
Epigenetics of Alzheimer’s Disease

 , , , , and
, , , , and 
Abstract
1. Introduction
2. Alzheimer’s Disease
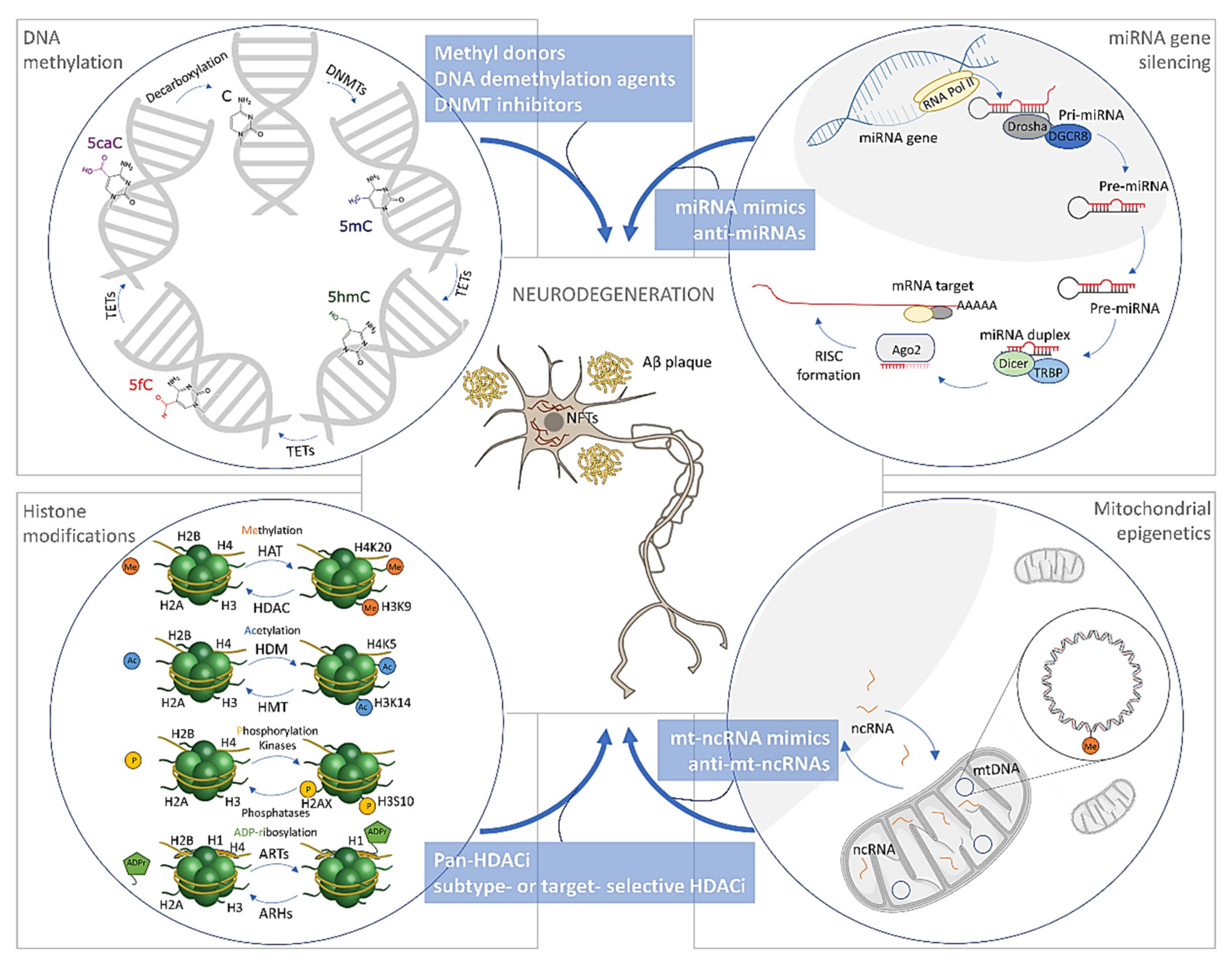
3. Epigenetic Alterations in Alzheimer’s Disease
3.1. DNA Methylation/Hydroxymethylation in Alzheimer’s Disease
3.1.1. DNA Methylation in Alzheimer’s Disease

{kind=link}
| Epigenetic Mechanism | Effect | Gene/Target Pathway Involved | Study Model 1 | Tissue/Study Design | Main Results | Ref. |
|---|---|---|---|---|---|---|
| 5mC | ↓ | Neurogenesis, neurodevelopment, amyloid neuropathies | AD (n = 31), Moderate AD (n = 32), Ctrl (n = 38) | CNS (PFC)/NGS | Identification of 1224 DMRs (enhancer regions) in AD, including enhancers in the DSCAML1 gene that targets BACE1. | [49] |
| 5mC | ↓↑ | WNT5B, ANK1, ARID5B | AD (n = 96/104) | CNS (ECx)/methylation array technology | Experiment-wide significant increase of 5mC in WNT5B (single CpG). Increased levels of 5mC in ANK1 (two probes). Decreased levels of 5mC in ARID5B (six probes). | [50] |
| 5mC | ↑ | ANKRD30B, ANK1 Cell adhesion, immunity | AD (n = 24), Neurotypical Ctrl (n = 49) | CNS (HPC, ECx, HPC, DLPFC, CB)/methylation array technology | Identification of 858 DMCs. Correlation between 5mC and gene expression levels. | [51] |
| 5mC | ↓ | B3GALT4, ZADH2 Cell survival, inflammation response | AD (n = 45), Ctrl (n = 39) | Blood/methylation array technology | Differential methylation of 477 DMCs, majority hypomethylated. Hypomethylation of B3GALT4 and ZADH2 associated with memory performance, and CSF levels of Aβ and tau. | [52] |
| 5mC | ↑ | HOXA3, GSTP1, CXXC1-3, BIN1 | AD (n = 18), Ctrl (n = 14) | CNS (FCx)/methylation array technology | Identification of 504 DMCs and 237 DMRs. Increased 5mC in pyramidal layer neurons in AD cases. 5mc pattern associated with oxidative stress. | [53] |
| 5mC | ↓ | KIAA0566 | NFT pathology stages I–VI (n = 17), Middle-aged cases (n = 3) | CNS (LC)/methylation array technology | Decreased levels of 5mC in KIAA0566 in NFT pathology AD cases. | [54] |
| 5mC | ↑ | HOXA gene cluster | Late-stage AD (n = 44), Middle stage AD (n = 43); Ctrl (n = 60) | CNS (PFC, STG)/methylation array technology | Identification of 208 DMCs in a 48kb HOXA gene cluster. | [55] |
| 5mC | ↓↑ | BRCA1 | AD cases (n = 30), Ctrl (n = 30) | CNS (ITG, CB, HPC, Ecx)/methylation array technology | Differential methylation of 8 DMRs in AD. Decreased levels of 5mC in BRCA1 in AD. BRCA1 5mC correlated with APOE e4 allele status. | [56] |
| 5mC | ↑ | Neuregulin receptor complex signaling pathway | AD cases (n = 10), Ctrl (n = 10) | CNS (TCx)/methylation array technology | Differential methylation of 161CpG positions associated with miRNA genes. | [57] |
| 5mC | ↑ | Neuron function and development, cholesterol/lipid metabolism | AD cases (n = 34), Ctrl (n = 34) | CNS (STG)/methylation array technology | Identification of 479 DMRs, majority hypermethylated. Overlap of hypermethylated DMRs and histone trimethylation marks. | [58] |
| 5mC | ↑ | ANK1 | Cohorts (n = 117/144/62) | CNS (Ecx, STG, PFC)/methylation array technology | Increased levels of 5mC in ANK1, associated with Braak stage. Strong correlation of top 100 DMCs between the cohorts. | [59] |
| 5mC | ↓↑ | ANK1, BIN1, RHBDF2 | Cohorts (n = 708/117) | CNS (DLPFC)/methylation array technology | Identification of 71 DMC associated AD pathology burden. Validation of 11 DMRs in an independent set. | [60] |
| 5mC | ↑ | Molecular functions associated with transcription, membrane transport, and protein metabolism | AD (n = 12), Ctrl (n = 12) | CNS (FCx)/methylation array technology | Identification of 948 DMCs in AD. | [61] |
| 5mC | ↓ | AS3MT, TBX15, WT1 | AD with psychosis (n = 29), AD without psychosis (n = 18) | CNS (PFC, Ecx, STG)/methylation array technology and pyrosequencing | Decreased levels of ASM3T 5mC (previously associated with SZ). Decreased levels of TBX1 and WT1 5mC (both previously associated with AD). | [62] |
| 5mC | ↓ | 5mC cell subtype localization | Early-AD (n = 5), Late-AD (n = 5), Ctrl (n = 5) | CNS (ITG)/immunohistochemistry | Decreased localization of extranuclear 5mC marks in neurofilament-positive pyramidal neurons and decreased localization of nuclear 5mC marks in astrocytes in AD cases compared to controls. | [63] |
| 5mC | ↑ | None stated | EOAD and LOAD (n = 29), Ctrl (n = 29) | CNS (MFG, MTG)/immunohistochemistry | Increased levels of 5mC in MFG and MTG of AD patients. Positive correlation of 5mC with 5hmC and AD markers (Aβ, tau, and ubiquitin loads). | [64] |
| 5mC | ↑ | None stated | AD (n = 7), Preclinical AD (n = 5), Ctrl (n = 5) | CNS (HPC/PHG, CB) mimunohistochemistry | Increased levels of 5mC in HPG of both AD patients and preclinical AD cases compared to control group subjects. | [65] |
| 5mC | ↓ | None stated | AD (n = 10), Ctrl (n = 10) | CNS (HPC)/immunohistochemistry | Decreased levels of 5mC in AD Negative correlation between 5mC and amyloid plaque load. | [66] |
3.1.2. DNA Hydroxymethylation in Alzheimer’s Disease
| Epigenetic Mechanism | Effect | Gene/Target Pathway Involved | Study Model 1 | Tissue/Study Design | Main Results | Ref. |
|---|---|---|---|---|---|---|
| 5hmC | ↓ | FBXL16, ANK1 | AD (n = 96/104) | CNS (ECx)/methylation array technology and pyrosequencing | Decreased levels of 5hmC in FBXL16 (four probes). Decreased levels of 5hmC in ANK1 (4 CpGs). | [50] |
| 5hmC | ↑↓ | BIN1 Signaling, energy metabolism, cell function processes, gene expression, protein degradation, and cell structure and stabilization | LOAD (n =3), Ctrl (n = 2) | CNS (HPC)/RRHP | Identification of 15.158 (DhMR), majority hyperhydroxymetylated. | [70] |
| 5hmC | ↑↓ | ABAT, CAMK1D, HTRA3, LRRN1 Long term memory and neurotrophin signaling pathway | AD (n = 20), MCI (n = 4), Ctrl (n = 6) | CNS (DLPFC)/NGS | Identification of 517 DhMRs, associated with neuritic plaques, and of 60 DhMRs, associated with neurofibrillary tangles. Correlation between 5hmC and gene expression. | [71] |
| 5hmC | ↑↓ | Neuron projection development, neurogenesis | AD (n = 3/2), Ctrl (n = 3/2) | CNS (PFC)/NGS | Identification of 7601 DhMR in the discovery set. Identification of 2351 DhMR in the replication set. | [72] |
| 5hmC | ↓↑ | 5hmC cell subtype localization | EOAD (n = 5), LOAD (n = 5), Ctrl (n = 5) | CNS (ITG)/immunohistochemistry | Decreased localization of nuclear 5hmC marks in AD cases compared to controls. No differences in localization of 5hmC in neurofilament-positive pyramidal neurons, disease-resistant calretinin-interneurons, microglia in AD cases compared to control subjects. | [63] |
| 5hmC | None | None | AD (n = 12; 10 sporadic + 2 familial), Ctrl (n = 14) | CNS (ECx, CB)/immunohistochemistry | No significant difference in 5hmC levels between studied groups. | [69] |
| 5hmC | ↓ | None stated | AD (n = 13), Ctrl (n = 8) | CNS (ECx, CB)/immunohistochemistry | Decreased levels of 5hmC in both ECx and CB of AD patients compared to control group subjects. | [68] |
| 5hmC | ↑ | 5hmC cell subtype localization | EOAD and LOAD (n = 29), Ctrl (n = 29) | CNS (MFG, MTG)/immunohistochemistry | Increased levels of 5hmC in MFG and MTG of AD patients. Positive correlation of 5hmC with 5mC and AD markers (Aβ, tau, and ubiquitin loads). Differences in cell subtype 5hmC distribution (lower levels in astrocytes and microglia, higher levels in neurons). | [64] |
| 5hmC | ↑ | None stated | preclinical AD (n = 5), AD (n = 7), Ctrl (n = 5) | CNS (HPG, CB)/immunohistochemistry | Increased levels of 5hmC in HPG of both AD patients and preclinical AD cases compared to control group subjects. | [65] |
| 5hmC | ↓ | 5hmC cell subtype localization | AD (n = 10), Ctrl (n = 10) monozygotic twins (AD twin and non-AD affected twin) | CNS (HPC)/immunohistochemistry | Decreased levels of 5hmC in AD. Decreased levels of 5hmC in CA3 HPC region glial cells and overall decrease in neuronal cells in AD. Negative correlation between 5hmC and amyloid plaque load. Decreased levels of in 5hmC of the AD twin compared to the non-AD affected twin. | [66] |
| 5hmC | None | TREM2 | AD (n = 12), Ctrl (n = 5) | CNS (HPC) 5hmC DNA immunoprecipitation/RT-qPCR | No significant difference in TREM2 5hmC levels between studied groups. | [73] |
3.1.3. Mitochondrial DNA Methylation in Alzheimer’s Disease
3.2. Histone Modifications in Alzheimer’s Disease
3.3. The microRNAs in Alzheimer’s Disease
3.3.1. miR-9
3.3.2. miR-29
3.3.3. miR-34
3.3.4. miR-107
3.3.5. miR-125
3.3.6. miR-132/-212
3.3.7. miR-146
3.3.8. mR-155
3.3.9. miR-181
3.3.10. miR-206
4. Treatment Opportunities through Epigenetics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hesson, L.B.; Pritchard, A.L. Genetics and epigenetics: A historical overview. In Clinical Epigenetics; Hesson, L., Pritchard, A., Eds.; Springer: Singapore, 2019; pp. 1–46. [Google Scholar]
- APA, Neurocognitive Disorders. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar]
- Hampel, H.; O’Bryant, S.E.; Durrleman, S.; Younesi, E.; Rojkova, K.; Escott-Price, V.; Corvol, J.-C.; Broich, K.; Dubois, B.; Lista, S. Alzheimer Precision Medicine Initiative. A Precision Medicine Initiative for Alzheimer’s Disease: The Road Ahead to Biomarker-Guided Integrative Disease Modeling. Climacteric 2017, 20, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Hickman, R.A.; Faustin, A.; Wisniewski, T. Alzheimer Disease and Its Growing Epidemic: Risk Factors, Biomarkers, and the Urgent Need for Therapeutics. Neurol. Clin. 2016, 34, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.; Wimo, A.; Guerchet, M.; Ali, G.-C.; Wu, Y.-T.; Prina, M. World Alzheimer Report 2015: The Global Impact of Dementia; Alzheimer’s Disease International: London, UK, 2015; Available online: https://www.alz.co.uk/research/WorldAlzheimerReport2015.pdf (accessed on 27 April 2020).
