
Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: The Human Herpesviruses Are Back!

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
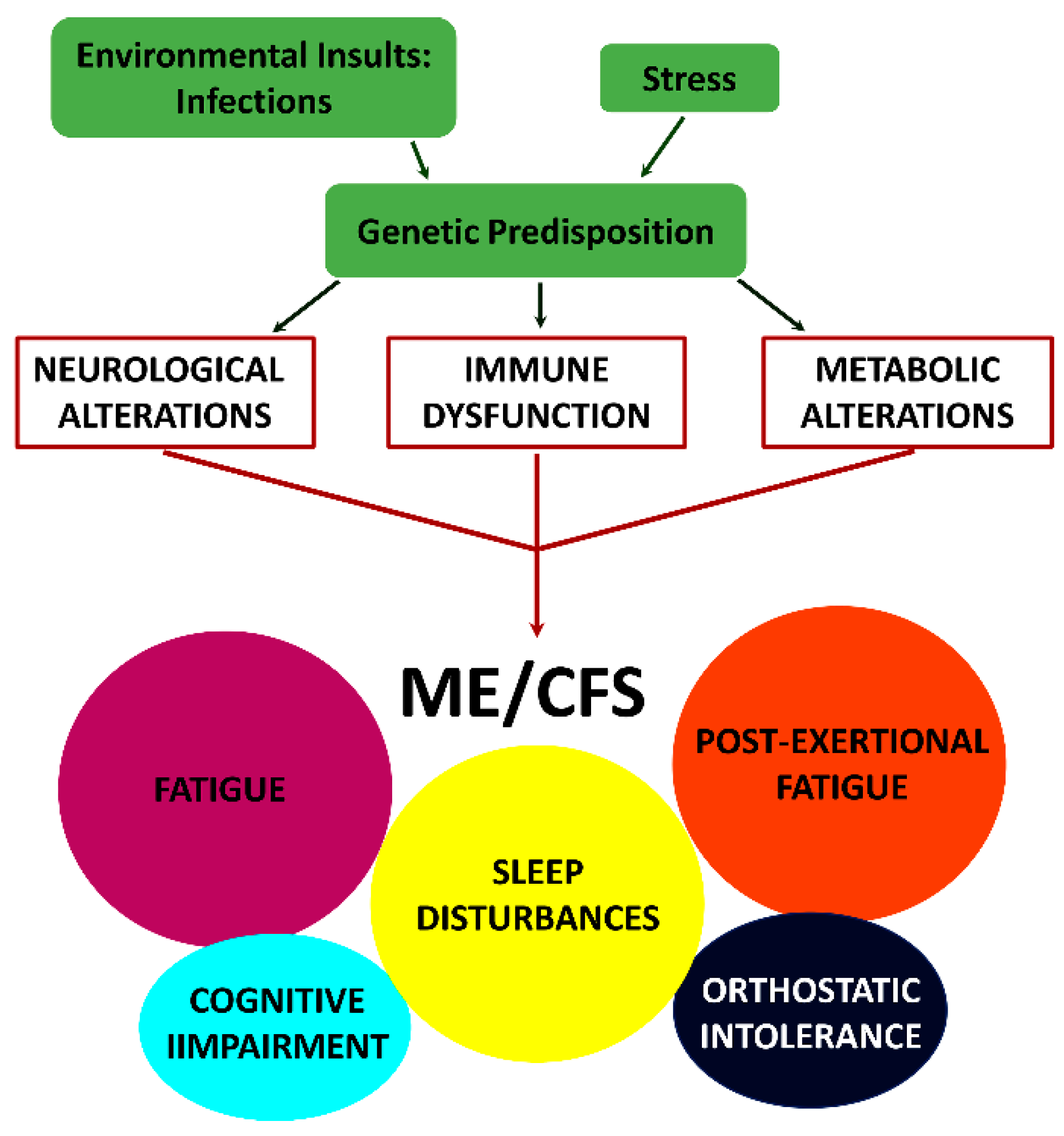
2. Hypotheses Regarding Mechanisms of Underlying Causes of ME/CFS
3. Herpesviruses and ME/CFS
3.1. Serology
3.2. Viral Load
4. “New” Data Suggesting That Some Herpesviruses, Particularly EBV and HHV-6, May Be Involved with the Symptomology of ME/CFS in a Subgroup of Patients
