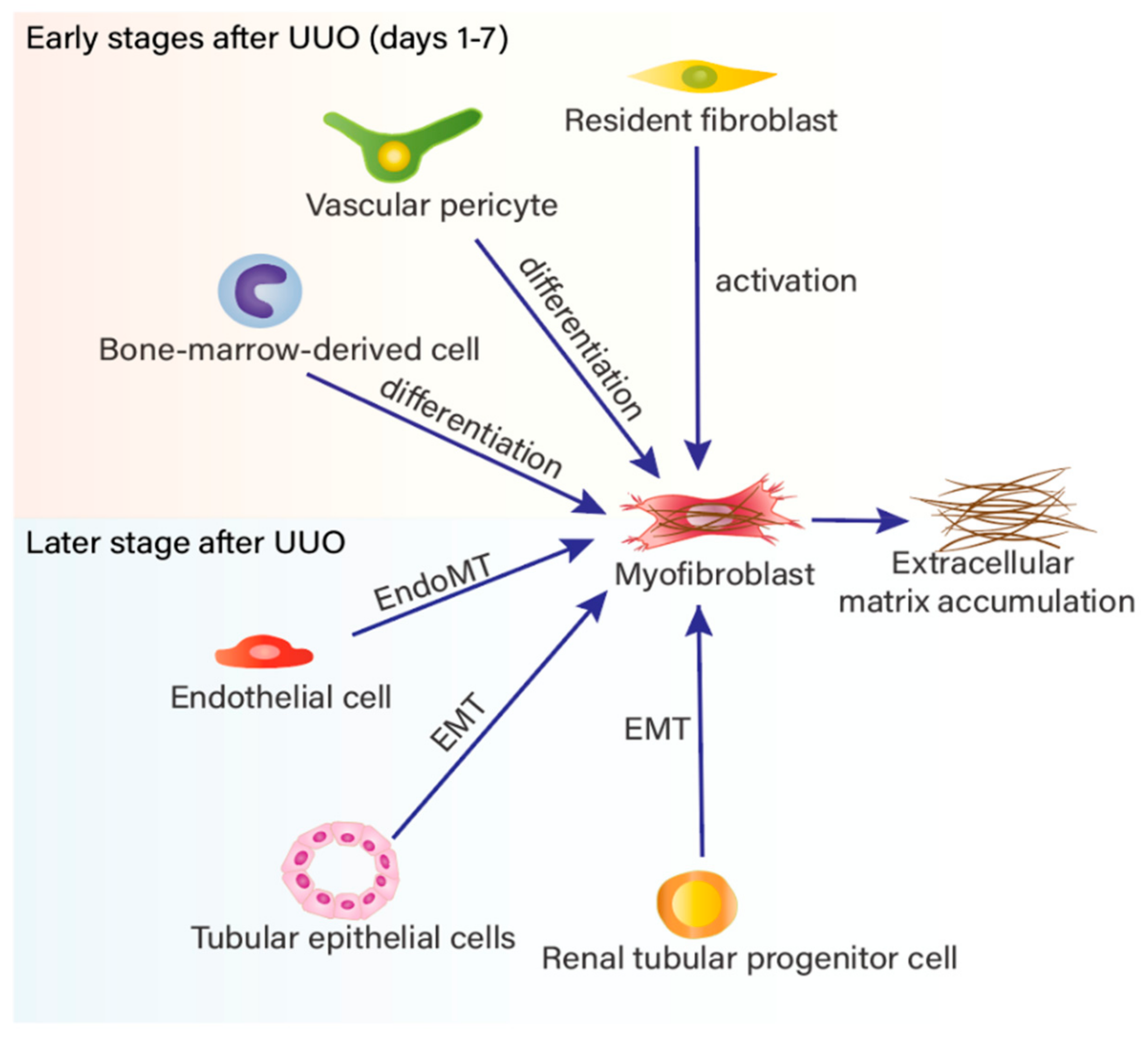
2. Myofibroblast Accumulation in Renal Fibrosis
3. Varied Phenotypes of CD8+ T Cells in Renal Inflammation and Fibrosis
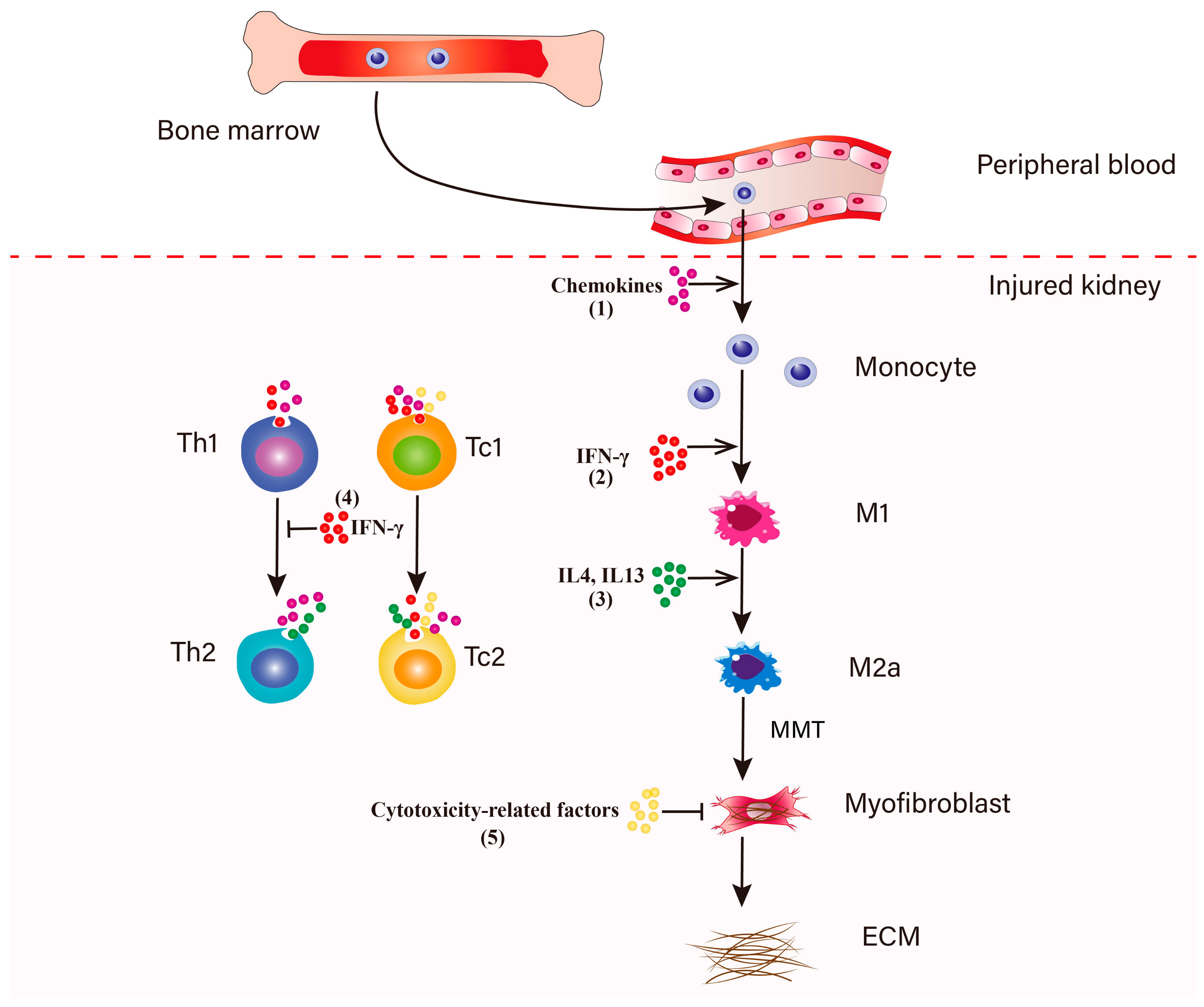
4. The Role of CD8+ T Cells in Renal Fibrosis
5. IFN-γ-Producing CD8+ T Cells in Renal Fibrosis
6. The Functions of CD8+ T Cell Subsets Which Identified by CD44, CD25 and CD62L on Myofibroblasts Accumulation in Renal Fibrosis
7. Cytotoxic Effect of CD8+ T Cells on Fibroblasts
8. Some Limitations of Existing Research and Perspective

9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kurzhagen, J.T.; Dellepiane, S.; Cantaluppi, V.; Rabb, H. AKI: An increasingly recognized risk factor for CKD development and progression. J. Nephrol. 2020, 33, 1171–1187. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, A.C.K.; Lan, H.Y. Chemokines in Renal Injury. J. Am. Soc. Nephrol. 2011, 22, 802–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratliff, B.B.; Rabadi, M.M.; Vasko, R.; Yasuda, K.; Goligorsky, M.S. Messengers without borders: Mediators of systemic inflammatory response in AKI. J. Am. Soc. Nephrol. 2013, 24, 529–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, H.; Ali, S.; McDonnell, B.J.; Burt, A.D.; Kirby, J.A. Chronic renal allograft dysfunction: The role of T cell-mediated tubular epithelial to mesenchymal cell transition. J. Am. Soc. Nephrol. 2004, 15, 390–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitagawa, K.; Wada, T.; Furuichi, K.; Hashimoto, H.; Ishiwata, Y.; Asano, M.; Takeya, M.; Kuziel, W.A.; Matsushima, K.; Mukaida, N.; et al. Blockade of CCR2 ameliorates progressive fibrosis in kidney. Am. J. Pathol. 2004, 165, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Eis, V.; Luckow, B.; Vielhauer, V.; Siveke, J.T.; Linde, Y.; Segerer, S.; de Lema, G.P.; Cohen, C.D.; Kretzler, M.; Mack, M.; et al. Chemokine Receptor CCR1 But Not CCR5 Mediates Leukocyte Recruitment and Subsequent Renal Fibrosis after Unilateral Ureteral Obstruction. J. Am. Soc. Nephrol. 2004, 15, 337–347. [Google Scholar] [CrossRef] [Green Version]
- Anders, H.J.; Vielhauer, V.; Frink, M.; Linde, Y.; Cohen, C.D.; Blattner, S.M.; Kretzler, M.; Strutz, F.; Mack, M.; Gröne, H.J.; et al. A chemokine receptor CCR-1 antagonist reduces renal fibrosis after unilateral ureter ligation. J. Clin. Investig. 2002, 109, 251–259. [Google Scholar] [CrossRef]
- Tapmeier, T.T.; Fearn, A.; Brown, K.; Chowdhury, P.; Sacks, S.H.; Sheerin, N.S.; Wong, W. Pivotal role of CD4+ T cells in renal fibrosis following ureteric obstruction. Kidney Int. 2010, 78, 351–362. [Google Scholar] [CrossRef] [Green Version]
- Niedermeier, M.; Reich, B.; Gomez, M.R.; Denzel, A.; Schmidbauer, K.; Göbel, N.; Talke, Y.; Schweda, F.; Mack, M. CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc. Natl. Acad. Sci. USA 2009, 106, 17892–17897. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Kou, P.; Zeng, Q.; Pei, G.; Li, Y.; Liang, H.; Xu, G.; Chen, S. CD4+ T Lymphocytes, Especially Th2 Cells, Contribute to the Progress of Renal Fibrosis. Am. J. Nephrol. 2012, 36, 386–396. [Google Scholar] [CrossRef]
- Peng, X.; Xiao, Z.; Zhang, J.; Li, Y.; Dong, Y.; Du, J. IL-17A produced by both γδ T and Th17 cells promotes renal fibrosis via RANTES-mediated leukocyte infiltration after renal obstruction. J. Pathol. 2015, 235, 79–89. [Google Scholar] [CrossRef]
- Dong, Y.; Yang, M.; Zhang, J.; Peng, X.; Cheng, J.; Cui, T.; Du, J. Depletion of CD8+ T Cells Exacerbates CD4+ T Cell-Induced Monocyte-to-Fibroblast Transition in Renal Fibrosis. J. Immunol. 2016, 196, 1874–1881. [Google Scholar] [CrossRef] [Green Version]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef]