- World Health Organization (WHO). Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 27 April 2020).
- Brookmeyer, R.; Abdalla, N.; Kawas, C.H.; Corrada, M.M. Forecasting the prevalence of preclinical and clinical Alzheimer’s disease in the United States. Alzheimers Dement. 2018, 14, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Niu, H.; Alvarez-Alvarez, I.; Guillen-Grima, F.; Aguinaga-Ontoso, I. Prevalence and incidence of Alzheimer’s disease in Europe: A meta-analysis. Neurologia 2017, 32, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, 1–23. [Google Scholar] [CrossRef]
- Schneider, J.A.; Arvanitakis, Z.; Leurgans, S.E.; Bennett, D.A. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann. Neurol. 2009, 66, 200–208. [Google Scholar] [CrossRef]
- Diniz, L.P.; Tortelli, V.; Matias, I.; Morgado, J.; Bergamo Araujo, A.P.; Melo, H.M.; Seixas da Silva, G.S.; Alves-Leon, S.V.; de Souza, J.M.; Ferreira, S.T.; et al. Astrocyte Transforming Growth Factor Beta 1 Protects Synapses against Abeta Oligomers in Alzheimer’s Disease Model. J. Neurosci. 2017, 37, 6797–6809. [Google Scholar] [CrossRef]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef]
- Carmona, S.; Hardy, J.; Guerreiro, R. The genetic landscape of Alzheimer disease. Handb. Clin. Neurol. 2018, 148, 395–408. [Google Scholar]
- Nikolac Perkovic, M.; Pivac, N. Genetic Markers of Alzheimer’s Disease. Adv. Exp. Med. Biol. 2019, 1192, 27–52. [Google Scholar] [PubMed]
- Wingo, T.S.; Lah, J.J.; Levey, A.I.; Cutler, D.J. Autosomal recessive causes likely in early-onset Alzheimer disease. Arch. Neurol. 2012, 69, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Nasreddine, Z.S.; Phillips, N.A.; Bedirian, V.; Charbonneau, S.; Whitehead, V.; Collin, I.; Cummings, J.L.; Chertkow, H. The Montreal Cognitive Assessment, MoCA: A brief screening tool for mild cognitive impairment. J. Am. Geriatr Soc. 2005, 53, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Folstein, M.F.; Folstein, S.E.; McHugh, P.R. Mini-mental state. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 1975, 12, 189–198. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Fodero-Tavoletti, M.T.; Okamura, N.; Furumoto, S.; Mulligan, R.S.; Connor, A.R.; McLean, C.A.; Cao, D.; Rigopoulos, A.; Cartwright, G.A.; O’Keefe, G.; et al. 18F-THK523: A novel in vivo tau imaging ligand for Alzheimer’s disease. Brain 2011, 134, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, M.N.; Lue, L.-F.; Fayard, D.; Shi, J. Increasing precision of clinical diagnosis of Alzheimer’s disease using a combined algorithm incorporating clinical and novel biomarker data. Neurol. Ther. 2017, 6, 83–95. [Google Scholar] [CrossRef]
- Prokhortchouk, E.; Defossez, P.A. The cell biology of DNA methylation in mammals. Biochim. Biophys. Acta 2008, 1783, 2167–2173. [Google Scholar] [CrossRef]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Illingworth, R.S.; Bird, A.P. CpG islands—A rough guide’. FEBS Lett. 2009, 583, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.J.; Evans, J.; Kim, K.; Chae, H.; Kim, S. Determining the effect of DNA methylation on gene expression in cancer cells. Methods Mol. Biol. 2014, 1101, 161–178. [Google Scholar] [PubMed]
- Du, Q.; Luu, P.L.; Stirzaker, C.; Clark, S.J. Methyl-CpG-binding domain proteins: Readers of the epigenome. Epigenomics 2015, 7, 1051–1073. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, S.; Subramaniam, S.; Shyy, J.Y.; Chien, S. Epigenetic regulation: A new frontier for biomedical engineers. Annu. Rev. Biomed. Eng. 2017, 19, 195–219. [Google Scholar] [CrossRef] [PubMed]
- Penn, N.W.; Suwalski, R.; O’Riley, C.; Bojanowski, K.; Yura, R. The presence of 5-hydroxymethylcytosine in animal deoxyribonucleic acid. Biochem. J. 1972, 126, 781–790. [Google Scholar] [CrossRef]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by mll partner tet1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef]
- Sherwani, S.I.; Khan, H.A. Role of 5-hydroxymethylcytosine in neurodegeneration. Gene 2015, 570, 17–24. [Google Scholar] [CrossRef]
- Globisch, D.; Münzel, M.; Müller, M.; Michalakis, S.; Wagner, M.; Koch, S.; Brückl, T.; Biel, M.; Carell, T. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE 2010, 5, e15367. [Google Scholar] [CrossRef]
- Hahn, M.A.; Qiu, R.; Wu, X.; Li, A.X.; Zhang, H.; Wang, J.; Jui, J.; Jin, S.G.; Jiang, Y.; Pfeifer, G.P.; et al. Dynamics of 5-hydroxymethylcytosine and chromatin marks in mammalian neurogenesis. Cell Rep. 2013, 3, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Efimova, O.A.; Koltsova, A.S.; Krapivin, M.I.; Tikhonov, A.V.; Pendina, A.A. Environmental epigenetics and genome flexibility: Focus on 5-hydroxymethylcytosine. Int. J. Mol. Sci. 2020, 21, 3223. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Xu, X. DNA methyltransferases, DNA methylation, and age-associated cognitive function. Int. J. Mol. Sci. 2018, 19, 1315. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Bernstein, A.; Chen, D.; Jin, P. 5-hydroxymethylcytosine: A new player in brain disorders? Exp. Neurol. 2015, 268, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.G.; Tulloch, J.; Chen, S.; Leong, L.; Saxton, A.D.; Kraemer, B.; Darvas, M.; Keene, C.D.; Shutes-David, A.; Todd, K.; et al. Redefining transcriptional regulation of the APOE gene and its association with Alzheimer’s disease. PLoS ONE 2020, 15, e0227667. [Google Scholar] [CrossRef]
- Tulloch, J.; Leong, L.; Thomson, Z.; Chen, S.; Lee, E.G.; Keene, C.D.; Millard, S.P.; Yu, C.E. Glia-specific APOE epigenetic changes in the Alzheimer’s disease brain. Brain Res. 2018, 1698, 179–186. [Google Scholar] [CrossRef]
- Shao, Y.; Shaw, M.; Todd, K.; Khrestian, M.; D’Aleo, G.; Barnard, P.J.; Zahratka, J.; Pillai, J.; Yu, C.E.; Keene, C.D.; et al. DNA methylation of TOMM40-APOE-APOC2 in Alzheimer’s disease. J. Hum. Genet. 2018, 63, 459–471. [Google Scholar] [CrossRef]
- Foraker, J.; Millard, S.P.; Leong, L.; Thomson, Z.; Chen, S.; Keene, C.D.; Bekris, L.M.; Yu, C.E. The APOE gene is differentially methylated in Alzheimer’s disease. J. Alzheimers Dis. 2015, 48, 745–755. [Google Scholar] [CrossRef]
- Mur, J.; McCartney, D.L.; Walker, R.M.; Campbell, A.; Bermingham, M.L.; Morris, S.W.; Porteous, D.J.; McIntosh, A.M.; Deary, I.J.; Evans, K.L.; et al. DNA methylation in APOE: The relationship with Alzheimer’s and with cardiovascular health. Alzheimers Dement. 2020, 6, 1–9. [Google Scholar] [CrossRef]
- Mise, A.; Yoshino, Y.; Yamazaki, K.; Ozaki, Y.; Sao, T.; Yoshida, T.; Mori, T.; Mori, Y.; Ochi, S.; Iga, J.I.; et al. TOMM40 and APOE gene expression and cognitive decline in Japanese Alzheimer’s disease subjects. J. Alzheimers Dis. 2017, 60, 1107–1117. [Google Scholar] [CrossRef]
- Nagata, T.; Kobayashi, N.; Ishii, J.; Shinagawa, S.; Nakayama, R.; Shibata, N.; Kuerban, B.; Ohnuma, T.; Kondo, K.; Arai, H.; et al. Association between DNA methylation of the BDNF promoter region and clinical presentation in Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. Extra 2015, 5, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Nicolia, V.; Ciraci, V.; Cavallaro, R.A.; Ferrer, I.; Scarpa, S.; Fuso, A. Gsk3beta 5′-flanking DNA methylation and expression in Alzheimer’s disease patients. Curr. Alzheimer Res. 2017, 14, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, Y.; Yoshino, Y.; Yamazaki, K.; Sao, T.; Mori, Y.; Ochi, S.; Yoshida, T.; Mori, T.; Iga, J.I.; Ueno, S.I. DNA methylation changes at TREM2 intron 1 and trem2 mRNA expression in patients with Alzheimer’s disease. J. Psychiatr. Res. 2017, 92, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.R.; Smith, R.G.; Burrage, J.; Troakes, C.; Al-Sarraj, S.; Kalaria, R.N.; Sloan, C.; Robinson, A.C.; Mill, J.; Lunnon, K. A cross-brain regions study of ANK1 DNA methylation in different neurodegenerative diseases. Neurobiol. Aging 2019, 74, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Marshall, L.; Oh, G.; Jakubowski, J.L.; Groot, D.; He, Y.; Wang, T.; Petronis, A.; Labrie, V. Epigenetic dysregulation of enhancers in neurons is associated with Alzheimer’s disease pathology and cognitive symptoms. Nat. Commun. 2019, 10, 2246–2260. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.R.; Smith, R.G.; Pishva, E.; Hannon, E.; Roubroeks, J.A.Y.; Burrage, J.; Troakes, C.; Al-Sarraj, S.; Sloan, C.; Mill, J.; et al. Parallel profiling of DNA methylation and hydroxymethylation highlights neuropathology-associated epigenetic variation in Alzheimer’s disease. Clin. Epigenet. 2019, 11, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Semick, S.A.; Bharadwaj, R.A.; Collado-Torres, L.; Tao, R.; Shin, J.H.; Deep-Soboslay, A.; Weiss, J.R.; Weinberger, D.R.; Hyde, T.M.; Kleinman, J.E.; et al. Integrated DNA methylation and gene expression profiling across multiple brain regions implicate novel genes in Alzheimer’s disease. Acta Neuropathol. 2019, 137, 557–569. [Google Scholar] [CrossRef]
- Madrid, A.; Hogan, K.J.; Papale, L.A.; Clark, L.R.; Asthana, S.; Johnson, S.C.; Alisch, R.S. DNA hypomethylation in blood links B3GALT4 and ZADH2 to Alzheimer’s disease. J. Alzheimers Dis. 2018, 66, 927–934. [Google Scholar] [CrossRef]
- Hernández, H.G.; Sandoval-Hernández, A.G.; Garrido-Gil, P.; Labandeira-Garcia, J.L.; Zelaya, M.V.; Bayon, G.F.; Fernandez, A.F.; Fraga, M.F.; Arboleda, G.; Arboleda, H. Alzheimer’s disease DNA methylome of pyramidal layers in frontal cortex: Laser-assisted microdissection study. Epigenomics 2018, 10, 1365–1382. [Google Scholar] [CrossRef]
- Andres-Benito, P.; Delgado-Morales, R.; Ferrer, I. Altered regulation of KIAA0566, and katanin signaling expression in the locus coeruleus with neurofibrillary tangle pathology. Front. Cell Neurosci. 2018, 12, 131–142. [Google Scholar] [CrossRef]
- Smith, R.G.; Hannon, E.; De Jager, P.L.; Chibnik, L.; Lott, S.J.; Condliffe, D.; Smith, A.R.; Haroutunian, V.; Troakes, C.; Al-Sarraj, S.; et al. Elevated DNA methylation across a 48-kb region spanning the HOXA gene cluster is associated with Alzheimer’s disease neuropathology. Alzheimers Dement. 2018, 14, 1580–1588. [Google Scholar] [CrossRef] [PubMed]
- Mano, T.; Nagata, K.; Nonaka, T.; Tarutani, A.; Imamura, T.; Hashimoto, T.; Bannai, T.; Koshi-Mano, K.; Tsuchida, T.; Ohtomo, R.; et al. Neuron-specific methylome analysis reveals epigenetic regulation and tau-related dysfunction of BRCA1 in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 9645–9654. [Google Scholar] [CrossRef] [PubMed]
- Villela, D.; Ramalho, R.F.; Silva, A.R.; Brentani, H.; Suemoto, C.K.; Pasqualucci, C.A.; Grinberg, L.T.; Krepischi, A.C.; Rosenberg, C. Differential DNA methylation of microRNA genes in temporal cortex from Alzheimer’s disease individuals. Neural Plast. 2016, 2016, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.T.; Roussos, P.; Garg, P.; Ho, D.J.; Azam, N.; Katsel, P.L.; Haroutunian, V.; Sharp, A.J. Genome-wide DNA methylation profiling in the superior temporal gyrus reveals epigenetic signatures associated with Alzheimer’s disease. Genome Med. 2016, 8, 5–20. [Google Scholar] [CrossRef]