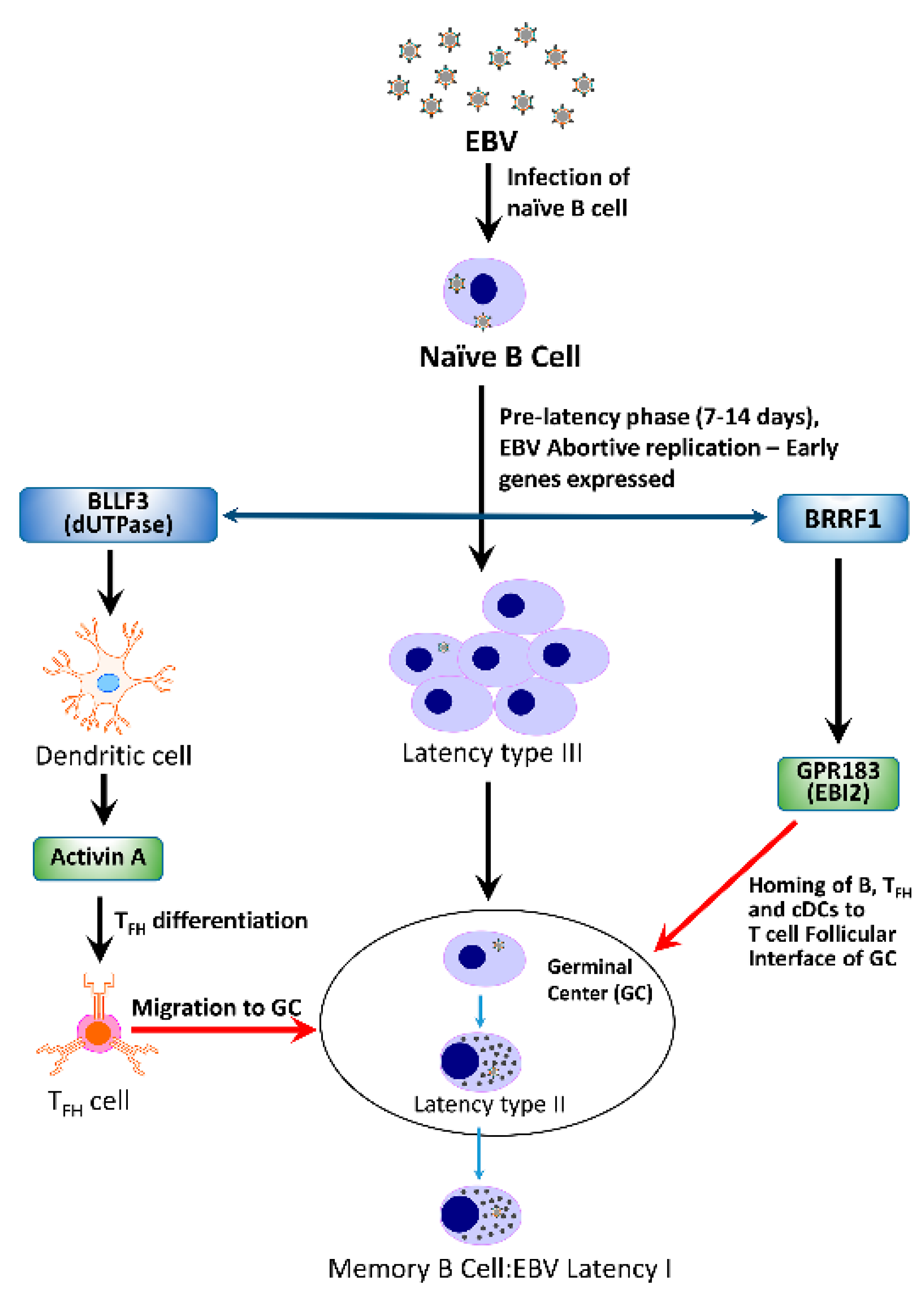
4.1. Abortive Lytic Replication (ALR)
4.2. EBV and the Immune System in ME/CFS
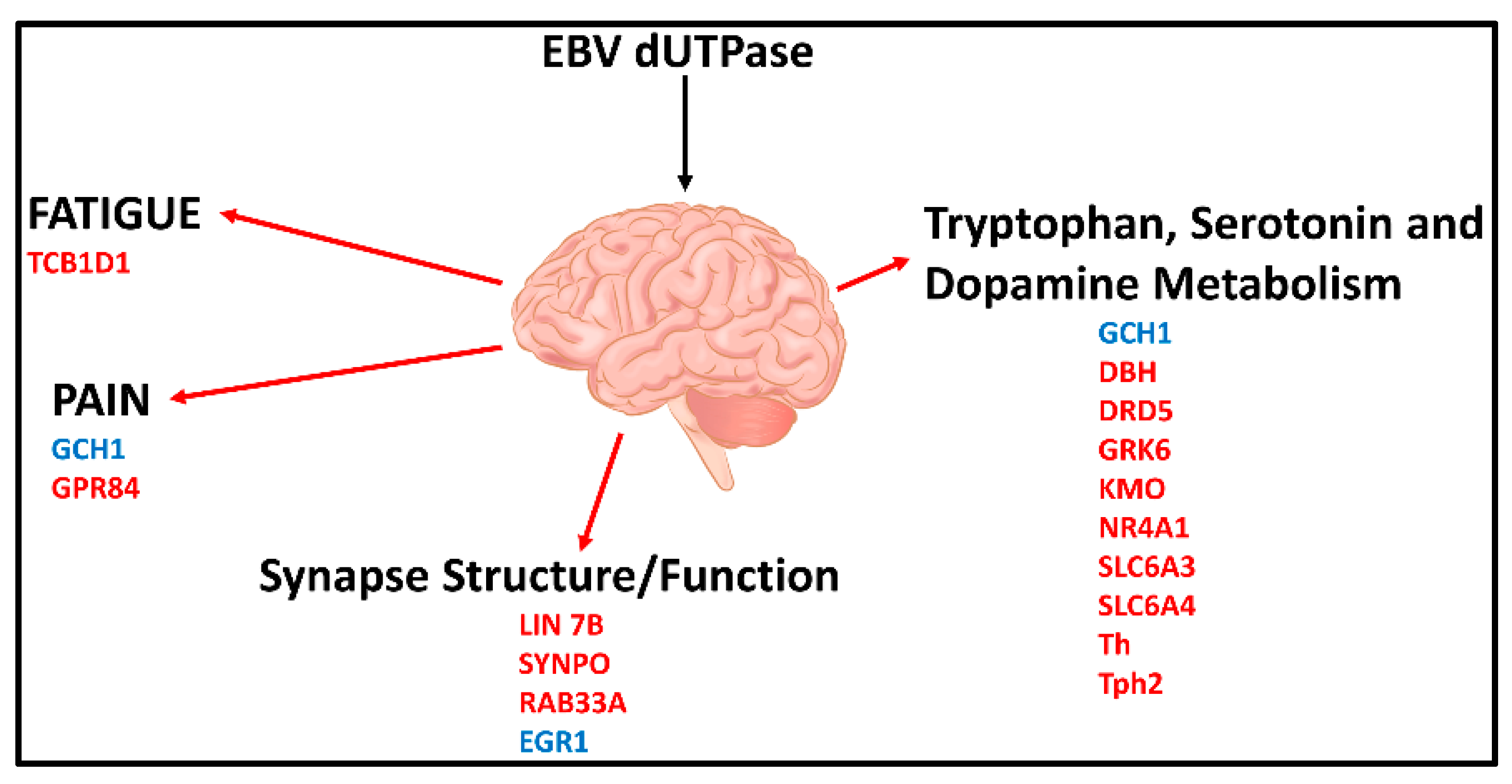
4.3. Can EBV Proteins Produced during ALR Contribute to the Symptomology of ME/CFS?
4.3.1. BRRF1 and Epstein-Barr Induced Gene 2 (EBI2)
4.3.2. BLLF3
BLLF3 in ME/CFS
BLLF3 Pro-Inflammatory Cytokines and Immune Dysfunction
BLLF3 in Autoimmune Disease
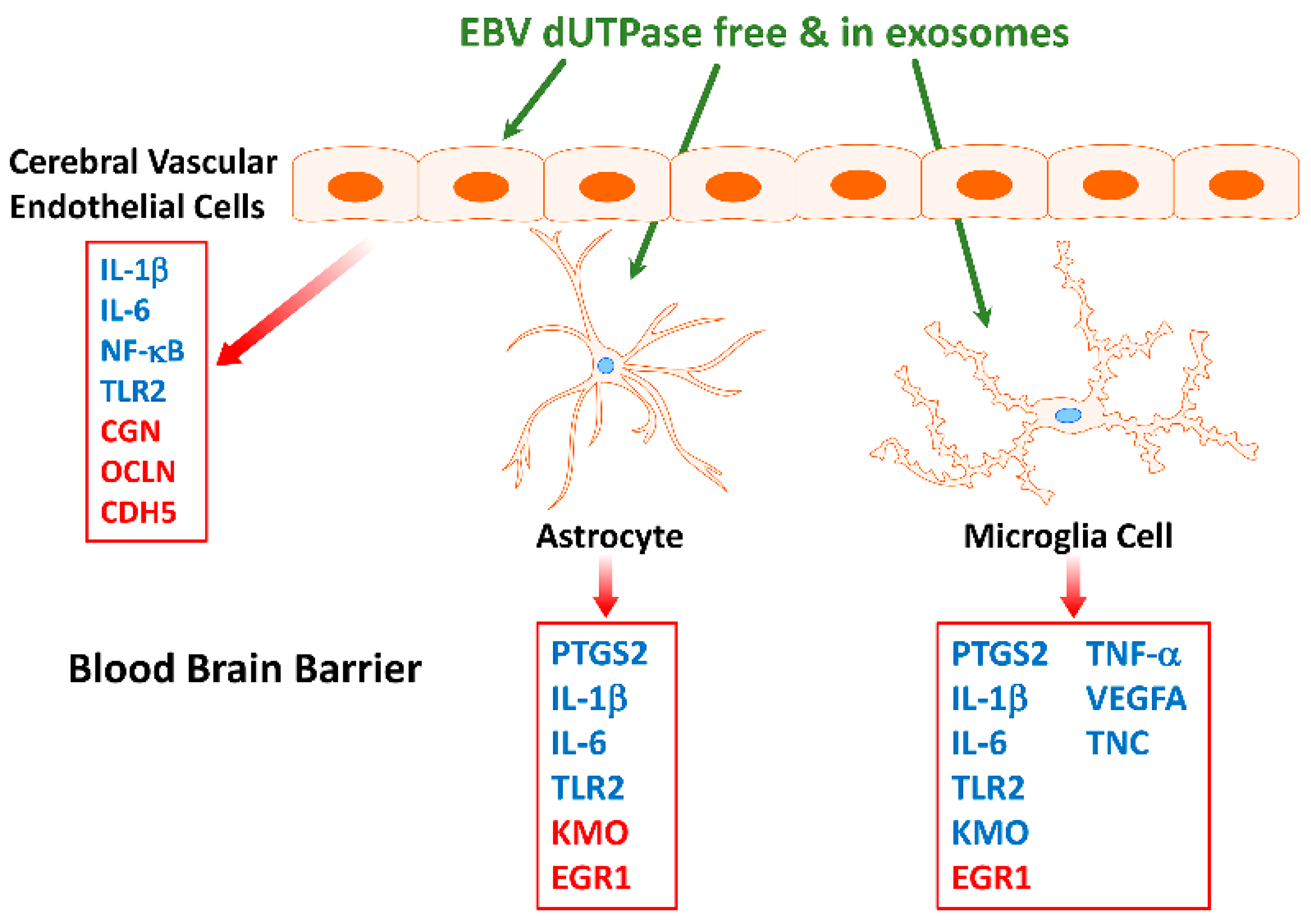
Neuroinflammation
5. Conclusions and Future Directions
Funding
Conflicts of Interest
References
- Fukuda, K.; Straus, S.E.; Hickie, I.; Sharpe, M.C.; Dobbins, J.G.; Komaroff, A. The chronic fatigue syndrome: A comprehensive approach to its definition and study. International Chronic Fatigue Syndrome Study Group. Ann. Intern. Med. 1994, 121, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, B.M.; Jain, A.K.; De Meirleir, D.L.; Peterson, D.L.; Klimas, N.G.; Lerner, A.M.; Bested, A.C.; Flor-Henry, P.; Joshi, P.; Powles, A.C.; et al. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Clinical Working Case Definition, Diagnostic and Treatment Protocols. J. CFS 2003, 11, 7–115. [Google Scholar] [CrossRef]
- Carruthers, B.M.; van de Sande, M.; De Meirleir, D.L.; Klimas, N.G.; Broderick, G.; Mitchell, T.; Staines, D.; Powles, A.C.; Speight, N.; Vallings, R.; et al. Myalgic encephalomyelitis: International consensus criteria. J. Intern. Med. 2011, 270, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Institute of Medicine (U.S.); Committee on the Diagnostic Criteria for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome; Institute of Medicine (U.S.). Board on the Health of Select Populations. Beyond Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Redefining an Illness; The National Academies Press: Washington, DC, USA, 2015.