- Do Valle Duraes, F.; Lafont, A.; Beibel, M.; Martin, K.; Darribat, K.; Cuttat, R.; Waldt, A.; Naumann, U.; Wieczorek, G.; Gaulis, S.; et al. Immune cell landscaping reveals a protective role for regulatory T cells during kidney injury and fibrosis. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Gondek, D.C.; Lu, L.F.; Quezada, S.A.; Sakaguchi, S.; Noelle, R.J. Cutting edge: Contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J. Immunol. 2005, 174, 1783–1786. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Sharma, R.; Gaskin, F.; Fu, S.M.; Ju, S.T. A novel role of IL-2 in organ-specific autoimmune inflammation beyond regulatory T cell checkpoint: Both IL-2 knockout and Fas mutation prolong lifespan of Scurfy mice but by different mechanisms. J. Immunol. 2007, 179, 8035–8041. [Google Scholar] [CrossRef] [Green Version]
- Strauss, L.; Bergmann, C.; Whiteside, T.L. Human circulating CD4+CD25highFoxp3+ regulatory T cells kill autologous CD8+ but not CD4+ responder cells by Fas-mediated apoptosis. J. Immunol. 2009, 182, 1469–1480. [Google Scholar] [CrossRef] [Green Version]
- Kuswanto, W.; Burzyn, D.; Panduro, M.; Wang, K.K.; Jang, Y.C.; Wagers, A.J.; Benoist, C.; Mathis, D. Poor Repair of Skeletal Muscle in Aging Mice Reflects a Defect in Local, Interleukin-33-Dependent Accumulation of Regulatory T Cells. Immunity 2016, 44, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Panduro, M.; Benoist, C.; Mathis, D. T(reg) cells limit IFN-γ production to control macrophage accrual and phenotype during skeletal muscle regeneration. Proc. Natl. Acad. Sci. USA 2018, 115, E2585–E2593. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Dwyer, G.K.; Zhao, Y.; Li, H.; Mathews, L.R.; Chakka, A.B.; Chandran, U.R.; Demetris, J.A.; Alcorn, J.F.; Robinson, K.M.; et al. IL-33-mediated IL-13 secretion by ST2+ Tregs controls inflammation after lung injury. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.L.; Kisseleva, T.; Brenner, D.A.; Duffield, J.S. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am. J. Pathol. 2008, 173, 1617–1627. [Google Scholar] [CrossRef] [Green Version]
- Hinz, B. Formation and Function of the Myofibroblast during Tissue Repair. J. Investig. Dermatol. 2007, 127, 526–537. [Google Scholar] [CrossRef]
- McAnulty, R.J. Fibroblasts and myofibroblasts: Their source, function and role in disease. Int. J. Biochem. Cell Biol. 2007, 39, 666–671. [Google Scholar] [CrossRef]
- Grgic, I.; Duffield, J.S.; Humphreys, B.D. The origin of interstitial myofibroblasts in chronic kidney disease. Pediatr. Nephrol. 2012, 27, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Yanagita, M. Resident fibroblasts in the kidney: A major driver of fibrosis and inflammation. Inflamm. Regen. 2017, 37, 17. [Google Scholar] [CrossRef]
- Wu, C.F.; Chiang, W.C.; Lai, C.F.; Chang, F.C.; Chen, Y.T.; Chou, Y.H.; Wu, T.H.; Linn, G.R.; Ling, H.; Wu, K.D.; et al. Transforming growth factor β-1 stimulates profibrotic epithelial signaling to activate pericyte-myofibroblast transition in obstructive kidney fibrosis. Am. J. Pathol. 2013, 182, 118–131. [Google Scholar] [CrossRef] [Green Version]
- Humphreys, B.D.; Lin, S.L.; Kobayashi, A.; Hudson, T.E.; Nowlin, B.T.; Bonventre, J.V.; Valerius, M.T.; McMahon, A.P.; Duffield, J.S. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 2010, 176, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Jinde, K.; Nikolic-Paterson, D.J.; Huang, X.R.; Sakai, H.; Kurokawa, K.; Atkins, R.C.; Lan, H.Y. Tubular phenotypic change in progressive tubulointerstitial fibrosis in human glomerulonephritis. Am. J. Kidney Dis. 2001, 38, 761–769. [Google Scholar] [CrossRef]