- Lunnon, K.; Smith, R.; Hannon, E.; De Jager, P.L.; Srivastava, G.; Volta, M.; Troakes, C.; Al-Sarraj, S.; Burrage, J.; Macdonald, R.; et al. Methylomic profiling implicates cortical deregulation of ank1 in alzheimer’s disease. Nat. Neurosci. 2014, 17, 1164–1170. [Google Scholar] [CrossRef]
- De Jager, P.L.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimer’s disease: Early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and oTher. loci. Nat. Neurosci. 2014, 17, 1156–1163. [Google Scholar] [CrossRef]
- Bakulski, K.M.; Dolinoy, D.C.; Sartor, M.A.; Paulson, H.L.; Konen, J.R.; Lieberman, A.P.; Albin, R.L.; Hu, H.; Rozek, L.S. Genome-wide DNA methylation differences between late-onset Alzheimer’s disease and cognitively normal controls in human frontal cortex. J. Alzheimers Dis. 2012, 29, 571–588. [Google Scholar] [CrossRef]
- Pishva, E.; Creese, B.; Smith, A.R.; Viechtbauer, W.; Proitsi, P.; van den Hove, D.L.A.; Ballard, C.; Mill, J.; Lunnon, K. Psychosis-associated DNA methylomic variation in Alzheimer’s disease cortex. Neurobiol. Aging 2020, 89, 83–88. [Google Scholar] [CrossRef]
- Phipps, A.J.; Vickers, J.C.; Taberlay, P.C.; Woodhouse, A. Neurofilament-labeled pyramidal neurons and astrocytes are deficient in DNA methylation marks in Alzheimer’s disease. Neurobiol. Aging 2016, 45, 30–42. [Google Scholar] [CrossRef]
- Coppieters, N.; Dieriks, B.V.; Lill, C.; Faull, R.L.; Curtis, M.A.; Dragunow, M. Global changes in DNA methylation and hydroxymethylation in alzheimer’s disease human brain. Neurobiol. Aging 2014, 35, 1334–1344. [Google Scholar] [CrossRef]
- Bradley-Whitman, M.A.; Lovell, M.A. Epigenetic changes in the progression of Alzheimer’s disease. Mech. Ageing Dev. 2013, 134, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Chouliaras, L.; Mastroeni, D.; Delvaux, E.; Grover, A.; Kenis, G.; Hof, P.R.; Steinbusch, H.W.; Coleman, P.D.; Rutten, B.P.; van den Hove, D.L. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol. Aging 2013, 34, 2091–2099. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.R.; Penzes, P. Ankyrins: Roles in synaptic biology and pathology. Mol. Cell Neurosci. 2018, 91, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Condliffe, D.; Wong, A.; Troakes, C.; Proitsi, P.; Patel, Y.; Chouliaras, L.; Fernandes, C.; Cooper, J.; Lovestone, S.; Schalkwyk, L.; et al. Cross-region reduction in 5-hydroxymethylcytosine in Alzheimer’s disease brain. Neurobiol. Aging 2014, 35, 1850–1854. [Google Scholar] [CrossRef] [PubMed]
- Lashley, T.; Gami, P.; Valizadeh, N.; Li, A.; Revesz, T.; Balazs, R. Alterations in global DNA methylation and hydroxymethylation are not detected in Alzheimer’s disease. Neuropathol. Appl. NeuroBiol. 2015, 41, 497–506. [Google Scholar] [CrossRef]
- Ellison, E.M.; Bradley-Whitman, M.A.; Lovell, M.A. Single-base resolution mapping of 5-hydroxymethylcytosine modifications in hippocampus of Alzheimer’s disease subjects. J. Mol. Neurosci. 2017, 63, 185–197. [Google Scholar] [CrossRef]
- Zhao, J.; Zhu, Y.; Yang, J.; Li, L.; Wu, H.; De Jager, P.L.; Jin, P.; Bennett, D.A. A genome-wide profiling of brain DNA hydroxymethylation in alzheimer’s disease. Alzheimers Dement. 2017, 13, 674–688. [Google Scholar] [CrossRef]
- Bernstein, A.I.; Lin, Y.; Street, R.C.; Lin, L.; Dai, Q.; Yu, L.; Bao, H.; Gearing, M.; Lah, J.J.; Nelson, P.T.; et al. 5-hydroxymethylation-associated epigenetic modifiers of Alzheimer’s disease modulate tau-induced neurotoxicity. Hum. Mol. Genet. 2016, 25, 2437–2450. [Google Scholar] [CrossRef]
- Celarain, N.; Sanchez-Ruiz de Gordoa, J.; Zelaya, M.V.; Roldan, M.; Larumbe, R.; Pulido, L.; Echavarri, C.; Mendioroz, M. TREM2 upregulation correlates with 5-hydroxymethycytosine enrichment in Alzheimer’s disease hippocampus. Clin. Epigenet. 2016, 8, 37–47. [Google Scholar] [CrossRef]
- Orre, M.; Kamphuis, W.; Osborn, L.M.; Jansen, A.H.P.; Kooijman, L.; Bossers, K.; Hol, E.M. Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol. Aging 2014, 35, 2746–2760. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta 2014, 1842, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Coskun, P.; Wyrembak, J.; Schriner, S.E.; Chen, H.W.; Marciniack, C.; Laferla, F.; Wallace, D.C. A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim. Biophys. Acta 2012, 1820, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Bioenergetic origins of complexity and disease. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hroudova, J.; Singh, N.; Fisar, Z. Mitochondrial dysfunctions in neurodegenerative diseases: Relevance to Alzheimer’s disease. Biomed. Res. Int. 2014, 2014, 175062–175071. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Mokranjac, D.; Neupert, W. Protein import into mitochondria. Biochem. Soc. Trans. 2005, 33, 1019–1023. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, M.; Larsson, N.G.; Gustafsson, C.M. DNA replication and transcription in mammalian mitochondria. Annu. Rev. Biochem. 2007, 76, 679–699. [Google Scholar] [CrossRef]
- Garrido, N.; Griparic, L.; Jokitalo, E.; Wartiovaara, J.; van der Bliek, A.M.; Spelbrink, J.N. Composition and dynamics of human mitochondrial nucleoids. Mol. Biol. Cell 2003, 14, 1583–1596. [Google Scholar] [CrossRef]
- Corral-Debrinski, M.; Horton, T.; Lott, M.T.; Shoffner, J.M.; McKee, A.C.; Beal, M.F.; Graham, B.H.; Wallace, D.C. Marked changes in mitochondrial DNA deletion levels in Alzheimer brains. Genomics 1994, 23, 471–476. [Google Scholar] [CrossRef]
- Hamblet, N.S.; Ragland, B.; Ali, M.; Conyers, B.; Castora, F.J. Mutations in mitochondrial-encoded cytochrome c oxidase subunits i, ii, and iii genes detected in Alzheimer’s disease using single-strand conformation polymorphism. Electrophoresis 2006, 27, 398–408. [Google Scholar] [CrossRef]
- Shoffner, J.M.; Brown, M.D.; Torroni, A.; Lott, M.T.; Cabell, M.F.; Mirra, S.S.; Beal, M.F.; Yang, C.C.; Gearing, M.; Salvo, R.; et al. Mitochondrial DNA variants observed in Alzheimer disease and Parkinson disease patients. Genomics 1993, 17, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.D.; Shoffner, J.M.; Kim, Y.L.; Jun, A.S.; Graham, B.H.; Cabell, M.F.; Gurley, D.S.; Wallace, D.C. Mitochondrial DNA sequence analysis of four Alzheimer’s and Parkinson’s disease patients. Am. J. Med. Genet. 1996, 61, 283–289. [Google Scholar] [CrossRef]
- Coskun, P.E.; Beal, M.F.; Wallace, D.C. Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc. Natl. Acad. Sci. USA 2004, 101, 10726–10731. [Google Scholar] [CrossRef] [PubMed]
- Podlesniy, P.; Figueiro-Silva, J.; Llado, A.; Antonell, A.; Sanchez-Valle, R.; Alcolea, D.; Lleo, A.; Molinuevo, J.L.; Serra, N.; Trullas, R. Low cerebrospinal fluid concentration of mitochondrial DNA in preclinical Alzheimer disease. Ann. Neurol. 2013, 74, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Podlesniy, P.; Llorens, F.; Puigros, M.; Serra, N.; Sepulveda-Falla, D.; Schmidt, C.; Hermann, P.; Zerr, I.; Trullas, R. Cerebrospinal fluid mitochondrial DNA in rapid and slow progressive forms of Alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 6298. [Google Scholar] [CrossRef] [PubMed]
- Cervera-Carles, L.; Alcolea, D.; Estanga, A.; Ecay-Torres, M.; Izagirre, A.; Clerigue, M.; Garcia-Sebastian, M.; Villanua, J.; Escalas, C.; Blesa, R.; et al. Cerebrospinal fluid mitochondrial DNA in the Alzheimer’s disease continuum. NeuroBiol. Aging 2017, 53, 192.e1–192.e4. [Google Scholar] [CrossRef] [PubMed]
- Iacobazzi, V.; Castegna, A.; Infantino, V.; Andria, G. Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol. Genet. Metab 2013, 110, 25–34. [Google Scholar] [CrossRef]
- Shmookler Reis, R.J.; Goldstein, S. Mitochondrial DNA in mortal and immortal human cells. Genome number, integrity, and methylation. J. Biol. Chem. 1983, 258, 9078–9085. [Google Scholar] [CrossRef]
- Pollack, Y.; Kasir, J.; Shemer, R.; Metzger, S.; Szyf, M. Methylation pattern of mouse mitochondrial DNA. Nucleic Acids Res. 1984, 12, 4811–4824. [Google Scholar] [CrossRef]
- Dawid, I.B. 5-methylcytidylic acid: Absence from mitochondrial DNA of frogs and HeLa cells. Science 1974, 184, 80–81. [Google Scholar] [CrossRef]
- Dzitoyeva, S.; Chen, H.; Manev, H. Effect of aging on 5-hydroxymethylcytosine in brain mitochondria. Neurobiol. Aging 2012, 33, 2881–2891. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, M.; Taniguchi, T.; Higashi, H.; Sugimura, H.; Sugano, K.; Kanno, T. Methylation of mitochondrial DNA is not a useful marker for cancer detection. Clin. Chem. 2004, 50, 1480–1481. [Google Scholar] [CrossRef] [PubMed]
- Devall, M.; Smith, R.G.; Jeffries, A.; Hannon, E.; Davies, M.N.; Schalkwyk, L.; Mill, J.; Weedon, M.; Lunnon, K. Regional differences in mitochondrial DNA methylation in human post-mortem brain tissue. Clin. Epigenet. 2017, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Sengupta, S.; Scaria, V. Comparative analysis of human mitochondrial methylomes shows distinct patterns of epigenetic regulation in mitochondria. Mitochondrion 2014, 18, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Shock, L.S.; Thakkar, P.V.; Peterson, E.J.; Moran, R.G.; Taylor, S.M. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 3630–3635. [Google Scholar] [CrossRef]
- Chestnut, B.A.; Chang, Q.; Price, A.; Lesuisse, C.; Wong, M.; Martin, L.J. Epigenetic regulation of motor neuron cell death through DNA methylation. J. Neurosci. 2011, 31, 16619–16636. [Google Scholar] [CrossRef]
- Bellizzi, D.; D’Aquila, P.; Scafone, T.; Giordano, M.; Riso, V.; Riccio, A.; Passarino, G. The control region of mitochondrial DNA shows an unusual CpG and non-CpG methylation pattern. DNA Res. 2013, 20, 537–547. [Google Scholar] [CrossRef]
- Blanch, M.; Mosquera, J.L.; Ansoleaga, B.; Ferrer, I.; Barrachina, M. Altered mitochondrial DNA methylation pattern in Alzheimer disease-related pathology and in Parkinson disease. Am. J. Pathol. 2016, 186, 385–397. [Google Scholar] [CrossRef]
- Stoccoro, A.; Siciliano, G.; Migliore, L.; Coppede, F. Decreased methylation of the mitochondrial d-loop region in late-onset Alzheimer’s disease. J. Alzheimers Dis. 2017, 59, 559–564. [Google Scholar] [CrossRef]
- Stoccoro, A.; Tannorella, P.; Migliore, L.; Coppede, F. Polymorphisms of genes required for methionine synthesis and DNA methylation influence mitochondrial DNA methylation. Epigenomics 2020, 12, 1003–1012. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yu, J.T.; Tan, M.S.; Jiang, T.; Tan, L. Epigenetic mechanisms in Alzheimer’s disease: Implications for pathogenesis and therapy. Ageing Res. Rev. 2013, 12, 1024–1041. [Google Scholar] [CrossRef] [PubMed]
- Suganuma, T.; Workman, J.L. Crosstalk among Histone Modifications. Cell 2008, 135, 604–607. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Torrellas, C. Epigenetics of Aging and Alzheimer’s disease: Implications for Pharmacogenomics and Drug Response. Int. J. Mol. Sci. 2015, 16, 30483–30543. [Google Scholar] [CrossRef] [PubMed]
