
Post-Translational Modifications That Drive Prostate Cancer Progression

Abstract
1. Introduction
2. Post-Translational Modifications
3. Post-Translational Modifications in Prostate Cancer
3.1. Phosphorylation
3.2. Glycosylation
3.3. Ubiquitination
3.4. SUMOylation
3.5. Acetylation
3.6. Lipidation
4. Therapeutic Potential of Post-Translational Modifications in Prostate Cancer
5. Conclusions
Funding
Conflicts of Interest
References
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [PubMed]
- Uo, T.; Sprenger, C.C.; Plymate, S.R. Androgen Receptor Signaling and Metabolic and Cellular Plasticity During Progression to Castration Resistant Prostate Cancer. Front. Oncol. 2020, 10, 580617. [Google Scholar] [CrossRef]
- Jasndeep, K.; Azhar, H.; Ayema, H.; Hassan, N.; Sundip, P. A Comprehensive Review of Pharmaceutical and Surgical Interventions of Prostate Cancer. Cureus 2020, 12, e11617. [Google Scholar]
- Braglia, L.; Zavatti, M.; Vinceti, M.; Martelli, A.M.; Marmiroli, S. Deregulated PTEN/PI3K/AKT/mTOR signaling in prostate cancer: Still a potential druggable target? Biochim. Biophys. Acta-Mol. Cell Res. 2020, 1867, 118731. [Google Scholar] [CrossRef]
- Park, S.; Kim, Y.S.; Kim, D.Y.; So, I.; Jeon, J.H. PI3K pathway in prostate cancer: All resistant roads lead to PI3K. Biochim. Biophys. Acta-Rev. Cancer 2018, 1870, 198–206. [Google Scholar] [CrossRef]
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-mTOR pathway and prostate cancer: At the crossroads of AR, MAPK, and WNT signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef] [PubMed]
- Canesin, G.; Krzyzanowska, A.; Hellsten, R.; Bjartell, A. Cytokines and Janus kinase/signal transducer and activator of transcription signaling in prostate cancer: Overview and therapeutic opportunities. Curr. Opin. Endocr. Metab. Res. 2020, 10, 36–42. [Google Scholar] [CrossRef]
- Guérin, O.; Fischel, J.L.; Ferrero, J.M.; Bozec, A.; Milano, G. EGFR targeting in hormone-refractory prostate cancer: Current appraisal and prospects for treatment. Pharmaceuticals 2010, 3, 2238–2247. [Google Scholar] [CrossRef]
- Deep, G.; Panigrahia, G.K. Hypoxia-induced signaling promotes prostate cancer progression: Exosomes role as messenger of hypoxic response in tumor microenvironment. Crit. Rev. Oncog. 2015, 20, 419–434. [Google Scholar] [CrossRef]
- Testa, U.; Castelli, G.; Pelosi, E. Cellular and Molecular Mechanisms Underlying Prostate Cancer Development: Therapeutic Implications. Medicines 2019, 6, 82. [Google Scholar] [CrossRef]
- Khoury, G.A.; Baliban, R.C.; Floudas, C.A. Proteome-wide post-translational modification statistics: Frequency analysis and curation of the swiss-prot database. Sci. Rep. 2011, 1, 90. [Google Scholar] [CrossRef] [PubMed]
- Oo, H.Z.; Seiler, R.; Black, P.C.; Daugaard, M. Post-translational modifications in bladder cancer: Expanding the tumor target repertoire. Urol. Oncol. Semin. Orig. Investig. 2020, 38, 858–866. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, S.; Tao, Y. Regulating tumor suppressor genes: Post-translational modifications. Signal Transduct. Target. Ther. 2020, 5, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Martín-Bernabé, A.; Balcells, C.; Tarragó-Celada, J.; Foguet, C.; Bourgoin-Voillard, S.; Seve, M.; Cascante, M. The importance of post-translational modifications in systems biology approaches to identify therapeutic targets in cancer metabolism. Curr. Opin. Syst. Biol. 2017, 3, 161–169. [Google Scholar] [CrossRef]
- Qian, M.; Yan, F.; Yuan, T.; Yang, B.; He, Q.; Zhu, H. Targeting post-translational modification of transcription factors as cancer therapy. Drug Discov. Today 2020, 25, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Theivendran, S.; Tang, J.; Lei, C.; Yang, Y.; Song, H.; Gu, Z.; Wang, Y.; Yang, Y.; Jin, L.; Yu, C. Post translational modification-assisted cancer immunotherapy for effective breast cancer treatment. Chem. Sci. 2020. [Google Scholar] [CrossRef]
- Zhao, Y.; Jensen, O.N. Modification-specific proteomics: Strategies for characterization of post-translational modifications using enrichment techniques. Proteomics 2009, 9, 4632–4641. [Google Scholar] [CrossRef]
- Spoel, S.H. Orchestrating the proteome with post-translational modifications. J. Exp. Bot. 2018, 69, 4499–4503. [Google Scholar] [CrossRef]
- Gao, Y.; Ha, Y.S.; Kwon, T.G.; Cho, Y.C.; Lee, S.; Lee, J.N. Characterization of Kinase Expression Related to Increased Migration of PC-3M Cells Using Global Comparative Phosphoproteome Analysis. Cancer Genom. Proteom. 2020, 17, 543–553. [Google Scholar] [CrossRef]
- Nguyen, E.V.; Pereira, B.A.; Lawrence, M.G.; Ma, X.; Rebello, R.J.; Chan, H.; Niranjan, B.; Wu, Y.; Ellem, S.; Guan, X.; et al. Proteomic profiling of human prostate cancer-associated fibroblasts (caf) reveals loxl2-dependent regulation of the tumor microenvironment. Mol. Cell. Proteom. 2019, 18, 1410–1427. [Google Scholar] [CrossRef]
- Lee, R.S.; Zhang, L.; Berger, A.; Lawrence, M.G.; Song, J.; Niranjan, B.; Davies, R.G.; Lister, N.L.; Sandhu, S.K.; Rubin, M.A.; et al. Characterization of the ERG-regulated Kinome in Prostate Cancer Identifies TNIK as a Potential Therapeutic Target. Neoplasia 2019, 21, 389–400. [Google Scholar] [CrossRef]
- Drake, J.M.; Paull, E.O.; Graham, N.A.; Lee, J.K.; Smith, B.A.; Titz, B.; Stoyanova, T.; Faltermeier, C.M.; Uzunangelov, V.; Carlin, D.E.; et al. Phosphoproteome Integration Reveals Patient-Specific Networks in Prostate Cancer. Cell 2016, 166, 1041–1054. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Pflug, B.R.; Lai, X.; Wang, M. Pyruvate dehydrogenase alpha 1 as a target of omega-3 polyunsaturated fatty acids in human prostate cancer through a global phosphoproteomic analysis. Proteomics 2016, 16, 2419–2431. [Google Scholar] [CrossRef]
- Drake, J.M.; Graham, N.A.; Stoyanova, T.; Sedghi, A.; Goldstein, A.S.; Cai, H.; Smith, D.A.; Zhang, H.; Komisopoulou, E.; Huang, J.; et al. Oncogene-specific activation of tyrosine kinase networks during prostate cancer progression. Proc. Natl. Acad. Sci. USA 2012, 109, 1643–1648. [Google Scholar] [CrossRef]
- Faltermeier, C.M.; Drake, J.M.; Clark, P.M.; Smith, B.A.; Zong, Y.; Volpe, C.; Mathis, C.; Morrissey, C.; Castor, B.; Huang, J.; et al. Functional screen identifies kinases driving prostate cancer visceral and bone metastasis. Proc. Natl. Acad. Sci. USA 2015, 113, E172–E181. [Google Scholar] [CrossRef] [PubMed]
- Ino, Y.; Arakawa, N.; Ishiguro, H.; Uemura, H.; Kubota, Y.; Hirano, H.; Toda, T. Phosphoproteome analysis demonstrates the potential role of THRAP3 phosphorylation in androgen-independent prostate cancer cell growth. Proteomics 2016, 16, 1069–1078. [Google Scholar] [CrossRef]
- Jiang, N.; Hjorth-Jensen, K.; Hekmat, O.; Iglesias-Gato, D.; Kruse, T.; Wang, C.; Wei, W.; Ke, B.; Yan, B.; Niu, Y.; et al. In vivo quantitative phosphoproteomic profiling identifies novel regulators of castration-resistant prostate cancer growth. Oncogene 2015. [Google Scholar] [CrossRef]
- Toughiri, R.; Li, X.; Du, Q.; Bieberich, C.J. Phosphorylation of NuMA by Aurora-A kinase in PC-3 prostate cancer cells affects proliferation, survival, and interphase NuMA localization. J. Cell. Biochem. 2013, 114, 823–830. [Google Scholar] [CrossRef]
- Lee, B.Y.; Hochgräfe, F.; Lin, H.M.; Castillo, L.; Wu, J.; Raftery, M.J.; Martin Shreeve, S.; Horvath, L.G.; Daly, R.J. Phosphoproteomic profiling identifies focal adhesion kinase as a mediator of docetaxel resistance in castrate-resistant prostate cancer. Mol. Cancer Ther. 2013, 13, 190–201. [Google Scholar] [CrossRef]
- Bai, R.; Luan, X.; Zhang, Y.; Robbe-Masselot, C.; Brockhausen, I.; Gao, Y. The expression and functional analysis of the sialyl-T antigen in prostate cancer. Glycoconj. J. 2020, 37, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Höti, N.; Lih, T.S.; Pan, J.; Zhou, Y.; Yang, G.; Deng, A.; Chen, L.; Dong, M.; Yang, R.B.; Tu, C.F.; et al. A Comprehensive analysis of fut8 overexpressing prostate cancer cells reveals the role of egfr in castration resistance. Cancers 2020, 12, 468. [Google Scholar] [CrossRef]
- Clark, D.J.; Schnaubelt, M.; Hoti, N.; Hu, Y.; Zhou, Y.; Gooya, M.; Zhang, H. Impact of Increased FUT8 Expression on the Extracellular Vesicle Proteome in Prostate Cancer Cells. J. Proteome Res. 2020, 19, 2195–2205. [Google Scholar] [CrossRef] [PubMed]
- Itkonen, H.M.; Urbanucci, A.; Martin, S.E.S.; Khan, A.; Mathelier, A.; Thiede, B.; Walker, S.; Mills, I.G. High OGT activity is essential for MYC-driven proliferation of prostate cancer cells. Theranostics 2019, 9, 2183–2197. [Google Scholar] [CrossRef] [PubMed]
- Höti, N.; Yang, S.; Hu, Y.; Shah, P.; Haffner, M.C.; Zhang, H. Overexpression of α (1,6) fucosyltransferase in the development of castration-resistant prostate cancer cells. Prostate Cancer Prostatic Dis. 2018, 21, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Glinskii, O.V.; Mooney, B.P.; Rittenhouse-Olson, K.; Pienta, K.J.; Glinsky, V.V. Cell surface Thomsen-Friedenreich proteome profiling of metastatic prostate cancer cells reveals potential link with cancer stem cell-like phenotype. Oncotarget 2017, 8, 98598–98608. [Google Scholar] [CrossRef]
- McCann, J.J.; Vasilevskaya, I.A.; Neupane, N.P.; Shafi, A.A.; McNair, C.; Dylgjeri, E.; Mandigo, A.C.; Schiewer, M.J.; Schrecengost, R.S.; Gallagher, P.; et al. USP22 functions as an oncogenic driver in prostate cancer by regulating cell proliferation and DNA repair. Cancer Res. 2020, 80, 430–443. [Google Scholar] [CrossRef]
- Gulati, T.; Huang, C.; Caramia, F.; Raghu, D.; Paul, P.J.; Goode, R.J.A.; Keam, S.P.; Williams, S.G.; Haupt, S.; Kleifeld, O.; et al. Proteotranscriptomic Measurements of E6-Associated Protein (E6AP) Targets in DU145 Prostate Cancer Cells. Mol. Cell. Proteom. 2018, 17, 1170–1183. [Google Scholar] [CrossRef]
- Theurillat, J.P.P.; Udeshi, N.D.; Errington, W.J.; Svinkina, T.; Baca, S.C.; Pop, M.; Wild, P.J.; Blattner, M.; Groner, A.C.; Rubin, M.A.; et al. Ubiquitylome analysis identifies dysregulation of effector substrates in SPOP-mutant prostate cancer. Science (80-) 2014, 346, 85–89. [Google Scholar] [CrossRef]
- Wen, D.; Xu, Z.; Xia, L.; Liu, X.; Tu, Y.; Lei, H.; Wang, W.; Wang, T.; Song, L.; Ma, C.; et al. Important role of SUMOylation of spliceosome factors in prostate cancer cells. J. Proteome Res. 2014, 13, 3571–3582. [Google Scholar] [CrossRef] [PubMed]
- Mariscal, J.; Vagner, T.; Kim, M.; Zhou, B.; Chin, A.; Zandian, M.; Freeman, M.R.; You, S.; Zijlstra, A.; Yang, W.; et al. Comprehensive palmitoyl-proteomic analysis identifies distinct protein signatures for large and small cancer-derived extracellular vesicles. J. Extracell. Vesicles 2020, 9. [Google Scholar] [CrossRef]
- Sharma, C.; Yang, W.; Steen, H.; Freeman, M.R.; Hemler, M.E. Antioxidant functions of DHHC3 suppress anti-cancer drug activities. Cell. Mol. Life Sci. 2020, 1–13. [Google Scholar] [CrossRef]
- Cui, L.; Liu, M.; Lai, S.; Hou, H.; Diao, T.; Zhang, D.; Wang, M.; Zhang, Y.; Wang, J. Androgen upregulates the palmitoylation of eIF3L in human prostate LNCaP cells. OncoTargets Ther. 2019, 12, 4451–4459. [Google Scholar] [CrossRef]
- Li, W.; Zhang, J.; Zou, L.; Cui, J.; Su, F.; Jin, J.; Xiao, F.; Liu, M.; Zhao, G. Palmitoylome profiling indicates that androgens regulate the palmitoylation of α‑tubulin in prostate cancer‑derived LNCaP cells and supernatants. Oncol. Rep. 2019. [Google Scholar] [CrossRef]
- Venkadakrishnan, V.B.; Ben-Salem, S.; Heemers, H.V. AR-dependent phosphorylation and phospho-proteome targets in prostate cancer. Endocr. Relat. Cancer 2020, 27, R193–R210. [Google Scholar] [CrossRef]
- Shah, K.; Bradbury, N.A.; Franklin, R.; Chicago, N. Kinase Modulation of Androgen Receptor Signaling: Implications for Prostate Cancer. Cancer Cell Microenviron. 2015, 2, 2. [Google Scholar] [CrossRef]
- Koryakina, Y.; Ta, H.Q.; Gioeli, D. Androgen receptor phosphorylation: Biological context and functional consequences. Endocr. Relat. Cancer 2014, 21, T131–T145. [Google Scholar] [CrossRef]
- Liu, Y.L.; Horning, A.M.; Lieberman, B.; Kim, M.; Lin, C.K.; Hung, C.N.; Chou, C.W.; Wang, C.M.; Lin, C.L.; Kirma, N.B.; et al. Spatial EGFR Dynamics and Metastatic Phenotypes Modulated by Upregulated EphB2 and Src Pathways in Advanced Prostate Cancer. Cancers 2019, 11, 1910. [Google Scholar] [CrossRef]
- Wang, C.; Ke, Y.; Liu, S.; Pan, S.; Liu, Z.; Zhang, H.; Fan, Z.; Zhou, C.; Liu, J.; Wang, F. Ectopic fibroblast growth factor receptor 1 promotes inflammation by promoting nuclear factor-κB signaling in prostate cancer cells. J. Biol. Chem. 2018, 293, 14839–14849. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Wang, F.; Matsubara, A.; Kan, M.; McKeehan, W.L. Fibroblast growth factor receptor 2 limits and receptor 1 accelerates tumorigenicity of prostate epithelial cells. Cancer Res. 1997, 57, 5369–5378. [Google Scholar] [PubMed]
- Teishima, J.; Hayashi, T.; Nagamatsu, H.; Shoji, K.; Shikuma, H.; Yamanaka, R.; Sekino, Y.; Goto, K.; Inoue, S.; Matsubara, A. Fibroblast Growth Factor Family in the Progression of Prostate Cancer. J. Clin. Med. 2019, 8, 183. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, Z.; Ke, Y.; Wang, F. Intrinsic FGFR2 and ectopic FGFR1 signaling in the prostate and prostate cancer. Front. Genet. 2019, 10. [Google Scholar] [CrossRef]
- Wang, J.; Yu, W.; Cai, Y.; Ren, C.; Ittmann, M.M. Altered fibroblast growth factor receptor 4 stability promotes prostate cancer progression. Neoplasia 2008, 10, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, N.; Varjosalo, M.; Meller, P.; Lohi, J.; Hyytiäinen, M.; Kilpinen, S.; Kallioniemi, O.; Ingvarsen, S.; Engelholm, L.H.; Taipale, J.; et al. fibroblast growth factor receptor 4 regulates tumor invasion by coupling fibroblast growth factor signaling to extracellular matrix degradation. Cancer Res. 2010, 70, 7851–7861. [Google Scholar] [CrossRef][Green Version]
- Kawada, M.; Inoue, H.; Masuda, T.; Ikeda, D. Insulin-like growth factor i secreted from prostate stromal cells mediates tumor-stromal cell interactions of prostate cancer. Cancer Res. 2006, 66, 4419–4425. [Google Scholar] [CrossRef][Green Version]
- Fan, W.Q.; Yanase, T.; Morinaga, H.; Okabe, T.; Nomura, M.; Daitoku, H.; Fukamizu, A.; Kato, S.; Takayanagi, R.; Nawata, H. Insulin-like growth factor 1/insulin signaling activates androgen signaling through direct interactions of Foxo1 with androgen receptor. J. Biol. Chem. 2007, 282, 7329–7338. [Google Scholar] [CrossRef]
- Dayyani, F.; Parikh, N.U.; Varkaris, A.S.; Song, J.H.; Moorthy, S.; Chatterji, T.; Maity, S.N.; Wolfe, A.R.; Carboni, J.M.; Gottardis, M.M.; et al. Combined Inhibition Of IGF-1R/IR And Src Family Kinases Enhances Antitumor Effects In Prostate Cancer By Decreasing Activated Survival Pathways. PLoS ONE 2012, 7, e51189. [Google Scholar] [CrossRef] [PubMed]
- Fahrenholtz, C.D.; Beltran, P.J.; Burnstein, K.L. Targeting IGF-IR with ganitumab inhibits tumorigenesis and increases durability of response to androgen-deprivation therapy in VCaP prostate cancer xenografts. Mol. Cancer Ther. 2013, 12, 394–404. [Google Scholar] [CrossRef]
- Maslova, K.; Kyriakakis, E.; Pfaff, D.; Frachet, A.; Frismantiene, A.; Bubendorf, L.; Ruiz, C.; Vlajnic, T.; Erne, P.; Resink, T.J.; et al. EGFR and IGF-1R in regulation of prostate cancer cell phenotype and polarity: Opposing functions and modulation by T-cadherin. FASEB J. 2014, 29, 494–507. [Google Scholar] [CrossRef] [PubMed]
- Chinni, S.R.; Sivalogan, S.; Dong, Z.; Trindade Filho, J.C.; Deng, X.; Bonfil, R.D.; Cher, M.L. CXCL12/CXCR4 signaling activates Akt-1 and MMP-9 expression in prostate cancer cells: The role of bone microenvironment-associated CXCL. Prostate 2006, 66, 32–48. [Google Scholar] [CrossRef]
- Chinni, S.R.; Yamamoto, H.; Dong, Z.; Sabbota, A.; Bonfil, R.D.; Cher, M.L. CXCL12/CXCR4 transactivates her2 in lipid rafts of prostate cancer cells and promotes growth of metastatic deposits in bone. Mol. Cancer Res. 2008, 6, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Chetram, M.A.; Odero-Marah, V.; Hinton, C.V. Loss of PTEN Permits CXCR4-Mediated Tumorigenesis through ERK1/2 in Prostate Cancer Cells. Mol. Cancer Res. 2011, 9, 90–102. [Google Scholar] [CrossRef]
- Kasina, S.; Macoska, J.A. The CXCL12/CXCR4 axis promotes ligand-independent activation of the androgen receptor. Mol. Cell. Endocrinol. 2012, 351, 249–263. [Google Scholar] [CrossRef]
- Cho, K.S.; Yoon, S.J.; Lee, J.Y.; Cho, N.H.; Choi, Y.D.; Song, Y.S.; Hong, S.J. Inhibition of tumor growth and histopathological changes following treatment with a chemokine receptor CXCR4 antagonist in a prostate cancer xenograft model. Oncol. Lett. 2013, 6, 933–938. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Srivastava, S.K.; Singh, S.; Arora, S.; Tyagi, N.; Andrews, J.; McClellan, S.; Carter, J.E.; Singh, A.P. CXCL12/CXCR4 signaling counteracts docetaxel-induced microtubule stabilization via p21-activated kinase 4-dependent activation of LIM domain kinase. Oncotarget 2014, 5, 11490–11500. [Google Scholar] [CrossRef]
- Begley, L.A.; Kasina, S.; Shah, R.B.; Macoska, J.A. Signaling mechanisms coupled to CXCL12/CXCR4-mediated cellular proliferation are PTEN-dependent. Am. J. Clin. Exp. Urol. 2015, 3, 91–99. [Google Scholar]
- Conley-LaComb, M.K.; Semaan, L.; Singareddy, R.; Li, Y.; Heath, E.I.; Kim, S.; Cher, M.L.; Chinni, S.R. Pharmacological targeting of CXCL12/CXCR4 signaling in prostate cancer bone metastasis. Mol. Cancer 2016, 15, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Shamaladevi, N.; Lyn, D.A.; Escudero, D.O.; Lokeshwar, B.L. CXC receptor-1 silencing inhibits androgen-independent prostate cancer. Cancer Res. 2009, 69, 8265–8274. [Google Scholar] [CrossRef]
- Singh, R.K.; Lokeshwar, B.L. The IL-8-regulated chemokine receptor CXCR7 stimulates EGFR signaling to promote prostate cancer growth. Cancer Res. 2011, 71, 3268–3277. [Google Scholar] [CrossRef] [PubMed]
- Sbrissa, D.; Semaan, L.; Govindarajan, B.; Li, Y.; Caruthers, N.J.; Stemmer, P.M.; Cher, M.L.; Sethi, S.; Vaishampayan, U.; Shisheva, A.; et al. A novel cross-talk between CXCR4 and PI4KIIIα in prostate cancer cells. Oncogene 2018, 38, 332–344. [Google Scholar] [CrossRef]
- Gioeli, D.; Mandell, J.W.; Petroni, G.R.; Frierson, H.F.; Weber, M.J. Activation of mitogen-activated protein kinase associated with prostate cancer progression. Cancer Res. 1999, 59, 279–284. [Google Scholar] [PubMed]