- Ng, Y.Y.; Huang, T.P.; Yang, W.C.; Chen, Z.P.; Yang, A.H.; Mu, W.; Nikolic-Paterson, D.J.; Atkins, R.C.; Lan, H.Y. Tubular epithelial-myofibroblast transdifferentiation in progressive tubulointerstitial fibrosis in 5/6 nephrectomized rats. Kidney Int. 1998, 54, 864–876. [Google Scholar] [CrossRef] [Green Version]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Potenta, S.E.; Sugimoto, H.; Zeisberg, M.; Kalluri, R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J. Am. Soc. Nephrol. 2008, 19, 2282–2287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Qu, X.; Bertram, J.F. Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am. J. Pathol. 2009, 175, 1380–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, P.M.-K.; Nikolic-Paterson, D.J.; Lan, H.-Y. Macrophages: Versatile players in renal inflammation and fibrosis. Nat. Rev. Nephrol. 2019, 15, 144–158. [Google Scholar] [CrossRef]
- Bucala, R.; Spiegel, L.A.; Chesney, J.; Hogan, M.; Cerami, A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol. Med. 1994, 1, 71–81. [Google Scholar] [CrossRef] [Green Version]
- Hong, K.M.; Belperio, J.A.; Keane, M.P.; Burdick, M.D.; Strieter, R.M. Differentiation of human circulating fibrocytes as mediated by transforming growth factor-beta and peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 2007, 282, 22910–22920. [Google Scholar] [CrossRef] [Green Version]
- Reich, B.; Schmidbauer, K.; Rodriguez Gomez, M.; Johannes Hermann, F.; Göbel, N.; Brühl, H.; Ketelsen, I.; Talke, Y.; Mack, M. Fibrocytes develop outside the kidney but contribute to renal fibrosis in a mouse model. Kidney Int. 2013, 84, 78–89. [Google Scholar] [CrossRef] [Green Version]
- Nikolic-Paterson, D.J.; Wang, S.; Lan, H.Y. Macrophages promote renal fibrosis through direct and indirect mechanisms. Kidney Int. 2014, 4, 34–38. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.M.; Wang, S.; Huang, X.R.; Yang, C.; Xiao, J.; Zhang, Y.; To, K.F.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory macrophages can transdifferentiate into myofibroblasts during renal fibrosis. Cell Death Dis. 2016, 7, e2495. [Google Scholar] [CrossRef]
- Meng, X.M.; Mak, T.S.; Lan, H.Y. Macrophages in Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 285–303. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef]
- Kramann, R.; Schneider, R.K.; DiRocco, D.P.; Machado, F.; Fleig, S.; Bondzie, P.A.; Henderson, J.M.; Ebert, B.L.; Humphreys, B.D. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 2015, 16, 51–66. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Meng, X.M.; Ng, Y.Y.; Ma, F.Y.; Zhou, S.; Zhang, Y.; Yang, C.; Huang, X.R.; Xiao, J.; Wang, Y.Y.; et al. TGF-β/Smad3 signalling regulates the transition of bone marrow-derived macrophages into myofibroblasts during tissue fibrosis. Oncotarget 2016, 7, 8809–8822. [Google Scholar] [CrossRef] [Green Version]
- Kaissling, B.; Le Hir, M. The renal cortical interstitium: Morphological and functional aspects. Histochem. Cell Biol. 2008, 130, 247–262. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Q.; Ma, R.; Yin, Y.; Lan, T.; Yu, M.; Ming, Y. Mesenchymal Stem Cells in Renal Fibrosis: The Flame of Cytotherapy. Stem Cells Int. 2019, 2019, 8387350. [Google Scholar] [CrossRef] [Green Version]
- Herzog, E.L.; Bucala, R. Fibrocytes in health and disease. Exp. Hematol. 2010, 38, 548–556. [Google Scholar] [CrossRef] [Green Version]
- Schnaper, H.W.; Jandeska, S.; Runyan, C.E.; Hubchak, S.C.; Basu, R.K.; Curley, J.F.; Smith, R.D.; Hayashida, T. TGF-beta signal transduction in chronic kidney disease. Front. Biosci. 2009, 14, 2448–2465. [Google Scholar] [CrossRef] [Green Version]
- Boor, P.; Floege, J. Chronic kidney disease growth factors in renal fibrosis. Clin. Exp. Pharm. Physiol. 2011, 38, 441–450. [Google Scholar] [CrossRef]