- Stoccoro, A.; Coppede, F. Role of epigenetics in Alzheimer’s disease pathogenesis. Neurodegener Dis. Manag. 2018, 8, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Weake, V.M.; Workman, J.L. Histone Ubiquitination: Triggering Gene Activity. Mol. Cell 2008, 29, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A. Targeting histone-modifications in Alzheimer’s disease. What is the evidence that this is a promising therapeutic avenue? Neuropharmacology 2014, 80, 95–102. [Google Scholar] [CrossRef]
- Messner, S.; Hottiger, M.O. Histone ADP-ribosylation in DNA repair, replication and transcription. Trends Cell Biol. 2011, 21, 534–542. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Mastroeni, D.; Grover, A.; Delvaux, E.; Whiteside, C.; Coleman, P.D.; Rogers, J. Epigenetic mechanisms in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 1161–1180. [Google Scholar] [CrossRef] [PubMed]
- Berson, A.; Nativio, R.; Berger, S.L.; Bonini, N.M. Epigenetic regulation in neurodegenerative diseases. Trends Neurosci. 2018, 41, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.; Hemberg, M.; Lewis, J.; Feany, M.B. Tau promotes neurodegeneration through global chromatin relaxation. Nat. Neurosci. 2014, 17, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Marzi, S.J.; Leung, S.K.; Ribarska, T.; Hannon, E.; Smith, A.R.; Pishva, E.; Poschmann, J.; Moore, K.; Troakes, C.; Al-Sarraj, S.; et al. A histone acetylome-wide association study of Alzheimer’s disease identifies disease-associated H3K27ac differences in the entorhinal cortex. Nat. Neurosci. 2018, 21, 1618–1627. [Google Scholar] [CrossRef] [PubMed]
- Narayan, P.J.; Lill, C.; Faull, R.; Curtis, M.A.; Dragunow, M. Increased acetyl and total histone levels in post-mortem Alzheimer’s 2 disease brain. Neurobiol. Dis. 2015, 74, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.W.; Turko, I.V. Histone post-translational modifications in frontal cortex from human donors with Alzheimer’s disease. Clin. Proteom. 2015, 12, 26–36. [Google Scholar] [CrossRef]
- Plagg, B.; Ehrlich, D.; Kniewallner, K.M.; Marksteiner, J.; Humpel, C. Increased Acetylation of Histone H4 at Lysine 12 (H4K12) in Monocytes of Transgenic Alzheimer’s Mice and in Human Patients. Curr. Alzheimer Res. 2015, 12, 752–760. [Google Scholar] [CrossRef]
- Schueller, E.; Paiva, I.; Blanc, F.; Wang, X.-L.; Cassel, J.-C.; Boutillie, A.-L.; Bousiges, O. Dysregulation of histone acetylation pathways in hippocampus and frontal cortex of Alzheimer’s disease patients. Eur. Neuropsychopharmacol. 2020, 33, 101–116. [Google Scholar] [CrossRef]
- Zhang, K.; Schrag, M.; Crofton, A.; Trivedi, R.; Vinters, H.; Kirsch, W. Targeted proteomics for quantification of histone acetylation in Alzheimer’s disease. Proteomics 2012, 12, 1261–1268. [Google Scholar] [CrossRef]
- Alsadany, M.A.; Shehata, H.H.; Mohamad, M.I. Histone deacetylases enzyme, copper, and IL-8 levels in patients with Alzheimer’s disease. Am. J. Alzheimers Dis. OTher. Demen. 2013, 28, 54–61. [Google Scholar] [CrossRef]
- Rao, J.S.; Keleshian, V.L.; Klein, S.; Rapoport, S.I. Epigenetic modifications in frontal cortex from Alzheimer’s disease and bipolar disorder patients. Transl. Psychiatry 2012, 2, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Myung, N.H.; Zhu, X.; Kruman, I.I.; Castellani, R.J.; Petersen, R.B.; Siedlak, S.L.; Perry, G.; Smith, M.A.; Lee, H. Evidence of DNA damage in Alzheimer disease: Phosphorylation of histone H2AX in astrocytes. Age 2008, 30, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, O.; Zhu, X.; Lee, H.G.; Raina, A.; Obrenovich, M.E.; Bowser, R.; Ghanbari, H.A.; Castellani, R.J.; Perry, G.; Smith, M.A. Ectopic localization of phosphorylated histone H3 in Alzheimer’s disease: A mitotic catastrophe? Acta Neuropathol. 2003, 105, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Love, S.; Barber, R.; Wilcock, G.K. Increased poly(ADP-ribosyl)ation of nuclear proteins in Alzheimer’s disease. Brain 1999, 122, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Wang, L.; Yu, C.; Yu, D.; Yu, G. Histone acetylation modifiers in the pathogenesis of Alzheimer’s disease. Front. Cell Neurosci. 2015, 9, 226–234. [Google Scholar] [CrossRef]
- MacBean, L.F.; Smith, A.R.; Lunnon, K. Exploring beyond the DNA sequence: A review of epigenomic studies of DNA and histone modifications in dementia. Curr. Genet. Med. Rep. 2020, 8, 79–92. [Google Scholar] [CrossRef]
- Klein, H.-U.; McCabe, C.; Gjoneska, E.; Sullivan, S.E.; Kaskow, B.J.; Tang, A.; Smith, R.V.; Xu, J.; Pfenning, A.R.; Bernstein, B.E.; et al. Epigenome-wide study uncovers large-scale changes in histone acetylation driven by tau pathology in aging and Alzheimer’s human brains. Nat. Neurosci. 2019, 22, 37–46. [Google Scholar] [CrossRef]
- Liu, X.; Jiao, B.; Shen, L. The epigenetics of Alzheimer’s disease: Factors and therapeutic implications. Front. Genet. 2018, 9, 579–589. [Google Scholar] [CrossRef]
- Ding, H.; Dolan, P.J.; Johnson, G.V. Histone deacetylase 6 interacts with the microtubule-associated protein tau. J. Neurochem. 2008, 106, 2119–2130. [Google Scholar] [CrossRef]
- Chouliaras, L.; Rutten, B.P.F.; Kenis, G.; Peerbooms, O.; Visser, P.J.; Verhey, F.; van Os, J.; Steinbusch, H.W.M.; van den Hove, D.L.A. Epigenetic regulation in the pathophysiology of Alzheimer’s disease. Prog. Neurobiol. 2010, 90, 498–510. [Google Scholar] [CrossRef]
- Konsoula, Z.; Barile, F.A. Epigenetic histone acetylation and deacetylation mechanisms in experimental models of neurodegenerative disorders. J. Pharmacol. Toxicol. Methods 2012, 66, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sheng, S.; Qin, C. The role of HDAC6 in Alzheimer’s Disease. J. Alzheimer Dis. 2013, 33, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Dai, X.-L.; Huang, H.-C.; Jiang, Z.-F. Targeting HDACs: A Promising therapy for Alzheimer’s disease. Oxid. Med. Cell. Longev. 2011, 2011, 143269–143274. [Google Scholar] [CrossRef] [PubMed]
- Julien, C.; Tremblay, C.; Emond, V.; Lebbadi, M.; Salem, N., Jr.; Bennett, D.A.; Calon, F. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2009, 68, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.; Barrup, M.; Kowall, N.W.; McKee, A. P3-260: Epigenetic modification in a monozygotic twin with Alzheimer’s disease. Alzheimers Dement. 2008, 4, 598. [Google Scholar] [CrossRef]
- Coneys, R.; Wood, I.C. Alzheimer’s disease: The potential of epigenetic treatments and current clinical candidates. Neurodegener Dis. Manag. 2020, 10, 543–558. [Google Scholar] [CrossRef]
- Gomes, A.Q.; Nolasco, S.; Soares, H. Non-coding RNAs: Multi-tasking molecules in the cell. Int. J. Mol. Sci. 2013, 14, 16010–16039. [Google Scholar] [CrossRef]
- Morris, K.V.; Mattick, J.S. The rise of regulatory RNA. Nat. Rev. Genet. 2014, 15, 423–437. [Google Scholar] [CrossRef]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef]
- Shin, C. Cleavage of the star strand facilitates assembly of some microRNAs into Ago2-containing silencing complexes in mammals. Mol. Cells 2008, 26, 308–313. [Google Scholar] [PubMed]
- Park, J.H.; Shin, C. MicroRNA-directed cleavage of targets: Mechanism and experimental approaches. BMB Rep. 2014, 47, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.M. An overview of microRNAs. Adv. Drug Deliv. Rev. 2015, 87, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, T.; Xiao, J. Circular RNAs: Promising biomarkers for human diseases. EBiomedicine 2018, 34, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, S.; Zoltowska, K.M.; Laskowska-Kaszub, K.; Wojda, U. microRNA diagnostic panel for Alzheimer’s disease and epigenetic trade-off between neurodegeneration and cancer. Ageing Res. Rev. 2019, 49, 125–143. [Google Scholar] [CrossRef]
- Burgos, K.; Malenica, I.; Metpally, R.; Courtright, A.; Rakela, B.; Beach, T.; Shill, H.; Adler, C.; Sabbagh, M.; Villa, S.; et al. Profiles of extracellular miRNA in cerebrospinal fluid and serum from patients with Alzheimer’s and Parkinson’s diseases correlate with disease status and features of pathology. PLoS ONE 2014, 9, e94839. [Google Scholar] [CrossRef]
- Cogswell, J.P.; Ward, J.; Taylor, I.A.; Waters, M.; Shi, Y.; Cannon, B.; Kelnar, K.; Kemppainen, J.; Brown, D.; Chen, C.; et al. Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J. Alzheimers Dis. 2008, 14, 27–41. [Google Scholar] [CrossRef]
- Geekiyanage, H.; Chan, C. MicroRNA-137/181c regulates serine palmitoyltransferase and in turn amyloid beta, novel targets in sporadic Alzheimer’s disease. J. Neurosci. 2011, 31, 14820–14830. [Google Scholar] [CrossRef]
- Hara, N.; Kikuchi, M.; Miyashita, A.; Hatsuta, H.; Saito, Y.; Kasuga, K.; Murayama, S.; Ikeuchi, T.; Kuwano, R. Serum microRNA miR-501-3p as a potential biomarker related to the progression of Alzheimer’s disease. Acta Neuropathol. Commun. 2017, 5, 10–19. [Google Scholar] [CrossRef]
- Riancho, J.; Vázquez-Higuera, J.L.; Pozueta, A.; Lage, C.; Kazimierczak, M.; Bravo, M.; Calero, M.; Gonalezález, A.; Rodríguez, E.; Lleó, A.; et al. MicroRNA profile in patients with Alzheimer’s disease: Analysis of miR-9-5p and miR-598 in raw and exosome enriched cerebrospinal fluid samples. J. Alzheimers Dis. 2017, 57, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, P.N.; Dua, P.; Hill, J.M.; Bhattacharjee, S.; Zhao, Y.; Lukiw, W.J. MicroRNA (miRNA) speciation in Alzheimer’s disease (AD) cerebrospinal fluid (CSF) and extracellular fluid (ECF). Int. J. Biochem. Mol. Biol. 2012, 3, 365–373. [Google Scholar] [PubMed]
- Sethi, P.; Lukiw, W.J. Micro-RNA abundance and stability in human brain: Specific alterations in Alzheimer’s disease temporal lobe neocortex. Neurosci. Lett. 2009, 459, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Alexandrov, P.N.; Zhao, Y.; Hill, J.M.; Bhattacharjee, S. Spreading of Alzheimer’s disease inflammatory signaling through soluble micro-RNA. Neuroreport 2012, 23, 621–626. [Google Scholar] [CrossRef]
- Lukiw, W.J. Micro-RNA speciation in fetal, adult and Alzheimer’s disease hippocampus. Neuroreport 2007, 18, 297–300. [Google Scholar] [CrossRef]
- Kiko, T.; Nakagawa, K.; Tsuduki, T.; Furukawa, K.; Arai, H.; Miyazawa, T. MicroRNAs in plasma and cerebrospinal fluid as potential markers for Alzheimer’s disease. J. Alzheimers Dis. 2014, 39, 253–259. [Google Scholar] [CrossRef]
- Lusardi, T.A.; Phillips, J.I.; Wiedrick, J.T.; Harrington, C.A.; Lind, B.; Lapidus, J.A.; Quinn, J.F.; Saugstad, J.A. MicroRNAs in human cerebrospinal fluid as biomarkers for Alzheimer’s disease. J. Alzheimers Dis. 2017, 55, 1223–1233. [Google Scholar] [CrossRef]
- Müller, M.; Jäkel, L.; Bruinsma, I.B.; Claassen, J.A.; Kuiperij, H.B.; Verbeek, M.M. MicroRNA-29a is a candidate biomarker for Alzheimer’s disease in cell-free cerebrospinal fluid. Mol. Neurobiol. 2016, 53, 2894–2899. [Google Scholar] [CrossRef]
- Geekiyanage, H.; Jicha, G.A.; Nelson, P.T.; Chan, C. Blood serum miRNA: Non-invasive biomarkers for Alzheimer’s disease. Exp. Neurol. 2012, 235, 491–496. [Google Scholar] [CrossRef]
- Lugli, G.; Cohen, A.M.; Bennett, D.A.; Shah, R.C.; Fields, C.J.; Hernandez, A.G.; Smalheiser, N.R. Plasma exosomal miRNAs in persons with and without Alzheimer disease: Altered expression and prospects for biomarkers. PLoS ONE 2015, 10, e0139233. [Google Scholar] [CrossRef]