- Monro, J.A.; Puri, B.K. A molecular neurobiological approach to understanding the aetiology of Chronic Fatigue Syndrome (Myalgic Encephalomyelitis or Systemic Exertion Intolerance Disease with treatment implications. Mol. Neurobiol. 2018, 55, 7377–7388. [Google Scholar] [CrossRef] [PubMed]
- Rasa, S.; Nora-Krukle, Z.; Henning, N.; Eliassen, E.; Shikova, E.; Harrer, T.; Scheibenbogen, C.; Murovska, M.; Prusty, B.K.; on behalf of the European Network on ME/CFS (EUROMEME). Chronic viral infections in myalgic encephalomyelitis/chronic fatigue syndrome. J. Transl. Med. 2018, 10, 268. [Google Scholar] [CrossRef]
- Hickie, I.; Davenport, T.; Wakefield, D.; Vollmer-Conna, U.; Cameron, B.; Vernon, S.D.; Reeves, W.C.; Lloyd, A.; for the Dubbo Infection Outcome Study Group. Post-infective and chronic fatigue syndromes precipitated by viral and non-viral pathogens: Prospective cohort study. BMJ 2006. [Google Scholar] [CrossRef]
- Katz, B.Z.; Shiraishi, Y.; Mears, C.J.; Binns, H.S.; Taylor, R. Chronic fatigue syndrome after infectious mononucleosis in adolescents. Pediatrics 2009, 124, 189–193. [Google Scholar] [CrossRef]
- Chu, L.; Valencia, I.J.; Garvet, D.W.; Montoya, J.G. Onset patterns and course of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Front. Pediatr. 2019, 7, 12. [Google Scholar] [CrossRef]
- Soto, N.E.; Straus, S.E. Chronic fatigue syndrome and herpesviruses: The fading evidence. Herpes 2000, 7, 46–50. [Google Scholar]
- Tobi, M.; Morag, A.; Ravid, Z.; Chowers, I.; Feld-Weiss, V.; Michaeli, Y.; Ben-Chetrit, E.; Shalit, M.; Knobler, H. Prolonged atypical illness associated with serological evidence of persistent Epstein-Barr virus infection. Lancet 1982, 1, 61–64. [Google Scholar] [CrossRef]
- DuBois, R.E.; Seeley, J.R.; Brus, I.; Sakamoto, K.; Ballow, M.; Harada, S.; Bechtold, T.A.; Pearson, G.; Purtilo, D.T. Chronic mononucleosis syndrome. South. Med. J. 1984, 77, 1376–1382. [Google Scholar] [CrossRef] [PubMed]
- Buchwald, D.; Sullivan, J.L.; Komaroff, A.L. Frequency of chronic active Epstein-Barr virus infection in a general medical practice. J. Am. Med. Assoc. 1987, 257, 2303–2307. [Google Scholar] [CrossRef]
- Holmes, G.P.; Kaplan, J.E.; Stewart, J.A.; Hunt, B.; Pinsky, P.F.; Schonberger, L.B. A cluster of patients with a chronic mononucleosis-like syndrome. Is Epstein-Barr virus the cause? J. Am. Med. Assoc. 1987, 257, 2297–2302. [Google Scholar] [CrossRef]
- Hellinger, W.C.; Smith, T.F.; Van Scoy, R.E.; Spitzer, P.G.; Forgacs, P.; Edson, R.S. Chronic fatigue syndrome and the diagnostic utility of antibody to Epstein-Barr early antigen. J. Am. Med Assoc. 1988, 260, 971–973. [Google Scholar] [CrossRef]
- Straus, S.E.; Dale, J.K.; Tobi, M.; Lawley, T.; Prebel, O.; Blaese, M.; Hallahan, C.; Henle, W. Acyclovir treatment of the Chronic Fatigue Syndrome. Lack of efficacy in a placebo-controlled trial. N. Engl. J. Med. 1988, 319, 1692–1698. [Google Scholar] [CrossRef]
- Gold, D.; Bowden, R.; Sixbey, J.; Riggs, R.; Katon, W.J.; Ashley, R.; Obrigewitch, R.; Corey, L. Chronic Fatigue. A prospective clinical and virologic study. J. Am. Med. Assoc. 1990, 264, 48–53. [Google Scholar] [CrossRef]
- Levine, P.H.; Jacobson, S.; Pocinki, A.G.; Cheney, P.; Peterson, D.; Connelly, R.R.; Weil, R.; Robinson, S.M.; Ablashi, G.V.; Salahuddin, S.Z.; et al. Clinical, epidemiological, and virological studies in four clusters of the Chronic Fatigue Syndrome. Arch. Intern. Med. 1992, 152, 1611–1616. [Google Scholar] [CrossRef]
- Buchwald, D.; Ashley, R.L.; Pearlman, T.; Kith, P.; Komaroff, A.L. Viral serologies in patients with Chronic Fatigue and Chronic Fatigue Syndrome. J. Med. Virol. 1996, 50, 25–30. [Google Scholar]
- Cameron, B.; Flamand, L.; Juwana, H.; Middeldrop, J.; Naing, Z.; Rawlinson, W.; Ablashi, D.; Lloyd, A. Serological and virological investigation of the role of the herpesviruses EBV, CMV and HHV-6 in post-infective fatigue syndrome. J. Med. Virol. 2010, 82, 1684–1688. [Google Scholar] [CrossRef]
- Loebel, M.; Eckey, M.; Sotzny, F.; Hahn, E.; Bauer, S.; Grabowski, P.; Zerweck, J.; Holenya, P.; Hanitsch, L.G.; Wittke, K.; et al. Serological profiling of the EBV immune response in Chronic Fatigue Syndrome using a peptide microarray. PLoS ONE 2017, 12, e0179124. [Google Scholar] [CrossRef]
- Blomberg, J.; Rizwan, M.; Bohlin-Wiener, A.; Elfaitouri, A.; Julin, P.; Zachrisson, O.; Rosen, A.; Gottfries, C.G. Antibodies to human herpesviruses in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Patients. Front. Immunol. 2019, 10, 1946. [Google Scholar] [CrossRef] [PubMed]
- Gartner, B.C.; Fischinger, J.M.; Roemer, K.; Mak, M.; Fleurent, B.; Mueller-Lantzsch, N. Evaluation of a recombinant line blot for diagnosis of Epstein-Barr virus compared with ELISA, using immunofluorescence as a reference. J Virol. Methods. 2001, 93, 89–96. [Google Scholar] [CrossRef]
- Shikova, E.; Reshkova, V.; Kumanova, A.; Raleva, S.; Alexandrova, D.; Capo, N.; Murovska, M.; on behalf of the European Network on ME/CFS (EUROMENE). Cytomegalovirus, Epstein-Barr virus and human herpesvirus-6 infections in patients with Myalgic encephalomyelitis/chronic fatigue syndrome. J. Med. Virol. 2020, 92, 3682–3688. [Google Scholar] [CrossRef] [PubMed]