- Hoshino, R.; Chatani, Y.; Yamori, T.; Tsuruo, T.; Oka, H.; Yoshida, O.; Shimada, Y.; Ari-I, S.; Wada, H.; Fujimoto, J.; et al. Constitutive activation of the 41-/43-kDa mitogen-activated protein kinase signaling pathway in human tumors. Oncogene 1999, 18, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Moro, L.; Arbini, A.A.; Marra, E.; Greco, M. Constitutive activation of MAPK/ERK inhibits prostate cancer cell proliferation through upregulation of BRCA. Int. J. Oncol. 2007, 30. [Google Scholar] [CrossRef]
- Wang, W.; Shen, T.; Dong, B.; Creighton, C.J.; Meng, Y.; Zhou, W.; Shi, Q.; Zhou, H.; Zhang, Y.; Moore, D.D.; et al. MAPK4 overexpression promotes tumor progression via noncanonical activation of AKT/mTOR signaling. J. Clin. Investig. 2019, 129, 1015–1029. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Hu, J. The role of JNK in prostate cancer progression and therapeutic strategies. Biomed. Pharmacother. 2020, 121, 109679. [Google Scholar] [CrossRef] [PubMed]
- Udhane, V.; Maranto, C.; Hoang, D.T.; Gu, L.; Erickson, A.; Devi, S.; Talati, P.G.; Banerjee, A.; Iczkowski, K.A.; Jacobsohn, K.; et al. Enzalutamide-induced feed-forward signaling loop promotes therapy-resistant prostate cancer growth providing an exploitable molecular target for JAK2 inhibitors. Mol. Cancer Ther. 2019, 19, 231–246. [Google Scholar] [CrossRef]
- Xu, L.J.; Ma, Q.; Zhu, J.; Li, J.; Xue, B.X.; Gao, J.; Sun, C.Y.; Zang, Y.C.; Zhou, Y.B.; Yang, D.R.; et al. Combined inhibition of JAK1,2/Stat3-PD-L1 signaling pathway suppresses the immune escape of Castration-resistant prostate cancer to NK cells in Hypoxia. Mol. Med. Rep. 2018, 17, 8111–8120. [Google Scholar] [CrossRef]
- Taddei, M.L.; Parri, M.; Angelucci, A.; Onnis, B.; Bianchini, F.; Giannoni, E.; Raugei, G.; Calorini, L.; Rucci, N.; Teti, A.; et al. Kinase-dependent and -independent roles of EphA2 in the regulation of prostate cancer invasion and metastasis. Am. J. Pathol. 2009, 174, 1492–1503. [Google Scholar] [CrossRef] [PubMed]
- Gelman, I.H.; Peresie, J.; Eng, K.H.; Foster, B.A. Differential requirement for Src family tyrosine kinases in the initiation, progression, and metastasis of prostate cancer. Mol. Cancer Res. 2014, 12, 1470–1479. [Google Scholar] [CrossRef]
- Slack, J.K.; Adams, R.B.; Rovin, J.D.; Bissonette, E.A.; Stoker, C.E.; Parsons, J.T. Alterations in the focal adhesion kinase/Src signal transduction pathway correlate with increased migratory capacity of prostate carcinoma cells. Oncogene 2001, 20, 1152–1163. [Google Scholar] [CrossRef]
- Johnson, T.R.; Khandrika, L.; Kumar, B.; Venezia, S.; Koul, S.; Chandhoke, R.; Maroni, P.; Donohue, R.; Meacham, R.B.; Koul, H.K. Focal adhesion kinase controls aggressive phenotype of androgen-independent prostate cancer. Mol. Cancer Res. 2008, 6, 1639–1648. [Google Scholar] [CrossRef]
- Guo, W.; Zhang, Z.; Li, G.; Lai, X.; Gu, R.; Xu, W.; Chen, H.; Xing, Z.; Chen, L.; Qian, J.; et al. Pyruvate Kinase M2 Promotes Prostate Cancer Metastasis Through Regulating ERK1/2-COX-2 Signaling. Front. Oncol. 2020, 10, 544288. [Google Scholar] [CrossRef]
- Hu, Z.; Gupta, J.; Zhang, Z.; Gerseny, H.; Berg, A.; Chen, Y.J.; Zhang, Z.; Du, H.; Brendler, C.B.; Xiao, X.; et al. Systemic delivery of oncolytic adenoviruses targeting transforming growth factor-β inhibits established bone metastasis in a prostate cancer mouse model. Hum. Gene Ther. 2012, 23, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Maolake, A.; Izumi, K.; Natsagdorj, A.; Iwamoto, H.; Kadomoto, S.; Makino, T.; Naito, R.; Shigehara, K.; Kadono, Y.; Hiratsuka, K.; et al. Tumor necrosis factor-α induces prostate cancer cell migration in lymphatic metastasis through CCR7 upregulation. Cancer Sci. 2018, 109, 1524–1531. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Lin, Y.; Lan, Y.; Lin, C.; Xuan, J.W.; Shen, M.M.; McKeehan, W.L.; Greenberg, N.M.; Wang, F. Role of epithelial cell fibroblast growth factor receptor substrate 2α in prostate development, regeneration and tumorigenesis. Development 2008, 135, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; You, P.; Chen, G.; Fu, X.; Zeng, X.; Wang, C.; Huang, Y.; An, L.; Wan, X.; Navone, N.; et al. Hyperactivated FRS2α-mediated signaling in prostate cancer cells promotes tumor angiogenesis and predicts poor clinical outcome of patients. Oncogene 2016, 35, 1750–1759. [Google Scholar] [CrossRef]
- Khan, M.I.; Johani, A.A.; Hamid, A.; Ateeq, B.; Manzar, N.; Adhami, V.M.; Lall, R.K.; Rath, S.; Sechi, M.; Siddiqui, I.A.; et al. Proproliferative function of adaptor protein GRB10 in prostate carcinoma. FASEB J. 2019, 33, 3198–3211. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Ci, X.; Wang, Y.; Choi, S.Y.C.; Sullivan, S.E.; Xue, H.; Wu, R.; Dong, X.; Haegert, A.M.; Collins, C.C.; et al. GRB10 sustains AR activity by interacting with PP2A in prostate cancer cells. Int. J. Cancer 2020. [Google Scholar] [CrossRef]
- Nunes-Xavier, C.E.; Mingo, J.; López, J.I.; Pulido, R. The role of protein tyrosine phosphatases in prostate cancer biology. Biochim. Biophys. Acta-Mol. Cell Res. 2019, 1866, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Drake, J.M.; Graham, N.A.; Lee, J.K.; Stoyanova, T.; Faltermeier, C.M.; Sud, S.; Titz, B.; Huang, J.; Pienta, K.J.; Graeber, T.G.; et al. Metastatic castration-resistant prostate cancer reveals intrapatient similarity and interpatient heterogeneity of therapeutic kinase targets. Proc. Natl. Acad. Sci. USA 2013, 110, E4762–E4769. [Google Scholar] [CrossRef]
- Lauc, G.; Krištić, J.; Zoldoš, V. Glycans—The third revolution in evolution. Front. Genet. 2014, 5, 145. [Google Scholar] [CrossRef]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Štambuk, T.; Klasić, M.; Zoldoš, V.; Lauc, G. N-glycans as functional effectors of genetic and epigenetic disease risk. Mol. Asp. Med. 2020, 100891. [Google Scholar] [CrossRef]
- Vojta, A.; Samaržija, I.; Bočkor, L.; Zoldoš, V. Glyco-genes change expression in cancer through aberrant methylation. Biochim. et Biophys. Acta (BBA)-Gen. Subj. 2016, 1860, 1776–1785. [Google Scholar] [CrossRef] [PubMed]
- Scott, E.; Munkley, J. Glycans as biomarkers in prostate cancer. Int. J. Mol. Sci. 2019, 20, 1389. [Google Scholar] [CrossRef]
- Munkley, J.; Mills, I.G.; Elliott, D.J. The role of glycans in the development and progression of prostate cancer. Nat. Rev. Urol. 2016, 13, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Vajaria, B.N.; Patel, K.R.; Begum, R.; Patel, P.S. Sialylation: An Avenue to Target Cancer Cells. Pathol. Oncol. Res. 2016, 22, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Munkley, J.; Oltean, S.; Vodák, D.; Wilson, B.T.; Livermore, K.E.; Zhou, Y.; Star, E.; Floros, V.I.; Johannessen, B.; Knight, B.; et al. The androgen receptor controls expression of the cancer-associated sTn antigen and cell adhesion through induction of ST6GalNAc1 in prostate cancer. Oncotarget 2015, 6, 34358–34374. [Google Scholar] [CrossRef]
- Kaushik, A.K.; Shojaie, A.; Panzitt, K.; Sonavane, R.; Venghatakrishnan, H.; Manikkam, M.; Zaslavsky, A.; Putluri, V.; Vasu, V.T.; Zhang, Y.; et al. Inhibition of the hexosamine biosynthetic pathway promotes castration-resistant prostate cancer. Nat. Commun. 2016, 7, 11612. [Google Scholar] [CrossRef]
- Wei, A.; Fan, B.; Zhao, Y.; Zhang, H.; Wang, L.; Yu, X.; Yuan, Q.; Yang, D.; Wang, S. ST6Gal-I overexpression facilitates prostate cancer progression via the PI3K/Akt/GSK-3β/β-catenin signaling pathway. Oncotarget 2016, 7, 65374–65388. [Google Scholar] [CrossRef]
- Kalaydina, R.V.; Zhou, H.; Markvicheva, E.; Burov, S.V.; Zulkernine, F.; Szewczuk, M.R. Impact of fucosylation on self-assembly of prostate and breast tumor spheroids by using cyclo-rgdfk (Tpp) peptide and image object detection. OncoTargets Ther. 2019, 12, 11153–11173. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Guillebon, A.D.; Hsu, J.W.; Barthel, S.R.; Dimitroff, C.J.; Lee, Y.F.; King, M.R. Human fucosyltransferase 6 enables prostate cancer metastasis to bone. Br. J. Cancer 2013, 109, 3014–3022. [Google Scholar] [CrossRef] [PubMed]
- Tsui, K.H.; Chang, P.L.; Feng, T.H.; Chung, L.C.; Sung, H.C.; Juang, H.H. Evaluating the function of matriptase and N-acetyl-glucosaminyltransferase V in prostate cancer metastasis. Anticancer Res. 2008, 28, 1993–1999. [Google Scholar] [PubMed]
- Munkley, J.; Vodak, D.; Livermore, K.E.; James, K.; Wilson, B.T.; Knight, B.; McCullagh, P.; McGrath, J.; Crundwell, M.; Harries, L.W.; et al. Glycosylation is an Androgen-Regulated Process Essential for Prostate Cancer Cell Viability. EBioMedicine 2016, 8, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Horak, P.; Tomasich, E.; Vaňhara, P.; Kratochvílová, K.; Anees, M.; Marhold, M.; Lemberger, C.E.; Gerschpacher, M.; Horvat, R.; Sibilia, M.; et al. TUSC3 Loss Alters the ER Stress Response and Accelerates Prostate Cancer Growth in vivo. Sci. Rep. 2014, 4, 3739. [Google Scholar] [CrossRef]
- Itkonen, H.M.; Gorad, S.S.; Duveau, D.Y.; Martin, S.E.S.; Barkovskaya, A.; Bathen, T.F.; Moestue, S.A.; Mills, I.G. Inhibition of O-GlcNAc transferase activity reprograms prostate cancer cell metabolism. Oncotarget 2016, 7, 12464–12476. [Google Scholar] [CrossRef]
- Tzeng, S.F.; Tsai, C.H.; Chao, T.K.; Chou, Y.C.; Yang, Y.C.; Tsai, M.H.; Cha, T.L.; Hsiao, P.W. O-Glycosylation–mediated signaling circuit drives metastatic castration-resistant prostate cancer. FASEB J. 2018, 32, 6869–6882. [Google Scholar] [CrossRef]
- Tsai, C.H.; Tzeng, S.F.; Chao, T.K.; Tsai, C.Y.; Yang, Y.C.; Lee, M.T.; Hwang, J.J.; Chou, Y.C.; Tsai, M.H.; Cha, T.L.; et al. Metastatic progression of prostate cancer is mediated by autonomous binding of galectin-4-O-glycan to cancer cells. Cancer Res. 2016, 76, 5756–5767. [Google Scholar] [CrossRef]
- Hagisawa, S.; Ohyama, C.; Takahashi, T.; Endoh, M.; Moriya, T.; Nakayama, J.; Arai, Y.; Fukuda, M. Expression of core 2 β1,6-N-acetylglucosaminyltransferase facilitates prostate cancer progression. Glycobiology 2005, 15, 1016–1024. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Yoneyama, T.; Tobisawa, Y.; Hatakeyama, S.; Yamamoto, H.; Kojima, Y.; Mikami, J.; Mori, K.; Hashimoto, Y.; Koie, T.; et al. Core 2 β-1, 6-N-acetylglucosaminyltransferase-1 expression in prostate biopsy specimen is an indicator of prostate cancer aggressiveness. Biochem. Biophys. Res. Commun. 2016, 470, 150–156. [Google Scholar] [CrossRef]
- Okamoto, T.; Yoneyama, M.S.; Hatakeyama, S.; Mori, K.; Yamamoto, H.; Koie, T.; Saitoh, H.; Yamaya, K.; Funyu, T.; Fukuda, M.; et al. Core2 O-glycan-expressing prostate cancer cells are resistant to NK cell immunity. Mol. Med. Rep. 2013. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, H.F.; Pace, K.E.; Cabrera, P.V.; White, R.; Porvari, K.; Kaija, H.; Vihko, P.; Baum, L.G. O-glycosylation regulates LNCaP prostate cancer cell susceptibility to apoptosis induced by galectin-1. Cancer Res. 2007, 67, 6155–6162. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Hatakeyama, S.; Yu, S.Y.; Bao, X.; Ohyama, C.; Khoo, K.H.; Fukuda, M.N.; Fukuda, M. Core3 O-glycan synthase suppresses tumor formation and metastasis of prostate carcinoma PC3 and LNCaP cells through down-regulation of α2β1 integrin complex. J. Biol. Chem. 2009, 284, 17157–17169. [Google Scholar] [CrossRef]
- Mikami, J.; Tobisawa, Y.; Yoneyama, T.; Hatakeyama, S.; Mori, K.; Hashimoto, Y.; Koie, T.; Ohyama, C.; Fukuda, M. I-branching N -acetylglucosaminyltransferase regulates prostate cancer invasiveness by enhancing α5β1 integrin signaling. Cancer Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Edwards, I.J. Proteoglycans in prostate cancer. Nat. Rev. Urol. 2012, 9, 196–206. [Google Scholar] [CrossRef]
- Martínez-Bosch, N.; Rodriguez-Vida, A.; Juanpere, N.; Lloreta, J.; Rovira, A.; Albanell, J.; Bellmunt, J.; Navarro, P. Galectins in prostate and bladder cancer: Tumorigenic roles and clinical opportunities. Nat. Rev. Urol. 2019, 16, 433–445. [Google Scholar] [CrossRef]
- Dondoo, T.O.; Fukumori, T.; Daizumoto, K.; Fukawa, T.; Kohzuki, M.; Kowada, M.; Kusuhara, Y.; Mori, H.; Nakatsuji, H.; Takahashi, M.; et al. Galectin-3 is implicated in tumor progression and resistance to anti-androgen drug through regulation of androgen receptor signaling in prostate cancer. Anticancer Res. 2017, 37, 125–134. [Google Scholar] [CrossRef][Green Version]
- Nakajima, K.; Kho, D.H.; Yanagawa, T.; Harazono, Y.; Hogan, V.; Chen, W.; Ali-Fehmi, R.; Mehra, R.; Raz, A. Galectin-3 cleavage alters bone remodeling: Different outcomes in breast and prostate cancer skeletal metastasis. Cancer Res. 2016, 76, 1391–1402. [Google Scholar] [CrossRef]
- Llop, E.; Ferrer-Batallé, M.; Barrabés, S.; Guerrero, P.E.; Ramírez, M.; Saldova, R.; Rudd, P.M.; Aleixandre, R.N.; Comet, J.; de Llorens, R.; et al. Improvement of Prostate Cancer Diagnosis by Detecting PSA Glycosylation-Specific Changes. Theranostics 2016, 6, 1190–1204. [Google Scholar] [CrossRef]
- Ferrer-Batallé, M.; Llop, E.; Ramírez, M.; Aleixandre, R.N.; Saez, M.; Comet, J.; de Llorens, R.; Peracaula, R. Comparative study of blood-based biomarkers, α2,3-sialic acid PSA and PHI, for high-risk prostate cancer detection. Int. J. Mol. Sci. 2017, 18, 845. [Google Scholar] [CrossRef]
- Chen, F.Z.; Zhao, X.K. Ubiquitin-proteasome pathway and prostate cancer. Oncol. Res. Treat. 2013, 36, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Voutsadakis, I.A.; Papandreou, C.N. The ubiquitin-proteasome system in prostate cancer and its transition to castration resistance. Urol. Oncol. Semin. Orig. Investig. 2012, 30, 752–761. [Google Scholar] [CrossRef]
- Zheng, N.; Shabek, N. Ubiquitin ligases: Structure, function, and regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef]
- Spratt, D.E.; Walden, H.; Shaw, G.S. RBR E3 ubiquitin ligases: New structures, new insights, new questions. Biochem. J. 2014, 458, 421–437. [Google Scholar] [CrossRef]
- Metzger, M.B.; Hristova, V.A.; Weissman, A.M. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 2012, 125, 531–537. [Google Scholar] [CrossRef]
- Chen, Z.; Lu, W. Roles of ubiquitination and SUMOylation on prostate cancer: Mechanisms and clinical implications. Int. J. Mol. Sci. 2015, 16, 4560–4580. [Google Scholar] [CrossRef]
- McClurg, U.L.; Robson, C.N. Deubiquitinating enzymes as oncotargets. Oncotarget 2015, 6, 9657–9668. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.T.; Zhou, X.; Chen, F.; Khan, M.A.; Fu, J.; Chen, H. Targeting the signalling pathways regulated by deubiquitinases for prostate cancer therapeutics. Cell Biochem. Funct. 2019, 37, 304–319. [Google Scholar] [CrossRef]
- Clark, A.; Burleson, M. SPOP and cancer: A systematic review. Am. J. Cancer Res. 2020, 10, 704. [Google Scholar] [PubMed]
- Wang, Z.; Song, Y.; Ye, M.; Dai, X.; Zhu, X.; Wei, W. The diverse roles of SPOP in prostate cancer and kidney cancer. Nat. Rev. Urol. 2020, 17, 339–350. [Google Scholar] [CrossRef]
- Blattner, M.; Liu, D.; Robinson, B.D.; Huang, D.; Poliakov, A.; Gao, D.; Nataraj, S.; Deonarine, L.D.; Augello, M.A.; Sailer, V.; et al. SPOP Mutation Drives Prostate Tumorigenesis In Vivo through Coordinate Regulation of PI3K/mTOR and AR Signaling. Cancer Cell 2017, 31, 436–451. [Google Scholar] [CrossRef]
- Qi, J.; Fan, L.; Hussain, A. Implications of ubiquitin ligases in castration-resistant prostate cancer. Curr. Opin. Oncol. 2015, 27, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Niu, Y.; Huang, H. Posttranslational regulation of androgen dependent and independent androgen receptor activities in prostate cancer. Asian J. Urol. 2020, 7, 203–218. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhao, D.; Yan, L.; Jiang, W.; Kim, J.S.; Gu, B.; Liu, Q.; Wang, R.; Xia, B.; Zhao, J.C.; et al. BMI1 regulates androgen receptor in prostate cancer independently of the polycomb repressive complex. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Qi, J.; Nakayama, K.; Cardiff, R.D.; Borowsky, A.D.; Kaul, K.; Williams, R.; Krajewski, S.; Mercola, D.; Carpenter, P.M.; Bowtell, D.; et al. Siah2-Dependent Concerted Activity of HIF and FoxA2 Regulates Formation of Neuroendocrine Phenotype and Neuroendocrine Prostate Tumors. Cancer Cell 2010, 18, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Tripathi, M.; Mishra, R.; Sahgal, N.; Fazil, L.; Ettinger, S.; Placzek, W.J.; Claps, G.; Chung, L.W.K.; Bowtell, D.; et al. The E3 ubiquitin ligase siah2 contributes to castration-resistant prostate cancer by regulation of androgen receptor transcriptional activity. Cancer Cell 2013, 23, 332–346. [Google Scholar] [CrossRef]
- Vatapalli, R.; Sagar, V.; Rodriguez, Y.; Zhao, J.C.; Unno, K.; Pamarthy, S.; Lysy, B.; Anker, J.; Han, H.; Yoo, Y.A.; et al. Histone methyltransferase DOT1L coordinates AR and MYC stability in prostate cancer. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Li, B.; Lu, W.; Yang, Q.; Yu, X.; Matusik, R.J.; Chen, Z. Skp2 regulates androgen receptor through ubiquitin-mediated degradation independent of Akt/mTOR pathways in prostate cancer. Prostate 2013, 74, 421–432. [Google Scholar] [CrossRef]
- Chang, K.H.; Li, R.; Kuri, B.; Lotan, Y.; Roehrborn, C.G.; Liu, J.; Vessella, R.; Nelson, P.S.; Kapur, P.; Guo, X.; et al. A gain-of-function mutation in dht synthesis in castration-resistant prostate cancer. Cell 2013, 154, 1074–1084. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Carracedo, A.; Salmena, L.; Song, S.J.; Egia, A.; Malumbres, M.; Pandolfi, P.P. Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell 2011, 144, 187–199. [Google Scholar] [CrossRef]
- Gao, D.; Inuzuka, H.; Tseng, A.; Wei, W. Akt finds its new path to regulate cell cycle through modulating Skp2 activity and its destruction by APC/Cdh1. Cell Div. 2009, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ue Luk, I.S.; Shrestha, R.; Xue, H.; Wang, Y.; Zhang, F.; Lin, D.; Haegert, A.; Wu, R.; Dong, X.; Collins, C.C.; et al. BIRC6 targeting as potential therapy for advanced, enzalutamide-resistant prostate cancer. Clin. Cancer Res. 2017. [Google Scholar] [CrossRef]
- Low, C.G.; Luk, I.S.U.; Lin, D.; Fazli, L.; Yang, K.; Xu, Y.; Gleave, M.; Gout, P.W.; Wang, Y. BIRC6 Protein, an Inhibitor of Apoptosis: Role in Survival of Human Prostate Cancer Cells. PLoS ONE 2013, 8, e55837. [Google Scholar] [CrossRef]
- Eigentler, A.; Tymoszuk, P.; Zwick, J.; Schmitz, A.A.; Pircher, A.; Kocher, F.; Schlicker, A.; Lesche, R.; Schäfer, G.; Theurl, I.; et al. The impact of Cand1 in prostate cancer. Cancers 2020, 12, 428. [Google Scholar] [CrossRef] [PubMed]