- Satoh, J.; Kino, Y.; Niida, S. MicroRNA-Seq data analysis pipeline to identify blood biomarkers for Alzheimer’s disease from public data. Biomark Insights 2015, 10, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Villa, C.; Ridolfi, E.; Fenoglio, C.; Ghezzi, L.; Vimercati, R.; Clerici, F.; Marcone, A.; Gallone, S.; Serpente, M.; Cantoni, C.; et al. Expression of the transcription factor Sp1 and its regulatory hsa-miR-29b in peripheral blood mononuclear cells from patients with Alzheimer’s disease. J. Alzheimers Dis. 2013, 35, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Banzhaf-Strathmann, J.; Benito, E.; May, S.; Arzberger, T.; Tahirovic, S.; Kretzschmar, H.; Fischer, A.; Edbauer, D. MicroRNA-125b induces tau hyperphosphorylation and cognitive deficits in Alzheimer’s disease. EMBO J. 2014, 33, 1667–1680. [Google Scholar] [CrossRef] [PubMed]
- Nunez-Iglesias, J.; Liu, C.C.; Morgan, T.E.; Finch, C.E.; Zhou, X.J. JoInt. genome-wide profiling of miRNA and mRNA expression in Alzheimer’s disease cortex reveals altered miRNA regulation. PLoS ONE 2010, 5, e8898. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Song, Y.; Zhou, X.; Deng, Y.; Liu, T.; Weng, G.; Yu, D.; Pan, S. DNA methyltransferase 3, a target of microRNA-29c, contributes to neuronal proliferation by regulating the expression of brain-derived neurotrophic factor. Mol. Med. Rep. 2015, 12, 1435–1442. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Xu, J.; Xu, J.; Cheng, J.; Jiao, D.; Zhou, C.; Dai, Y.; Chen, Q. Lower serum levels of miR-29c-3p and miR-19b-3p as biomarkers for Alzheimer’s disease. Tohoku J. Exp. Med. 2017, 242, 129–136. [Google Scholar] [CrossRef]
- Gui, Y.; Liu, H.; Zhang, L.; Lv, W.; Hu, X. Altered microRNA profiles in cerebrospinal fluid exosome in Parkinson disease and Alzheimer disease. Oncotarget 2015, 6, 37043–37053. [Google Scholar] [CrossRef]
- Yang, G.; Song, Y.; Zhou, X.; Deng, Y.; Liu, T.; Weng, G.; Yu, D.; Pan, S. MicroRNA-29c targets beta-site amyloid precursor protein-cleaving enzyme 1 and has a neuroprotective role in vitro and in vivo. Mol. Med. Rep. 2015, 12, 3081–3088. [Google Scholar] [CrossRef]
- Lei, X.; Lei, L.; Zhang, Z.; Cheng, Y. Downregulated miR-29c correlates with increased BACE1 expression in sporadic Alzheimer’s disease. Int. J. Clin. Exp. Pathol. 2015, 8, 1565–1574. [Google Scholar]
- Sorensen, S.; Nygaard, A.; Christensen, T. miRNA expression profiles in cerebrospinal fluid and blood of patients with Alzheimer’s disease and oTher. types of dementia—An exploratory study. Transl. Neurodegener. 2016, 5, 6–18. [Google Scholar] [CrossRef]
- Cosín-Tomás, M.; Antonell, A.; Lladó, A.; Alcolea, D.; Fortea, J.; Ezquerra, M.; Lleó, A.; Martí, M.J.; Pallàs, M.; Sanchez-Valle, R.; et al. Plasma miR-34a-5p and miR-545-3p as early biomarkers of Alzheimer’s disease: Potential and limitations. Mol. Neurobiol. 2017, 54, 5550–5562. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Maes, O.C.; Chertkow, H.M.; Wang, E. MicroRNA expression in Alzheimer blood mononuclear cells. Gene Regul. Syst. Biol. 2007, 1, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Pogue, A.I.; Lukiw, W.J. Up-regulated pro-inflammatory microRNAs (miRNAs) in Alzheimer’s disease (AD) and age-related macular degeneration (AMD). Cell Mol. NeuroBiol. 2018, 38, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Agostini, M.; Tucci, P.; Killick, R.; Candi, E.; Sayan, B.S.; di Val Cervo, P.R.; Nicotera, P.; McKeon, F.; Knight, R.A.; Mak, T.W.; et al. Neuronal differentiation by TAp73 is mediated by microRNA-34a regulation of synaptic protein targets. Proc. Natl. Acad. Sci. USA 2011, 108, 21093–21098. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Bhattacharjee, S.; Jones, B.M.; Dua, P.; Alexandrov, P.N.; Hill, J.M.; Lukiw, W.J. Regulation of TREM2 expression by NF-κB-sensitive miRNA-34a. Neuroreport 2013, 24, 318–323. [Google Scholar] [CrossRef]
- Leidinger, P.; Backes, C.; Deutscher, S.; Schmitt, K.; Mueller, S.; Frese, K.; Haas, J.; Ruprecht, K.; Paul, F.; Stahler, C.; et al. A blood based 12-miRNA signature of Alzheimer disease patients. Genome Biol. 2013, 14, 78–94. [Google Scholar] [CrossRef]
- Wang, T.; Chen, K.; Li, H.; Dong, S.; Su, N.; Liu, Y.; Cheng, Y.; Dai, J.; Yang, C.; Xiao, S. The feasibility of utilizing plasma MiRNA107 and BACE1 messenger RNA gene expression for clinical diagnosis of amnestic mild cognitive impairment. J. Clin. Psychiatry 2015, 76, 135–141. [Google Scholar] [CrossRef]
- Wang, W.X.; Rajeev, B.W.; Stromberg, A.J.; Ren, N.; Tang, G.; Huang, Q.; Rigoutsos, I.; Nelson, P.T. The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J. Neurosci. 2008, 28, 1213–1223. [Google Scholar] [CrossRef]
- Moncini, S.; Lunghi, M.; Valmadre, A.; Grasso, M.; Vescovo, V.D.; Riva, P.; Denti, M.A.; Venturin, M. The miR-15/107 family of microRNA genes regulates CDK5R1/p35 with implications for Alzheimer’s disease pathogenesis. Mol. Neurobiol. 2017, 54, 4329–4342. [Google Scholar] [CrossRef]
- Müller, M.; Kuiperij, H.B.; Claassen, J.A.; Kusters, B.; Verbeek, M.M. MicroRNAs in Alzheimer’s disease: Differential expression in hippocampus and cell-free cerebrospinal fluid. Neurobiol. Aging 2014, 35, 152–158. [Google Scholar] [CrossRef]
- Galimberti, D.; Villa, C.; Fenoglio, C.; Serpente, M.; Ghezzi, L.; Cioffi, S.M.; Arighi, A.; Fumagalli, G.; Scarpini, E. Circulating miRNAs as potential biomarkers in Alzheimer’s disease. J. Alzheimers Dis. 2014, 42, 1261–1267. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Yu, J.T.; Tan, M.S.; Liu, Q.Y.; Wang, H.F.; Zhang, W.; Jiang, T.; Tan, L. Genome-wide serum microRNA expression profiling identifies serum biomarkers for Alzheimer’s disease. J. Alzheimers Dis. 2014, 40, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Dangla-Valls, A.; Molinuevo, J.L.; Altirriba, J.; Sanchez-Valle, R.; Alcolea, D.; Fortea, J.; Rami, L.; Balasa, M.; Muñoz-García, C.; Ezquerra, M.; et al. CSF microRNA profiling in Alzheimer’s disease: A screening and validation study. Mol. Neurobiol. 2017, 54, 6647–6654. [Google Scholar] [CrossRef] [PubMed]
- McKeever, P.M.; Schneider, R.; Taghdiri, F.; Weichert, A.; Multani, N.; Brown, R.A.; Boxer, A.L.; Karydas, A.; Miller, B.; Robertson, J.; et al. MicroRNA expression levels are altered in the cerebrospinal fluid of patients with young-onset Alzheimer’s disease. Mol. Neurobiol. 2018, 55, 8826–8841. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Liu, L.; Meng, J. MicroRNA-125b promotes neurons cell apoptosis and tau phosphorylation in Alzheimer’s disease. Neurosci. Lett. 2017, 661, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Cha, D.; Mengel, D.; Mustapic, M.; Liu, W.; Selkoe, D.J.; Kapogiannis, D.; Galasko, D.; Rissman, R.A.; Bennett, D.A.; Walsh, D.M. miR-212 and miR-132 are downregulated in neurally derived plasma exosomes of Alzheimer’s patients. Front. Neurosci. 2019, 13, 1208–1212. [Google Scholar] [CrossRef]
- Annese, A.; Manzari, C.; Lionetti, C.; Picardi, E.; Horner, D.S.; Chiara, M.; Caratozzolo, M.F.; Tullo, A.; Fosso, B.; Pesole, G.; et al. Whole transcriptome profiling of late-onset Alzheimer’s disease patients provides insights into the molecular changes involved in the disease. Sci. Rep. 2018, 8, 4282–4297. [Google Scholar] [CrossRef]
- Hebert, S.S.; Wang, W.-X.; Zhu, Q.; Nelson, P.T. A study of small RNAs from cerebral neocortex of pathology-verified Alzheimer’s disease, dementia with Lewy bodies, hippocampal sclerosis, frontotemporal lobar dementia, and non-demented human controls. J. Alzheimers Dis. 2013, 35, 335–348. [Google Scholar] [CrossRef]
- Lau, P.; Bossers, K.; Janky, R.S.; Salta, E.; Frigerio, C.S.; Barbash, S.; Rothman, R.; Sierksma, A.S.; Thathiah, A.; Greenberg, D.; et al. Alteration of the microRNA network during the progression of Alzheimer’s disease. EMBO Mol. Med. 2013, 5, 1613–1634. [Google Scholar] [CrossRef]
- Pichler, S.; Gu, W.; Hartl, D.; Gasparoni, G.; Leidinger, P.; Keller, A.; Meese, E.; Mayhaus, M.; Hampel, H.; Riemenschneider, M. The miRNome of Alzheimer’s disease: Consistent downregulation of the miR-132/212 cluster. Neurobiol. Aging 2017, 50, 1–10. [Google Scholar] [CrossRef]
- Smith, P.Y.; Hernandez-Rapp, J.; Jolivette, F.; Lecours, C.; Bisht, K.; Goupil, C.; Dorval, V.; Parsi, S.; Morin, F.; Planel, E.; et al. miR-132/212 deficiency impairs tau metabolism and promotes pathological aggregation in vivo. Hum. Mol. Genet. 2015, 24, 6721–6735. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.B.; Mufson, E.J.; Counts, S.E. Evidence for a neuroprotective microRNA pathway in amnestic mild cognitive impairment. Front. Neurosci. 2015, 9, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.K.; Veremeyko, T.; Patel, N.; Lemere, C.A.; Walsh, D.M.; Esau, C.; Vanderburg, C.; Krichevsky, A.M. De-repression of FOXO3a death axis by microRNA-132 and -212 causes neuronal apoptosis in Alzheimer’s disease. Hum. Mol. Genet. 2013, 22, 3077–3092. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Zhou, H.; Zhang, R.; Song, M.; Yu, L.; Wang, L.; Liu, Z.; Zhang, Q.; Cui, D.; Wang, X.; et al. Serum miR-206 and miR-132 as potential circulating biomarkers for mild cognitive impairment. J. Alzheimers Dis. 2015, 45, 721–731. [Google Scholar] [CrossRef]
- Dong, H.; Li, J.; Huang, L.; Chen, X.; Li, D.; Wang, T.; Hu, C.; Xu, J.; Zhang, C.; Zen, K.; et al. Serum microRNA profiles serve as novel biomarkers for the diagnosis of Alzheimer’s disease. Dis. Markers 2015, 2015, 625659–625681. [Google Scholar] [CrossRef]
- Wu, H.Z.Y.; Thalamuthu, A.; Cheng, L.; Fowler, C.; Masters, C.L.; Sachdev, P.; Mather, K.A. The Australian Imaging Biomarkers and Lifestyle Flagship Study of Ageing. Differential blood miRNA expression in brain amyloid imaging-defined Alzheimer’s disease and controls. Alzheimers Res. Ther. 2020, 12, 59–70. [Google Scholar] [CrossRef]
- Denk, J.; Boelmans, K.; Siegismund, C.; Lassner, D.; Arlt, S.; Jahn, H. MicroRNA profiling of CSF reveals potential biomarkers to detect Alzheimer`s disease. PLoS ONE 2015, 10, e0126423. [Google Scholar] [CrossRef]
- Cui, J.G.; Li, Y.Y.; Zhao, Y.; Bhattacharjee, S.; Lukiw, W.J. Differential regulation of interleukin-1 receptor-associated kinase-1 (IRAK-1) and IRAK-2 by microRNA-146a and NFkappaB in stressed human astroglial cells and in Alzheimer disease. J. Biol. Chem. 2010, 285, 38951–38960. [Google Scholar] [CrossRef]
- Zhao, Y.; Alexandrov, P.N.; Jaber, V.; Lukiw, W.J. Deficiency in the ubiquitin conjugating enzyme UBE2A in Alzheimer’s disease (AD) is linked to deficits in a natural circular miRNA-7 sponge (circRNA; ciRS-7). Genes 2016, 7, 116. [Google Scholar] [CrossRef]
- Lukiw, W.J.; Zhao, Y.; Cui, J.G. An NF-kappaB-sensitive microRNA-146a-mediated inflammatory circuit in Alzheimer disease and in stressed human brain cells. J. Biol. Chem. 2008, 283, 31315–31322. [Google Scholar] [CrossRef]