- Yang, J.; Dai, C.; Liu, Y. Hepatocyte growth factor suppresses renal interstitial myofibroblast activation and intercepts Smad signal transduction. Am. J. Pathol. 2003, 163, 621–632. [Google Scholar] [CrossRef] [Green Version]
- Dai, C.; Liu, Y. Hepatocyte growth factor antagonizes the profibrotic action of TGF-beta1 in mesangial cells by stabilizing Smad transcriptional corepressor TGIF. J. Am. Soc. Nephrol. 2004, 15, 1402–1412. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y. Hepatocyte growth factor in kidney fibrosis: Therapeutic potential and mechanisms of action. Am. J. Physiol. Ren. Physiol. 2004, 287, F7–F16. [Google Scholar] [CrossRef] [Green Version]
- Luo, D.D.; Phillips, A.; Fraser, D. Bone morphogenetic protein-7 inhibits proximal tubular epithelial cell Smad3 signaling via increased SnoN expression. Am. J. Pathol. 2010, 176, 1139–1147. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, M.; Hanai, J.; Sugimoto, H.; Mammoto, T.; Charytan, D.; Strutz, F.; Kalluri, R. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003, 9, 964–968. [Google Scholar] [CrossRef]
- Wang, J.; Tian, J.; Sun, J.; Gao, M.; Dong, Y. Two identified subsets of CD8 T cells in obstructed kidneys play different roles in inflammation and fibrosis. Aging 2020, 12, 17528–17540. [Google Scholar] [CrossRef]
- Savinko, T.; Matikainen, S.; Saarialho-Kere, U.; Lehto, M.; Wang, G.; Lehtimaki, S.; Karisola, P.; Reunala, T.; Wolff, H.; Lauerma, A.; et al. IL-33 and ST2 in atopic dermatitis: Expression profiles and modulation by triggering factors. J. Investig. Derm. 2012, 132, 1392–1400. [Google Scholar] [CrossRef] [Green Version]
- Gharaee-Kermani, M.; Kasina, S.; Moore, B.B.; Thomas, D.; Mehra, R.; Macoska, J.A. CXC-type chemokines promote myofibroblast phenoconversion and prostatic fibrosis. PLoS ONE 2012, 7, e49278. [Google Scholar] [CrossRef] [Green Version]
- Scotton, C.J.; Chambers, R.C. Molecular targets in pulmonary fibrosis: The myofibroblast in focus. Chest 2007, 132, 1311–1321. [Google Scholar] [CrossRef]
- Kurts, C.; Panzer, U.; Anders, H.J.; Rees, A.J. The immune system and kidney disease: Basic concepts and clinical implications. Nat. Rev. Immunol. 2013, 13, 738–753. [Google Scholar] [CrossRef]
- Peng, X.; Zhang, J.; Xiao, Z.; Dong, Y.; Du, J. CX3CL1-CX3CR1 Interaction Increases the Population of Ly6C(-)CX3CR1(hi) Macrophages Contributing to Unilateral Ureteral Obstruction-Induced Fibrosis. J. Immunol. 2015, 195, 2797–2805. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Bai, Y.; Li, J.; Li, L.; Dong, Y. CD11c+ CD8+ T Cells Reduce Renal Fibrosis Following Ureteric Obstruction by Inducing Fibroblast Apoptosis. Int. J. Mol. Sci. 2017, 18, 1. [Google Scholar] [CrossRef] [Green Version]
- Jose, M.D.; Le Meur, Y.; Atkins, R.C.; Chadban, S.J. Blockade of macrophage colony-stimulating factor reduces macrophage proliferation and accumulation in renal allograft rejection. Am. J. Transpl. 2003, 3, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Le Meur, Y.; Tesch, G.H.; Hill, P.A.; Mu, W.; Foti, R.; Nikolic-Paterson, D.J.; Atkins, R.C. Macrophage accumulation at a site of renal inflammation is dependent on the M-CSF/c-fms pathway. J. Leukoc. Biol. 2002, 72, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.K.; Ma, F.Y.; Nikolic-Paterson, D.J.; Thomas, M.C.; Hurst, L.A.; Tesch, G.H. Antibody blockade of c-fms suppresses the progression of inflammation and injury in early diabetic nephropathy in obese db/db mice. Diabetologia 2009, 52, 1669–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, F.Y.; Ikezumi, Y.; Nikolic-Paterson, D.J. Macrophage signaling pathways: A novel target in renal disease. Semin. Nephrol. 2010, 30, 334–344. [Google Scholar] [CrossRef]