- Korzeniewski, N.; Hohenfellner, M.; Duensing, S. CAND1 promotes PLK4-mediated centriole overduplication and is frequently disrupted in prostate cancer. Neoplasia 2012, 14, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, N.; Shrestha, H.; Ryu, T.; Kim, H.; Simkhada, S.; Cho, Y.C.; Park, S.Y.; Cho, S.; Lee, K.Y.; Lee, J.H.; et al. δ-Catenin Increases the Stability of EGFR by Decreasing c-Cbl Interaction and Enhances EGFR/Erk1/2 Signaling in Prostate Cancer. Mol. Cells 2018, 41, 320–330. [Google Scholar]
- Dallavalle, C.; Albino, D.; Civenni, G.; Merulla, J.; Ostano, P.; Mello-Grand, M.; Rossi, S.; Losa, M.; D’Ambrosio, G.; Sessa, F.; et al. MicroRNA-424 impairs ubiquitination to activate STAT3 and promote prostate tumor progression. J. Clin. Investig. 2016, 126, 4585–4602. [Google Scholar] [CrossRef] [PubMed]
- Vitari, A.C.; Leong, K.G.; Newton, K.; Yee, C.; Oĝrourke, K.; Liu, J.; Phu, L.; Vij, R.; Ferrando, R.; Couto, S.S.; et al. COP1 is a tumour suppressor that causes degradation of ETS transcription factors. Nature 2011. [Google Scholar] [CrossRef]
- Li, N.; Xue, W.; Yuan, H.; Dong, B.; Ding, Y.; Liu, Y.; Jiang, M.; Kan, S.; Sun, T.; Ren, J.; et al. AKT-mediated stabilization of histone methyltransferase WHSC1 promotes prostate cancer metastasis. J. Clin. Investig. 2017, 127, 1284–1302. [Google Scholar] [CrossRef]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Fan, P.; Chang, S.W.; Tsao, Y.P.; Huang, H.P.; Chen, S.L. NRIP/DCAF6 stabilizes the androgen receptor protein by displacing DDB2 from the CUL4A-DDB1 E3 ligase complex in prostate cancer. Oncotarget 2017, 8, 21501–21515. [Google Scholar] [CrossRef] [PubMed]
- Jiao, M.; Qi, M.; Zhang, F.; Hu, J.; Feng, T.; Zhao, M.; Li, X.; Liu, H.; Teng, W.; Zhang, J.; et al. CUL4B regulates cancer stem-like traits of prostate cancer cells by targeting BMI1 via miR200b/c. Prostate 2019, 79, 1294–1303. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Qi, M.; Li, X.; Hu, J.; Zhang, J.; Jiao, M.; Bai, X.; Peng, X.; Han, B. CUL4B/miR-33b/C-MYC axis promotes prostate cancer progression. Prostate 2019, 79, 480–488. [Google Scholar] [CrossRef]
- Kuchay, S.; Giorgi, C.; Simoneschi, D.; Pagan, J.; Missiroli, S.; Saraf, A.; Florens, L.; Washburn, M.P.; Collazo-Lorduy, A.; Castillo-Martin, M.; et al. PTEN counteracts FBXL2 to promote IP3R3- and Ca2+-mediated apoptosis limiting tumour growth. Nature 2017, 546, 554–558. [Google Scholar] [CrossRef]
- Stankiewicz, E.; Mao, X.; Mangham, D.C.; Xu, L.; Yeste-Velasco, M.; Fisher, G.; North, B.; Chaplin, T.; Young, B.; Wang, Y.; et al. Identification of FBXL4 as a Metastasis Associated Gene in Prostate Cancer. Sci. Rep. 2017, 7, 5124. [Google Scholar] [CrossRef]
- Moro, L.; Simoneschi, D.; Kurz, E.; Arbini, A.A.; Jang, S.; Guaragnella, N.; Giannattasio, S.; Wang, W.; Chen, Y.A.; Pires, G.; et al. Epigenetic silencing of the ubiquitin ligase subunit FBXL7 impairs c-SRC degradation and promotes epithelial-to-mesenchymal transition and metastasis. Nat. Cell Biol. 2020, 1–13. [Google Scholar] [CrossRef]
- Chen, X.; Sahasrabuddhe, A.A.; Szankasi, P.; Chung, F.; Basrur, V.; Rangnekar, V.M.; Pagano, M.; Lim, M.S.; Elenitoba-Johnson, K.S.J. Fbxo45-mediated degradation of the tumor-suppressor Par-4 regulates cancer cell survival. Cell Death Differ. 2014, 21, 1535–1545. [Google Scholar] [CrossRef]
- Hebbar, N.; Burikhanov, R.; Shukla, N.; Qiu, S.; Zhao, Y.; Elenitoba-Johnson, K.S.J.; Rangnekar, V.M. A naturally generated decoy of the prostate apoptosis response-4 protein overcomes therapy resistance in tumors. Cancer Res. 2017, 77, 4039–4050. [Google Scholar] [CrossRef]
- Li, Z.; Sun, Y.; Chen, X.; Squires, J.; Nowroozizadeh, B.; Liang, C.; Huang, J. P53 mutation directs AURKA overexpression via miR-25 and FBXW7 in prostatic small cell neuroendocrine carcinoma. Mol. Cancer Res. 2015, 13, 584–591. [Google Scholar] [CrossRef]
- Hong, Z.; Zhang, W.; Ding, D.; Huang, Z.; Yan, Y.; Cao, W.; Pan, Y.; Hou, X.; Weroha, S.J.; Karnes, R.J.; et al. DNA Damage Promotes TMPRSS2-ERG Oncoprotein Destruction and Prostate Cancer Suppression via Signaling Converged by GSK3β and WEE. Mol. Cell 2020, 79, 1008–1023.e4. [Google Scholar] [CrossRef]
- Yuan, W.C.; Lee, Y.R.; Huang, S.F.; Lin, Y.M.; Chen, T.Y.; Chung, H.C.; Tsai, C.H.; Chen, H.Y.; Chiang, C.T.; Lai, C.K.; et al. A Cullin3-KLHL20 Ubiquitin Ligase-Dependent Pathway Targets PML to Potentiate HIF-1 Signaling and Prostate Cancer Progression. Cancer Cell 2011, 20, 214–228. [Google Scholar] [CrossRef]
- Arai, S.; Varkaris, A.; Nouri, M.; Chen, S.; Xie, L.; Balk, S.P. March5 mediates noxa-dependent mcl1 degradation driven by kinase inhibitors and integrated stress response activation. eLife 2020, 9. [Google Scholar] [CrossRef]
- Giridhar, P.V.; Williams, K.; VonHandorf, A.P.; Deford, P.L.; Kasper, S. Constant degradation of the androgen receptor by MDM2 conserves prostate cancer stem cell integrity. Cancer Res. 2019, 79, 1124–1137. [Google Scholar] [CrossRef]
- Lin, H.K.; Wang, L.; Hu, Y.C.; Altuwaijri, S.; Chang, C. Phosphorylation-dependent ubiquitylation and degradation of androgen receptor by Akt require Mdm2 E3 ligase. EMBO J. 2002, 21, 4037–4048. [Google Scholar] [CrossRef] [PubMed]
- Feng, F.Y.; Zhang, Y.; Kothari, V.; Evans, J.R.; Jackson, W.C.; Chen, W.; Johnson, S.B.; Luczak, C.; Wang, S.; Hamstra, D.A. MDM2 Inhibition Sensitizes Prostate Cancer Cells to Androgen Ablation and Radiotherapy in a p53-Dependent Manner. Neoplasia 2016, 18, 213–222. [Google Scholar] [CrossRef]
- Kaczorowski, A.; Tolstov, Y.; Falkenstein, M.; Vasioukhin, V.; Prigge, E.S.; Geisler, C.; Kippenberger, M.; Nientiedt, C.; Ratz, L.; Kuryshev, V.; et al. Rearranged ERG confers robustness to prostate cancer cells by subverting the function of p53. Urol. Oncol. Semin. Orig. Investig. 2020, 38, 736.e1–736.e10. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, H.; Li, M.; Rayburn, E.R.; Agrawal, S.; Zhang, R. Stabilization of E2F1 protein by MDM2 through the E2F1 ubiquitination pathway. Oncogene 2005, 24, 7238–7247. [Google Scholar] [CrossRef]
- Li, Y.; Xie, N.; Gleave, M.E.; Rennie, P.S.; Dong, X. AR-v7 protein expression is regulated by protein kinase and phosphatase. Oncotarget 2015, 6, 33743–33754. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, Y.; He, J.; Deng, R.; Huang, X.; Guo, Y.; Li, L.; Xie, R.; Yu, J. LncRNA UCA1 maintains the low-tumorigenic and nonmetastatic status by stabilizing E-cadherin in primary prostate cancer cells. Mol. Carcinog. 2020. [Google Scholar] [CrossRef]
- Chopra, H.; Khan, Z.; Contreras, J.; Wang, H.; Sedrak, A.; Zhu, Y. Activation of p53 and destabilization of androgen receptor by combinatorial inhibition of MDM2 and MDMX in prostate cancer cells. Oncotarget 2018. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Ueno, A.; Ueda, T.; Nakagawa, H.; Taniguchi, H.; Kayukawa, N.; Fujihara-Iwata, A.; Hongo, F.; Okihara, K.; Ukimura, O. CNPY2 inhibits MYLIP-mediated AR protein degradation in prostate cancer cells. Oncotarget 2018, 9, 17645–17655. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, S.; Miyajima, N.; Bohgaki, M.; Tsukiyama, T.; Shigemura, M.; Nonomura, K.; Hatakeyama, S. Ubiquitylation of ε-COP by PIRH2 and regulation of the secretion of PSA. Mol. Cell. Biochem. 2008. [Google Scholar] [CrossRef]
- Logan, I.R.; Gaughan, L.; McCracken, S.R.C.; Sapountzi, V.; Leung, H.Y.; Robson, C.N. Human PIRH2 Enhances Androgen Receptor Signaling through Inhibition of Histone Deacetylase 1 and Is Overexpressed in Prostate Cancer. Mol. Cell. Biol. 2006, 26, 6502–6510. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, W.; Ji, W.; Liu, X.; Ouyang, G.; Xiao, W. The Von Hippel-Lindau protein suppresses androgen receptor activity. Mol. Endocrinol. 2014, 28, 239–248. [Google Scholar] [CrossRef]
- Cook, K.M.; Kataria, N.; Martinez, C.A.; Kerr, B.; Zaiter, S.S.; Morgan, M.; McAlpine, S.R. C-terminal HSP90 inhibitors block the HIF-1 hypoxic response by degrading HIF-1α through the oxygen-dependent degradation pathway. Cell. Physiol. Biochem. 2019, 53, 480–495. [Google Scholar] [CrossRef]
- Wei, M.; Jiao, D.; Han, D.; Wu, J.; Wei, F.; Zheng, G.; Guo, Z.; Xi, W.; Yang, F.; Xie, P.; et al. Knockdown of RNF2 induces cell cycle arrest and apoptosis in prostate cancer cells through the upregulation of TXNIP. Oncotarget 2017. [Google Scholar] [CrossRef]
- Su, W.; Han, H.H.; Wang, Y.; Zhang, B.; Zhou, B.; Cheng, Y.; Rumandla, A.; Gurrapu, S.; Chakraborty, G.; Su, J.; et al. The Polycomb Repressor Complex 1 Drives Double-Negative Prostate Cancer Metastasis by Coordinating Stemness and Immune Suppression. Cancer Cell 2019, 36, 139–155.e10. [Google Scholar] [CrossRef]
- Xu, K.; Shimelis, H.; Linn, D.E.; Jiang, R.; Yang, X.; Sun, F.; Guo, Z.; Chen, H.; Li, W.; Chen, H.; et al. Regulation of Androgen Receptor Transcriptional Activity and Specificity by RNF6-Induced Ubiquitination. Cancer Cell 2009, 15, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Xu, J.; Siddiqui, J.; Feng, F.; Sun, Y. Depletion of SAG/RBX2 E3 ubiquitin ligase suppresses prostate tumorigenesis via inactivation of the PI3K/AKT/mTOR axis. Mol. Cancer 2016, 15, 1–14. [Google Scholar] [CrossRef]
- Xiao, Y.; Jiang, Y.; Song, H.; Liang, T.; Li, Y.; Yan, D.; Fu, Q.; Li, Z. RNF7 knockdown inhibits prostate cancer tumorigenesis by inactivation of ERK1/2 pathway. Sci. Rep. 2017, 7, 43683. [Google Scholar] [CrossRef]
- Chen, C.; Zhou, Z.; Liu, R.; Li, Y.; Azmi, P.B.; Seth, A.K. The WW domain containing E3 ubiquitin protein ligase 1 upregulates ErbB2 and EGFR through RING finger protein. Oncogene 2008, 27, 6845–6855. [Google Scholar] [CrossRef]
- Jääskeläinen, T.; Makkonen, H.; Visakorpi, T.; Kim, J.; Roeder, R.G.; Palvimo, J.J. Histone H2B ubiquitin ligases RNF20 and RNF40 in androgen signaling and prostate cancer cell growth. Mol. Cell. Endocrinol. 2012, 350, 87–98. [Google Scholar] [CrossRef]
- Chen, L.; Siddiqui, S.; Bose, S.; Mooso, B.; Asuncion, A.; Bedolla, R.G.; Vinall, R.; Tepper, C.G.; Gandour-Edwards, R.; Shi, X.B.; et al. Nrdp1-mediated regulation of ErbB3 expression by the androgen receptor in androgen-dependent but not castrate-resistant prostate cancer cells. Cancer Res. 2010, 70, 5994–6003. [Google Scholar] [CrossRef]
- Zhi, X.; Zhao, D.; Wang, Z.; Zhou, Z.; Wang, C.; Chen, W.; Liu, R.; Chen, C. E3 ubiquitin ligase RNF126 promotes cancer cell proliferation by targeting the tumor suppressor p21 for ubiquitin-mediated degradation. Cancer Res. 2013, 73, 385–394. [Google Scholar] [CrossRef]
- Yu, X.; Ai, J.; Cai, L.; Jing, Y.; Wang, D.; Dong, J.; Pascal, L.E.; Zhang, J.; Luo, R.; Wang, Z. Regulation of tumor suppressor EAF2 polyubiquitination by ELL1 and SIAH2 in prostate cancer cells. Oncotarget 2016, 7, 29245–29254. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Liu, Y.; Xia, X.; Shao, Z.; Huang, C.; He, J.; Jiang, L.; Tang, D.; Liu, J.; Huang, H. Targeting GRP78-dependent AR-V7 protein degradation overcomes castration-resistance in prostate cancer therapy. Theranostics 2020, 10, 3366–3381. [Google Scholar] [CrossRef]
- Ren, D.; Dai, Y.; Yang, Q.; Zhang, X.; Guo, W.; Ye, L.; Huang, S.; Chen, X.; Lai, Y.; Du, H.; et al. Wnt5a induces and maintains prostate cancer cells dormancy in bone. J. Exp. Med. 2018, 216, 428–449. [Google Scholar] [CrossRef] [PubMed]
- Christian, P.A.; Fiandalo, M.V.; Schwarze, S.R. Possible role of death receptor-mediated apoptosis by the E3 ubiquitin ligases Siah2 and POSH. Mol. Cancer 2011, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Liu, S.; Li, B.; Xie, Y.; Izban, M.G.; Ballard, B.R.; Sathyanarayana, S.A.; Adunyah, S.E.; Matusik, R.J.; Chen, Z. SKP2 loss destabilizes EZH2 by promoting TRAF6-mediated ubiquitination to suppress prostate cancer. Oncogene 2017, 36, 1364–1373. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lu, Y.; Wang, L.; Mizokami, A.; Keller, E.T.; Zhang, J.; Fu, J. Skp2 is associated with paclitaxel resistance in prostate cancer cells. Oncol. Rep. 2016, 36, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Bauzon, F.; Fu, H.; Lu, Z.; Cui, J.; Nakayama, K.; Nakayama, K.I.; Locker, J.; Zhu, L. Skp2 Deletion Unmasks a p27 Safeguard that Blocks Tumorigenesis in the Absence of pRb and p53 Tumor Suppressors. Cancer Cell 2013, 24, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Shim, E.H.; Johnson, L.; Noh, H.L.; Kim, Y.J.; Sun, H.; Zeiss, C.; Zhang, H. Expression of the F-box protein SKP2 induces hyperplasia, dysplasia, and low-grade carcinoma in the mouse prostate. Cancer Res. 2003, 63, 1583–1588. [Google Scholar] [PubMed]
- Lu, W.; Liu, S.; Li, B.; Xie, Y.; Adhiambo, C.; Yang, Q.; Ballard, B.R.; Nakayama, K.I.; Matusik, R.J.; Chen, Z. SKP2 inactivation suppresses prostate tumorigenesis by mediating JARID1B ubiquitination. Oncotarget 2015. [Google Scholar] [CrossRef]
- Tsai, Y.S.; Lai, C.L.; Lai, C.H.; Chang, K.H.; Wu, K.; Tseng, S.F.; Fazli, L.; Gleave, M.; Xiao, G.; Gandee, L.; et al. The role of homeostatic regulation between tumor suppressor DAB2IP and oncogenic Skp2 in prostate cancer growth. Oncotarget 2014, 5, 6425–6436. [Google Scholar] [CrossRef]
- Yang, W.L.; Jin, G.; Li, C.F.; Jeong, Y.S.; Moten, A.; Xu, D.; Feng, Z.; Chen, W.; Cai, Z.; Darnay, B.; et al. Cycles of ubiquitination and deubiquitination critically regulate growth factor-mediated activation of Akt signaling. Sci. Signal. 2013, 6, ra3. [Google Scholar] [CrossRef]
- Arbini, A.A.; Guerra, F.; Greco, M.; Marra, E.; Gandee, L.; Xiao, G.; Lotan, Y.; Gasparre, G.; Hsieh, J.T.; Moro, L. Mitochondrial DNA depletion sensitizes cancer cells to PARP inhibitors by translational and post-translational repression of BRCA2. Oncogenesis 2013, 2, e82. [Google Scholar] [CrossRef]
- Lin, H.K.; Chen, Z.; Wang, G.; Nardella, C.; Lee, S.W.; Chan, C.H.; Yang, W.L.; Wang, J.; Egia, A.; Nakayama, K.I.; et al. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature 2010, 464, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Ruan, D.; He, J.; Li, C.F.; Lee, H.J.; Liu, J.; Lin, H.K.; Chan, C.H. Skp2 deficiency restricts the progression and stem cell features of castration-resistant prostate cancer by destabilizing Twist. Oncogene 2017, 36, 4299–4310. [Google Scholar] [CrossRef]
- Liu, J.; Peng, Y.; Shi, L.; Wan, L.; Inuzuka, H.; Long, J.; Guo, J.; Zhang, J.; Yuan, M.; Zhang, S.; et al. Skp2 dictates cell cycle-dependent metabolic oscillation between glycolysis and TCA cycle. Cell Res. 2020. [Google Scholar] [CrossRef]
- Wang, F.; Chan, C.H.; Chen, K.; Guan, X.; Lin, H.K.; Tong, Q. Deacetylation of FOXO3 by SIRT1 or SIRT2 leads to Skp2-mediated FOXO3 ubiquitination and degradation. Oncogene 2012. [Google Scholar] [CrossRef] [PubMed]
- Inuzuka, H.; Gao, D.; Finley, L.W.S.; Yang, W.; Wan, L.; Fukushima, H.; Chin, Y.R.; Zhai, B.; Shaik, S.; Lau, A.W.; et al. Acetylation-dependent regulation of Skp2 function. Cell 2012, 150, 179–193. [Google Scholar] [CrossRef]
- Šimečková, Š.; Kahounová, Z.; Fedr, R.; Remšík, J.; Slabáková, E.; Suchánková, T.; Procházková, J.; Bouchal, J.; Kharaishvili, G.; Král, M.; et al. High Skp2 expression is associated with a mesenchymal phenotype and increased tumorigenic potential of prostate cancer cells. Sci. Rep. 2019, 9, 5695. [Google Scholar] [CrossRef]
- Das, R.; Gregory, P.A.; Fernandes, R.C.; Denis, I.; Wang, Q.; Townley, S.L.; Zhao, S.G.; Hanson, A.R.; Pickering, M.A.; Armstrong, H.K.; et al. MicroRNA-194 promotes prostate cancer metastasis by inhibiting SOCS2. Cancer Res. 2017, 77, 1021–1034. [Google Scholar] [CrossRef] [PubMed]
- Paul, I.; Batth, T.S.; Iglesias-Gato, D.; Al-Araimi, A.; Al-Haddabi, I.; Alkharusi, A.; Norstedt, G.; Olsen, J.V.; Zadjali, F.; Flores-Morales, A. The ubiquitin ligase Cullin5SOCS2 regulates NDR1/STK38 stability and NF-κB transactivation. Sci. Rep. 2017, 7, 42800. [Google Scholar] [CrossRef]
- Guan, B.; Pungaliya, P.; Li, X.; Uquillas, C.; Mutton, L.N.; Rubin, E.H.; Bieberich, C.J. Ubiquitination by TOPORS regulates the prostate tumor suppressor NKX3. J. Biol. Chem. 2008, 283, 4834–4840. [Google Scholar] [CrossRef]
- Singh, R.; Karri, D.; Shen, H.; Shao, J.; Dasgupta, S.; Huang, S.; Edwards, D.P.; Ittmann, M.M.; O’Malley, B.W.; Yi, P. TRAF4-mediated ubiquitination of NGF receptor TrkA regulates prostate cancer metastasis. J. Clin. Investig. 2018, 128, 3129–3143. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, A.; Song, J.; Thakur, N.; Itoh, S.; Marcusson, A.; Bergh, A.; Heldin, C.H.; Landström, M. TGF-β promotes PI3K-AKT signaling and prostate cancer cell migration through the TRAF6-mediated ubiquitylation of p85α. Sci. Signal. 2017, 10, eaal4186. [Google Scholar] [CrossRef]
- Sundar, R.; Gudey, S.K.; Heldin, C.H.; Landström, M. TRAF6 promotes TGFβ-induced invasion and cell-cycle regulation via Lys63-linked polyubiquitination of Lys178 in TGFβ type I receptor. Cell Cycle 2015, 14, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Gudey, S.K.; Sundar, R.; Mu, Y.; Wallenius, A.; Zang, G.; Bergh, A.; Heldin, C.H.; Landström, M. TRAF6 stimulates the tumor-promoting effects of TGFβ type I receptor through polyubiquitination and activation of presenilin. Sci. Signal. 2014, 7, ra2. [Google Scholar] [CrossRef] [PubMed]
- Linares, J.F.; Duran, A.; Reina-Campos, M.; Aza-Blanc, P.; Campos, A.; Moscat, J.; Diaz-Meco, M.T. Amino Acid Activation of mTORC1 by a PB1-Domain-Driven Kinase Complex Cascade. Cell Rep. 2015, 12, 1339–1352. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, A.; Von Bulow, V.; Hamidi, R.; Winssinger, N.; Barluenga, S.; Heldin, C.H.; Landström, M. Polyubiquitination of transforming growth factor β(TGFβ)- associated kinase 1 mediates nuclear factor-κB activation in response to different inflammatory stimuli. J. Biol. Chem. 2012, 287, 123–133. [Google Scholar] [CrossRef]
- Pan, Y.; Zhang, R.; Chen, H.; Chen, W.; Wu, K.; Lv, J. Expression of tripartite motif-containing proteactiin 11 (TRIM11) is associated with the progression of human prostate cancer and is downregulated by microRNA-5193. Med Sci. Monit. 2019, 25, 98–106. [Google Scholar] [CrossRef]
- Qi, L.; Lu, Z.; Sun, Y.H.; Song, H.T.; Xu, W.K. TRIM16 suppresses the progression of prostate tumors by inhibiting the Snail signaling pathway. Int. J. Mol. Med. 2016, 38, 1734–1742. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Kollipara, R.K.; Humphries, C.G.; Ma, S.H.; Hutchinson, R.; Li, R.; Siddiqui, J.; Tomlins, S.A.; Raj, G.V.; Kittler, R. The ubiquitin ligase TRIM25 targets ERG for degradation in prostate cancer. Oncotarget 2016, 7, 64921–64931. [Google Scholar] [CrossRef]