- Swanink, C.M.; van der Meer, J.W.; Vancoulen, J.H.; Bleijenberg, G.; Fennis, J.F.; Galama, J.M. Epstein-Barr virus (EBV) and the chronic fatigue syndrome: Normal virus load in blood and normal immunological reactivity in the EBV regression assay. Clin. Infect. Dis. 1995, 20, 1390–1392. [Google Scholar] [CrossRef]
- Loebel, M.; Strohschein, K.; Giannini, C.; Koelsch, U.; Bauer, S.; Doebis, C.; Thomas, S.; Unterwalder, N.; von Baehr, V.; Reinke, P.; et al. Deficient EBV-specific B-and T-cell response in patients with Chronic Fatigue Syndrome. PLoS ONE 2014, 9, e85387. [Google Scholar] [CrossRef]
- Kristiansen, M.S.; Stabursvik, J.; O’Leary, E.C.; Pedersen, M.; Asprusten, T.T.; Leegaard, T.; Osnes, L.T.; Tjade, T.; Skovlund, E.; Godang, K.; et al. Clinical symptoms and markers of disease mechanisms in adolescent chronic fatigue following Epstein-Barr virus infections: An exploratory cross-sectional study. Brain Behav. Immun. 2019, 80, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Laichalk, L.L.; Thorley-Lawson, D.A. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J. Virol. 2005, 79, 1296–1307. [Google Scholar] [CrossRef]
- Al Tabaa, Y.; Tuaillon, E.; Bollore, K.; Foulongne, V.; Petitjean, G.; Seigneurin, J.M.; Duperray, C.; Desgranges, C.; Vendrell, J.P. Functional Epstein-Barr virus reservoir in plasma cells derived from infected blood memory B cells. Blood 2009, 113, 604–611. [Google Scholar] [CrossRef]
- Al Tabaa, Y.; Tuaillon, E.; Jeziorski, E.; Ouedraogo, D.E.; Bollore, K.; Rubbo, P.A.; Foulongne, V.; Rodiere, M.; Vendrell, J.P. B-cell polyclonal activation and Epstein-Barr viral abortive lytic cycle are two key features in acute infectious mononucleosis. J Clin. Virol. 2011, 52, 33–37. [Google Scholar] [CrossRef]
- Altmann, M.; Hammerschmidt, W. Epstein-Barr virus provides a new paradigm: A requirement for the immediate inhibition of apoptosis. PLoS Biol. 2005, 3, e404. [Google Scholar] [CrossRef]
- Wen, W.; Iwakiri, D.; Yamamoto, K.; Maruo, S.; Kanda, T.; Takada, K. Epstein-Barr virus BZLF1 gene, a switch from latency to lytic replication, is expressed as an immediate-early gene after primary infection of B lymphocytes. J. Virol. 2007, 81, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Shannon-Lowe, C.; Adland, E.; Bell, A.I.; Delecluse, H.J.; Rickman, A.B.; Rowe, M. Features distinguishing Epstein-Barr virus infections of epithelial cells and B cells: Viral genome expression, genome maintenance, and genome amplification. J. Virol. 2009, 83, 7749–7760. [Google Scholar] [CrossRef] [PubMed]
- Kalla, M.; Hammerschmidt, W. Human B cells on their route to latent infection-early but transient expression of lytic genes of Epstein-Barr virus. Eur. J. Cell Biol. 2012, 91, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Martel-Renoir, D.; Grunewald, V.; Touitou, R.; Schwaab, G.; Joab, I. Qualitative analysis of the expression of Epstein-Barr virus lytic genes in nasopharyngeal carcinoma biopsies. J. Gen. Virol. 1995, 76, 1401–1408. [Google Scholar] [CrossRef] [PubMed]
- Ramayanti, O.; Juwana, H.; Verkuijlen, S.M.W.; Adham, M.; Pegtel, M.D.; Greijer, A.E.; Middeldoro, J.M. Epstein-Barr virus mRNA profiles and viral DNA methylation status in nasopharyngeal brushing from nasopharyngeal carcinoma patients reflect tumor origin. Int. J. Cancer 2017, 140, 149–162. [Google Scholar] [CrossRef]
- Caves, E.A.; Cook, S.A.; Lee, N.; Stoltz, D.; Watkins, S.; Shair, K.H.Y. Air-liquid interface method to study Epstein-Barr virus pathogenesis in nasopharyngeal epithelial cells. mSphere 2018, 3, e00152. [Google Scholar] [CrossRef]
- Guo, X.; Li, T.; Li, F.; Xu, Y.; Wang, H.; Cheng, W.; Tang, J.; Zhou, G.; Chen, H.; Ng, M.; et al. Intermittent abortive reactivation of Epstein-Barr virus during the progression of nasopharyngeal cancer as indicated by elevated antibody levels. Oral Oncol. 2019, 93, 85–90. [Google Scholar] [CrossRef]
- Xue, S.A.; Labrecque, L.G.; Lu, Q.L.; Ong, K.; Lampert, I.A.; Kazembe, P.; Molyneux, E.; Broadhead, R.L.; Borgstein, E.; Griffin, B.E. Promiscuous expression of Epstein-Barr virus genes in Burkitt’s lymphoma from the central African country Malawi. Int. J. Cancer 2002, 99, 636–643. [Google Scholar] [CrossRef]
- Strong, M.J.; Xu, G.; Coco, J.; Baribault, C.; Vinay, D.S.; Lacey, M.R.; Strong, A.L.; Lehman, T.A.; Seddon, M.B.; Lin, Z.; et al. differences in gastric carcinoma microenvironment stratify according to EBV infection intensity: Implications for possible Immune adjuvant therapy. PLoS Pathog. 2013, 9, e1003341. [Google Scholar] [CrossRef]
- Borozan, I.; Zapatka, M.; Frappier, L.; Ferretti, V. Analysis of Epstein-Barr virus genomes and expression profiles in gastric carcinoma. J. Virol. 2018, 92, e01239-17. [Google Scholar] [CrossRef]
- Ma, S.D.; Hegde, S.; Young, K.H.; Sullivan, R.; Rajesh, D.; Zhou, Y.; Jankowska-Gan, E.; Burlington, W.J.; Sun, X.; Gulley, M.L.; et al. A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J. Virol. 2011, 85, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.D.; Yu, X.; Mertz, J.E.; Gumperz, J.E.; Reinheim, E.; Zhou, Y.; Tang, W.; Burlington, W.J.; Gulley, M.L.; Kenney, S.C. An Epstein-Barr virus (EBV) mutant with enhanced BZLF1 expression causes lymphomas with abortive lytic EBV infection in a humanized mouse model. J. Virol. 2012, 86, 7976–7987. [Google Scholar] [CrossRef] [PubMed]