- Ricardo, S.D.; van Goor, H.; Eddy, A.A. Macrophage diversity in renal injury and repair. J. Clin. Investig. 2008, 118, 3522–3530. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-Y.; Jiang, H.; Pan, J.; Huang, X.-R.; Wang, Y.-C.; Huang, H.-F.; To, K.-F.; Nikolic-Paterson, D.J.; Lan, H.-Y.; Chen, J.-H. Macrophage-to-Myofibroblast Transition Contributes to Interstitial Fibrosis in Chronic Renal Allograft Injury. J. Am. Soc. Nephrol. 2017, 28, 2053–2067. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.; Zhang, H.; Wang, C.; Li, H.; Zhang, Q.; Bai, J. M2A and M2C Macrophage Subsets Ameliorate Inflammation and Fibroproliferation in Acute Lung Injury Through Interleukin 10 Pathway. Shock 2017, 48, 119–129. [Google Scholar] [CrossRef]
- Zhang, M.Z.; Wang, X.; Wang, Y.; Niu, A.; Wang, S.; Zou, C.; Harris, R.C. IL-4/IL-13-mediated polarization of renal macrophages/dendritic cells to an M2a phenotype is essential for recovery from acute kidney injury. Kidney Int. 2017, 91, 375–386. [Google Scholar] [CrossRef] [Green Version]
- Lv, W.; Booz, G.W.; Wang, Y.; Fan, F.; Roman, R.J. Inflammation and renal fibrosis: Recent developments on key signaling molecules as potential therapeutic targets. Eur. J. Pharmacol. 2018, 820, 65–76. [Google Scholar] [CrossRef]
- Strutz, F.; Okada, H.; Lo, C.W.; Danoff, T.; Carone, R.L.; Tomaszewski, J.E.; Neilson, E.G. Identification and characterization of a fibroblast marker: FSP1. J. Cell Biol. 1995, 130, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.M.; Ng, Y.Y.; Hill, P.A.; Nikolic-Paterson, D.J.; Mu, W.; Atkins, R.C.; Lan, H.Y. Transforming growth factor-beta regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int. 1999, 56, 1455–1467. [Google Scholar] [CrossRef] [Green Version]
- Sui, H.; Zhu, L.; Deng, W.; Li, Q. Epithelial-mesenchymal transition and drug resistance: Role, molecular mechanisms, and therapeutic strategies. Oncol. Res. Treat. 2014, 37, 584–589. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Piera-Velazquez, S.; Li, Z.; Jimenez, S.A. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 2011, 179, 1074–1080. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Mendoza, F.A.; Jimenez, S.A. Endothelial to Mesenchymal Transition (EndoMT) in the Pathogenesis of Human Fibrotic Diseases. J. Clin. Med. 2016, 5, 45. [Google Scholar] [CrossRef]
- Xavier, S.; Vasko, R.; Matsumoto, K.; Zullo, J.A.; Chen, R.; Maizel, J.; Chander, P.N.; Goligorsky, M.S. Curtailing endothelial TGF-β signaling is sufficient to reduce endothelial-mesenchymal transition and fibrosis in CKD. J. Am. Soc. Nephrol. 2015, 26, 817–829. [Google Scholar] [CrossRef] [Green Version]
- Tang, P.M.; Zhou, S.; Li, C.J.; Liao, J.; Xiao, J.; Wang, Q.M.; Lian, G.Y.; Li, J.; Huang, X.R.; To, K.F.; et al. The proto-oncogene tyrosine protein kinase Src is essential for macrophage-myofibroblast transition during renal scarring. Kidney Int. 2018, 93, 173–187. [Google Scholar] [CrossRef] [Green Version]
- Floege, J.; Eitner, F.; Alpers, C.E. A new look at platelet-derived growth factor in renal disease. J. Am. Soc. Nephrol. 2008, 19, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Wynes, M.W.; Frankel, S.K.; Riches, D.W. IL-4-induced macrophage-derived IGF-I protects myofibroblasts from apoptosis following growth factor withdrawal. J. Leukoc. Biol. 2004, 76, 1019–1027. [Google Scholar] [CrossRef] [Green Version]
- Diao, W.; Chen, W.; Cao, W.; Yuan, H.; Ji, H.; Wang, T.; Chen, W.; Zhu, X.; Zhou, H.; Guo, H.; et al. Astaxanthin protects against renal fibrosis through inhibiting myofibroblast activation and promoting CD8(+) T cell recruitment. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1360–1370. [Google Scholar] [CrossRef]