- Takayama, K.I.; Suzuki, T.; Tanaka, T.; Fujimura, T.; Takahashi, S.; Urano, T.; Ikeda, K.; Inoue, S. TRIM25 enhances cell growth and cell survival by modulating p53 signals via interaction with G3BP2 in prostate cancer. Oncogene 2018, 37, 2165–2180. [Google Scholar] [CrossRef]
- Cohen, M.; Amir, S.; Golan, M.; Ben-Neriah, Y.; Mabjeesh, N.J. β-TrCP upregulates HIF-1 in prostate cancer cells. Prostate 2019, 79, 403–413. [Google Scholar] [CrossRef]
- Zhao, D.; Lu, X.; Wang, G.; Lan, Z.; Liao, W.; Li, J.; Liang, X.; Chen, J.R.; Shah, S.; Shang, X.; et al. Synthetic essentiality of chromatin remodelling factor CHD1 in PTEN-deficient cancer. Nat. Cell Biol. 2017, 542, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Shaik, S.; Wan, L.; Tron, A.E.; Wang, Z.; Sun, L.; Inuzuka, H.; Wei, W. SCFβ-TRCP targets MTSS1 for ubiquitination-mediated destruction to regulate cancer cell proliferation and migration. Oncotarget 2013, 4, 2339–2353. [Google Scholar] [CrossRef] [PubMed]
- Svensson, C.; Ceder, J.; Iglesias-Gato, D.; Chuan, Y.C.; Pang, S.T.; Bjartell, A.; Martinez, R.M.; Bott, L.; Helczynski, L.; Ulmert, D.; et al. REST mediates androgen receptor actions on gene repression and predicts early recurrence of prostate cancer. Nucleic Acids Res. 2014, 42, 999–1015. [Google Scholar] [CrossRef]
- Shrestha, H.; Yuan, T.; He, Y.; Moon, P.G.; Shrestha, N.; Ryu, T.; Park, S.Y.; Cho, Y.C.; Lee, C.H.; Baek, M.C.; et al. Investigation of the molecular mechanism of δ-catenin ubiquitination: Implication of β-TrCP-1 as a potential E3 ligase. Biochim. Biophys. Acta-Mol. Cell Res. 2016, 1863, 2311–2321. [Google Scholar] [CrossRef]
- Gluschnaider, U.; Hidas, G.; Cojocaru, G.; Yutkin, V.; Ben-Neriah, Y.; Pikarsky, E. β-TrCP inhibition reduces prostate cancer cell growth via upregulation of the aryl hydrocarbon receptor. PLoS ONE 2010, 5, e9060. [Google Scholar] [CrossRef]
- Bhatia, N.; Thiyagarajan, S.; Elcheva, I.; Saleem, M.; Dlugosz, A.; Mukhtar, H.; Spiegelman, V.S. Gli2 is targeted for ubiquitination and degradation by β-TrCP ubiquitin ligase. J. Biol. Chem. 2006, 281, 19320–19326. [Google Scholar] [CrossRef]
- Wan, X.; Yang, S.; Huang, W.; Wu, D.; Chen, H.; Wu, M.; Li, J.; Li, T.; Li, Y. UHRF1 overexpression is involved in cell proliferation and biochemical recurrence in prostate cancer after radical prostatectomy. J. Exp. Clin. Cancer Res. 2016, 35, 34. [Google Scholar] [CrossRef] [PubMed]
- Babbio, F.; Pistore, C.; Curti, L.; Castiglioni, I.; Kunderfranco, P.; Brino, L.; Oudet, P.; Seiler, R.; Thalman, G.N.; Roggero, E.; et al. The SRA protein UHRF1 promotes epigenetic crosstalks and is involved in prostate cancer progression. Oncogene 2012, 31, 4878–4887. [Google Scholar] [CrossRef]
- Di Sante, G.; Pestell, T.G.; Casimiro, M.C.; Bisetto, S.; Powell, M.J.; Lisanti, M.P.; Cordon-Cardo, C.; Castillo-Martin, M.; Bonal, D.M.; Debattisti, V.; et al. Loss of sirt1 promotes prostatic intraepithelial neoplasia, reduces mitophagy, and delays park2 translocation to mitochondria. Am. J. Pathol. 2015, 185, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Fan, L.; Jeon, H.Y.; Zhang, F.; Cui, X.; Mickle, M.B.; Peng, G.; Hussain, A.; Fazli, L.; Gleave, M.E.; et al. p300-Mediated Acetylation of Histone Demethylase JMJD1A Prevents Its Degradation by Ubiquitin Ligase STUB1 and Enhances Its Activity in Prostate Cancer. Cancer Res. 2020, 80, 3074–3087. [Google Scholar] [CrossRef]
- Seo, J.H.; Agarwal, E.; Bryant, K.G.; Caino, M.C.; Kim, E.T.; Kossenkov, A.V.; Tang, H.Y.; Languino, L.R.; Gabrilovich, D.I.; Cohen, A.R.; et al. Syntaphilin ubiquitination regulates mitochondrial dynamics and tumor cell movements. Cancer Res. 2018, 78, 4215–4228. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lou, W.; Yang, J.C.; Liu, L.; Armstrong, C.M.; Lombard, A.P.; Zhao, R.; Noel, O.D.V.; Tepper, C.G.; Chen, H.W.; et al. Proteostasis by STUB1/HSP70 complex controls sensitivity to androgen receptor targeted therapy in advanced prostate cancer. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Cheng, L.; Zang, J.; Dai, H.J.; Li, F.; Guo, F. Ubiquitin ligase CHIP functions as an oncogene and activates the AKT signaling pathway in prostate cancer. Int. J. Oncol. 2018, 53, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, H.; Zhang, X.; Wang, P.; Wang, H.; Huang, F.; Zhou, C.; Zhou, J.; Li, S. PC-1 works in conjunction with E3 ligase CHIP to regulate androgen receptor stability and activity. Oncotarget 2016, 7, 81377–81388. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Brautigan, D.L.; Larner, J.M. Aurora kinase A promotes AR degradation via the E3 ligase CHIP. Mol. Cancer Res. 2017, 15, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Brautigan, D.L.; Parsons, S.J.; Larner, J.M. Androgen receptor degradation by the E3 ligase CHIP modulates mitotic arrest in prostate cancer cells. Oncogene 2014, 33, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Biswas, K.; Sarkar, S.; Said, N.; Brautigan, D.L.; Larner, J.M. Aurora B Kinase Promotes CHIP-Dependent Degradation of HIF1α in Prostate Cancer Cells. Mol. Cancer Ther. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.T.; Zeng, L.F.; He, Q.Y.; Tao, W.A.; Zha, Z.G.; Hu, C.D. The E3 ubiquitin ligase CHIP mediates ubiquitination and proteasomal degradation of PRMT5. Biochim. et Biophys. Acta (BBA)-Bioenerg. 2016, 1863, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.F.; Deb, S.; Paul, I.; Chatterjee, A.; Mandal, T.; Chatterjee, U.; Ghosh, M.K. The chaperone-assisted E3 ligase C terminus of Hsc70-interacting protein (CHIP) targets PTEN for proteasomal degradation. J. Biol. Chem. 2012, 287, 15996–16006. [Google Scholar] [CrossRef]
- Sun, Y.; Jia, X.; Gao, Q.; Liu, X.; Hou, L. The ubiquitin ligase UBE4A inhibits prostate cancer progression by targeting interleukin-like EMT inducer (ILEI). IUBMB Life 2017, 69, 16–21. [Google Scholar] [CrossRef]
- Bian, P.; Dou, Z.; Jia, Z.; Li, W.; Pan, D. Activated Wnt/β-Catenin signaling contributes to E3 ubiquitin ligase EDD-conferred docetaxel resistance in prostate cancer. Life Sci. 2020, 254, 116816. [Google Scholar] [CrossRef]
- Gamell, C.; Bandilovska, I.; Gulati, T.; Kogan, A.; Lim, S.C.; Kovacevic, Z.; Takano, E.A.; Timpone, C.; Agupitan, A.D.; Litchfield, C.; et al. E6AP Promotes a Metastatic Phenotype in Prostate Cancer. iScience 2019, 22, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Raghu, D.; Paul, P.J.; Gulati, T.; Deb, S.; Khoo, C.; Russo, A.; Gallo, E.; Blandino, G.; Chan, A.L.; Takano, E.; et al. E6AP promotes prostate cancer by reducing p27 expression. Oncotarget 2017, 8, 42939–42948. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hu, X.; Chen, J.; Fu, G. Impact of E6-associated protein on the proliferation and invasion of prostate cancer cells in bone metastasis. Int. J. Clin. Exp. Pathol. 2015, 8, 6571–6575. [Google Scholar]
- Srinivasan, S.; Nawaz, Z. E3 ubiquitin protein ligase, E6-associated protein (E6-AP) regulates PI3K-Akt signaling and prostate cell growth. Biochim. Biophys. Acta-Gene Regul. Mech. 2011, 1809, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.J.; Raghu, D.; Chan, A.L.; Gulati, T.; Lambeth, L.; Takano, E.; Herold, M.J.; Hagekyriakou, J.; Vessella, R.L.; Fedele, C.; et al. Restoration of tumor suppression in prostate cancer by targeting the E3 ligase E6AP. Oncogene 2016, 35, 6235–6245. [Google Scholar] [CrossRef]
- Zhang, L.; Anglesio, M.S.; O’Sullivan, M.; Zhang, F.; Yang, G.; Sarao, R.; Nghiem, M.P.; Cronin, S.; Hara, H.; Melnyk, N.; et al. The E3 ligase HACE1 is a critical chromosome 6q21 tumor suppressor involved in multiple cancers. Nat. Med. 2007, 13, 1060–1069. [Google Scholar] [CrossRef]
- Lee, H.J.; Li, C.F.; Ruan, D.; He, J.; Montal, E.D.; Lorenz, S.; Girnun, G.D.; Chan, C.H. Non-proteolytic ubiquitination of Hexokinase 2 by HectH9 controls tumor metabolism and cancer stem cell expansion. Nat. Commun. 2019, 10, 2625. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Peng, G.; Sahgal, N.; Fazli, L.; Gleave, M.; Zhang, Y.; Hussain, A.; Qi, J. Regulation of c-Myc expression by the histone demethylase JMJD1A is essential for prostate cancer cell growth and survival. Oncogene 2016, 35, 2441–2452. [Google Scholar] [CrossRef]
- Qu, H.; Liu, H.; Jin, Y.; Cui, Z.; Han, G. HUWE1 upregulation has tumor suppressive effect in human prostate cancer cell lines through c-Myc. Biomed. Pharmacother. 2018, 106, 309–315. [Google Scholar] [CrossRef]
- Le Clorennec, C.; Lazrek, Y.; Dubreuil, O.; Larbouret, C.; Poul, M.A.; Mondon, P.; Melino, G.; Pèlegrin, A.; Chardès, T. The anti-HER3 (ErbB3) therapeutic antibody 9F7-F11 induces HER3 ubiquitination and degradation in tumors through JNK1/2-dependent ITCH/AIP4 activation. Oncotarget 2016, 7, 37013–37029. [Google Scholar] [CrossRef]
- Fukushima, T.; Yoshihara, H.; Furuta, H.; Kamei, H.; Hakuno, F.; Luan, J.; Duan, C.; Saeki, Y.; Tanaka, K.; Iemura, S.I.; et al. Nedd4-induced monoubiquitination of IRS-2 enhances IGF signalling and mitogenic activity. Nat. Commun. 2015, 6, 6780. [Google Scholar] [CrossRef]
- Li, H.; Xu, L.L.; Masuda, K.; Raymundo, E.; McLeod, D.G.; Dobi, A.; Srivastava, S. A feedback loop between the androgen receptor and a NEDD4-binding protein, PMEPA1, in prostate cancer cells. J. Biol. Chem. 2008, 283, 28988–28995. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Choi, B.K.; Mujoo, K.; Fan, X.; Fa, M.; Mukherjee, S.; Owiti, N.; Zhang, N.; An, Z. The E3 ubiquitin ligase NEDD4 negatively regulates HER3/ErbB3 level and signaling. Oncogene 2014, 34, 1105–1115. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, D.; Liu, B.; Jin, X.; Wang, X.; Pan, J.; Tu, W.; Shao, Y. IMP3 accelerates the progression of prostate cancer through inhibiting PTEN expression in a SMURF1-dependent way. J. Exp. Clin. Cancer Res. 2020, 39, 1–12. [Google Scholar] [CrossRef]
- Gang, X.; Wang, G.; Huang, H. Androgens regulate SMAD ubiquitination regulatory factor-1 expression and prostate cancer cell invasion. Prostate 2015, 75, 561–572. [Google Scholar] [CrossRef]
- Chen, C.; Sun, X.; Guo, P.; Dong, X.Y.; Sethi, P.; Zhou, W.; Zhou, Z.; Petros, J.; Frierson, H.F.; Vessella, R.L.; et al. Ubiquitin E3 ligase WWP1 as an oncogenic factor in human prostate cancer. Oncogene 2007. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, Z.; Chen, C. WW domain-containing E3 ubiquitin protein ligase 1 targets p63 transcription factor for ubiquitin-mediated proteasomal degradation and regulates apoptosis. Cell Death Differ. 2008, 15, 1941–1951. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Sun, X.; Guo, P.; Dong, X.Y.; Sethi, P.; Cheng, X.; Zhou, J.; Ling, J.; Simons, J.W.; Lingrel, J.B.; et al. Human Kruppel-like factor 5 is a target of the E3 ubiquitin ligase WWP1 for proteolysis in epithelial cells. J. Biol. Chem. 2005, 280, 41553–41561. [Google Scholar] [CrossRef]
- Goto, Y.; Kojima, S.; Kurozumi, A.; Kato, M.; Okato, A.; Matsushita, R.; Ichikawa, T.; Seki, N. Regulation of E3 ubiquitin ligase-1 (WWP1) by microRNA-452 inhibits cancer cell migration and invasion in prostate cancer. Br. J. Cancer 2016, 114, 1135–1144. [Google Scholar] [CrossRef] [PubMed]
- Bawa-Khalfe, T.; Yang, F.M.; Ritho, J.; Lin, H.K.; Cheng, J.; Yeh, E.T.H. SENP1 regulates PTEN stability to dictate prostate cancer development. Oncotarget 2017. [Google Scholar] [CrossRef]
- Wen, M.; Kwon, Y.; Wang, Y.; Mao, J.H.; Wei, G. Elevated expression of UBE2T exhibits oncogenic properties in human prostate cancer. Oncotarget 2015, 6, 25226–25239. [Google Scholar] [CrossRef] [PubMed]
- Vila, I.K.; Yao, Y.; Kim, G.; Xia, W.; Kim, H.; Kim, S.J.; Park, M.K.; Hwang, J.P.; González-Billalabeitia, E.; Hung, M.C.; et al. A UBE2O-AMPKα2 Axis that Promotes Tumor Initiation and Progression Offers Opportunities for Therapy. Cancer Cell 2017, 31, 208–224. [Google Scholar] [CrossRef] [PubMed]
- Vere, G.; Kealy, R.; Kessler, B.M.; Pinto-Fernandez, A. Ubiquitomics: An overview and future. Biomolecules 2020, 10, 1453. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 enzymes: More than just middle men. Cell Res. 2016, 26, 423–440. [Google Scholar] [CrossRef] [PubMed]
- Vlachostergios, P.J.; Papandreou, C.N. The role of the small ubiquitin-related modifier (SUMO) pathway in prostate cancer. Biomolecules 2012, 2, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Bawa, T.; Lee, P.; Gong, L.; Yeh, E.T.H. Role of desumoylation in the development of prostate cancer. Neoplasia 2006, 8, 667–676. [Google Scholar] [CrossRef]
- Nishida, T.; Yasuda, H. PIAS1 and PIASxα function as SUMO-E3 ligases toward androgen receptor and repress androgen receptor-dependent transcription. J. Biol. Chem. 2002, 277, 41311–41317. [Google Scholar] [CrossRef]
- Zheng, Z.; Cai, C.; Omwancha, J.; Chen, S.Y.; Baslan, T.; Shemshedini, L. SUMO-3 enhances androgen receptor transcriptional activity through a sumoylation-independent mechanism in prostate cancer cells. J. Biol. Chem. 2006, 281, 4002–4012. [Google Scholar] [CrossRef]
- Yang, N.; Liu, S.; Qin, T.; Liu, X.; Watanabe, N.; Mayo, K.H.; Li, J.; Li, X. SUMO3 modification by PIAS1 modulates androgen receptor cellular distribution and stability. Cell Commun. Signal. 2019, 17, 1–16. [Google Scholar] [CrossRef]
- Sutinen, P.; Malinen, M.; Heikkinen, S.; Palvimo, J.J. SUMOylation modulates the transcriptional activity of androgen receptor in a target gene and pathway selective manner. Nucleic Acids Res. 2014, 42, 8310–8319. [Google Scholar] [CrossRef]
- Rytinki, M.; Kaikkonen, S.; Sutinen, P.; Paakinaho, V.; Rahkama, V.; Palvimo, J.J. Dynamic SUMOylation Is Linked to the Activity Cycles of Androgen Receptor in the Cell Nucleus. Mol. Cell. Biol. 2012, 32, 4195–4205. [Google Scholar] [CrossRef]
- Mukherjee, S.; Cruz-Rodríguez, O.; Bolton, E.; Iñiguez-Lluhi, J.A. The in vivo role of androgen receptor SUMOylation as revealed by androgen insensitivity syndrome and prostate cancer mutations targeting the proline/glycine residues of synergy control motifs. J. Biol. Chem. 2012, 287, 31195–31206. [Google Scholar] [CrossRef]
- Huang, J.; Yan, J.; Zhang, J.; Zhu, S.; Wang, Y.; Shi, T.; Zhu, C.; Chen, C.; Liu, X.; Cheng, J.; et al. SUMO1 modification of PTEN regulates tumorigenesis by controlling its association with the plasma membrane. Nat. Commun. 2012, 3, 911. [Google Scholar] [CrossRef]
- Ashikari, D.; Takayama, K.; Tanaka, T.; Suzuki, Y.; Obinata, D.; Fujimura, T.; Urano, T.; Takahashi, S.; Inoue, S. Androgen induces G3BP2 and SUMO-mediated p53 nuclear export in prostate cancer. Oncogene 2017, 36, 6272–6281. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.M.; Yang, W.H. Loss of SUMOylation on ATF3 inhibits proliferation of prostate cancer cells by modulating CCND1/2 activity. Int. J. Mol. Sci. 2013, 14, 8367–8380. [Google Scholar] [CrossRef]
- Sutinen, P.; Rahkama, V.; Rytinki, M.; Palvimo, J.J. Nuclear mobility and activity of FOXA1 with Androgen Receptor Are Regulated by SUMOylation. Mol. Endocrinol. 2014, 28, 1719–1728. [Google Scholar] [CrossRef]
- Belaguli, N.S.; Zhang, M.; Brunicardi, F.C.; Berger, D.H. Forkhead Box Protein A2 (FOXA2) Protein Stability and Activity Are Regulated by Sumoylation. PLoS ONE 2012, 7, e48019. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.R.; Liu, Y.; Wan, X.D.; Li, J.L.; Wu, M.; Zhang, Q.M.; Wu, D.L.; Zhao, X.; Wang, T.R. Sumoylation Negatively Regulates CSR1-Dependent Prostate Cancer Cell Death. Cell. Physiol. Biochem. 2018, 46, 1861–1867. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Na, Y.; Lee, M.H.; Seo, J.S.; Lee, Y.H.; Choi, K.C.; Choi, H.K.; Yoon, H.G. SUMOylation of TBL1 and TBLR1 promotes androgen-independent prostate cancer cell growth. Oncotarget 2016, 7, 41110–41122. [Google Scholar] [CrossRef][Green Version]
- Choi, H.J.; Park, J.H.; Park, J.H.; Lee, K.B.; Oh, S.M. Pc2-mediated SUMOylation of WWOX is essential for its suppression of DU145 prostate tumorigenesis. FEBS Lett. 2015, 589, 3977–3988. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.K.; Ji, M.L.; Hye, J.N.; Hee, J.C.; Jung, W.Y.; Lee, J.S.; Mi, H.K.; Kim, S.-I.; Chin, H.C.; Keun, I.K.; et al. SUMOylation of pontin chromatin-remodeling complex reveals a signal integration code in prostate cancer cells. Proc. Natl. Acad. Sci. USA 2007, 104, 20793–20798. [Google Scholar] [CrossRef]
- Xie, Y.; Liu, S.; Lu, W.; Yang, Q.; Williams, K.D.; Binhazim, A.A.; Carver, B.S.; Matusik, R.J.; Chen, Z. Slug regulates E-cadherin repression via p19Arf in prostate tumorigenesis. Mol. Oncol. 2014, 8, 1355–1364. [Google Scholar] [CrossRef]
- Gudey, S.K.; Sundar, R.; Heldin, C.H.; Bergh, A.; Landström, M. Pro-invasive properties of Snail1 are regulated by sumoylation in response to TGFβ stimulation in cancer. Oncotarget 2017, 8, 97703–97726. [Google Scholar] [CrossRef]
- Puhr, M.; Hoefer, J.; Neuwirt, H.; Eder, I.E.; Kern, J.; Schäfer, G.; Geley, S.; Heidegger, I.; Klocker, H.; Culig, Z. PIAS1 is a crucial factor for prostate cancer cell survival and a valid target in docetaxel resistant cells. Oncotarget 2014, 5, 12043–12056. [Google Scholar] [CrossRef]
- Toropainen, S.; Malinen, M.; Kaikkonen, S.; Rytinki, M.; Jääskeläinen, T.; Sahu, B.; Jänne, O.A.; Palvimo, J.J. SUMO ligase PIAS1 functions as a target gene selective androgen receptor coregulator on prostate cancer cell chromatin. Nucleic Acids Res. 2015, 43, 848–861. [Google Scholar] [CrossRef]
- Puhr, M.; Hoefer, J.; Eigentler, A.; Dietrich, D.; Van Leenders, G.; Uhl, B.; Hoogland, M.; Handle, F.; Schlick, B.; Neuwirt, H.; et al. PIAS1 is a determinant of poor survival and acts as a positive feedback regulator of AR signaling through enhanced AR stabilization in prostate cancer. Oncogene 2016. [Google Scholar] [CrossRef]
- Kaikkonen, S.; Jääskeläinen, T.; Karvonen, U.; Rytinki, M.M.; Makkonen, H.; Gioeli, D.; Paschal, B.M.; Palvimo, J.J. SUMO-specific protease 1 (SENP1) reverses the hormone-augmented SUMOylation of androgen receptor and modulates gene responses in prostate cancer cells. Mol. Endocrinol. 2009, 23, 292–307. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, H.; Wang, H.; Xiao, F.; Seth, P.; Xu, W.; Jia, Q.; Wu, C.; Yang, Y.; Wang, L. SUMO-specific cysteine protease 1 promotes epithelial mesenchymal transition of prostate cancer cells via regulating SMAD4 deSUMOylation. Int. J. Mol. Sci. 2017, 18, 808. [Google Scholar] [CrossRef] [PubMed]
- Bawa-Khalfe, T.; Cheng, J.; Lin, S.H.; Ittmann, M.M.; Yeh, E.T.H. SENP1 induces prostatic intraepithelial neoplasia through multiple mechanisms. J. Biol. Chem. 2010, 285, 25859–25866. [Google Scholar] [CrossRef]