- Okuno, Y.; Murata, T.; Sato, Y.; Muramatsu, H.; Ito, Y.; Watanabe, T.; Okuno, T.; Murakami, N.; Yoshida, K.; Sawada, A.; et al. Defective Epstein-Barr virus in chronic active infection and haematological malignancy. Nat. Microbiol. 2019, 4, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Okuno, Y.; Sato, Y.; Watanabe, T.; Kimura, H. Oncogenesis of CAEBV revealed: Intragenic deletions in the viral genome and leaky expression of lytic genes. Rev. Med. Virol. 2019, 30, e2095. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Hirai, Y.; Miyake, T.; Hamada, T.; Yamasaki, O.; Mrizane, S.; Fujimoto, W.; Iwatsuki, K. Epstein-Barr virus reactivation is induced, but abortive, in cutaneous lesions of systemic hydroa vacciniforme and hypersensitivity to mosquito bites. J. Dermatol. Sci. 2016, 82, 153–158. [Google Scholar] [CrossRef]
- Prusty, B.K.; Gulve, N.; Chowdhury, S.R.; Schuster, M.; Strempel, S.; Descamps, V.; Rudel, T. HHV-6 encoded small non-coding RNAs define an immediate and early stage in viral replication. Genom. Med. 2018, 3, 25. [Google Scholar] [CrossRef]
- Halpin, P.; Williams, M.V.; Klimas, N.G.; Fletcher, M.A.; Barnes, Z.; Ariza, M.E. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome and Gulf War Illness patients exhibit increased humoral responses to the Herpesviruses dUTPases: Implications in disease pathophysiology. J. Med. Virol. 2017, 89, 1636–1645. [Google Scholar] [CrossRef]
- Brenu, E.W.; van Driel, M.L.; Staines, D.R.; Ashton, K.J.; Ramos, S.B.; Kaene, J.; Klimas, N.G.; Marshall-Gradisnik, S.M. Immunological abnormalities as potential biomarkers in Chronic Fatigue Syndrome/Myalgic Encephalomyelitis. J. Transl. Med. 2011, 9, 81. [Google Scholar] [CrossRef]
- Brenu, E.W.; Huth, T.K.; Hardcastle, S.L.; Fuller, K.; Kaur, M.; Johnston, S.; Ramos, S.B.; Staines, D.R.; Marshall-Gradisnik, S.M. Role of adaptive and innate immune cells in chronic fatigue syndrome/Myalgic encephalomyelitis. Int. Immunol. 2013, 26, 233–242. [Google Scholar] [CrossRef]
- Curriu, M.; Carrillo, J.; Massanella, M.; Rigau, J.; Alegre, J.; Puig, J.; Garcia-Quintana, A.M.; Castro-Marrero, J.; Negredo, E.; Clotet, B.; et al. Screening NK-, B- and T-cell phenotype and function in patients suffering from Chronic Fatigue Syndrome. J. Transl. Med. 2013, 11, 68. [Google Scholar] [CrossRef]
- Ramos, S.; Brenu, E.; Broadley, S.; Kwiatek, R.; Ng, J.; Nguyen, T.; Freeman, S.; Staines, D.; Marshall-Gradisnik, S.M. Regulatory T, natural killer T and γδ T cells in multiple sclerosis and chronic fatigue syndrome/Myalgic encephalomyelitis: A comparison. Asian Pac. J. Allergy Immunol. 2016, 34, 300–305. [Google Scholar] [PubMed]
- Sepulveda, N.; Carneiro, J.; Lacerda, E.; Nacul, L. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome as a hyper-regulated immune system driven by an interplay between regulatory T cells and chronic human herpesvirus infections. Front. Immunol. 2019, 10, 2684. [Google Scholar] [CrossRef] [PubMed]
- Hong, G.K.; Delecluse, H.J.; Gruffat, H.; Morrison, T.E.; Feng, W.H.; Sergeant, A.; Kenney, S.C. The BRRF1 early gene of Epstein-Barr virus encodes a transcription factor that enhances induction of lytic infection by BRLF1. J. Virol. 2004, 78, 4983–4992. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hagemeier, S.R.; Barlow, E.A.; Kleman, A.A.; Kenney, S.C. The Epstein-Barr virus BRRF1 protein, Na, induces lytic infection in TRAF2- and p53-dependent manner. J. Virol. 2011, 85, 4318–4329. [Google Scholar]
- Cornaby, C.; Jafek, J.L.; Birrell, C.; Mayhew, V.; Syndergaard, L.; Mella, J.; Cheney, W.; Poole, B.D. EBI2 expression in B lymphocytes is controlled by the Epstein-Barr virus transcription factor, BRRF1 (Na), during viral infection. J. Gen. Virol. 2017, 98, 435–446. [Google Scholar]
- Kerr, J.R.; Petty, R.; Burke, B.; Gough, J.; Fear, D.; Sinclair, L.I.; Mattey, D.L.; Richards, S.C.M.; Montgomery, J.; Baldwin, D.A.; et al. Gene expression subtypes in patients with chronic fatigue syndrome/Myalgic encephalomyelitis. J. Infect. Dis. 2008, 197, 1171–1184. [Google Scholar] [CrossRef]
- Zhang, L.; Gough, J.; Christmas, D.; Mattey, D.L.; Richards, S.C.M.; Main, J.; Enlander, D.; Honeybourne, D.; Ayres, J.G.; Nutt, D.J.; et al. Microbial infections in eight genomic subtypes of chronic fatigue syndrome/Myalgic encephalomyelitis. J. Clin. Pathol. 2010, 63, 156–164. [Google Scholar] [CrossRef]
- Kerr, J.R. Epstein-Barr virus induced gene-2 upregulation identifies a particular subtype of chronic fatigue syndrome/Myalgic encephalomyelitis. Front. Pediatrics 2019, 7, 59. [Google Scholar] [CrossRef]
- Kerr, J. Early growth response gene upregulation in Epstein-Barr virus (EBV) associated Myalgic encephalomyelitis/chronic fatigue syndrome. Biomolecules 2020, 10, 1484. [Google Scholar] [CrossRef]
- Pereira, J.P.; Kelly, L.M.; Cyster, J.G. Finding the right niche: B-cell migration in the early phases of T-dependent antibody responses. Int. Immunol. 2010, 22, 413–419. [Google Scholar] [CrossRef]
- Rutkowska, A.; O’Sullivan, S.A.; Christen, I.; Zhang, J.; Sailer, A.W.; Dev, K.K. The EBI2 signaling pathway plays a role in cellular crosstalk between astrocytes and macrophages. Sci. Rep. 2016, 6, 25520. [Google Scholar] [CrossRef] [PubMed]