- Honjo, T.; Chyu, K.Y.; Dimayuga, P.C.; Lio, W.M.; Yano, J.; Trinidad, P.; Zhao, X.; Zhou, J.; Cercek, B.; Shah, P.K. Immunization with an ApoB-100 Related Peptide Vaccine Attenuates Angiotensin-II Induced Hypertension and Renal Fibrosis in Mice. PLoS ONE 2015, 10, e0131731. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.D.; Suresh, R.; Vakil, V.; Gomer, R.H.; Pilling, D. Pivotal Advance: Th-1 cytokines inhibit, and Th-2 cytokines promote fibrocyte differentiation. J. Leukoc. Biol. 2008, 83, 1323–1333. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Zhang, Z.; Yan, J.; Wang, Y.; Hu, Z.; Mitch, W.E.; Wang, Y. The IL-4 receptor α has a critical role in bone marrow–derived fibroblast activation and renal fibrosis. Kidney Int. 2017, 92, 1433–1443. [Google Scholar] [CrossRef]
- Srikiatkhachorn, A.; Braciale, T.J. Virus-specific CD8+ T lymphocytes downregulate T helper cell type 2 cytokine secretion and pulmonary eosinophilia during experimental murine respiratory syncytial virus infection. J. Exp. Med. 1997, 186, 421–432. [Google Scholar] [CrossRef]
- Das, G.; Sheridan, S.; Janeway, C.A., Jr. The source of early IFN-gamma that plays a role in Th1 priming. J. Immunol. 2001, 167, 2004–2010. [Google Scholar] [CrossRef] [Green Version]
- Uzonna, J.E.; Joyce, K.L.; Scott, P. Low dose Leishmania major promotes a transient T helper cell type 2 response that is down-regulated by interferon gamma-producing CD8+ T cells. J. Exp. Med. 2004, 199, 1559–1566. [Google Scholar] [CrossRef]
- Oldroyd, S.D.; Thomas, G.L.; Gabbiani, G.; El Nahas, A.M. Interferon-γ inhibits experimental renal fibrosis. Kidney Int. 1999, 56, 2116–2127. [Google Scholar] [CrossRef] [Green Version]
- Poosti, F.; Bansal, R.; Yazdani, S.; Prakash, J.; Beljaars, L.; van den Born, J.; de Borst, M.H.; van Goor, H.; Hillebrands, J.-L.; Poelstra, K. Interferon gamma peptidomimetic targeted to interstitial myofibroblasts attenuates renal fibrosis after unilateral ureteral obstruction in mice. Oncotarget 2016, 7, 54240–54252. [Google Scholar] [CrossRef] [Green Version]
- Kanai, R.; Nakashima, A.; Doi, S.; Kimura, T.; Yoshida, K.; Maeda, S.; Ishiuchi, N.; Yamada, Y.; Ike, T.; Doi, T.; et al. Interferon-γ enhances the therapeutic effect of mesenchymal stem cells on experimental renal fibrosis. Sci. Rep. 2021, 11, 850. [Google Scholar] [CrossRef]
- Teixeira, L.K.; Fonseca, B.P.; Vieira-de-Abreu, A.; Barboza, B.A.; Robbs, B.K.; Bozza, P.T.; Viola, J.P. IFN-gamma production by CD8+ T cells depends on NFAT1 transcription factor and regulates Th differentiation. J. Immunol. 2005, 175, 5931–5939. [Google Scholar] [CrossRef] [Green Version]
- Pernis, A.; Gupta, S.; Gollob, K.J.; Garfein, E.; Coffman, R.L.; Schindler, C.; Rothman, P. Lack of interferon gamma receptor beta chain and the prevention of interferon gamma signaling in TH1 cells. Science 1995, 269, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Bach, E.A.; Szabo, S.J.; Dighe, A.S.; Ashkenazi, A.; Aguet, M.; Murphy, K.M.; Schreiber, R.D. Ligand-induced autoregulation of IFN-gamma receptor beta chain expression in T helper cell subsets. Science 1995, 270, 1215–1218. [Google Scholar] [CrossRef] [PubMed]
- Poosti, F.; Bansal, R.; Yazdani, S.; Prakash, J.; Post, E.; Klok, P.; van den Born, J.; de Borst, M.H.; van Goor, H.; Poelstra, K.; et al. Selective delivery of IFN-γ to renal interstitial myofibroblasts: A novel strategy for the treatment of renal fibrosis. FASEB J. 2015, 29, 1029–1042. [Google Scholar] [CrossRef] [PubMed]