- Wang, Q.; Xia, N.; Li, T.; Xu, Y.; Zou, Y.; Zuo, Y.; Fan, Q.; Bawa-Khalfe, T.; Yeh, E.T.H.; Cheng, J. SUMO-specific protease 1 promotes prostate cancer progression and metastasis. Oncogene 2013. [Google Scholar] [CrossRef]
- Zhu, H.; Ren, S.; Bitler, B.G.; Aird, K.M.; Tu, Z.; Skordalakes, E.; Zhu, Y.; Yan, J.; Sun, Y.; Zhang, R. SPOP E3 Ubiquitin Ligase Adaptor Promotes Cellular Senescence by Degrading the SENP7 deSUMOylase. Cell Rep. 2015, 13, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Tao, Y.; Li, M.; Che, T.; Qu, J. Protein acetylation and deacetylation: An important regulatory modification in gene transcription (Review). Exp. Ther. Med. 2020, 20, 2923–2940. [Google Scholar] [CrossRef] [PubMed]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 156–174. [Google Scholar] [CrossRef]
- Drazic, A.; Myklebust, L.M.; Ree, R.; Arnesen, T. The world of protein acetylation. Biochim. Biophys. Acta-Proteins Proteomics. 2016, 1864, 1372–1401. [Google Scholar] [CrossRef] [PubMed]
- Cang, S.; Xu, X.; Ma, Y.; Liu, D.; Chiao, J.W. Hypoacetylation, hypomethylation, and dephosphorylation of H2B histones and excessive histone deacetylase activity in DU-145 prostate cancer cells. J. Hematol. Oncol. 2016, 9, 3. [Google Scholar] [CrossRef]
- Cang, S.; Feng, J.; Konno, S.; Han, L.; Liu, K.; Sharma, S.C.; Choudhury, M.; Chiao, J.W. Deficient histone acetylation and excessive deacetylase activity as epigenomic marks of prostate cancer cells. Int. J. Oncol. 2009, 35, 1417–1422. [Google Scholar] [CrossRef]
- Seligson, D.B.; Horvath, S.; Shi, T.; Yu, H.; Tze, S.; Grunstein, M.; Kurdistani, S.K. Global histone modification patterns predict risk of prostate cancer recurrence. Nature 2005. [Google Scholar] [CrossRef] [PubMed]
- Bianco-Miotto, T.; Chiam, K.; Buchanan, G.; Jindal, S.; Day, T.K.; Thomas, M.; Pickering, M.A.; O’Loughlin, M.A.; Ryan, N.K.; Raymond, W.A.; et al. Global levels of specific histone modifications and an epigenetic gene signature predict prostate cancer progression and development. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2611–2622. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.A.; Watson, C.J.; McCrohan, A.M.; Woodfine, K.; Tosetto, M.; McDaid, J.; Gallagher, E.; Betts, D.; Baugh, J.; O’Sullivan, J.; et al. Generation of an epigenetic signature by chronic hypoxia in prostate cells. Hum. Mol. Genet. 2009, 18, 3594–3604. [Google Scholar] [CrossRef]
- Valdés-Mora, F.; Gould, C.M.; Colino-Sanguino, Y.; Qu, W.; Song, J.Z.; Taylor, K.M.; Buske, F.A.; Statham, A.L.; Nair, S.S.; Armstrong, N.J.; et al. Acetylated histone variant H2A.Z is involved in the activation of neo-enhancers in prostate cancer. Nat. Commun. 2017, 8, 1–17. [Google Scholar] [CrossRef]
- Pathak, R.; Philizaire, M.; Mujtaba, S. Dichotomy in the epigenetic mark lysine acetylation is critical for the proliferation of prostate cancer cells. Cancers 2015, 7, 1622–1642. [Google Scholar] [CrossRef]
- Wen, S.; He, Y.; Wang, L.; Zhang, J.; Quan, C.; Niu, Y.; Huang, H. Aberrant activation of super enhancer and choline metabolism drive antiandrogen therapy resistance in prostate cancer. Oncogene 2020, 1–16. [Google Scholar] [CrossRef]
- Takeda, D.Y.; Spisák, S.; Seo, J.H.; Bell, C.; O’Connor, E.; Korthauer, K.; Ribli, D.; Csabai, I.; Solymosi, N.; Szállási, Z.; et al. A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer. Cell 2018, 174, 422–432.e13. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, Z.; Guo, J.; Li, Y.; Bavarva, J.H.; Qian, C.; Brahimi-Horn, M.C.; Tan, D.; Liu, W. Inactivation of androgen-induced regulator ARD1 inhibits androgen receptor acetylation and prostate tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 3053–3058. [Google Scholar] [CrossRef] [PubMed]
- DePaolo, J.S.; Wang, Z.; Guo, J.; Zhang, G.; Qian, C.; Zhang, H.; Zabaleta, J.; Liu, W. Acetylation of androgen receptor by ARD1 promotes dissociation from HSP90 complex and prostate tumorigenesis. Oncotarget 2016, 7, 71417–71428. [Google Scholar] [CrossRef]
- Fu, M.; Rao, M.; Wang, C.; Sakamaki, T.; Wang, J.; Di Vizio, D.; Zhang, X.; Albanese, C.; Balk, S.; Chang, C.; et al. Acetylation of Androgen Receptor Enhances Coactivator Binding and Promotes Prostate Cancer Cell Growth. Mol. Cell. Biol. 2003, 23, 8563–8575. [Google Scholar] [CrossRef]
- Jia, L.; Berman, B.P.; Jariwala, U.; Yan, X.; Cogan, J.P.; Walkers, A.; Chen, T.; Buchanan, G.; Frenkel, B.; Coetzee, G.A. Genomic androgen receptor-occupied regions with different functions, defined by histone acetylation, coregulators and transcriptional capacity. PLoS ONE 2008, 3, e3645. [Google Scholar] [CrossRef]
- Urbanucci, A.; Barfeld, S.J.; Kytölä, V.; Itkonen, H.M.; Coleman, I.M.; Vodák, D.; Sjöblom, L.; Sheng, X.; Tolonen, T.; Minner, S.; et al. Androgen Receptor Deregulation Drives Bromodomain-Mediated Chromatin Alterations in Prostate Cancer. Cell Rep. 2017, 19, 2045–2059. [Google Scholar] [CrossRef]
- Shao, G.; Liu, Y.; Ma, T.; Zhang, L.; Yuan, M.; Zhao, S. GCN5 inhibition prevents IL-6-induced prostate cancer metastases through PI3K/PTEN/Akt signaling by inactivating Egr-1. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Sun, H.; Zou, R.; Zhou, T.; Wang, S.; Sun, S.; Tong, C.; Luo, H.; Li, Y.; Li, Z.; et al. MDC1 functionally identified as an androgen receptor co-activator participates in suppression of prostate cancer. Nucleic Acids Res. 2015, 43, 4893–4908. [Google Scholar] [CrossRef]
- Lv, L.; Li, D.; Zhao, D.; Lin, R.; Chu, Y.; Zhang, H.; Zha, Z.; Liu, Y.; Li, Z.; Xu, Y.; et al. Acetylation Targets the M2 Isoform of Pyruvate Kinase for Degradation through Chaperone-Mediated Autophagy and Promotes Tumor Growth. Mol. Cell 2011, 42, 719–730. [Google Scholar] [CrossRef]
- Ding, L.; Chen, S.; Liu, P.; Pan, Y.; Zhong, J.; Regan, K.M.; Wang, L.; Yu, C.; Rizzardi, A.; Cheng, L.; et al. CBP loss cooperates with PTEN haploinsufficiency to drive prostate cancer: Implications for epigenetic therapy. Cancer Res. 2014, 74, 2050–2061. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; He, D.; Cheng, L.; Huang, C.; Zhang, Y.; Rao, X.; Kong, Y.; Li, C.; Zhang, Z.; Liu, J.; et al. p300/CBP inhibition enhances the efficacy of programmed death-ligand 1 blockade treatment in prostate cancer. Oncogene 2020, 39, 3939–3951. [Google Scholar] [CrossRef]
- Jin, L.; Garcia, J.; Chan, E.; De La Cruz, C.; Segal, E.; Merchant, M.; Kharbanda, S.; Raisner, R.; Haverty, P.M.; Modrusan, Z.; et al. Therapeutic targeting of the CBP/p300 bromodomain blocks the growth of castration-resistant prostate cancer. Cancer Res. 2017, 77, 5564–5575. [Google Scholar] [CrossRef]
- Gang, X.; Yang, Y.; Zhong, J.; Jiang, K.; Pan, Y.; Jeffrey Karnes, R.; Zhang, J.; Xu, W.; Wang, G.; Huang, H. P300 acetyltransferase regulates fatty acid synthase expression, lipid metabolism and prostate cancer growth. Oncotarget 2016, 7, 15135–15149. [Google Scholar] [CrossRef]
- Zhong, J.; Ding, L.; Bohrer, L.R.; Pan, Y.; Liu, P.; Zhang, J.; Sebo, T.J.; Karnes, R.J.; Tindall, D.J.; Van Deursen, J.; et al. P300 acetyltransferase regulates androgen receptor degradation and pten-deficient prostate tumorigenesis. Cancer Res. 2014, 74, 1870–1880. [Google Scholar] [CrossRef]
- Knowell, A.E.; Patel, D.; Morton, D.J.; Sharma, P.; Glymph, S.; Chaudhary, J. Id4 dependent acetylation restores mutant-p53 transcriptional activity. Mol. Cancer 2013, 12, 161. [Google Scholar] [CrossRef] [PubMed]
- Ianculescu, I.; Wu, D.Y.; Siegmund, K.D.; Stallcup, M.R. Selective roles for cAMP response element-binding protein binding protein and p300 protein as coregulators for androgen-regulated gene expression in advanced prostate cancer cells. J. Biol. Chem. 2012, 287, 4000–4013. [Google Scholar] [CrossRef] [PubMed]
- Santer, F.R.; Höschele, P.P.S.; Su, J.O.; Erb, H.H.H.; Bouchal, J.; Cavarretta, I.T.; Parson, W.; Meyers, D.J.; Cole, P.A.; Culig, Z. Inhibition of the acetyltransferases p300 and CBP reveals a targetable function for p300 in the survival and invasion pathways of prostate cancer cell lines. Mol. Cancer Ther. 2011, 10, 1644–1655. [Google Scholar] [CrossRef]
- Bouchal, J.; Santer, F.R.; Höschele, P.P.S.; Tomastikova, E.; Neuwirt, H.; Culig, Z. Transcriptional coactivators p300 and CBP stimulate estrogen receptor-beta signaling and regulate cellular events in prostate cancer. Prostate 2011. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.O.; Chun, J.Y.; Nadiminty, N.; Lou, W.; Feng, S.; Gao, A.C. Interleukin-4 activates androgen receptor through CBP/p. Prostate 2009, 69, 126–132. [Google Scholar] [CrossRef]
- Heemers, H.V.; Debes, J.D.; Tindall, D.J. The role of the transcriptional coactivator p300 in prostate cancer progression. Adv. Exp. Med. Biol. 2008, 617, 535–540. [Google Scholar] [CrossRef]
- Yuan, Z.L.; Guan, Y.J.; Chatterjee, D.; Chin, Y.E. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science (80-) 2005, 307, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.J.; Zhang, J.; Ellis, L.M.; Semenza, G.L.; Evans, D.B.; Watowich, S.S.; Gallick, G.E. HIF-1α, STAT3, CBP/p300 and Ref-1/APE are components of a transcriptional complex that regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas. Oncogene 2005, 24, 3110–3120. [Google Scholar] [CrossRef] [PubMed]
- Debes, J.D.; Sebo, T.J.; Lohse, C.M.; Murphy, L.M.; Haugen, D.A.L.; Tindall, D.J. p300 in Prostate Cancer Proliferation and Progression. Cancer Res. 2003, 63, 7638–7640. [Google Scholar] [PubMed]
- Debes, J.D.; Schmidt, L.J.; Huang, H.; Tindall, D.J. p300 mediates androgen-independent transactivation of the androgen receptor by interleukin. Cancer Res. 2002, 62, 5632–5636. [Google Scholar]
- Fu, M.; Wang, C.; Reutens, A.T.; Wang, J.; Angeletti, R.H.; Siconolfi-Baez, L.; Ogryzko, V.; Avantaggiati, M.L.; Pestell, R.G. p300 and p300/cAMP-response element-binding protein-associated factor acetylate the androgen receptor at sites governing hormone-dependent transactivation. J. Biol. Chem. 2000, 275, 20853–20860. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Lee, D.H. KAT5 Negatively regulates the proliferation of prostate cancer LNCaP cells via the caspase 3-dependent apoptosis pathway. Anim. Cells Syst. 2019, 23, 253–259. [Google Scholar] [CrossRef]
- Xie, X.; Xu, Z.; Wang, C.; Fang, C.; Zhao, J.; Xu, L.; Qian, X.; Dai, J.; Sun, F.; Xu, D.; et al. Tip60 is associated with resistance to X-ray irradiation in prostate cancer. FEBS Open Bio. 2018, 8, 271–278. [Google Scholar] [CrossRef]
- Gao, C.; Bourke, E.; Scobie, M.; Famme, M.A.; Koolmeister, T.; Helleday, T.; Eriksson, L.A.; Lowndes, N.F.; Brown, J.A.L. Rational design and validation of a Tip60 histone acetyltransferase inhibitor. Sci. Rep. 2014, 4, 5372. [Google Scholar] [CrossRef]
- Judes, G.; Rifaï, K.; Ngollo, M.; Daures, M.; Bignon, Y.J.; Penault-Llorca, F.; Bernard-Gallon, D. A bivalent role of TIP60 histone acetyl transferase in human cancer. Epigenomics 2015, 7, 1351–1363. [Google Scholar] [CrossRef]
- Lee, M.T.; Leung, Y.K.; Chung, I.; Tarapore, P.; Ho, S.M. Estrogen receptor β (ERβ1) transactivation is differentially modulated by the transcriptional coregulator Tip60 in a cis-acting element-dependent manner. J. Biol. Chem. 2013, 288, 25038–25052. [Google Scholar] [CrossRef]
- He, W.; Zhang, M.G.; Wang, X.J.; Zhong, S.; Shao, Y.; Zhu, Y.; Shen, Z.J. KAT5 and KAT6B are in positive regulation on cell proliferation of prostate cancer through PI3K-AKT signaling. Int. J. Clin. Exp. Pathol. 2013, 6, 2864–2871. [Google Scholar] [PubMed]
- Coffey, K.; Blackburn, T.J.; Cook, S.; Golding, B.T.; Griffin, R.J.; Hardcastle, I.R.; Hewitt, L.; Huberman, K.; McNeill, H.V.; Newell, D.R.; et al. Characterisation of a Tip60 Specific Inhibitor, NU9056, in Prostate Cancer. PLoS ONE 2012, 7, e45539. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Yokomizo, A.; Masubuchi, D.; Tada, Y.; Inokuchi, J.; Eto, M.; Uchiumi, T.; Fujimoto, N.; Naito, S. Tip60 promotes prostate cancer cell proliferation by translocation of androgen receptor into the nucleus. Prostate 2010. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, B.; Cai, L.; Choi, H.J.; Ohgi, K.A.; Tran, C.; Chen, C.; Chung, C.H.; Huber, O.; Rose, D.W.; et al. Transcriptional regulation of a metastasis suppressor gene by Tip60 and β-catenin complexes. Nature 2005. [Google Scholar] [CrossRef] [PubMed]
- Halkidou, K.; Gnanapragasam, V.J.; Mehta, P.B.; Logan, I.R.; Brady, M.E.; Cook, S.; Leung, H.Y.; Neal, D.E.; Robson, C.N. Expression of Tip60, an androgen receptor coactivator, and its role in prostate cancer development. Oncogene 2003, 22, 2466–2477. [Google Scholar] [CrossRef]
- Gaughan, L.; Logan, I.R.; Cook, S.; Neal, D.E.; Robson, C.N. Tip60 and histone deacetylase 1 regulate androgen receptor activity through changes to the acetylation status of the receptor. J. Biol. Chem. 2002, 277, 25904–25913. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Yu, J.; Abdulkadir, S.A.; Chakravarti, D. KAT8 regulates androgen signaling in prostate cancer cells. Mol. Endocrinol. 2016, 30, 925–936. [Google Scholar] [CrossRef]
- Jaganathan, A.; Chaurasia, P.; Xiao, G.Q.; Philizaire, M.; Lv, X.; Yao, S.; Burnstein, K.L.; Liu, D.P.; Levine, A.C.; Mujtaba, S. Coactivator MYST1 regulates nuclear factor-κB and androgen receptor functions during proliferation of prostate cancer cells. Mol. Endocrinol. 2014, 28, 872–885. [Google Scholar] [CrossRef]
- Katoh, H.; Qin, Z.S.; Liu, R.; Wang, L.; Li, W.; Li, X.; Wu, L.; Du, Z.; Lyons, R.; Liu, C.G.; et al. FOXP3 orchestrates H4K16 acetylation and H3K4 trimethylation for activation of multiple genes by recruiting MOF and causing displacement of PLU-1. Mol. Cell 2011, 44, 770–784. [Google Scholar] [CrossRef]
- Shankar, E.; Pandey, M.; Verma, S.; Abbas, A.; Candamo, M.; Kanwal, R.; Shukla, S.; MacLennan, G.T.; Gupta, S. Role of class I histone deacetylases in the regulation of maspin expression in prostate cancer. Mol. Carcinog. 2020, 59, 955–966. [Google Scholar] [CrossRef]
- Ma, J.B.; Bai, J.Y.; Zhang, H.B.; Jia, J.; Shi, Q.; Yang, C.; Wang, X.; He, D.; Guo, P. KLF5 inhibits STAT3 activity and tumor metastasis in prostate cancer by suppressing IGF1 transcription cooperatively with HDAC. Cell Death Dis. 2020, 11, 466. [Google Scholar] [CrossRef]
- Kim, N.H.; Kim, S.N.; Kim, Y.K. Involvement of HDAC1 in E-cadherin expression in prostate cancer cells; its implication for cell motility and invasion. Biochem. Biophys. Res. Commun. 2011, 404, 915–921. [Google Scholar] [CrossRef]
- Gaughan, L.; Logan, I.R.; Neal, D.E.; Robson, C.N. Regulation of androgen receptor and histone deacetylase 1 by Mdm2-mediated ubiquitylation. Nucleic Acids Res. 2005, 33, 13–26. [Google Scholar] [CrossRef]
- Welsbie, D.S.; Xu, J.; Chen, Y.; Borsu, L.; Scher, H.I.; Rosen, N.; Sawyers, C.L. Histone deacetylases are required for androgen receptor function in hormone-sensitive and castrate-resistant prostate cancer. Cancer Res. 2009, 69, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Tse, A.K.W.; Li, P.; Ma, Q.; Xiang, S.; Nicosia, S.V.; Seto, E.; Zhang, X.; Bai, W. Inhibition of androgen receptor activity by histone deacetylase 4 through receptor SUMOylation. Oncogene 2011, 30, 2207–2218. [Google Scholar] [CrossRef]
- Ren, G.; Zhang, G.; Dong, Z.; Liu, Z.; Li, L.; Feng, Y.; Su, D.; Zhang, Y.; Huang, B.; Lu, J. Recruitment of HDAC4 by transcription factor YY1 represses HOXB13 to affect cell growth in AR-negative prostate cancers. Int. J. Biochem. Cell Biol. 2009, 41, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Corno, C.; Arrighetti, N.; Ciusani, E.; Corna, E.; Carenini, N.; Zaffaroni, N.; Gatti, L.; Perego, P. Synergistic Interaction of Histone Deacetylase 6- and MEK-Inhibitors in Castration-Resistant Prostate Cancer Cells. Front. Cell Dev. Biol. 2020, 8, 610. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Huang, Y.; Liang, X.; Jiang, F.; He, Y.; Li, T.; Xu, G.; Zhao, H.; Yang, W.; Jiang, G.; et al. Metastatic prostate cancer-associated P62 inhibits autophagy flux and promotes epithelial to mesenchymal transition by sustaining the level of HDAC6. Prostate 2018, 78, 426–434. [Google Scholar] [CrossRef]
- Ai, J.; Wang, Y.; Dar, J.A.; Liu, J.; Liu, L.; Nelson, J.B.; Wang, Z. HDAC6 regulates androgen receptor hypersensitivity and nuclear localization via modulating Hsp90 acetylation in castration-resistant prostate cancer. Mol. Endocrinol. 2009, 23, 1963–1972. [Google Scholar] [CrossRef]
- Bakin, R.E.; Jung, M.O. Cytoplasmic sequestration of HDAC7 from mitochondrial and nuclear compartments upon initiation of apoptosis. J. Biol. Chem. 2004, 279, 51218–51225. [Google Scholar] [CrossRef]
- Deubzer, H.E.; Schier, M.C.; Oehme, I.; Lodrini, M.; Haendler, B.; Sommer, A.; Witt, O. HDAC11 is a novel drug target in carcinomas. Int. J. Cancer 2012, 132, 2200–2208. [Google Scholar] [CrossRef]
- Muscolini, M.; Castiello, L.; Palermo, E.; Zevini, A.; Ferrari, M.; Olagnier, D.; Hiscott, J. SIRT1 Modulates the Sensitivity of Prostate Cancer Cells to Vesicular Stomatitis Virus Oncolysis. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhang, Q.; Meng, Q.; Zong, C.; Liang, L.; Yang, X.; Lin, R.; Liu, Y.; Zhou, Y.; Zhang, H.; et al. Mesenchymal stem cells overexpressing Sirt1 inhibit prostate cancer growth by recruiting natural killer cells and macrophages. Oncotarget 2016, 7, 71112–71122. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Sharad, S.; Petrovics, G.; Mohamed, A.; Dobi, A.; Sreenath, T.L.; Srivastava, S.; Biswas, R. Loss of miR-449a in ERG-associated prostate cancer promotes the invasive phenotype by inducing SIRT1. Oncotarget 2016, 7, 22791–22806. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.; Wang, Z.; Chen, W.Y. SIRT1 and LSD1 competitively regulate KU70 functions in DNA repair and mutation acquisition in cancer cells. Oncotarget 2016, 7, 50195–50214. [Google Scholar] [CrossRef]
- Cui, Y.; Li, J.; Zheng, F.; Ouyang, Y.; Chen, X.; Zhang, L.; Chen, Y.; Wang, L.; Mu, S.; Zhang, H. Effect of SIRT1 gene on epithelial-mesenchymal transition of human prostate cancer PC-3 cells. Med. Sci. Monit. 2016, 22, 380–386. [Google Scholar] [CrossRef]
- Sun, L.; Kokura, K.; Izumi, V.; Koomen, J.M.; Seto, E.; Chen, J.; Fang, J. MPP 8 and SIRT 1 crosstalk in E-cadherin gene silencing and epithelial–mesenchymal transition. EMBO Rep. 2015, 16, 689–699. [Google Scholar] [CrossRef]
- Baptista, T.; Graça, I.; Sousa, E.J.; Oliveira, A.I.; Costa, N.R.; Costa-Pinheiro, P.; Amado, F.; Henrique, R.; Jerónimo, C. Regulation of histone H2A.Z expression is mediated by sirtuin 1 in prostate cancer. Oncotarget 2013, 4, 1673–1685. [Google Scholar] [CrossRef]