- Guedes, J.R.; Santana, I.; Cunha, C.; Duro, D.; Almeida, M.R.; Cardoso, A.M.; de Lima, M.C.; Cardoso, A.L. MicroRNA deregulation and chemotaxis and phagocytosis impairment in Alzheimer’s disease. Alzheimers Dement. 2016, 3, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Siedlecki-Wullich, D.; Catala-Solsona, J.; Fabregas, C.; Hernandez, I.; Clarimon, J.; Lleo, A.; Boada, M.; Saura, C.A.; Rodríguez-Álvarez, J.; Miñano-Molina, A.J. Altered microRNAs related to synaptic function as potential plasma biomarkers for Alzheimer’s disease. Alzheimers Res. Ther. 2019, 11, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Kenny, A.; McArdle, H.; Calero, M.; Rabano, A.; Madden, S.F.; Adamson, K.; Forster, R.; Spain, E.; Prehn, J.H.M.; Henshall, D.C.; et al. Elevated plasma microRNA-206 levels predict cognitive decline and progression to dementia from mild cognitive impairment. Biomolecules 2019, 9, 734. [Google Scholar] [CrossRef] [PubMed]
- van Harten, A.C.; Mulders, J.; Scheltens, P.; van der Flier, W.M.; Oudejans, C.B. Differential expression of microRNA in cerebrospinal fluid as a potential novel biomarker for Alzheimer’s disease. J. Alzheimers Dis. 2015, 47, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Millan, M.J. Linking deregulation of non-coding RNA to the core pathophysiology of Alzheimer’s disease: An integrative review. Prog. NeuroBiol. 2017, 156, 1–68. [Google Scholar] [CrossRef]
- Das, U.; Wang, L.; Ganguly, A.; Saikia, J.M.; Wagner, S.L.; Koo, E.H.; Roy, S. Visualizing APP and BACE-1 approximation in neurons yields insight into the amyloidogenic pathway. Nat. Neurosci. 2016, 19, 55–64. [Google Scholar] [CrossRef]
- Long, J.M.; Ray, B.; Lahiri, D.K. MicroRNA-339-5p down-regulates protein expression of beta-site amyloid precursor protein-cleaving enzyme 1 (BACE1) in human primary brain cultures and is reduced in brain tissue specimens of Alzheimer disease subjects. J. Biol. Chem. 2014, 289, 5184–5198. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Zhang, Y.; Xing, H.; Guo, S.; Zheng, Z.; Wang, H.; Xu, D. MicroRNA-135b has a neuroprotective role via targeting of beta-site APP-cleaving enzyme 1. Exp. Ther. Med. 2016, 12, 809–814. [Google Scholar] [CrossRef]
- Santa-Maria, I.; Alaniz, M.E.; Renwick, N.; Cela, C.; Fulga, T.A.; Van Vactor, D.; Tuschl, T.; Clark, L.N.; Shelanski, M.L.; McCabe, B.D.; et al. Dysregulation of microRNA-219 promotes neurodegeneration through post-transcriptional regulation of tau. J. Clin. Investig. 2015, 125, 681–686. [Google Scholar] [CrossRef]
- Zhou, Y.; Deng, J.; Chu, X.; Zhao, Y.; Guo, Y. Role of post-transcriptional control of calpain by miR-124-3p in the development of Alzheimer’s disease. J. Alzheimers Dis. 2019, 67, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Remenyi, J.; Hunter, C.J.; Cole, C.; Ando, H.; Impey, S.; Monk, C.E.; Martin, K.J.; Barton, G.J.; Hutvagner, G.; Arthur, J.S. Regulation of the miR-212/132 locus by MSK1 and CREB in response to neurotrophins. Biochem. J. 2010, 428, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, H.; Numakawa, T.; Kumamaru, E.; Adachi, N.; Mizuno, H.; Ninomiya, M.; Kunugi, H.; Hashido, K. Glucocorticoid attenuates brain-derived neurotrophic factor-dependent upregulation of glutamate receptors via the suppression of microRNA-132 expression. Neuroscience 2010, 165, 1301–1311. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Espejo, S.; Santos-Zorrozua, B.; Álvarez-González, P.; Lopez-Lopez, E.; Garcia-Orad, Á. A systematic review of microRNA expression as biomarker of late-onset Alzheimer’s disease. Mol. Neurobiol. 2019, 56, 8376–8391. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, Y.; Simkin, A.; Gascon, E.; Gao, F.B. MicroRNA-9: Functional evolution of a conserved small regulatory RNA. RNA Biol. 2011, 8, 557–564. [Google Scholar] [CrossRef]
- Holohan, K.N.; Lahiri, D.K.; Schneider, B.P.; Foroud, T.; Saykin, A.J. Functional microRNAs in Alzheimer’s disease and cancer: Differential regulation of common mechanisms and pathways. Front. Genet. 2012, 3, 323–339. [Google Scholar] [CrossRef]
- Shaik, M.M.; Tamargo, I.A.; Abubakar, M.B.; Kamal, M.A.; Greig, N.H.; Gan, S.H. The role of microRNAs in Alzheimer’s disease and their therapeutic potentials. Genes 2018, 9, 174. [Google Scholar] [CrossRef]
- Chang, F.; Zhang, L.H.; Xu, W.P.; Jing, P.; Zhan, P.Y. microRNA-9 attenuates amyloidβ-induced synaptotoxicity by targeting calcium/calmodulin-dependent protein kinase kinase 2. Mol. Med. Rep. 2014, 9, 1917–1922. [Google Scholar] [CrossRef]
- Mairet-Coello, G.; Courchet, J.; Pieraut, S.; Courchet, V.; Maximov, A.; Polleux, F. The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Aβ oligomers through Tau phosphorylation. Neuron 2013, 78, 94–108. [Google Scholar] [CrossRef]
- Bettens, K.; Brouwers, N.; Engelborghs, S.; Van Miegroet, H.; De Deyn, P.P.; Theuns, J.; Sleegers, K.; Van Broeckhoven, C. APP and BACE1 miRNA genetic variability has no major role in risk for Alzheimer disease. Hum. Mutat. 2009, 30, 1207–1213. [Google Scholar] [CrossRef]
- Zong, Y.; Wang, H.; Dong, W.; Quan, X.; Zhu, H.; Xu, Y.; Huang, L.; Ma, C.; Qin, C. miR-29c regulates BACE1 protein expression. Brain Res. 2011, 1395, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Shioya, M.; Obayashi, S.; Tabunoki, H.; Arima, K.; Saito, Y.; Ishida, T.; Satoh, J. Aberrant microRNA expression in the brains of neurodegenerative diseases: miR-29a decreased in Alzheimer disease brains targets neurone navigator 3. Neuropathol. Appl. Neurobiol. 2010, 36, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Kole, A.J.; Swahari, V.; Hammond, S.M.; Deshmukh, M. miR-29b is activated during neuronal maturation and targets BH3-only genes to restrict apoptosis. Genes Dev. 2011, 25, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001, 15, 2922–2933. [Google Scholar]
- Rohn, T.T.; Vyas, V.; Hernandez-Estrada, T.; Nichol, K.E.; Christie, L.A.; Head, E. Lack of pathology in a triple transgenic mouse model of Alzheimer’s disease after overexpression of the anti-apoptotic protein Bcl-2. J. Neurosci. 2008, 28, 3051–3059. [Google Scholar] [CrossRef]
- Howard, S.; Bottino, C.; Brooke, S.; Cheng, E.; Giffard, R.G.; Sapolsky, R. Neuroprotective effects of bcl-2 overexpression in hippocampal cultures: Interactions with pathways of oxidative damage. J. Neurochem. 2002, 83, 914–923. [Google Scholar] [CrossRef]
- Li, L.H.; Tu, Q.Y.; Deng, X.H.; Xia, J.; Hou, D.R.; Guo, K.; Zi, X.H. Mutant presenilin2 promotes apoptosis through the p53/miR-34a axis in neuronal cells. Brain Res. 2017, 1662, 57–64. [Google Scholar] [CrossRef]
- Wang, X.; Liu, P.; Zhu, H.; Xu, Y.; Ma, C.; Dai, X.; Huang, L.; Liu, Y.; Zhang, L.; Qin, C. miR-34a, a microRNA up-regulated in a double transgenic mouse model of Alzheimer’s disease, inhibits BCL2 translation. Brain Res. Bull. 2009, 80, 268–273. [Google Scholar] [CrossRef]
- Dickson, J.R.; Kruse, C.; Montagna, D.R.; Finsen, B.; Wolfe, M.S. Alternative polyadenylation and miR-34 family members regulate tau expression. J. Neurochem. 2013, 127, 739–749. [Google Scholar] [CrossRef]
- Nelson, P.T.; Wang, W.X. MiR-107 is reduced in Alzheimer’s disease brain neocortex: Validation study. J. Alzheimers Dis. 2010, 21, 75–79. [Google Scholar] [CrossRef]
- Van Damme, P.; Van Hoecke, A.; Lambrechts, D.; Vanacker, P.; Bogaert, E.; van Swieten, J.; Carmeliet, P.; Van Den Bosch, L.; Robberecht, W. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J. Cell Biol. 2008, 181, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.X.; Wilfred, B.R.; Madathil, S.K.; Tang, G.; Hu, Y.; Dimayuga, J.; Stromberg, A.J.; Huang, Q.; Saatman, K.E.; Nelson, P.T. miR-107 regulates granulin/progranulin with implications for traumatic brain injury and neurodegenerative disease. Am. J. Pathol. 2010, 177, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Hennessey, T.; Flynt, A.; Lai, E.; Beal, M.F.; Lin, M.T. MicroRNA-related cofilin abnormality in Alzheimer’s disease. PLoS ONE 2010, 5, e15546. [Google Scholar] [CrossRef] [PubMed]
- Moncini, S.; Salvi, A.; Zuccotti, P.; Viero, G.; Quattrone, A.; Barlati, S.; De Petro, G.; Venturin, M.; Riva, P. The role of miR-103 and miR-107 in regulation of CDK5R1 expression and in cellular migration. PLoS ONE 2011, 6, e20038. [Google Scholar] [CrossRef] [PubMed]
- Rademakers, R.; Sleegers, K.; Theuns, J.; Van den Broeck, M.; Bel Kacem, S.; Nilsson, L.-G.; Adolfsson, R.; van Duijn, C.M.; Van Broeckhoven, C.; Cruts, M. Association of cyclin-dependent kinase 5 and neuronal activators p35 and p39 complex in early-onset Alzheimer’s disease. Neurobiol. Aging 2005, 26, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Augustin, R.; Endres, K.; Reinhardt, S.; Kuhn, P.H.; Lichtenthaler, S.F.; Hansen, J.; Wurst, W.; Trümbach, D. Computational identification and experimental validation of microRNAs binding to the Alzheimer-related gene ADAM10. BMC Med. Genet. 2012, 13, 35. [Google Scholar] [CrossRef]
- Hansen, K.F.; Sakamoto, K.; Aten, S.; Snider, K.H.; Loeser, J.; Hesse, A.M.; Page, C.E.; Pelz, C.; Arthur, J.S.; Impey, S.; et al. Targeted deletion of miR-132/-212 impairs memory and alters the hippocampal transcriptome. Learn. Mem. 2016, 23, 61–71. [Google Scholar] [CrossRef]
- Lukiw, W.J. NF-κB-regulated micro RNAs (miRNAs) in primary human brain cells. Exp. Neurol. 2012, 235, 484–490. [Google Scholar] [CrossRef]
- Li, Y.Y.; Cui, J.G.; Dua, P.; Pogue, A.I.; Bhattacharjee, S.; Lukiw, W.J. Differential expression of miRNA-146a-regulated inflammatory genes in human primary neural, astroglial and microglial cells. Neurosci. Lett. 2011, 499, 109–113. [Google Scholar] [CrossRef]
- Fernandes, A.; Ribeiro, A.R.; Monteiro, M.; Garcia, G.; Vaz, A.R.; Brites, D. Secretome from SH-SY5Y APPSwe cells trigger time-dependent CHME3 microglia activation phenotypes, ultimately leading to miR-21 exosome shuttling. Biochimie 2018, 155, 67–82. [Google Scholar] [CrossRef]
- Li, J.J.; Wang, B.; Kodali, M.C.; Chen, C.; Kim, E.; Patters, B.J.; Lan, L.; Kumar, S.; Wang, X.; Yue, J.; et al. In vivo evidence for the contribution of peripheral circulating inflammatory exosomes to neuroinflammation. J. Neuroinflamm. 2018, 15, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Lee, J.E. miR-155 is involved in Alzheimer’s disease by regulating T lymphocyte function. Front. Aging Neurosci. 2015, 7, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Schonrock, N.; Ke, Y.D.; Humphreys, D.; Staufenbiel, M.; Ittner, L.M.; Preiss, T.; Götz, J. Neuronal microRNA deregulation in response to Alzheimer’s disease amyloid-beta. PLoS ONE 2010, 5, e11070. [Google Scholar] [CrossRef] [PubMed]
- Schonrock, N.; Humphreys, D.T.; Preiss, T.; Götz, J. Target gene repression mediated by miRNAs miR-181c and miR-9 both of which are down-regulated by amyloid-β. J. Mol. Neurosci. 2012, 46, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, E.R.; Kawamoto, E.M.; Taub, D.D.; Lal, A.; Abdelmohsen, K.; Zhang, Y.; Wood, W.H., 3rd; Lehrmann, E.; Camandola, S.; Becker, K.G.; et al. Evidence for miR-181 involvement in neuroinflammatory responses of astrocytes. Glia 2013, 61, 1018–1028. [Google Scholar] [CrossRef]
- Lee, S.T.; Chu, K.; Jung, K.H.; Kim, J.H.; Huh, J.Y.; Yoon, H.; Park, D.-K.; Lim, J.-Y.; Kim, J.-M.; Daejong, J.; et al. miR-206 regulates brain-derived neurotrophic factor in Alzheimer disease model. Ann. Neurol. 2012, 72, 269–277. [Google Scholar] [CrossRef]