- Mittrücker, H.-W.; Visekruna, A.; Huber, M. Heterogeneity in the Differentiation and Function of CD8+ T Cells. Arch. Immunol. Et. Ther. Exp. 2014, 62, 449–458. [Google Scholar] [CrossRef]
- Salgame, P.; Abrams, J.S.; Clayberger, C.; Goldstein, H.; Convit, J.; Modlin, R.L.; Bloom, B.R. Differing lymphokine profiles of functional subsets of human CD4 and CD8 T cell clones. Science 1991, 254, 279–282. [Google Scholar] [CrossRef]
- Maggi, E.; Giudizi, M.G.; Biagiotti, R.; Annunziato, F.; Manetti, R.; Piccinni, M.P.; Parronchi, P.; Sampognaro, S.; Giannarini, L.; Zuccati, G.; et al. Th2-like CD8+ T cells showing B cell helper function and reduced cytolytic activity in human immunodeficiency virus type 1 infection. J. Exp. Med. 1994, 180, 489–495. [Google Scholar] [CrossRef]
- Maggi, E.; Manetti, R.; Annunziato, F.; Cosmi, L.; Giudizi, M.G.; Biagiotti, R.; Galli, G.; Zuccati, G.; Romagnani, S. Functional characterization and modulation of cytokine production by CD8+ T cells from human immunodeficiency virus-infected individuals. Blood 1997, 89, 3672–3681. [Google Scholar] [CrossRef]
- Vukmanovic-Stejic, M.; Vyas, B.; Gorak-Stolinska, P.; Noble, A.; Kemeny, D.M. Human Tc1 and Tc2/Tc0 CD8 T-cell clones display distinct cell surface and functional phenotypes. Blood 2000, 95, 231–240. [Google Scholar] [CrossRef]
- Dobrzanski, M.J.; Reome, J.B.; Hollenbaugh, J.A.; Dutton, R.W. Tc1 and Tc2 effector cell therapy elicit long-term tumor immunity by contrasting mechanisms that result in complementary endogenous type 1 antitumor responses. J. Immunol. 2004, 172, 1380–1390. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Cao, Q.; Zheng, D.; Sun, Y.; Wang, C.; Yu, X.; Wang, Y.; Lee, V.W.S.; Zheng, G.; Tan, T.K.; et al. Discrete functions of M2a and M2c macrophage subsets determine their relative efficacy in treating chronic kidney disease. Kidney Int. 2013, 84, 745–755. [Google Scholar] [CrossRef] [Green Version]
- Tau, G.Z.; Cowan, S.N.; Weisburg, J.; Braunstein, N.S.; Rothman, P.B. Regulation of IFN-gamma signaling is essential for the cytotoxic activity of CD8(+) T cells. J. Immunol. 2001, 167, 5574–5582. [Google Scholar] [CrossRef] [Green Version]
- Breuer, D.A.; Pacheco, M.C.; Washington, M.K.; Montgomery, S.A.; Hasty, A.H.; Kennedy, A.J. CD8+ T cells regulate liver injury in obesity-related nonalcoholic fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G211–G224. [Google Scholar] [CrossRef]
- Ayano, M.; Tsukamoto, H.; Kohno, K.; Ueda, N.; Tanaka, A.; Mitoma, H.; Akahoshi, M.; Arinobu, Y.; Niiro, H.; Horiuchi, T.; et al. Increased CD226 Expression on CD8+ T Cells Is Associated with Upregulated Cytokine Production and Endothelial Cell Injury in Patients with Systemic Sclerosis. J. Immunol. 2015, 195, 892–900. [Google Scholar] [CrossRef] [Green Version]
- Drakopanagiotakis, F.; Xifteri, A.; Polychronopoulos, V.; Bouros, D. Apoptosis in lung injury and fibrosis. Eur. Respir. J. 2008, 32, 1631–1638. [Google Scholar] [CrossRef] [Green Version]
- Davis, P.A.; Corless, D.J.; Aspinall, R.; Wastell, C. Effect of CD4(+) and CD8(+) cell depletion on wound healing. Br. J. Surg. 2001, 88, 298–304. [Google Scholar] [CrossRef]
- Iwahori, K. Cytotoxic CD8(+) Lymphocytes in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1224, 53–62. [Google Scholar] [CrossRef]
- Chen, Z.; Han, Y.; Gu, Y.; Liu, Y.; Jiang, Z.; Zhang, M.; Cao, X. CD11c(high)CD8+ regulatory T cell feedback inhibits CD4 T cell immune response via Fas ligand-Fas pathway. J. Immunol. 2013, 190, 6145–6154. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.; Wang, C.T.; Ma, T.T.; Li, Z.Y.; Zhou, L.N.; Mu, B.; Leng, F.; Shi, H.S.; Li, Y.O.; Wei, Y.Q. Immunotherapy targeting fibroblast activation protein inhibits tumor growth and increases survival in a murine colon cancer model. Cancer Sci. 2010, 101, 2325–2332. [Google Scholar] [CrossRef]