- Lovaas, J.D.; Zhu, L.; Chiao, C.Y.; Byles, V.; Faller, D.V.; Dai, Y. SIRT1 enhances matrix metalloproteinase-2 expression and tumor cell invasion in prostate cancer cells. Prostate 2013. [Google Scholar] [CrossRef]
- Byles, V.; Zhu, L.; Lovaas, J.D.; Chmilewski, L.K.; Wang, J.; Faller, D.V.; Dai, Y. SIRT1 induces EMT by cooperating with EMT transcription factors and enhances prostate cancer cell migration and metastasis. Oncogene 2012, 31, 4619–4629. [Google Scholar] [CrossRef]
- Nakane, K.; Fujita, Y.; Terazawa, R.; Atsumi, Y.; Kato, T.; Nozawa, Y.; Deguchi, T.; Ito, M. Inhibition of cortactin and SIRT1 expression attenuates migration and invasion of prostate cancer DU145 cells. Int. J. Urol. 2012. [Google Scholar] [CrossRef]
- Powell, M.J.; Casimiro, M.C.; Cordon-Cardo, C.; He, X.; Yeow, W.S.; Wang, C.; McCue, P.A.; McBurney, M.W.; Pestell, R.G. Disruption of a Sirt1-dependent autophagy checkpoint in the prostate results in prostatic intraepithelial neoplasia lesion formation. Cancer Res. 2011, 71, 964–975. [Google Scholar] [CrossRef]
- Kang, H.; Suh, J.Y.; Jung, Y.S.; Jung, J.W.; Kim, M.K.; Chung, J.H. Peptide switch is essential for Sirt1 deacetylase activity. Mol. Cell 2011, 44, 203–213. [Google Scholar] [CrossRef]
- Jung-Hynes, B.; Nihal, M.; Zhong, W.; Ahmad, N. Role of sirtuin histone deacetylase SIRT1 in prostate cancer: A target for prostate cancer management via its inhibition? J. Biol. Chem. 2009. [Google Scholar] [CrossRef] [PubMed]
- Jung-Hynes, B.; Ahmad, N. Role of p53 in the anti-proliferative effects of Sirt1 inhibition in prostate cancer cells. Cell Cycle 2009, 8, 1478–1483. [Google Scholar] [CrossRef]
- Kojima, K.; Ohhashi, R.; Fujita, Y.; Hamada, N.; Akao, Y.; Nozawa, Y.; Deguchi, T.; Ito, M. A role for SIRT1 in cell growth and chemoresistance in prostate cancer PC3 and DU145 cells. Biochem. Biophys. Res. Commun. 2008, 373, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Ngo, D.; Forman, L.W.; Qin, D.C.; Jacob, J.; Faller, D.V. Sirtuin 1 is required for antagonist-induced transcriptional repression of androgen-responsive genes by the androgen receptor. Mol. Endocrinol. 2007, 21, 1807–1821. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Liu, M.; Sauve, A.A.; Jiao, X.; Zhang, X.; Wu, X.; Powell, M.J.; Yang, T.; Gu, W.; Avantaggiati, M.L.; et al. Hormonal Control of Androgen Receptor Function through SIRT1. Mol. Cell. Biol. 2006, 26, 8122–8135. [Google Scholar] [CrossRef]
- Yang, Y.; Hou, H.; Haller, E.M.; Nicosia, S.V.; Bai, W. Suppression of FOXO1 activity by FHL2 through SIRT1-mediated deacetylation. EMBO J. 2005, 24, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Damodaran, S.; Damaschke, N.; Gawdzik, J.; Yang, B.; Shi, C.; Allen, G.O.; Huang, W.; Denu, J.; Jarrard, D. Dysregulation of Sirtuin 2 (SIRT2) and histone H3K18 acetylation pathways associates with adverse prostate cancer outcomes. BMC Cancer 2017, 17, 874. [Google Scholar] [CrossRef]
- Sawant Dessai, A.; Palestino Dominguez, M.; Chen, U.-I.; Hasper, J.; Prechtl, C.; Yu, C.; Katsuta, E.; Dai, T.; Zhu, B.; Jung, S.Y.; et al. Transcriptional repression of SIRT3 potentiates mitochondrial aconitase activation to drive aggressive prostate cancer to the bone. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Fu, W.; Li, H.; Fu, H.; Zhao, S.; Shi, W.; Sun, M.; Li, Y. The SIRT3 and SIRT6 Promote Prostate Cancer Progression by Inhibiting Necroptosis-Mediated Innate Immune Response. J. Immunol. Res. 2020, 2020, 1–12. [Google Scholar] [CrossRef]
- Li, R.; Quan, Y.; Xia, W. SIRT3 inhibits prostate cancer metastasis through regulation of FOXO3A by suppressing Wnt/β-catenin pathway. Exp. Cell Res. 2018, 364, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Quan, Y.; Wang, N.; Chen, Q.; Xu, J.; Cheng, W.; Di, M.; Xia, W.; Gao, W.Q. SIRT3 inhibits prostate cancer by destabilizing oncoprotein c-MYC through regulation of the PI3K/Akt pathway. Oncotarget 2015, 6, 26494–26507. [Google Scholar] [CrossRef]
- Li, T.; Li, Y.; Liu, T.; Hu, B.; Li, J.; Liu, C.; Liu, T.; Li, F. Mitochondrial PAK6 inhibits prostate cancer cell apoptosis via the PAK6-SIRT4-ANT2 complex. Theranostics 2020, 10, 2571–2586. [Google Scholar] [CrossRef]
- Guan, J.; Jiang, X.; Gai, J.; Sun, X.; Zhao, J.; Li, J.; Li, Y.; Cheng, M.; Du, T.; Fu, L.; et al. Sirtuin 5 regulates the proliferation, invasion and migration of prostate cancer cells through acetyl-CoA acetyltransferase. J. Cell. Mol. Med. 2020, 24, 14039–14049. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Seto, E.; Zhang, J. E2F1 enhances glycolysis through suppressing Sirt6 transcription in cancer cells. Oncotarget 2015, 6, 11252–11263. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xie, Q.R.; Wang, B.; Shao, J.; Zhang, T.; Liu, T.; Huang, G.; Xia, W. Inhibition of SIRT6 in prostate cancer reduces cell viability and increases sensitivity to chemotherapeutics. Protein Cell 2013, 4, 702–710. [Google Scholar] [CrossRef]
- Ding, M.; Jiang, C.Y.; Zhang, Y.; Zhao, J.; Han, B.M.; Xia, S.J. SIRT7 depletion inhibits cell proliferation and androgen-induced autophagy by suppressing the AR signaling in prostate cancer. J. Exp. Clin. Cancer Res. 2020, 39, 1–18. [Google Scholar] [CrossRef]
- Haider, R.; Massa, F.; Kaminski, L.; Clavel, S.; Djabari, Z.; Robert, G.; Laurent, K.; Michiels, J.F.; Durand, M.; Ricci, J.E.; et al. Sirtuin 7: A new marker of aggressiveness in prostate cancer. Oncotarget 2017, 8, 77309–77316. [Google Scholar] [CrossRef]
- Malik, S.; Villanova, L.; Tanaka, S.; Aonuma, M.; Roy, N.; Berber, E.; Pollack, J.R.; Michishita-Kioi, E.; Chua, K.F. SIRT7 inactivation reverses metastatic phenotypes in epithelial and mesenchymal tumors. Sci. Rep. 2015, 5, 9841. [Google Scholar] [CrossRef] [PubMed]
- Morton, D.J.; Patel, D.; Joshi, J.; Hunt, A.; Knowell, A.E.; Chaudhary, J. ID4 regulates transcriptional activity of wild type and mutant p53 via K373 acetylation. Oncotarget 2016, 8, 2536–2549. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Tenniswood, M. Site-specific acetylation of p53 directs selective transcription complex assembly. J. Biol. Chem. 2007, 282, 4765–4771. [Google Scholar] [CrossRef] [PubMed]
- Kai, L.; Samuel, S.K.; Levenson, A.S. Resveratrol enhances p53 acetylation and apoptosis in prostate cancer by inhibiting MTA1/NuRD complex. Int. J. Cancer 2010, 126, 1538–1548. [Google Scholar] [CrossRef]
- Dhar, S.; Kumar, A.; Li, K.; Tzivion, G.; Levenson, A.S. Resveratrol regulates PTEN/Akt pathway through inhibition of MTA1/HDAC unit of the NuRD complex in prostate cancer. Biochim. Biophys. Acta-Mol. Cell Res. 2015, 1853, 265–275. [Google Scholar] [CrossRef]
- Li, J.; Zhang, B.; Liu, M.; Fu, X.; Ci, X.; Jun, A.; Fu, C.; Dong, G.; Wu, R.; Zhang, Z.; et al. Klf5 is crucial for androgen-ar signaling to transactivate genes and promote cell proliferation in prostate cancer cells. Cancers 2020, 12, 748. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gomez-Pinillos, A.; Loder, C.; Carrillo-De Santa Pau, E.; Qiao, R.; Unger, P.D.; Kurek, R.; Oddoux, C.; Melamed, J.; Gallagher, R.E.; et al. KLF6 loss of function in human prostate cancer progression is implicated in resistance to androgen deprivation. Am. J. Pathol. 2012, 181, 1007–1016. [Google Scholar] [CrossRef][Green Version]
- Zhang, B.; Ci, X.; Tao, R.; Ni, J.J.; Xuan, X.; King, J.L.; Xia, S.; Li, Y.; Frierson, H.F.; Lee, D.K.; et al. Klf5 acetylation regulates luminal differentiation of basal progenitors in prostate development and regeneration. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, B.; Xiang, L.; Xia, S.; Kucuk, O.; Deng, X.; Boise, L.H.; Dong, J.T. TGF-β causes docetaxel resistance in prostate cancer via the induction of Bcl-2 by acetylated KLF5 and protein stabilization. Theranostics 2020, 10, 7656–7670. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, B.; Wu, Q.; Ci, X.; Zhao, R.; Zhang, Z.; Xia, S.; Su, D.; Chen, J.; Ma, G.; et al. Interruption of KLF5 acetylation converts its function from tumor suppressor to tumor promoter in prostate cancer cells. Int. J. Cancer 2015. [Google Scholar] [CrossRef] [PubMed]
- Xing, C.; Fu, X.; Sun, X.; Guo, P.; Li, M.; Dong, J.T. Different Expression Patterns and Functions of Acetylated and Unacetylated Klf5 in the Proliferation and Differentiation of Prostatic Epithelial Cells. PLoS ONE 2013, 8, e65538. [Google Scholar] [CrossRef]
- Li, D.; Yea, S.; Dolios, G.; Martignetti, J.A.; Narla, G.; Wang, R.; Walsh, M.J.; Friedman, S.L. Regulation of Krüppel-like Factor 6 Tumor Suppressor Activity by Acetylation. Cancer Res. 2005, 65, 9216–9225. [Google Scholar] [CrossRef]
- Jiang, H.; Zhang, X.; Chen, X.; Aramsangtienchai, P.; Tong, Z.; Lin, H. Protein Lipidation: Occurrence, Mechanisms, Biological Functions, and Enabling Technologies. Chem. Rev. 2018, 118, 919–988. [Google Scholar] [CrossRef]
- De Piano, M.; Manuelli, V.; Zadra, G.; Otte, J.; Edqvist, P.H.D.; Pontén, F.; Nowinski, S.; Niaouris, A.; Grigoriadis, A.; Loda, M.; et al. Lipogenic signalling modulates prostate cancer cell adhesion and migration via modification of Rho GTPases. Oncogene 2020, 39, 3666–3679. [Google Scholar] [CrossRef]
- De Piano, M.; Manuelli, V.; Zadra, G.; Loda, M.; Muir, G.; Chandra, A.; Morris, J.; Van Hemelrijck, M.; Wells, C.M. Exploring a role for fatty acid synthase in prostate cancer cell migration. Small GTPases 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sabbisetti, V.; Di Napoli, A.; Seeley, A.; Amato, A.M.; O’Regan, E.; Ghebremichael, M.; Loda, M.; Signoretti, S. p63 promotes cell survival through fatty acid synthase. PLoS ONE 2009, 4, e5877. [Google Scholar] [CrossRef] [PubMed]
- Little, J.L.; Wheeler, F.B.; Fels, D.R.; Koumenis, C.; Kridel, S.J. Inhibition of fatty acid synthase induces endoplasmic reticulum stress in tumor cells. Cancer Res. 2007, 67, 1262–1269. [Google Scholar] [CrossRef] [PubMed]
- Coleman, D.T.; Bigelow, R.; Cardelli, J.A. Inhibition of fatty acid synthase by luteolin post-transcriptionally down-regulates c-Met expression independent of proteosomal/lysosomal degradation. Mol. Cancer Ther. 2009, 8, 214–224. [Google Scholar] [CrossRef]
- Han, C.; Yu, G.; Mao, Y.; Song, S.; Li, L.; Zhou, L.; Wang, Z.; Liu, Y.; Li, M.; Xu, B. LPCAT1 enhances castration resistant prostate cancer progression via increased mRNA synthesis and PAF production. PLoS ONE 2020, 15, e0240801. [Google Scholar] [CrossRef] [PubMed]
- Seshacharyulu, P.; Rachagani, S.; Muniyan, S.; Siddiqui, J.A.; Cruz, E.; Sharma, S.; Krishnan, R.; Killips, B.J.; Sheinin, Y.; Lele, S.M.; et al. FDPS cooperates with PTEN loss to promote prostate cancer progression through modulation of small GTPases/AKT axis. Oncogene 2019, 38, 5265–5280. [Google Scholar] [CrossRef]
- Virtanen, S.S.; Sandholm, J.; Yegutkin, G.; Kalervo Väänänen, H.; Härkönen, P.L. Inhibition of GGTase-I and FTase disrupts cytoskeletal organization of human PC-3 prostate cancer cells. Cell Biol. Int. 2010, 34, 815–826. [Google Scholar] [CrossRef]
- Weissenrieder, J.S.; Reilly, J.E.; Neighbors, J.D.; Hohl, R.J. Inhibiting geranylgeranyl diphosphate synthesis reduces nuclear androgen receptor signaling and neuroendocrine differentiation in prostate cancer cell models. Prostate 2019, 79, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Reilly, J.E.; Neighbors, J.D.; Hohl, R.J. Targeting protein geranylgeranylation slows tumor development in a murine model of prostate cancer metastasis. Cancer Biol. Ther. 2017. [Google Scholar] [CrossRef]
- Reilly, J.E.; Neighbors, J.D.; Tong, H.; Henry, M.D.; Hohl, R.J. Targeting geranylgeranylation reduces adrenal gland tumor burden in a murine model of prostate cancer metastasis. Clin. Exp. Metastasis 2015, 32, 555–566. [Google Scholar] [CrossRef]
- Moon, H.; Hill, M.M.; Roberts, M.J.; Gardiner, R.A.; Brown, A.J. Statins: Protectors or pretenders in prostate cancer? Trends Endocrinol. Metab. 2014, 25, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Cui, X.X.; Gao, Z.; Zhao, Y.; Lin, Y.; Shih, W.J.; Huang, M.T.; Liu, Y.; Rabson, A.; Reddy, B.; et al. Atorvastatin and celecoxib in combination inhibits the progression of androgen-dependent LNCaP xenograft prostate tumors to androgen independence. Cancer Prev. Res. 2010, 3, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Tao, W.; Wang, Y.; Bikow, J.; Lu, B.; Keating, A.; Verma, S.; Parker, T.G.; Han, R.; Wen, X.Y. Rosuvastatin, identified from a zebrafish chemical genetic screen for antiangiogenic compounds, suppresses the growth of prostate cancer. Eur. Urol. 2010, 58, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Kim, J.; Adam, R.M.; Solomon, K.R.; Freeman, M.R. Cholesterol targeting alters lipid raft composition and cell survival in prostate cancer cells and xenografts. J. Clin. Investig. 2005, 115, 959–968. [Google Scholar] [CrossRef]
- Yokomizo, A.; Shiota, M.; Kashiwagi, E.; Kuroiwa, K.; Tatsugami, K.; Inokuchi, J.; Takeuchi, A.; Naito, S. Statins reduce the androgen sensitivity and cell proliferation by decreasing the androgen receptor protein in prostate cancer cells. Prostate 2011. [Google Scholar] [CrossRef]
- Hoque, A.; Chen, H.; Xu, X.C. Statin induces apoptosis and cell growth arrest in prostate cancer cells. Cancer Epidemiol. Biomarkers Prev. 2008, 17, 88–94. [Google Scholar] [CrossRef]
- Parikh, A.; Childress, C.; Deitrick, K.; Lin, Q.; Rukstalis, D.; Yang, W. Statin-induced autophagy by inhibition of geranylgeranyl biosynthesis in prostate cancer PC3 cells. Prostate 2010, 70, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Smith, D.A.; Memarzadeh, S.; Lowell, C.A.; Cooper, J.A.; Witte, O.N. Differential transformation capacity of Src family kinases during the initiation of prostate cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 6579–6584. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Ingram, L.; Kim, S.; Beharry, Z.; Cooper, J.A.; Cai, H. Paracrine Fibroblast Growth Factor Initiates Oncogenic Synergy with Epithelial FGFR/Src Transformation in Prostate Tumor Progression. Neoplasia 2018, 20, 233–243. [Google Scholar] [CrossRef]
- Kim, S.; Yang, X.; Li, Q.; Wu, M.; Costyn, L.; Beharry, Z.; Bartlett, M.G.; Cai, H. Myristoylation of Src kinase mediates Src-induced and high-fat diet–accelerated prostate tumor progression in mice. J. Biol. Chem. 2017, 292, 18422–18433. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Alsaidan, O.A.; Goodwin, O.; Li, Q.; Sulejmani, E.; Han, Z.; Bai, A.; Albers, T.; Beharry, Z.; Zheng, Y.G.; et al. Blocking myristoylation of Src inhibits its kinase activity and suppresses prostate cancer progression. Cancer Res. 2017, 77, 6950–6962. [Google Scholar] [CrossRef]
- Adam, R.M.; Mukhopadhyay, N.K.; Kim, J.; Di Vizio, D.; Cinar, B.; Boucher, K.; Solomon, K.R.; Freeman, M.R. Cholesterol sensitivity of endogenous and myristoylated Akt. Cancer Res. 2007, 67, 6238–6246. [Google Scholar] [CrossRef]
- Razandi, M.; Pedram, A.; Levin, E.R. Heat Shock Protein 27 Is Required for Sex Steroid Receptor Trafficking to and Functioning at the Plasma Membrane. Mol. Cell. Biol. 2010, 30, 3249–3261. [Google Scholar] [CrossRef]
- Zhou, B.; Liu, L.; Reddivari, M.; Zhang, X.A. The palmitoylation of metastasis suppressor KAI1/CD82 is important for its motility- and invasiveness-inhibitory activity. Cancer Res. 2004, 64, 7455–7463. [Google Scholar] [CrossRef]
- Li, Q.; Alsaidan, O.A.; Ma, Y.; Kim, S.; Liu, J.; Albers, T.; Liu, K.; Beharry, Z.; Zhao, S.; Wang, F.; et al. Pharmacologically targeting the myristoylation of the scaffold protein FRS2 inhibits FGF/FGFR-mediated oncogenic signaling and tumor progression. J. Biol. Chem. 2018, 293, 6434–6448. [Google Scholar] [CrossRef] [PubMed]
- Pottier, C.; Fresnais, M.; Gilon, M.; Jérusalem, G.; Longuespée, R.; Sounni, N.E. Tyrosine kinase inhibitors in cancer: Breakthrough and challenges of targeted therapy. Cancers 2020, 12, 731. [Google Scholar] [CrossRef]
- Ojemuyiwa, M.A.; Madan, R.A.; Dahut, W.L. Tyrosine kinase inhibitors in the treatment of prostate cancer: Taking the next step in clinical development. Expert Opin. Emerg. Drugs 2014, 19, 459–470. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.S.; De Giorgi, U.; Rodrigues, D.N.; Massard, C.; Bracarda, S.; Font, A.; Arija, J.A.A.; Shih, K.C.; Radavoi, G.D.; Xu, N.; et al. Randomized phase II study evaluating AKT blockade with ipatasertib, in combination with abiraterone, in patients with metastatic prostate cancer with and without PTEN loss. Clin. Cancer Res. 2019, 25, 928–936. [Google Scholar] [CrossRef]
- Ramroop, J.R.; Stein, M.N.; Drake, J.M. Impact of phosphoproteomics in the era of precision medicine for prostate cancer. Front. Oncol. 2018, 8, 28. [Google Scholar] [CrossRef]
- Yang, W.; Freeman, M.R.; Kyprianou, N. Personalization of prostate cancer therapy through phosphoproteomics. Nat. Rev. Urol. 2018, 15, 483–497. [Google Scholar] [CrossRef]
- Yamada, Y.; Beltran, H. Clinical and Biological Features of Neuroendocrine Prostate Cancer. Curr. Oncol. Rep. 2021, 23, 1–10. [Google Scholar] [CrossRef]
- Beltran, H.; Oromendia, C.; Danila, D.C.; Montgomery, B.; Hoimes, C.; Szmulewitz, R.Z.; Vaishampayan, U.; Armstrong, A.J.; Stein, M.; Pinski, J.; et al. A phase II trial of the aurora kinase a inhibitor alisertib for patients with castration-resistant and neuroendocrine prostate cancer: Efficacy and biomarkers. Clin. Cancer Res. 2019, 25, 43–51. [Google Scholar] [CrossRef]
- VanDeusen, H.R.; Ramroop, J.R.; Morel, K.L.; Bae, S.Y.; Sheahan, A.V.; Sychev, Z.; Lau, N.A.; Cheng, L.C.; Tan, V.M.; Li, Z.; et al. Targeting RET Kinase in Neuroendocrine Prostate Cancer. Mol. Cancer Res. 2020, 18, 1176–1188. [Google Scholar] [CrossRef]
- Mateo, J.; McKay, R.; Abida, W.; Aggarwal, R.; Alumkal, J.; Alva, A.; Feng, F.; Gao, X.; Graff, J.; Hussain, M.; et al. Accelerating precision medicine in metastatic prostate cancer. Nat. Cancer 2020, 1, 1041–1053. [Google Scholar] [CrossRef]
- Ku, S.Y.; Gleave, M.E.; Beltran, H. Towards precision oncology in advanced prostate cancer. Nat. Rev. Urol. 2019, 16, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Haven, N.; Meeting, A. Proof-of-Concept with PROTACs in Prostate Cancer. Cancer Discov. 2020, 10, 1084. [Google Scholar] [CrossRef]
- van der Steen, T.; Tindall, D.J.; Huang, H. Posttranslational modification of the androgen receptor in prostate cancer. Int. J. Mol. Sci. 2013, 14, 14833–14859. [Google Scholar] [CrossRef] [PubMed]