- Tian, N.; Cao, Z.; Zhang, Y. MiR-206 decreases brain-derived neurotrophic factor levels in a transgenic mouse model of Alzheimer’s disease. Neurosci. Bull. 2014, 30, 191–197. [Google Scholar] [CrossRef]
- Wang, C.N.; Wang, Y.J.; Wang, H.; Song, L.; Chen, Y.; Wang, J.L.; Ye, Y.; Jiang, B. The anti-dementia effects of donepezil involve miR-206-3p in the hippocampus and cortex. Biol. Pharm Bull. 2017, 40, 465–472. [Google Scholar] [CrossRef]
- Chuang, D.-M.; Leng, Y.; Marinova, Z.; Kim, H.-J.; Chiu, C.-T. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009, 32, 591–601. [Google Scholar] [CrossRef]
- Cuadrado-Tejedor, M.; Oyarzabal, J.; Pascual Lucas, M.; Franco, R.; Garcia-Osta, A. Epigenetic drugs in Alzheimer’s disease. Biomol. Concepts 2013, 4, 433–445. [Google Scholar] [CrossRef]
- Esposito, M.; Sherr, G.L. Epigenetic modifications in Alzheimer’s neuropathology and therapeutics. Front. Neurosci. 2019, 13, 476–490. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.B.; McNamara, P.; Heo, S.; Turner, A.; Lane, W.S.; Chakravarti, D. Regulation of histone acetylation and transcription by INHAT, a human cellular complex containing the set oncoprotein. Cell 2001, 104, 119–130. [Google Scholar] [CrossRef]
- Tsujio, I.; Zaidi, T.; Xu, J.; Kotula, L.; Grundke-Iqbal, I.; Iqbal, K. Inhibitors of protein phosphatase-2A from human brain structures, immunocytological localization and activities towards dephosphorylation of the Alzheimer type hyperphosphorylated tau. FEBS Lett. 2005, 579, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Tanimukai, H.; Grundke-Iqbal, I.; Iqbal, K. Upregulation of inhibitors of protein phosphatase-2A in Alzheimer’s disease. Am. J. Pathol. 2005, 166, 1761–1771. [Google Scholar] [CrossRef]
- Chai, G.S.; Feng, Q.; Wang, Z.H.; Hu, Y.; Sun, D.-S.; Li, X.-G.; Ke, D.; Li, H.-L.; Liu, G.-P.; Wang, J.-Z. Downregulating ANP32A rescues synapse and memory loss via chromatin remodeling in Alzheimer model. Mol. Neurodegener. 2017, 12, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.; Hossain, G.S.; Kocerha, J. The potential for microRNA therapeutics and clinical research. Front. Genet. 2019, 10, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Alexandrov, P.N.; Lukiw, W.J. Anti-microRNAs as novel therapeutic agents in the clinical management of Alzheimer’s disease. Front. Neurosci. 2016, 10, 59–66. [Google Scholar] [CrossRef]
- Paul, S.; Bravo Vázquez, L.A.; Pérez Uribe, S.; Roxana Reyes-Pérez, P.; Sharma, A. current status of microRNA-based therapeutic approaches in neurodegenerative disorders. Cells 2020, 9, 1698. [Google Scholar] [CrossRef]
- Kim, H.; Kim, J.S. A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 2014, 15, 321–334. [Google Scholar] [CrossRef]
- Barman, N.C.; Khan, N.M.; Islam, M.; Nain, Z.; Roy, R.K.; Haque, A.; Barman, S.K. CRISPR-Cas9: A promising genome editing therapeutic tool for Alzheimer’s disease-a narrative review. Neurol. Ther. 2020, 9, 419–434. [Google Scholar] [CrossRef]
- Cota-Coronado, A.; Díaz-Martínez, N.F.; Padilla-Camberos, E.; Díaz-Martínez, N.E. Editing the central nervous system through CRISPR/Cas9 systems. Front. Mol. Neurosci. 2019, 1, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Rohn, T.T.; Kim, N.; Isho, N.F.; Mack, J.M. The potential of CRISPR/Cas9 gene editing as a treatment strategy for Alzheimer’s disease. J. Alzheimers Dis. Parkinsonism 2018, 8, 439. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, A.A.; Lim, W.A.; Qi, L.S. Beyond editing: Repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat. Rev. Mol. Cell Biol. 2016, 17, 5–15. [Google Scholar] [CrossRef]
- Kearns, N.A.; Pham, H.; Tabak, B.; Genga, R.M.; Silverstein, N.J.; Garber, M.; Maehr, R. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat. Methods 2015, 12, 401–403. [Google Scholar] [CrossRef] [PubMed]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef]
- Cano-Rodriguez, D.; Gjaltema, R.A.; Jilderda, L.J.; Jellema, P.; Dokter-Fokkens, J.; Ruiters, M.H.; Rots, M.G. Writing of H3K4Me3 overcomes epigenetic silencing in a sustained but context-dependent manner. Nat. Commun. 2016, 7, 12284. [Google Scholar] [CrossRef]
- Kwon, D.Y.; Zhao, Y.T.; Lamonica, J.M.; Zhou, Z. Locus-specific histone deacetylation using a synthetic CRISPR-Cas9-based HDAC. Nat. Commun. 2017, 8, 15315. [Google Scholar] [CrossRef]
- Lei, Y.; Zhang, X.; Su, J.; Jeong, M.; Gundry, M.C.; Huang, Y.H.; Zhou, Y.; Li, W.; Goodell, M.A. Targeted DNA methylation in vivo using an engineered dCas9-MQ1 fusion protein. Nat. Commun. 2017, 8, 16026. [Google Scholar] [CrossRef]
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA Methylation in the Mammalian Genome. Cell 2016, 167, 233–247 e217. [Google Scholar] [CrossRef]
- McDonald, J.I.; Celik, H.; Rois, L.E.; Fishberger, G.; Fowler, T.; Rees, R.; Kramer, A.; Martens, A.; Edwards, J.R.; Challen, G.A. Reprogrammable CRISPR/Cas9-based system for inducing site-specific DNA methylation. Biol. Open 2016, 5, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Vojta, A.; Dobrinic, P.; Tadic, V.; Bockor, L.; Korac, P.; Julg, B.; Klasic, M.; Zoldos, V. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016, 44, 5615–5628. [Google Scholar] [CrossRef] [PubMed]
- Xiong, T.; Meister, G.E.; Workman, R.E.; Kato, N.C.; Spellberg, M.J.; Turker, F.; Timp, W.; Ostermeier, M.; Novina, C.D. Targeted DNA methylation in human cells using engineered dCas9-methyltransferases. Sci. Rep. 2017, 7, 6732. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.R.; Cui, Y.; Lubecka, K.; Stefanska, B.; Irudayaraj, J. CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget 2016, 7, 46545–46556. [Google Scholar] [CrossRef] [PubMed]
- Morita, S.; Noguchi, H.; Horii, T.; Nakabayashi, K.; Kimura, M.; Okamura, K.; Sakai, A.; Nakashima, H.; Hata, K.; Nakashima, K.; et al. Targeted DNA demethylation in vivo using dCas9-peptide repeat and scFv-TET1 catalytic domain fusions. Nat. Biotechnol. 2016, 34, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tao, Y.; Gao, X.; Zhang, L.; Li, X.; Zou, W.; Ruan, K.; Wang, F.; Xu, G.L.; Hu, R. A CRISPR based approach for targeted DNA demethylation. Cell Discov. 2016, 2, 16009. [Google Scholar] [CrossRef] [PubMed]
- Shechner, D.M.; Hacisuleyman, E.; Younger, S.T.; Rinn, J.L. Multiplexable, locus-specific targeting of long RNAs with CRISPR-Display. Nat. Methods 2015, 12, 664–670. [Google Scholar] [CrossRef]
- Manev, H.; Dzitoyeva, S. Progress in mitochondrial epigenetics. Biomol. Concepts 2013, 4, 381–389. [Google Scholar] [CrossRef]
- Mattsson, N. CSF biomarkers in neurodegenerative diseases. Clin. Chem. Lab. Med. 2011, 49, 345–352. [Google Scholar] [CrossRef]
- Johansson, P.; Mattsson, N.; Hansson, O.; Wallin, A.; Johansson, J.O.; Andreasson, U.; Zetterberg, H.; Blennow, K.; Svensson, J. Cerebrospinal fluid biomarkers for Alzheimer’s disease: Diagnostic performance in a homogeneous mono-center population. J. Alzheimers Dis. 2011, 24, 537–546. [Google Scholar] [CrossRef]
- Toraño, E.G.; García, M.G.; Fernández-Morera, J.L.; Niño-García, P.; Fernández, A.F. The impact of external factors on the epigenome: In utero and over lifetime. Biomed. Res. Int. 2016, 2568635. [Google Scholar]
- Kurdyukov, S.; Bullock, M. DNA methylation analysis: Choosing the right method. Biology 2016, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Bai, S.F.; Yan, J.Q. Blood circulating miRNAs as biomarkers of Alzheimer’s disease: A systematic review and meta-analysis. Biomark Med. 2019, 13, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Podlesniy, P.; Llorens, F.; Golanska, E.; Sikorska, B.; Liberski, P.; Zerr, I.; Trullas, R. Mitochondrial DNA differentiates Alzheimer’s disease from Creutzfeldt-Jakob disease. Alzheimers Dement. 2016, 12, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Sheng, B.; Wang, X.; Su, B.; Lee, H.G.; Casadesus, G.; Perry, G.; Zhu, X. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J. Neurochem. 2012, 120, 419–429. [Google Scholar] [CrossRef] [PubMed]

| Epigenetic Mechanism | Effect | Gene/Target Pathway Involved | Study Model | Tissue/Study Design | Main Results | Ref. |
|---|---|---|---|---|---|---|
| Acetylation (H3K27) | ↑ | CR1, GPR22, KMO, PIM3, PSEN1, RGCC | AD (n = 24), Ctrl (n = 23) | CNS (Ecx)/ChIP-seq | Acetylated peaks identified close to the genes involved in tau and amyloid neuropathologies. | [118] |
| Acetylation (H3, H4, acetyl histone H3, acetyl histone H4) | ↑ | Genes implicated in AD development and synaptic plasticity. BACE1, PSEN1 | AD (n = 14), Ctrl (n = 17) | CNS (ITG, MTG)/immunohistochemistry and tissue microarrays | Significant positive correlations found between ubiquitin load and histone modifications. | [119] |
| Acetylation (H4K12, H4K16) | ↑ | None stated | AD (n = 6), Ctrl (n = 6) | CNS (FCx)/LC-MS/MS | First study to report changes in methylation of H2BK108 and H4R55, and ubiquitination of H2BK120 in FCx of AD subjects. | [120] |
| AD (n = 34), MCI (n = 15), Ctrl (n = 31) | Monocytes/quantification fluorometric kit | Significantly elevated acetylation of H4K12 in human patients with MCI but not in patients with AD. | [121] | |||
| Acetylation (HDAC1, HDAC2) | ↓ | None stated | AD (n = 8), Ctrl (n = 7) | CNS (FCx, HPC)/Western blot | HDAC1 and HDAC2 are decreased in FCX, while HDAC1 is decreased in HPC of AD patients. | [122] |
| Acetylation (H3K9K14, H2B) | ↑ | None stated | AD (n = 8), Ctrl (n = 7) | CNS (FCx, HPC)/Western blot | Increased histone levels associated with the cytoplasmic fraction and pointing to a dysregulation in histone catabolism in FCx. | [122] |
| Acetylation (H3K18/K23) | ↓ | None stated | AD (n = 11), Ctrl (n = 4) | CNS (TCx)/LC-MS/MS and Western blot | Histone acetylation significantly lower in AD temporal lobe than in aged controls. | [123] |
| Acetylation (HDACs) | ↑ | None stated | AD (n = 25), Ctrl (n = 25) | Plasma/colorimetric HDAC activity assay | Plasma levels of HDACs might be used as peripheral biomarkers of AD. | [124] |
| Methylation (H2BK108, H4R55) | ↓ | None stated | AD (n = 6), Ctrl (n = 6) | CNS (FCx)/LC-MS/MS | First study to report changes in methylation of H2BK108, methylation of H4R55, and ubiquitination of H2BK120 in FCx of AD subjects | [120] |
| Phosphorylation (H3) | ↑ | None stated | AD (n = 10), BD (n = 10), Ctrl (n = 10) | CNS (FCx)/ELISA | Increased H3 phosphorylation in AD and BD subjects indicate an onset of apoptosis and cell death. | [125] |
| Phosphorylation (H2AX) | ↑ | None stated | AD (n = 13), Ctrl (n = 13) | CNS (HPC, TCx)/immunohistochemistry | Increased DNA damage in the astrocytes of AD brains, as evidenced by the astrocytic nuclear accumulation of γH2AX. | [126] |
| Phosphorylation (H3) | ↑ | None stated | AD (n = 17), Ctrl (n = 9) | CNS (HPC)/immunohistochemistry | Study implicates that neurons in AD are mitotically activated. This is in line with other studies that indicate that the cell cycle is activated in AD neurons. | [127] |