- De Palma, R.; D’Aiuto, E.; Vettori, S.; Cuoppolo, P.; Abbate, G.; Valentini, G. Peripheral T cells from patients with early systemic sclerosis kill autologous fibroblasts in co-culture: Is T-cell response aimed to play a protective role? Rheumatology 2010, 49, 1257–1266. [Google Scholar] [CrossRef] [Green Version]
- Hammerich, L.; Bangen, J.M.; Govaere, O.; Zimmermann, H.W.; Gassler, N.; Huss, S.; Liedtke, C.; Prinz, I.; Lira, S.A.; Luedde, T.; et al. Chemokine receptor CCR6-dependent accumulation of γδ T cells in injured liver restricts hepatic inflammation and fibrosis. Hepatology 2014, 59, 630–642. [Google Scholar] [CrossRef] [Green Version]
- Golstein, P.; Griffiths, G.M. An early history of T cell-mediated cytotoxicity. Nat. Rev. Immunol. 2018, 18, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Voskoboinik, I.; Whisstock, J.C.; Trapani, J.A. Perforin and granzymes: Function, dysfunction and human pathology. Nat. Rev. Immunol. 2015, 15, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Pribila, J.T.; Quale, A.C.; Mueller, K.L.; Shimizu, Y. Integrins and T cell-mediated immunity. Annu. Rev. Immunol. 2004, 22, 157–180. [Google Scholar] [CrossRef] [PubMed]
- Keizer, G.D.; Borst, J.; Visser, W.; Schwarting, R.; de Vries, J.E.; Figdor, C.G. Membrane glycoprotein p150,95 of human cytotoxic T cell clone is involved in conjugate formation with target cells. J. Immunol. 1987, 138, 3130–3136. [Google Scholar]
- Beyer, M.; Wang, H.; Peters, N.; Doths, S.; Koerner-Rettberg, C.; Openshaw, P.J.; Schwarze, J. The beta2 integrin CD11c distinguishes a subset of cytotoxic pulmonary T cells with potent antiviral effects in vitro and in vivo. Respir. Res. 2005, 6, 70. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.-R.; Byeon, Y.; Kim, D.; Park, S.-G. Recent insights of T cell receptor-mediated signaling pathways for T cell activation and development. Exp. Mol. Med. 2020, 52, 750–761. [Google Scholar] [CrossRef]
- Scheper, W.; Kelderman, S.; Fanchi, L.F.; Linnemann, C.; Bendle, G.; de Rooij, M.A.J.; Hirt, C.; Mezzadra, R.; Slagter, M.; Dijkstra, K.; et al. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat. Med. 2019, 25, 89–94. [Google Scholar] [CrossRef]
- Mooney, J.E.; Rolfe, B.E.; Osborne, G.W.; Sester, D.P.; van Rooijen, N.; Campbell, G.R.; Hume, D.A.; Campbell, J.H. Cellular plasticity of inflammatory myeloid cells in the peritoneal foreign body response. Am. J. Pathol. 2010, 176, 369–380. [Google Scholar] [CrossRef]
- Bertrand, S.; Godoy, M.; Semal, P.; Van Gansen, P. Transdifferentiation of macrophages into fibroblasts as a result of Schistosoma mansoni infection. Int. J. Dev. Biol. 1992, 36, 179–184. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, M.; Wang, J.; Zang, J.; An, Y.; Dong, Y. The Mechanism of CD8+ T Cells for Reducing Myofibroblasts Accumulation during Renal Fibrosis. Biomolecules 2021, 11, 990. https://doi.org/10.3390/biom11070990
Gao M, Wang J, Zang J, An Y, Dong Y. The Mechanism of CD8+ T Cells for Reducing Myofibroblasts Accumulation during Renal Fibrosis. Biomolecules. 2021; 11(7):990. https://doi.org/10.3390/biom11070990
Chicago/Turabian StyleGao, Min, Jing Wang, Jianghua Zang, Yina An, and Yanjun Dong. 2021. "The Mechanism of CD8+ T Cells for Reducing Myofibroblasts Accumulation during Renal Fibrosis" Biomolecules 11, no. 7: 990. https://doi.org/10.3390/biom11070990
APA StyleGao, M., Wang, J., Zang, J., An, Y., & Dong, Y. (2021). The Mechanism of CD8+ T Cells for Reducing Myofibroblasts Accumulation during Renal Fibrosis. Biomolecules, 11(7), 990. https://doi.org/10.3390/biom11070990