- Rutkowska, A.; Shimshek, D.R.; Sailer, A.W.; Dev, K.K. EBI2 regulates pro-inflammatory signaling and cytokine release in astrocytes. Neuropharmacology 2018, 133, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Padgett, D.A.; Hotchkiss, A.K.; Pyter, L.M.; Nelson, R.J.; Yang, E.; Yeh, P.E.; Litsky, M.; Williams, M.; Glaser, R. Epstein-Barr Virus-Encoded dUTPase Modulates Immune Function and Induces Sickness Behavior in Mice. J. Med. Virol. 2004, 74, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Glaser, R.; Litsky, M.L.; Padgett, D.A.; Baiocchi, R.A.; Yang, E.V.; Chen, M.; Yeh, P.E.; Green-Church, K.B.; Caligiuri, M.A.; Williams, M. EBV-encoded dUTPase induces immune dysregulation: Implications for the pathophysiology of EBV-associated malignant disease. Virology 2006, 346, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Waldman, W.J.; Williams, M.V.; Lemeshow, S.A.; Binkley, P.; Guttridge, D.; Kiecolt-Glaser, J.K.; Knight, D.A.; Guttridge, K.; Glaser, R. Epstein-Barr virus encoded dUTPase enhances proinflammatory cytokine production by macrophages in contact with endothelial cells: Evidence for depression-induced atherosclerotic risk. Brain Behav. Immun. 2008, 22, 215–223. [Google Scholar] [CrossRef]
- Ariza, M.E.; Glaser, R.; Kaumaya, P.T.P.; Jones, C.; Williams, M. The Epstein-Barr Virus (EBV)-Encoded dUTPase Activates NF-κB through the TLR2 and MyD88-dependent Signaling Pathway. J. Immunol. 2009, 18, 851–859. [Google Scholar] [CrossRef]
- Ariza, M.E.; Williams, M.V. A human endogenous retrovirus K dUTPase triggers a TH1, TH17 cytokine response: Does it play a role in psoriasis? J. Investig. Dermatol. 2011, 131, 2419–2428. [Google Scholar] [CrossRef]
- Lai, O.Y.; Chen, H.; Michaud, H.A.; Hayashi, G.; Kuebler, P.J.; Hultman, G.H.; Ariza, M.E.; Williams, M.V.; Batista, M.D.; Nixon, D.F.; et al. Protective effect of human endogenous retrovirus K dUTPase variants on psoriasis susceptibility. J. Investig. Dermatol. 2012, 132, 1833–1840. [Google Scholar] [CrossRef]
- Lerner, A.M.; Ariza, M.E.; Williams, M.; Jason, L.; Beqaj, S.; Fitzgerald, J.T.; Lemeshow, S.; Glaser, R. Antibody to Epstein-Barr virus deoxyuridine triphosphate nucleotidohydrolase and deoxyribonucleotide polymerase in a Chronic Fatigue Syndrome subset. PLoS ONE 2012, 7, e47891. [Google Scholar]
- Binkley, P.; Cooke, G.E.; Lesinski, A.; Taylor, M.; Chen, M.; Laskowski, B.; Waldman, W.J.; Ariza, M.E.; Williams, M.V.; Knight, D.A.; et al. Evidence for the role of viral infections in the pathogenesis of acute coronary events. PLoS ONE 2013, 8, e54008. [Google Scholar] [CrossRef]
- Ariza, M.E.; Rivailler, P.; Glaser, R.; Chen, M.; Williams, M.V. Epstein-Barr virus encoded dUTPase containing exosomes modulate innate and adaptive immune responses in human dendritic cells and peripheral blood mononuclear cells. PLoS ONE 2013, 8, e69827. [Google Scholar] [CrossRef] [PubMed]
- Aubrecht, T.G.; Zachary, M.; Weil, Z.M.; Ariza, M.E.; Williams, M.; Reader, B.; Glaser, R.; Sheridan, J.; Nelson, R.J. Epstein-Barr virus (EBV)-encoded dUTPase and chronic restraint induce impaired learning and memory and sickness responses. Physiol. Behav. 2014, 137, 18–24. Physiol. Behav. 2014, 137, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Ariza, M.E.; Glaser, R.; Williams, M.V. Human herpesviruses encoded dUTPases: A family of proteins that modulate dendritic cells function and innate immunity. Front. Microbiol. 2014, 5, 504. [Google Scholar] [CrossRef] [PubMed]
- Aubrecht, T.G.; Salloum, B.A.; Ariza, M.E.; Williams, M.; Reader, B.; Glaser, R.; Sheridan, J.; Nelson, R.J. Restraint induces sickness responses independent of injection with Epstein-Barr virus-encoded dUTPase. J. Behav. Brain Sci. 2014, 4, 491–505. [Google Scholar] [CrossRef]
- Aubrecht, T.G.; Weil, Z.M.; Salloum, B.A.; Ariza, M.E.; Williams, M.; Reader, B.; Glaser, R.; Sheridan, J.; Nelson, R.J. Chronic physical stress does not interact with Epstein-Barr virus (EBV) encoded dUTPase to alter the sickness response. J. Behav. Brain Sci. 2015, 5, 513–523. [Google Scholar] [CrossRef]
- Young, N.A.; Williams, M.V.; Jarjour, W.N.; Bruss, M.S.; Bolon, B.; Parikh, S.; Satoskar, A.; Ariza, M.E. Epstein-Barr virus (EBV) encoded dUTPase exacerbates the immune pathology of lupus nephritis in vivo. Int. J. Immunol. Immunother. 2016, 3, 23. [Google Scholar] [CrossRef]
- Ariza, M.E.; Williams, M.V. EBV-dUTPase modulates host immune responses, potentially altering the tumor microenvironment and promoting lymphomagenesis. J. Curr. Res. HIV/AIDS 2016, 2016, 1–9. [Google Scholar]
- Williams, M.V.; Cox, B.; Ariza, M.E. Herpesviruses-encoded dUTPases: A new family of Pathogen-Associated Molecular Pattern (PAMP) proteins with Implications in human disease. Pathogens 2016, 6, 2. [Google Scholar] [CrossRef]
- Broderick, G.; Fuite, J.; Kreitz, A.; Vernon, S.D.; Klimas, N.; Fletcher, M.A. A formal analysis of cytokine networks in chronic fatigue syndrome. Brain Behav. Immun. 2010, 24, 1209–1217. [Google Scholar] [CrossRef]
- Hornig, M.; Montoya, J.G.; Klimas, N.G.; Levine, S.; Felsenstein, D.; Bateman, L.; Peterson, D.L.; Gottschalk, C.G.; Schultz, A.F.; Che, X.; et al. Distinct plasma immune signature signatures in ME/CFS are present early in the course of illness. Sci. Adv. 2015, 1, e1400121. [Google Scholar]