| Ubiquitination (H2BK120) | ↑ | None stated | AD (n = 6), Ctrl (n = 6) | CNS (FCx)/LC-MS/MS | First to report changes in methylation of H2BK108, methylation of H4R55, and ubiquitination of H2BK120 in AD FCx. | [120] |
| Poly(ADP-ribosyl)ation | ↑ | Gnenes coding for nuclear proteins: MAP2, GFAP, CD68 | AD (n = 20), Ctrl (n = 10) | CNS (FCx, TCx)/immunohistochemistry | Findings indicate that there is enhanced PARP activity in AD. | [128] |
| Epigenetic Mechanism | Effect | Study Model 1 | Tissue Study/Design | Ref. |
|---|---|---|---|---|
| miR-9-5p | ↓ | AD (n = 69), PD (n = 67), Ctrl (n = 78) | CSF/miRNA-seq | [151] |
| Braak III-VI (n = 20), Braak 0-I (n = 7) | CNS (HPC, MFG, CB)/RT-qPCR | [152] | ||
| AD (n = 7), Ctrl (n = 7) | CNS (neocortex)/RT-qPCR | [153] | ||
| AD (n = 27), Ctrl (n = 18) | CNS (TCx)/miRNA-seq/RT-qPCR | [154] | ||
| ↑ | AD (n = 10/18), Ctrl (n = 10/18) | CSF/RT-qPRC | [155] | |
| AD (n = 6), Ctrl (n = 6) | CNS (TCx)/array and RT-qPCR | [156] | ||
| AD (n = 6), Ctrl (n = 6) | CNS (TCx)/array | [157] | ||
| AD (n = 5), Ctrl (n = 5) | CNS (TCx)/RT-qPCR | [158] | ||
| AD (n = 5), Ctrl (n = 5) | CNS (HPC)/array | [159] | ||
| miR-29a(-3p) | ↓ | AD (n = 10), Ctrl (n = 10) | CSF/RT-qPCR | [160] |
| AD (n = 50/16); Ctrl (n = 49/16) | CSF/array and RT-qPCR | [161] | ||
| AD (n = 7), Ctrl (n = 7) | CNS (neocortex)/RT-qPCR | [153] | ||
| ↑ | AD (n = 18), Ctrl (n = 20) | CSF/RT-qPCR | [162] | |
| Braak III-VI (n = 20), Braak 0-I (n = 7) | CNS (HPC, MFG, CB)/RT-qPCR | [152] | ||
| miR-29b(-3p) | ↓ | Probable AD (n = 7), aMCI/Probable Early AD (n = 7), Ctrl (n = 7) | Serum/RT-qPCR | [163] |
| AD (n = 35), Ctrl (n = 35) | Plasma (exosomes)/miRNA-seq | [164] | ||
| AD (n = 48), Ctrl (n = 22) | Blood/omiRas and DIANA miRPath | [165] | ||
| AD (n = 28), Ctrl (n = 25) | PBMC/RT-qPCR | [166] | ||
| AD (n = 10), Ctrl (n = 5) | CNS (FCx)/RT-qPCR | [167] | ||
| AD (n = 5), Ctrl (n = 5) | CNS (PCx) array | [168] | ||
| ↑ | Braak III-VI (n = 20), Braak 0-I (n = 7) | CNS (HPC, MFG, CB)/RT-qPCR | [152] | |
| miR-29c(-3p) | ↓ | AD (n = 30), Ctrl (n = 30) | Blood/RT-qPCR | [169] |
| AD (n = 20), Ctrl (n = 20) | Serum/miRNA-seq & RT-qPCR | [170] | ||
| AD (n = 28), PD (n = 47), Ctrl (n = 27) | CSF (exosomes)/array and RT-qPCR | [171] | ||
| AD (n = 30), Ctrl (n = 30) | CSF/RT-qPCR | [172] | ||
| AD (n = 31), Ctrl (n = 29) | CNS (FCx)/RT-qPCR | [173] | ||
| AD (n = 5), Ctrl (n = 5) | CNS (PCx)/array | [168] | ||
| ↑ | AD (n = 10), VD (n = 4), FTD (n = 4), DLB (n = 2) | CSF/RT-qPCR | [174] | |
| miR-34a(-5p) | ↓ | AD (n = 10), Ctrl (n = 10) | Plasma/RT-qPCR | [160] |
| AD (n = 21/15), preclinical AD (n = 21/15), Ctrl (n = 21/15), PD (n = 21/0) | Plasma/RT-qPCR | [175] | ||
| AD (n = 10), Ctrl (n = 10) | CSF/RT-qPCR | [160] | ||
| ↑ | AD (n = 16), Ctrl (n = 16) | PBMC/array and RT-qPCR | [176] | |
| AD (n = 5), Ctrl (n = 5) | CNS (TCx)/RT-qPCR | [158] | ||
| AD (n = 26), Ctrl (n = 19) | CNS (TCx)/array | [177] | ||
| AD (n = 29), Ctrl (n = 20) | CNS (HPC)/RT-qPCR | [178] | ||
| AD (n = 3), Ctrl (n = 3) | CNS (HPC)/array | [179] | ||
| miR-107 | ↓ | AD (n = 48/106), MCI (n = 18/0), MS (n = 16/0), PD (n = 9/0), DEP (n = 15/0), BD (n = 15/0), SCH (n = 14/0), Ctrl (n = 22/22) | Blood/miRNA-seq and RT-qPCR | [180] |
| AD (n = 97), aMCI (n = 116), Ctrl (n = 81) | Plasma/RT-qPCR | [181] | ||
| AD (n = 27), Ctrl (n = 18) | CNS (TCx)/miRNA-seq and RT-qPCR | [154] | ||
| AD (n = 6), MCI (n = 6), Ctrl (n = 11) | CNS (TCx)/array | [182] | ||
| AD (n = 12), Ctrl (n = 12) | CNS (HPC, TCx, CB)/RT-qPCR | [183] | ||
| AD (n = 10), Ctrl (n = 11) | CNS (HPC)/RT-qPCR | [184] | ||
| miR-125b | ↓ | AD (n = 69), PD (n = 67), Ctrl (n = 78) | Serum/miRNA-seq | [151] |
| AD (n = 22), FTD (n = 10), Ctrl (n = 26) | Serum/array and RT-qPCR | [185] | ||
| AD (n = 10), Ctrl (n = 10) | Plasma/RT-qPCR | [160] | ||
| AD (n = 35), Ctrl (n = 35) | Plasma (exosomes)/miRNA-seq | [164] | ||
| Probable AD (n = 105), Ctrl (n = 150) | Serum/RT-qPCR | [186] | ||
| AD (n = 22), FTD (n = 10), Ctrl (n = 26) | CSF/array and RT-qPCR | [185] | ||
| AD (n = 50/16), Ctrl (n = 49/16) | CSF/array and RT-qPCR | [161] | ||
| ↑ | AD (n = 6), Ctrl (n = 6) | CSF/array and RT-qPCR | [156] | |
| AD (n = 10/37), Ctrl (n = 10/32) | CSF/array and RT-qPCR | [187] | ||
| AD (n = 10); Ctrl (n = 10) | CSF/RT-qPCR | [160] | ||
| YOAD (n = 17/17), LOAD (n = 13/13), Ctrl (n = 12/12) | CSF (exosomes)/RT-qPCR | [188] | ||
| miR-125b-3p | ↓ | AD (n = 20), Ctrl (n = 20) | Serum/miRNA-seq and RT-qPCR | [170] |
| ↑ | AD (n = 6), Ctrl (n = 6) | CNS (TCx)/array and RT-qPCR | [156] | |
| AD (n = 27), Ctrl (n = 18) | CNS (TCx)/miRNA-seq and RT-qPCR | [154] | ||
| AD (n = 5), Ctrl (n = 5) | CNS (TCx)/RT-qPCR | [158] | ||
| AD (n = 26), Ctrl (n = 19) | CNS (TCx)/array | [177] | ||
| AD (n = 6), Ctrl (n = 6) | CNS (TCx) array | [157] | ||
| Braak III-VI (n = 20), Braak 0-I (n = 7) | CNS (HPC, MFG, CB)/RT-qPCR | [152] | ||
| AD (n = 3), Ctrl n = 3) | CNS (HPC)/array | [179] | ||
| AD (n = 10), Ctrl (n = 5) | CNS (FCx)/RT-qPCR | [167] | ||
| AD (n = 9), MCI (n = 8), Ctrl (n = 10) | CNS/RT-qPCR | [189] | ||
| miR-132(-3p) | ↓ | AD (n = 31), AD-MCI (n = 16), Ctrl (n = 16) | Plasma (exosomes)/array and RT-qPCR | [190] |
| AD (n = 11), Ctrl (n = 8) | CNS/array and RT-qPCR | [190] | ||
| AD (n = 6), PD (n = 6), Ctrl (n = 6) | CNS (HPC, TCx, FCx)/miRNA-seq and RT-qPCR | [191] | ||
| Braak III-VI (n = 20), Braak 0-I (n = 7) | CNS (HPC, MFG, CB)/RT-qPCR | [152] | ||
| AD (n = 27), Ctrl (n = 18) | CNS (TCx)/miRNA-seq and RT-qPCR | [154] | ||
| AD (n = 5), DLB (n = 4), FTD (n = 5), HS-aging (n = 4), Ctrl (n = 2) | CNS (TCx)/RNA deep seq and RT-qPCR | [192] | ||
| HPC: AD (n = 41), Ctrl (n = 23); FCx: AD (n = 21), Ctrl (n = 28), TCx: AD (n = 8), Ctrl (n = 8) | CNS (HPC, FCx, TCx)/RNA deep seq and RT-qPCR | [193] | ||
| FCx: AD (n = 225/8), Ctrl (n = 87/8), TCx: AD (n = 39/8), Ctrl (n = 25/8) | CNS (FCx, TCx)/array and RT-qPCR | [194] | ||
| HPC: AD (n = 10), Ctrl (n = 13); FCx: AD (n = 7), Ctrl (n = 5); TCx: AD (n = 8/11), MCI (n = 0/10), Ctrl (n = 8/11) | CNS (HPC, FCx, TCx)/RT-qPCR | [195] | ||
| AD (n = 3/10), MCI (n = 0/10), Ctrl (n = 3/12) | CNS (FCx)/array and RT-qPCR | [196] | ||
| Braak IV (n = 18), Braak III/IV (n = 14), Ctrl (n = 18) | CNS (TCx)/RT-qPCR | [197] | ||
| ↑ | MCI (n = 66), Ctrl (n = 76) | Serum/RT-qPCR | [198] | |
| miR-146a(-5p) | ↓ | AD (n = 127), MCI (n = 30), VD (n = 30) | Serum/miRNA-seq and RT-qPCR | [199] |
| AD (n = 10), Ctrl (n = 10) | Plasma/RT-qPCR | [160] | ||
| AD (n = 40), Ctrl (n = 31); Validation cohort: publicly available dataset of miRNA data | Blood/miRNA-seq | [200] | ||
| AD (n = 10), Ctrl (n = 10) | CSF/RT-qPCR | [160] | ||
| AD (n = 50/16), Ctrl (n = 49/16) | CSF/array and RT-qPCR | [161] | ||
| AD (n = 20), Ctrl (n = 20) | CSF/RT-qPCR | [184] | ||
| AD (n = 60), MCI-AD (n = 39), FTD (n = 37), DLB (n = 37), Ctrl (n = 40) | CSF/RT-qPCR | [162] | ||
| Braak III-VI (n = 20), Braak 0-I (n = 7) | CNS (HPC, MFG, CB)/RT-qPCR | [152] | ||
| AD (n = 27), Ctrl (n = 18) | CNS (TCx)/miRNA-seq and RT-qPCR | [154] | ||
| AD (n = 10), Ctrl (n = 11) | CNS (HPC)/array | [184] | ||
| Braak III-VI (n = 20), Braak 0-I (n = 7) | CNS (HPC, MFG, CB)/RT-qPCR | [152] | ||
| AD (n = 27), Ctrl (n = 18) | CNS (TCx)/miRNA-seq and RT-qPCR | [154] | ||
| ↑ | AD (n = 20), Ctrl (n = 20) | Serum/miRNA-seq and RT-qPCR | [170] | |
| AD (n = 6), Ctrl (n = 6) | CSF/array and RT-qPCR | [156] | ||
| AD (n = 22), Ctrl (n = 28) | CSF/RT-qPCR | [201] | ||
| AD (n = 6), Ctrl (n = 6) | CNS (TCx)/array and RT-qPCR | [156] | ||
| AD (n = 36), Ctrl (n = 30) | CNS (HPC, TCx)/array | [202] | ||
| AD (n = 5), Ctrl (n = 5) | CNS (TCx)/RT-qPCR | [158] | ||
| AD (n = 26), Ctrl (n = 19) | CNS (TCx)/array | [177] | ||
| AD (n = 6), Ctrl (n = 6) | CNS (TCx)/array | [157] | ||
| AD (n = 12), Ctrl (n = 6) | CNS/array | [203] | ||
| AD (n = 23), Ctrl (n = 23) | CNS (TCx)/array | [204] | ||
| AD (n = 3), Ctrl (n = 3) | CNS (HPC)/array | [179] | ||
| miR-155 | ↑ | AD (n = 36), MCI (n = 52), Ctrl (n = 6) | PBMC/RT-qPCR | [205] |
| AD (n = 16), Ctrl (n = 16) | PBMC/array and RT-qPCR | [176] | ||
| AD (n = 6), Ctrl (n = 6) | CNS (TCx)/array and RT-qPCR | [156] | ||
| AD (n = 12), Ctrl (n = 6) | CNS/array | [203] | ||
| AD (n = 5), Ctrl (n = 5) | CNS (TCx)/RT-qPCR | [158] | ||
| AD (n = 3), Ctrl (n = 3) | CNS (HPC)/array | [179] | ||
| Braak III-VI (n = 20), Braak 0-I (n = 7) | CNS (HPC, MFG, CB)/RT-qPCR | [152] | ||
| AD (n = 27), Ctrl (n = 18) | CNS (TCx)/miRNA-seq and RT-qPCR | [154] | ||
| AD (n = 26), Ctrl (n = 19) | CNS (TCx)/array | [177] | ||
| miR-181c(-5p) | ↓ | Probable AD (n = 7), aMCI/Probable Early AD (n = 7), Ctrl (n = 7) | Serum/RT-qPCR | [163] |
| Probable AD (n = 105), Ctrl (n = 150) | Serum/RT-qPCR | [186] | ||
| AD (n = 7), Ctrl (n = 7) | CNS (neocortex)/RT-qPCR | [153] | ||
| AD (n = 5), Ctrl (n = 5) | CNS (PCx)/array | [168] | ||
| ↑ | AD (n = 56/0), MCI (n = 26/0), FTD (n = 0/27), Ctrl (n = 14/24) | Plasma/RT-qPCR | [206] | |
| miR-206 | ↑ | AD (n = 25), MCI (n = 30), Ctrl (n = 31); Longitudinal cohort: MCI-MCI-Dementia (n = 6), Ctrl-MCI-Dementia (n = 6), Ctrl-MCI-MCI (n = 6) | Plasma/array and RT-qPCR | [207] |
| MCI (n = 66), Ctrl (n = 76) | Serum/RT-qPCR | [198] | ||
| AD (n = 10/18), Ctrl (n = 10/18) | CSF/RT-qPRC | [155] | ||
| AD (n = 19/19); Ctrl (n = 19/19) | CSF/array and RT-qPCR | [208] | ||
| miR-212(-3p) | ↓ | AD (n = 31), AD-MCI (n = 16), Ctrl (n = 16) | Plasma (exosomes)/array and RT-qPCR | [190] |
| AD (n = 11), Ctrl (n = 8) | CNS/Array & RT-qPCR | [190] | ||
| AD (n = 6), PD (n = 6), Ctrl (n = 6) | CNS (HPC, FCx, TCx)/miRNA-seq and RT-qPCR | [191] | ||
| Braak III-VI (n = 20), Braak 0-I (n = 7) | CNS (HPC, MFG, CB)/RT-qPCR | [152] | ||
| AD (n = 3/10), MCI (n = 10), Ctrl (n = 3/12) | CNS (FCx)/array and RT-qPCR | [196] | ||
| Braak IV (n = 18), Braak III/IV (n = 14), Ctrl (n = 18) | CNS (TCx)/RT-qPCR | [197] | ||
| FCX: AD (n = 225/8), Ctrl (n = 87/8); TCx: AD (n = 39/8), Ctrl (n = 25/8) | CNS (FCx, TCx)/array and RT-qPCR | [194] | ||
| AD (n = 27), Ctrl (n = 18) | CNS (TCx)/miRNA-seq and RT-qPCR | [154] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikolac Perkovic, M.; Videtic Paska, A.; Konjevod, M.; Kouter, K.; Svob Strac, D.; Nedic Erjavec, G.; Pivac, N. Epigenetics of Alzheimer’s Disease. Biomolecules 2021, 11, 195. https://doi.org/10.3390/biom11020195
Nikolac Perkovic M, Videtic Paska A, Konjevod M, Kouter K, Svob Strac D, Nedic Erjavec G, Pivac N. Epigenetics of Alzheimer’s Disease. Biomolecules. 2021; 11(2):195. https://doi.org/10.3390/biom11020195
Chicago/Turabian StyleNikolac Perkovic, Matea, Alja Videtic Paska, Marcela Konjevod, Katarina Kouter, Dubravka Svob Strac, Gordana Nedic Erjavec, and Nela Pivac. 2021. "Epigenetics of Alzheimer’s Disease" Biomolecules 11, no. 2: 195. https://doi.org/10.3390/biom11020195
APA StyleNikolac Perkovic, M., Videtic Paska, A., Konjevod, M., Kouter, K., Svob Strac, D., Nedic Erjavec, G., & Pivac, N. (2021). Epigenetics of Alzheimer’s Disease. Biomolecules, 11(2), 195. https://doi.org/10.3390/biom11020195