- Coffey, K.; Robson, C.N. Regulation of the androgen receptor by post-translational modifications. J. Endocrinol. 2012, 215, 221–237. [Google Scholar] [CrossRef]
- Kumar, A.; Zhang, K.Y.J. Advances in the development of SUMO specific protease (SENP) inhibitors. Comput. Struct. Biotechnol. J. 2015, 13, 204–211. [Google Scholar] [CrossRef]
- Yang, Y.; Xia, Z.; Wang, X.; Zhao, X.; Sheng, Z.; Ye, Y.; He, G.; Zhou, L.; Zhu, H.; Xu, N.; et al. Small-molecule inhibitors targeting protein sumoylation as novel anticancer compounds. Mol. Pharmacol. 2018, 94, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Rana, Z.; Diermeier, S.; Hanif, M.; Rosengren, R.J. Understanding failure and improving treatment using HDAC inhibitors for prostate cancer. Biomedicines 2020, 8, 22. [Google Scholar] [CrossRef]
- He, Z.X.; Wei, B.F.; Zhang, X.; Gong, Y.P.; Ma, L.Y.; Zhao, W. Current development of CBP/p300 inhibitors in the last decade. Eur. J. Med. Chem. 2021, 209, 112861. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.F.; Campos, D.; Reis, C.A.; Gomes, C. Targeting Glycosylation: A New Road for Cancer Drug Discovery. Trends Cancer 2020, 6, 757–766. [Google Scholar] [CrossRef]
- Leslie, N.R.; Kriplani, N.; Hermida, M.A.; Alvarez-Garcia, V.; Wise, H.M. The PTEN protein: Cellular localization and post-translational regulation. Biochem. Soc. Trans. 2016, 44, 273–278. [Google Scholar] [CrossRef]
- Cocchiola, R.; Rubini, E.; Altieri, F.; Chichiarelli, S.; Paglia, G.; Romaniello, D.; Carissimi, S.; Giorgi, A.; Giamogante, F.; Macone, A.; et al. STAT3 post-translational modifications drive cellular signaling pathways in prostate cancer cells. Int. J. Mol. Sci. 2019, 20, 1815. [Google Scholar] [CrossRef]
- Lin, W.Y.; Luo, J.; Sun, Y.; Lin, C.Y.; Li, G.; Niu, Y.; Chang, C. ASC-J9 ® suppresses prostate cancer cell invasion via altering the sumoylation-phosphorylation of STAT3. Cancer Lett. 2018, 425, 21–30. [Google Scholar] [CrossRef]
- Padmanabhan, A.; Rao, V.; De Marzo, A.M.; Bieberich, C.J. Regulating NKX3.1 stability and function: Post-translational modifications and structural determinants. Prostate 2016, 76, 523–533. [Google Scholar] [CrossRef] [PubMed]



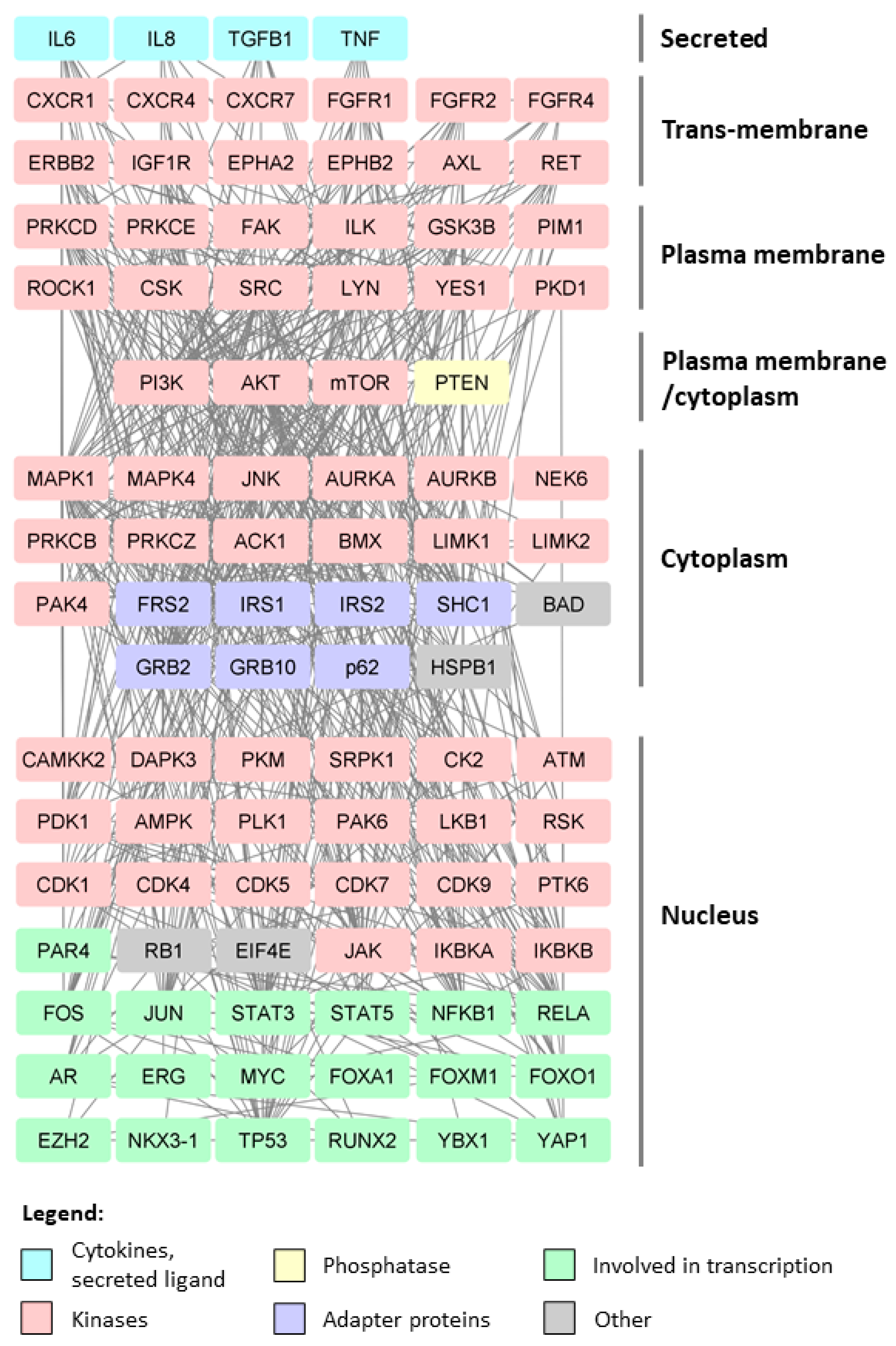{kind=link}
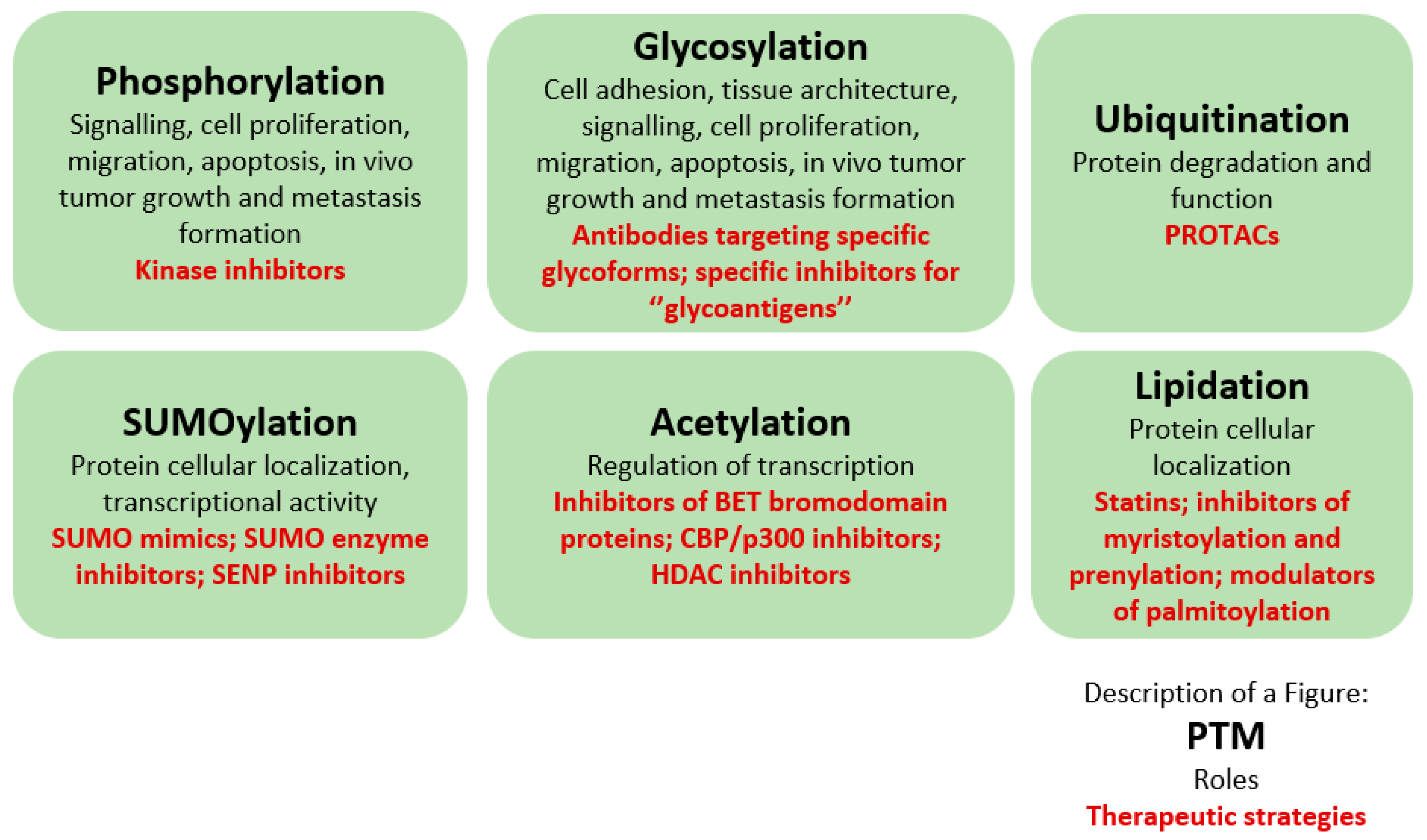{kind=link}
| PTM | Experimental Setting | Main Findings | Ref. |
|---|---|---|---|
| Phosphorylation | Comparative phosphoproteomics of differentially expressed kinases between the highly aggressive PC-3 and PC-3M cells. | PAK2, SLK, MST4, MAP2K2, and ARAF are kinases that are potentially associated with increased migration in PC-3M cells. | [19] |
| (Phospho)proteomic profiling of human prostate cancer (PCa)-associated fibroblasts. | PCa-associated fibroblasts-derived LOXL2 is an important mediator of intercellular communication within the prostate tumor microenvironment. | [20] | |
| Characterization of the ERG-regulated kinome. | TNIK is suggested as a potential therapeutic target. | [21] | |
| Phosphoproteome of treatment naive and metastatic CRPC tissue samples integrated with genomic and transcriptomic data. | Six major signaling pathways with phosphorylation of several key residues are significantly enriched in CRPC tumors; clinically relevant information (kinase target potential based on patient-specific networks) potentially suitable for patient stratification and targeted therapies in late stage PCa is provided. | [22] | |
| Analysis of global phosphoproteomic changes induced by fish oil in human PCa. | Pyruvate dehydrogenase alpha 1 is a target of omega-3 polyunsaturated fatty acids in human PCa. | [23] | |
| Phosphoproteomics data from mouse model of PCa progression [24] integrated with gene expression analysis and literature mining. | A total of 125 wild type kinases implicated in human PCa metastasis were selected for screen for in vivo metastatic ability; the RAF family, MERTK, and NTRK2 kinases drive PCa bone and visceral metastasis, and are highly expressed in human metastatic PCa tissues, potentially representing important therapeutic targets. | [25] | |
| Comparative phosphoproteome analysis of a PCa cell line, LNCaP, and an LNCaP-derived androgen-independent cell line, LNCaP-AI. | The phosphorylation level of THRAP3 is significantly lower in LNCaP-AI cells; nonphosphorylatable mutant form of THRAP3 and the phosphorylation-mimic form differ significantly in protein binding repertoire; many of the differentially interacting proteins were identified as being involved in RNA splicing and processing. | [26] | |
| Quantitative proteomic approach to compare protein phosphorylation in orthotopic xenograft tumors grown in either intact or castrated mice. | Changes in phosphorylation of YAP1 and PAK2 and their elevated levels in CRPC identified; YAP2 and PAK2 regulate cell colony formation and invasion in androgen-independent cells; PAK2 influences cell proliferation and mitotic timing; pharmacologic inhibitors of PAK2 and YAP1 are able to inhibit the growth of androgen-independent PC-3 xenografts. | [27] | |
| Phosphotyrosine peptide enrichment and quantitative mass spectrometry (MS) in oncogene(non-TK)-driven mouse model of PCa progression. | Elevated TK signaling (EGFR, EPHA2, JAK2, ABL1, and steroid receptor coactivator (SRC) tyrosine kinase activation) is recorded. | [24] | |
| Proteome analysis of Aurora-A substrates using small molecule inhibitor and reverse in-gel kinase assay in PC-3 cells. | NuMA becomes hypo-phosphorylated in vivo upon Aurora-A inhibition; mutation of three of these phospho-sites significantly diminishes cell proliferation and increases the rate of apoptosis; NuMA T1804A mutant mislocalizes to the cytoplasm in interphase nuclei in a punctate pattern. | [28] | |
| Phosphoproteomics of metastatic docetaxel-resistant PCa cell lines (DU145-Rx and PC-3-Rx). | Increased phosphorylation of FAK mediates chemoresistance in CRPC. | [29] | |
| Glycosylation | Proteomics analysis to determine the O-glycan profiles of PCa cells metastasized to bone (PC-3), brain (DU145), lymph node (LNCaP), and vertebra (VCaP) in comparison to immortalized RWPE-1 cells derived from normal prostatic tissue. | PCa cells exhibit an elevation of simple/short O-glycans, with a reduction of complex O-glycans, increased O-glycan sialylation, and decreased fucosylation. Core 1 sialylation is increased in all PCa cells. The expression of sialyl-3T antigen, which is the product of ST3Gal-I is increased. ST3Gal-I is associated with PC-3 cell proliferation, migration and apoptosis. Downregulation of ST3Gal-I reduces the tumor size in xenograft mouse model. | [30] |
| Comprehensive proteomic approaches of FUT8 overexpressing PCa cells. | Upregulation of EGFR and its downstream signaling; increased cell survival in androgen-depleted conditions. | [31] | |
| Extracellular vesicles (EV)-derived glycoproteins upon overexpression of FUT8 in PCa cells. | Reduced number of vesicles secreted by PCa cells; increase in the abundance of proteins associated with cell motility and PCa metastasis; altered glycans on select EV-derived glycoproteins. | [32] | |
| O-GlcNAc chromatin consensus motif imposed by OGT used as a bait for MS; combination with MYC chromatin immunoprecipitation (ChIP)-MS in PCa cells. | OGT is an essential mediator in androgen-independency, which is the major mechanism of PCa progression. | [33] | |
| Proteomics of androgen-dependent and androgen-resistant LAPC4 cells. | FUT8 is significantly overexpressed in the androgen-resistant LAPC4 cells; overexpression of FUT8 might be responsible for the decreased PSA expression in prostate cancer specimens. | [34] | |
| Cell surface Thomsen–Friedenreich (TF) antigen proteome profiling of metastatic PCa cells. | CD44, α2 integrin, β1 integrin, CD49f, CD133, CD59, EphA2, CD138, transferrin receptor and profilin express TF antigen; TF antigen positive prostate cancer cells form significantly more and larger prostaspheres under both non-differentiating and differentiating conditions and express higher levels of stem cell markers. | [35] | |
| Ubiquitination | Overexpression or depletion of USP22 in PCa cells and analysis of the ubiquitylome. | Depletion of USP22 sensitizes cells to genotoxic insult; analysis of the USP22-sensitive ubiquitylome identified the nucleotide excision repair protein, XPC, as a critical mediator of the USP22-mediated response to genotoxic insult. | [36] |
| Knockdown of E6AP in DU145 cells and analysis of a proteome. | Clusterin is a novel target of E6AP; concomitant knockdown of clusterin and E6AP partially restores cell growth. | [37] | |
| Changes in the ubiquitin landscape induced by prostate cancer–associated mutations of SPOP in immortalized prostate epithelial cells expressing endogenous SPOP. | DEK and TRIM24 are effector substrates consistently upregulated by SPOP mutants with decreases in ubiquitination and proteasomal degradation resulting from heteromeric complexes of wild type and mutant SPOP protein; DEK stabilization promotes prostate epithelial cell invasion. | [38] | |
| SUMOylation | Quantitative proteomics to identify SUMOylated proteins in SUMO stably transfected PC-3 cells. | More than 900 putative target proteins of SUMO are identified; mutation of newly identified SUMO modification sites of USP39 further promotes the proliferation-enhancing effect of USP39 on PCa cells. | [39] |
| Palmitoylation | Palmitoyl-proteomic analysis of large and small cancer-derived PCa EVs [40]. | STEAP1, STEAP2, and ABCC4 are identified as PCa-specific palmitoyl-proteins abundant in both EV populations; their localization in EVs is reduced upon inhibition of palmitoylation in the producing cells. | [40] |
| Palmitoyl proteomic analysis of breast and PCa cell lines, ±DHHC3 ablation. | Putative substrates include 22–28 antioxidant/redox-regulatory proteins and DHHC3 ablation elevates oxidative stress; DHHC3 ablation, in combination with chemotherapeutic drug treatment, elevates oxidative stress, with a greater than additive effect, and enhances the anti-growth effects of the chemotherapeutic agents; DHHC3 ablation synergizes with PARP inhibitor PJ-34, to decrease cell proliferation and increase oxidative stress. | [41] | |
| Proteomic experiments using clickable palmitate probe (Alk-C16) between three individual pairs of androgen-treated and non-treated LNCaP cells. | Androgen treatment significantly increased the palmitoylation level of eIF3L, which may be used as a biomarker for the diagnosis of early-stage PCa. | [42] | |
| LNCaP cells metabolically-labeled with Alk-C16, a palmitate probe and treated with R1881, an androgen, or DMSO after which palmitoylome profiling was performed. | Androgen treatment significantly increases the palmitoylation level of α-tubulin and Ras-related protein Rab-7a (Rab7a), which are essential for cell proliferation; in the supernatant of LNCaP cells, the palmitoylation level of α-tubulin is also increased following androgen treatment, which may represent a biomarker for early-stage PCa. | [43] |
Sialylation O-linked glycans: in vitro proliferation, migration, apoptosis; tumor size in mouse model [30]; cell adhesion [99]; N-linked glycans: in vitro proliferation, migration, invasion [101]. | Fucosylation Self-assembly of spheroids [102]; EGFR signaling; cell survival in androgen-depleted conditions [31]; vesicles secreted by PCa cells [32]; PSA expression [34]; metastasis to bone [103]. | Biosynthesis of1,6 GlcNAc-Branched N-glycans  In vitro invasion; tumor growth in xenograft models [104]. | Mannose Trimming of N-glycans Essential for cell viability [105]. |
| Regulation of N-glycosylation Substrate Specificity In vitro proliferation, migration and invasion; xenograft growth in a PTEN negative background; ER structure and stress response; Akt signaling [106]. | O-Linked N-Acetylgalactosamine Addition Essential for cell viability [105]. | O-Linked N-Acetylglucosamine Addition Essential process in androgen-independency [33]; metabolism [107]. | Generation of the Common Core 1 O-glycan Structure Castration resistance and metastasis [108,109]. |
Core-2-branched O-linked glycosylation Tumor growth in mouse model [110,111]; cell adhesion [110]; resistance to NK cell immunity [112]; LNCaP susceptibility to apoptosis induced by Galectin-1 [113]. | Core-3 O-linked glycan formation Tumor formation andmetastasis of PC-3 and LNCaP cells through downregulation of α2β1 integrin complex [114]. | I-branching Migration and invasion; integrin signaling via indirect mechanisms; in DU145 cells appears to largely occur on glycolipids and partially on O-glycans [115]. | Legend: N-acetylglucosamine N-acetylglucosamine N-acetylgalactosamine N-acetylgalactosamine Galactose Galactose Mannose Mannose Sialic acid Sialic acid Fucose Fucose |
| (Component of) E3 Ligase | Description | Affected Protein(s) and/or Signaling Pathways | Effects on Processes |
|---|---|---|---|
| RING type | |||
| AMFR | RING-type E3 ubiquitin transferase, component of a complex that participates in the final step of ER-associated degradation | 3βHSD1 [140] | DHT synthesis necessary to activate the AR [140] |
| APC/C | Multi-subunit cullin-RING E3 ubiquitin ligase that regulates progression through the metaphase to anaphase of the cell cycle | Cyclin A2, Geminin, PLK1, Aurora A, and CDC20 [141]; SKP2 [142] | PTEN loss but not phosphatase inactivation results in hypersensitivity to pharmacological inhibition of APC-CDH1 targets PLK1 and Aurora A [141]; cell cycle [142] |
| BIRC6 | Consists of a BIR and a ubiquitin-conjugating (UBC) domain with chimeric E2/E3 ubiquitin ligase activity; through its BIR domain binds to active caspases; through its UBC domain, facilitates proteasomal degradation of pro-apoptotic proteins | GPCR and matrisome signaling; prosurvival genes [143] | Implicated in advanced, Enzalutamide (Enz)-resistant PCa [143]; role in PCa progression and treatment resistance [144] |
| BMI1 | Contains a RING motif; it does not have E3 ubiquitin ligase activities; forms a complex with RING1B to ubiquitinate H2A-K119 and repress the expression levels of polycomb repressive complex 1 (PRC1) targets | AR [135] | PRC1-independent role in MDM2-mediated AR protein degradation; tumor growth of xenografts that have developed resistance to surgical castration and Enz treatment [135] |
| CAND1 | F-box protein exchange factor; key assembly factor of SCF E3 ubiquitin ligase complexes | p21 [145]; PLK4 [146] | In vitro cell viability, proliferation, apoptosis [145]; centriole overduplication [146] |
| c-CBL | RING domain E3 ligase | EGFR [147] | EGFR/Erk1/2 signaling-mediated PCa [147] |
| COP1 | RING-type E3 ubiquitin transferase | STAT3 [148]; ETS transcription factors [149] | Tumorigenesis; proliferation and cancer stem-like properties in prostate epithelial cells [148,149] |
| CRL4/Cdt2 | Proliferating cell nuclear antigen (PCNA)-dependent E3 ubiquitin ligase | WHSC1 [150] | Interaction with key intracellular signaling molecules, AKT, RICTOR, and Rac1, to drive PCa metastasis [150] |
| CUL3 | Cullin–RING-based E3 ubiquitin ligase | Mutated in a subset of PCa indicating possible driving roles [151] | |
| CUL4A | Cullin family of ubiquitin ligase proteins | AR [152] | AR protein homeostasis [152] |
| CUL4B | Scaffold protein that assembles the Cullin4B-RING E3 ligase complex | BMI1 [153], c-MYC [154] | Cancer stem-like traits of PCa cells [153]; PCa progression [154] |
| FBXL2 | F-box protein; the receptor subunit of one of 69 human SCF ubiquitin ligase complexes | IP3R3 [155] | Ca2+-mediated apoptosis and tumor growth [155] |
| FBXL4 | Member of the F-box protein family; part of a modular E3 SCF ubiquitin ligase complexes | Potentially ERLEC1 [156] | PCa progression and metastasis [156] |
| FBXL7 | F-box protein that functions as substrate receptor for SCF | c-SRC [157] | Epithelial-to-mesenchymal transition (EMT) and metastasis [157] |
| FBXO45 | Substrate-specific adaptor subunit of SCF E3 ubiquitin ligase complex | PAR4 [158,159] | Cell survival [158,159]; therapy resistance [159] |
| FBXW7 | F-box and WD repeat domain containing 7 | AURKA [160] | Pathogenesis of prostatic small cell neuroendocrine carcinoma [160] |
| FBW7 | F-box protein; a substrate receptor for SCF-type E3 ligase | Dual phosphorylated ERG [161] | Driving of prostate oncogenesis [161] |
| KLHL20 | Substrate-binding subunit of Cullin3 ligase | PML, HIF-1α [162] | PCa progression [162] |
| MARCH5 | RING-finger E3 ligase | MCL1 [163] | Apoptosis in response to a BH3 mimetic agent targeting BCLXL [163] |
| MDM2 | The RING domain E3 ubiquitin ligase; key regulator of p53 tumor suppressor protein activity and stability | AR [164,165]; p53 [166,167]; E2F1 [168]; AR-v7 [169]; E-cadherin [170]; activation of p53 and destabilization of AR by combinatorial inhibition of MDM2 and MDMX [171] | Phosphorylation-dependent ubiquitination and degradation of AR by AKT [165]; stem cell integrity [164]; survival and proliferation of genomically unstable tumor cells [167]; prolongs the half-life of the E2F1 protein by inhibiting its ubiquitination (MDM2 displaces SCFSKP2); influences cell proliferation [168] |
| MYCBP2 | Atypical E3 ubiquitin-protein ligase, which mediates ubiquitination of threonine and serine, instead of lysine residues | AR, MYC [138] | Tumorigenicity of AR-positive PCa cells [138] |
| MYLIP | E3 ubiquitin-protein ligase whose activity depends on E2 enzymes of the UBE2D family | AR [172] | AR activity [172] |
| PIRH2 | Ring finger protein with ubiquitin ligase activity | Epsilon-COP [173]; HDAC1 [174] | Regulation of the secretion of PSA [173]; AR signaling [174] |
| pVHL | Substrate recognition subunit of the VHL-Elongin B/C E3 ligase complex that targets the HIF-1/2 for proteasomal degradation under normoxia conditions | AR (enhanced AR de-ubiquitination instead of inducing AR ubiquitination) [175]; HIF-1α [176] | Suppression of AR activity [175]; HIF-1 hypoxic response [176] |
| RNF2 | Also known as RING1b or RING2; catalytic subunit of PRC1 | TXNIP [177]; CCL2 [178] | Cell cycle arrest and apoptosis [177]; metastasis in mice inoculated intracardially with PC-3M cells [178] |
| RNF6 | RING finger-type E3 ligase | Poly- and mono-ubiquitination of AR [179] | Promotes AR transcriptional activity and specificity [179] |
| RNF7 | RING component of CRL (Cullin-RING ligase) | PHLPP1 and DEPTOR (PI3K/AKT/mTOR axis) [180]; p21, p27, NOXA; ERK1/2 signaling [181] | Proliferation in monolayer and soft agar; clonogenic survival; migration [180]; PCa tumorigenesis [181] |
| RNF11 | RING finger-type E3 ligase | ErbB2 and EGFR [182] | Growth arrest [182] |
| RNF20 and RNF40 | Histone H2B ubiquitin E3 ligases | AR, several cell cycle promoters [183] | Proliferation (due to changed expression of several cell cycle promoters) and modulation of AR transcriptional activity in intact cells [183] |
| RNF41 | Ring Finger Protein 41, E3 ligase | ErbB3 [184] | AR-independent proliferation [184] |
| RNF126 | E3 ligase that contributes to BAG6-mediated quality control | p21 [185] | Proliferation [185] |
| SIAH2 | E3 RING finger ubiquitin ligase; member of the seven in absentia homolog (SIAH) family | EAF2 [186]; AR [137]; AR-V7 [187]; HIF-1α and FOXA2 [136]; Wnt/β-catenin signaling [188] | Apoptosis [186]; lipid metabolism, cell motility, proliferation, cell growth under androgen-deprivation condition in vitro and in vivo, PCa regression upon castration [137]; castration-resistance in PCa therapy [187]; formation of neuroendocrine phenotype and neuroendocrine prostate tumors [136]; inducing and maintaining PCa cells dormancy in bone [188]; death receptor-mediated apoptosis [189] |
| SKP2 | F-box protein; crucial component of the SCF (Skp1-Cullin1-F-box) type of E3 ubiquitin ligase complexes | EZH2 [190]; p27 [191,192,193]; JARID1B [194]; DAB2IP [195]; AKT [196]; BRCA2 [197]; ATF4, p27, p21 [198]; Twist [199]; AR [139]; IDH1/2 [200]; FOXO3 [201]; E-cadherin [202] | TRAF6-mediated ubiquitination of EZH2; progression of PCa and CRPC through upregulation and activation of progenitor genes, as well as AR-target genes [190]; paclitaxel resistance [191]; tumorigenesis [192,193,194,195,196]; proliferation, survival, glucose uptake [196]; homologous recombination and sensitivity to the PARP inhibitor rucaparib [197]; oncogenic-stress-driven senescence [198]; progression and stem cell features of CRPC [199]; cell cycle-dependent metabolic oscillation between glycolysis and TCA cycle [200]; cell migration [202]; high expression is associated with a mesenchymal phenotype and increased tumorigenic potential [203] |
| SOCS2 | Probable substrate recognition component of a SCF-like ECS (Elongin BC-CUL2/5-SOCS-box protein) E3 ubiquitin ligase complex | FLT3 and JAK2 [204]; NDR1 stability; NF-κB transactivation [205] | Metastasis formation [204]; SOCS2-deficiency leads to hyper-activation of NF-κB and downstream pathological implications [205] |
| TOPORS | RING domain containing E3 ligase | NKX3.1 [206] | Tumor progression [206] |
| TRAF4 | RING domain E3 ubiquitin ligase | TrkA [207] | Metastasis formation [207] |
| TRAF6 | RING domain E3 ubiquitin ligase | p85a [208]; TGFβ type I receptor [209,210]; PS1 [210]; mTOR [211]; AKT [196]; TAK1 [212]; EZH2 [190] | PI3K/AKT signaling; migration [208]; tumor-promoting effects of TGFβ type I receptor [209,210]; activation of mTOR; regulation of autophagy and cell proliferation [211]; proliferation, survival, glucose uptake, in vivo tumor growth [196]; activation of NF-κB signaling downstream of several receptors [212] |
| TRIM11 | E3 ubiquitin-protein ligase; the TRIM motif contains a RING domain | Cell proliferation in vitro and the progression of PCa [213] | |
| TRIM16 | It lacks a RING domain found in other TRIM proteins, but can dimerize with other TRIM proteins and has E3 ubiquitin ligase activity | SNAIL signaling pathway [214] | Progression of prostate tumors [214] |
| TRIM25 | RING domain E3 ubiquitin ligase | ERG [215]; G3BP2 [216] | Driving of prostate carcinogenesis [215]; cell growth and survival by modulating p53 signals [216] |
| β-TrCP | Substrate recognition subunit for the SCFβ-TrCP E3 ligases | HIF-1α [217], Twist [199]; CHD1 [218]; MTSS1 [219]; REST [220]; δ-catenin [221]; AhR [222]; Gli2 [223] | Progression and stem cell features of CRPC [199]; transcription of the pro-tumorigenic TNF–NF-κB gene network [218]; proliferation and migration [219]; AR activity [220]; cell growth [222] |
| UHRF1 | Ubiquitin Like with PHD And Ring Finger Domains 1; E3 ubiquitin ligase | Cell proliferation and biochemical recurrence after radical prostatectomy [224]; epigenetic crosstalk and PCa progression [225] | |
| RBR type | |||
| PRKN | Parkin RBR E3 Ubiquitin Protein Ligase | Participates in removal of damaged mitochondria via mitophagy [226] | |
| U-box type | |||
| CHIP | U-box type chaperone associated E3 ligase | JMJD1A [227]; SNPH [228]; AR/AR-V7 [229]; AKT signaling pathway [230]; AR [231,232,233]; HIF-1α [234]; PRMT5 [235]; PTEN [236] | AR activity [227]; mitochondrial dynamics, tumor chemotaxis, invasion, and metastasis in vivo [228]; anti-androgen resistance [229]; in vitro migration and invasion [230]; mitotic arrest [233]; potential role in PCa oncogenesis through PRMT5 [235] |
| UBE4A | Ubiquitin-protein ligase that probably functions as an E3 ligase; may also function as an E4 ligase complementing actions of another E3 ubiquitin ligase | Interleukin-like EMT inducer (ILEI) [237] | In vitro migration and invasion [237] |
| HECT type | |||
| EDD | E3 ubiquitin-protein ligase, which is a component of the N-end rule pathway | Wnt/β-Catenin signaling [238] | Sensitivity of hormone-refractory PCa to docetaxel in vitro and in vivo [238] |
| E6AP | The founding member of the HECT (Homologous to E6AP Carboxyl Terminus) domain E3 ligases | NDRG1 [239], p27 [240]; PI3K, AKT [241,242], mTOR [241] | Acquisition of mesenchymal features, migration, ability for anchorage-independent growth [239]; tumor growth [240]; proliferation and invasion in bone metastasis [241]; cell growth, proliferation, apoptosis [242]; cellular senescence in vivo, radiation-induced cell death [243] |
| HACE1 | HECT domain and ankyrin repeat-containing ubiquitin ligase | HACE1 is a critical chromosome 6q21 tumor suppressor involved in prostate cancer [244] | |
| HECTD4 | Probable HECT domain E3 ubiquitin-protein ligase | AR, MYC [138] | Tumorigenicity of AR-positive PCa cells [138] |
| HUWE1 | WWE domain-containing protein 1, E3 ubiquitin protein ligase | HK2 [245]; c-MYC [246,247] | Metabolism and cancer stem cell expansion [245]; survival [246]; proliferation [246,247] and migration in vitro, and explant growth in vivo [247] |
| ITCH/AIP4 | HECT-type E3 ubiquitin transferase Itchy homolog | ErbB3 [248] | ErbB3 ubiquitination and degradation in cancer cells through JNK1/2-dependent ITCH/AIP4 activation [248] |
| Nedd4 | Comprised of a catalytic C-terminal HECT domain and N-terminal C2 domain and WW domains responsible for cellular localization and substrate recognition | IRS-2 [249]; AR [250]; ErbB3 levels and signaling [251] | IGF signaling and mitogenic activity [249]; cancer cell proliferation in vitro and in vivo; sensitization of cancer cells for growth inhibition by an anti-ErbB3 antibody [251] |
| SMURF1 | SMAD specific E3 ubiquitin protein ligase 1 | PTEN [252] | PCa progression [252]; invasion [253] |
| WWP1 | WW domain-containing E3 ubiquitin protein ligase-1 | TGFβ [254]; p63 [255]; KLF5 [256] | Migration and invasion [257]; 22Rv1 cells colony formation; PC-3 cells proliferation and TGFβ-mediated growth inhibition [254]; apoptosis [255] |
| WWP2 | WW Domain Containing E3 Ubiquitin Protein Ligase 2 | SUMO1-modified PTEN [258] | PCa development [258] |
| Enzyme | Involvement(s) in Prostate Cancer | Ref. |
|---|---|---|
| KATs | ||
| KAT2A | KAT2A inhibition prevents interleukin (IL) 6-induced PCa metastases through PI3K/PTEN/AKT signaling by inactivating Egr-1 | [307] |
| Association between AR and histone acetyltransferase KAT2A increases histone H3 acetylation level on cis-regulatory elements of AR target genes | [308] | |
| KAT2B | Promotes PKM2 acetylation and decreases PKM2 protein level through degradation through chaperone-mediated autophagy; promotes tumor growth | [309] |
| CBP (KAT3A) | CBP loss cooperates with PTEN haploinsufficiency to drive PCa | [310] |
| p300 (KAT3B) | p300-mediated acetylation of histone demethylase JMJD1A prevents its degradation by CHIP and enhances its activity | [227] |
| p300/CBP inhibition enhances the efficacy of programmed death-ligand 1 blockade treatment | [311] | |
| Therapeutic targeting of the CBP/p300 bromodomain blocks the growth of CRPC | [312] | |
| p300 regulates fatty acid synthase expression, lipid metabolism and PCa growth | [313] | |
| p300 regulates AR degradation and PTEN-deficient prostate tumorigenesis | [314] | |
| The assembly of a macromolecular complex involving CBP/p300 results in acetylation of p53 at K373, a critical PTM required for its biological activity | [315] | |
| SKP2 is acetylated by p300 at K68 and K71, which promotes its cytoplasmic retention, and cytoplasmic SKP2 enhances cellular migration through ubiquitination and destruction of E-cadherin | [202] | |
| p300 is the dominant coregulator of the CBP/p300 pair for androgen-regulated gene expression in C4-2B cells; p300 is required at an early stage of chromatin remodeling and transcription complex assembly after binding of AR to the gene but before many critical histone modifications occur | [316] | |
| Function in the survival and invasion pathways of PCa cell lines | [317] | |
| p300 and CBP stimulate estrogen receptor-beta (ER-β) signaling and regulate cellular events in PCa | [318] | |
| IL-4 activates AR through enhanced expression of CBP/p300 and its histone acetyltransferase activity | [319] | |
| p300 modulates nuclear morphology in PCa and is required for androgen depletion independent activation of the AR | [320] | |
| p300 mediates STAT3 acetylation on Lys685, which mediates STAT3 dimerization and is reversible by type I HDAC | [321] | |
| CBP/p300 is a component of a transcriptional complex that regulates SRC-dependent hypoxia-induced expression of VEGF | [322] | |
| The downregulation of p300 inhibits PCa cell proliferation both at the basal level and on IL6 stimulation | [323] | |
| p300 mediates androgen-independent transactivation of the AR by IL6 | [324] | |
| p300 and p300/CBP acetylate the AR at sites governing hormone-dependent transactivation | [325] | |
| Tip60 (KAT5) | Negatively regulates the proliferation of LNCaP cells via the caspase 3-dependent apoptosis pathway | [326] |
| Associated with resistance to X-ray irradiation | [327] | |
| Inhibition by TH1834 increases the effect of ionizing radiation in PC-3 and DU145 cells, induces apoptosis and increases unrepaired DNA damage | [328] | |
| Interacts with ER-β to regulate endogenous gene expression such as CXCL12 and cyclin D2 | [329,330] | |
| KAT5 and KAT6B positively regulate cell proliferation through PI3K/AKT signaling | [331] | |
| Inhibition by NU9056 induces a decrease of AR, PSA, p21 and p53 levels in LNCaP cells, which might explain the increase of apoptosis and the decrease of proliferation | [332] | |
| Overexpression increases the acetylation of the AR and its localization in the nucleus and promotes cell proliferation | [333] | |
| Tip60 and β-catenin complexes regulate expression of metastasis suppressor gene KAI1 | [334] | |
| A possible role for Tip60 in the molecular pathway leading to the development of androgen-independent PCa following long-term androgen deprivation therapy | [335] | |
| Tip60 and HDAC1 regulate AR activity through changes to the acetylation status of the receptor | [336] | |
| MYST1 (KAT8) | Regulates androgen signaling in PCa cells | [337] |
| Regulates NF-κB and AR functions during proliferation of PCa cells | [338] | |
| FOXP3 induces H4K16 acetylation and H3K4 trimethylation and activation of multiple genes by recruiting KAT8 and causing displacement of PLU-1 | [339] | |
| KDACs | ||
| Class I | Maspin induction is a critical epigenetic event altered by class I HDACs in the restoration of balance to delay proliferation and migration ability of PCa cells | [340] |
| HDAC1 | KLF5 inhibits STAT3 activity and tumor metastasis in PCa by suppressing IGF1 transcription cooperatively with HDAC1 | [341] |
| Involved in E-cadherin expression in PCa cells | [342] | |
| Ubiquitination of the AR and HDAC1 may constitute an additional mechanism for regulating AR function; HDAC1 and MDM2 function co-operatively to reduce AR mediated transcription that is attenuated by the HAT activity of the AR co-activator Tip60 | [343] | |
| HDAC3 | Genetic knockdown of either HDAC1 or HDAC3 can suppress expression of AR-regulated genes, recapitulating the effect of HDAC inhibitor treatment | [344] |
| HDAC4 | Positive regulator of AR SUMOylation, revealing a deacetylase-independent mechanism of HDAC action in PCa cells | [345] |
| Recruitment of HDAC4 by transcription factor YY1 represses HOXB13 to affect cell growth in AR-negative PCa | [346] | |
| HDAC6 | Synergistic interaction with MEK-inhibitors in CRPC cells | [347] |
| Metastatic prostate cancer-associated p62 inhibits autophagy flux and promotes EMT by sustaining the level of HDAC6 | [348] | |
| Regulates AR hypersensitivity and nuclear localization via modulating Hsp90 acetylation in CRPC | [349] | |
| HDAC7 | HDAC7 localizes to the mitochondrial inner membrane space of prostate epithelial cells and exhibits cytoplasmic relocalization in response to initiation of the apoptotic cascade, which highlights a link between HDACs, mitochondria, and programmed cell death | [350] |
| HDAC11 | HDAC11 depletion is sufficient to cause cell death and to inhibit metabolic activity in PC-3 cells | [351] |
| SIRT1 | Modulates the sensitivity of PCa cells to vesicular stomatitis virus oncolysis | [352] |
| Mesenchymal stem cells overexpressing SIRT1 inhibit PCa growth by recruiting NK cells and macrophages | [353] | |
| Loss of miR-449a in ERG-associated PCa promotes the invasive phenotype by inducing SIRT1 | [354] | |
| SIRT1 and LSD1 competitively regulate KU70 functions in DNA repair and mutation acquisition | [355] | |
| The silencing of SIRT1 gene in PC-3 cells suppresses the movement, migration, and invasion, possibly via reversing the EMT process | [356] | |
| Loss of Sirt1 promotes prostatic intraepithelial neoplasia, reduces mitophagy, and delays Park2 translocation to mitochondria | [226] | |
| Existence of SIRT1 and MPP8 crosstalk in E-cadherin gene silencing and EMT | [357] | |
| Regulation of histone H2A.Z expression is mediated by SIRT1 in PCa | [358] | |
| Enhances matrix metalloproteinase-2 expression and tumor cell invasion of PCa cells | [359] | |
| SIRT1 induces EMT by cooperating with EMT transcription factors and enhances PCa cell migration and metastasis | [360] | |
| Inhibition of cortactin and SIRT1 expression attenuates migration and invasion of DU145 cells | [361] | |
| Deacetylation of FOXO3 by SIRT1 or SIRT2 leads to SKP2-mediated FOXO3 ubiquitination and degradation | [201] | |
| Disruption of a SIRT1-dependent autophagy checkpoint in the prostate results in prostatic intraepithelial neoplasia lesion formation | [362] | |
| Inhibition of SIRT1 activity increases the chemosensitivity of androgen-refractory PCa cells | [363] | |
| SIRT1 inhibition at the activity level as well as via shRNA results in a significant inhibition in the growth and viability of human PCa cells; inhibition of SIRT1 causes an increase in FOXO1 acetylation and transcriptional activation in PCa cells | [364] | |
| SIRT1 inhibition causes a decrease in cell growth, cell viability and the colony formation ability and an increase in FOXO1 acetylation and subsequent transcriptional activation regardless of p53 status; SIRT1 inhibition results in an increase in senescence in PC-3-p53 (wild type p53) cells whereas it results in an increase in apoptosis in PC-3 (lack p53) cells | [365] | |
| Upregulation of SIRT1 expression may play an important role in promoting cell growth and chemoresistance in androgen-refractory PC-3 and DU145 cells | [366] | |
| Required for antagonist-induced transcriptional repression of androgen-responsive genes by the AR | [367] | |
| SIRT1 is a regulator of AR expression and function | [368] | |
| FOXO1 activity in PCa cells is inhibited by deacetylation by SIRT1 | [369] | |
| SIRT2 | Dysregulation of SIRT2 and histone H3K18 acetylation pathways associates with adverse PCa outcomes | [370] |
| SIRT3 | Transcriptional repression of SIRT3 potentiates mitochondrial aconitase activation to drive aggressive PCa to the bone | [371] |
| SIRT3 and SIRT6 promote PCa progression by inhibiting necroptosis-mediated innate immune response | [372] | |
| Inhibits PCa metastasis through regulation of FOXO3A by suppressing Wnt/β-catenin pathway | [373] | |
| Inhibits PCa by destabilizing c-MYC through regulation of the PI3K/AKT pathway | [374] | |
| Inactivation of SIRT3 leads to elevated SKP2 acetylation, which leads to increased SKP2 stability through impairment of the CDH1-mediated proteolysis pathway resulting in increase of SKP2 oncogenic function; cells expressing an acetylation-mimetic mutant display enhanced cellular proliferation and tumorigenesis in vivo | [202] | |
| SIRT4 | Mitochondrial PAK6 inhibits PCa cell apoptosis via the PAK6-SIRT4-ANT2 complex | [375] |
| SIRT5 | SIRT 5 regulates the proliferation, invasion, and migration of PCa cells through acetyl-CoA acetyltransferase 1 | [376] |
| SIRT6 | E2F1 enhances glycolysis through suppressing Sirt6 transcription in cancer cells | [377] |
| Inhibition of SIRT6 reduces cell viability and increases sensitivity to chemotherapeutics | [378] | |
| SIRT7 | SIRT7 depletion inhibits cell proliferation and androgen-induced autophagy by suppressing the AR signaling in PCa | [379] |
| Promotes PCa cell aggressiveness and chemoresistance | [380] | |
| SIRT7 inactivation reverses metastatic phenotypes | [381] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samaržija, I. Post-Translational Modifications That Drive Prostate Cancer Progression. Biomolecules 2021, 11, 247. https://doi.org/10.3390/biom11020247
Samaržija I. Post-Translational Modifications That Drive Prostate Cancer Progression. Biomolecules. 2021; 11(2):247. https://doi.org/10.3390/biom11020247
Chicago/Turabian StyleSamaržija, Ivana. 2021. "Post-Translational Modifications That Drive Prostate Cancer Progression" Biomolecules 11, no. 2: 247. https://doi.org/10.3390/biom11020247
APA StyleSamaržija, I. (2021). Post-Translational Modifications That Drive Prostate Cancer Progression. Biomolecules, 11(2), 247. https://doi.org/10.3390/biom11020247