- Russell, L.; Broderick, G.; Taylor, R.; Fernandes, H.; Harvey, J.; Barnes, Z.; Smylie, A.L.; Collado, F.; Balbin, E.G.; Katz, B.Z.; et al. Illness progression in chronic fatigue syndrome: A shifting immune baseline. BMC Immunol. 2016, 17, 3. [Google Scholar] [CrossRef] [PubMed]
- Montoya, J.G.; Holmes, T.H.; Anderson, J.N.; Maecker, H.T.; Rosenberg-Hassonc, Y.; Valencia, I.J.; Chub, L.; Younger, J.W.; Tato, C.M.; Davisc, M.M. Cytokine signature associated with disease severity in chronic fatigue syndrome patients. Proc. Nat. Acad. Sci. USA 2017, 114, E7150–E7158. [Google Scholar] [CrossRef]
- Blundell, S.; Ray, K.K.; Buckland, M.; White, P.D. Chronic fatigue syndrome and circulating cytokines: A systemic review. Brain Behav. Immun. 2015, 50, 186–195. [Google Scholar] [CrossRef]
- Strawbridge, R.; Sartor, M.L.; Scott, F.; Cleare, A.J. Inflammatory proteins are altered in chronic fatigue syndrome-a systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2019, 107, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Yang, Y.; Wang, D.; Li, C.; Qu, Y.; Guo, J.; Shi, T.; Bo, W.; Sun, Z.; Asakawa, T. The clinical value of cytokine in chronic fatigue syndrome. J. Transl. Med. 2019, 17, 213. [Google Scholar] [CrossRef] [PubMed]
- Corbitt, M.; Eaton-Fitch, N.; Staines, D.; Cabanes, H.; Marshall-Gradisnk, S. A systemic review of cytokines in chronic fatigue syndrome/Myalgic encephalomyelitis/systemic exertion intolerance disease (CFS/ME/SEID). BMC Neurol. 2019, 19, 207. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R.; Kelley, K.W. Twenty years of research on cytokine-induced sickness behavior. Brain Behav. Immun. 2007, 21, 153–160. [Google Scholar] [CrossRef]
- Castro-Marrero, J.; Serrano-Pertierra, E.; Oliveira-Rodriguez, M.; Zaragoza, M.C.; Martinez-Martinez, A.; Blanco-Lopez, M.C.; Alegre, J. Circulating extracellular vesicle as potential biomarkers in chronic fatigue syndrome/Myalgic encephalomyelitis: An exploratory pilot study. J. Extracell. Vesicles 2018, 7, 1453730. [Google Scholar] [CrossRef]
- Maes, M.; Mihaylova, I.; Kubera, M.; Leunis, J.C.; Twisk, F.N.; Geffard, M. IgM-mediated autoimmune responses directed against anchorage epitopes are greater in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) than in major depression. Metab. Brain Dis. 2012, 27, 415–423. [Google Scholar] [CrossRef]
- Elfaitouri, A.; Herrmann, B.; Bolin-Wiener, A.; Wang, Y.; Gottfries, C.G.; Zachrisson, O.; Pipkorn, R.; Ronnblom, L.; Blomberg, J. Epitopes of microbial and human heat shock protein 60 and their recognition in Myalgic Encephalomyelitis. PLoS ONE 2013, 8, e81155. [Google Scholar] [CrossRef]
- Vernon, D.; Reeves, W.C. Evaluation of autoantibodies to common and neuronal cell antigens in Chronic Fatigue Syndrome. J. Autoimmune Dis. 2005, 2, 5. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Ouchi, Y.; Nakatsuka, D.; Tahara, T.; Mizuno, K.; Tajima, S.; Onoe, H.; Yoshikawa, E.; Tsukada, H.; Iwase, M.; et al. Reduction of [11C](+)3-MPB binding in brain of chronic fatigue syndrome with serum autoantibody against muscarinic cholinergic receptor. PLoS ONE 2012, 7, e51515. [Google Scholar] [CrossRef] [PubMed]
- Loebel, M.; Grabowski, P.; Heidecke, H.; Bauer, S.; Hanitsch, L.G.; Wittke, K.; Meisel, C.; Reinke, P.; Volk, H.D.; Fluge, O.; et al. Antibodies to β adrenergic and muscarinic cholinergic receptors in patients with Chronic Fatigue Syndrome. Brain Behav. Immun. 2016, 52, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Konstantinov, K.; von Mikecz, A.; Buchwald, D.; Jones, J.; Gerace, L.; Tan, E.M. Autoantibodies to nuclear envelope antigens in Chronic Fatigue Syndrome. J. Clin. Investig. 1996, 98, 1888–1896. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Hernandez, O.D.; Cuccia, M.; Bozzini, S.; Bassi, N.; Moscavitzh, S.; Diaz-Gallo, L.M.; Blank, M.; Agmon-Levin, N.; Shoenfield, Y. Autoantibodies, polymorphisms in the serotonin pathway, and human leukocyte antigen class II alleles in Chronic Fatigue Syndrome. Ann. N. Y. Acad. Sci. 2009, 1173, 589–599. [Google Scholar] [CrossRef]
- Klein, R.; Berg, P.A. High incidence of antibodies to 5-hydroxytryptamine, gangliosides and phospholipids in patients with chronic fatigue and fibromyalgia syndrome and their relatives: Evidence for a clinical entity of both disorders. Eur. J. Med. Res. 1995, 1, 21–26. [Google Scholar]
- Von Mikecz, A.; Konstantinov, K.; Buchwald, D.; Gerace, L.; Tan, E.M. High frequency of autoantigens to insoluble cellular antigens in patients with Chronic Fatigue Syndrome. Arthritis Rheum. 1997, 40, 295–305. [Google Scholar] [CrossRef]
- Sotzny, F.; Blanco, J.; Capelli, E.; Castro-Marrero, J.; Steiner, S.; Murovska, M.; Scheibenbogen, C.; on behalf of the European Network on ME/CFS (EUROMEME). Myalgic Encephalomyelitis/Chronic Fatigue Syndrome-Evidence for an autoimmune disease. Autoimmun. Rev. 2018, 17, 601–609. [Google Scholar] [CrossRef]
- Blomberg, J.; Gottfries, C.G.; Elfaitouri, A.; Rizwan, M.; Roseri, A. Infection elicited autoimmunity and Myalgic encephalomyelitis/Chronic Fatigue Syndrome: An explanatory model. Front. Immunol. 2018, 9, 229. [Google Scholar] [CrossRef]
- Bradley, A.S.; Ford, B.; Bansal, A.S. Altered functional B cell subset populations in patients with chronic fatigue syndrome compared to healthy controls. Clin. Exp. Immunol. 2012, 172, 73–80. [Google Scholar] [CrossRef]
- Mensah, F.; Bansal, A.; Berkovitz, S.; Sharma, A.; Reddy, V.; Leandro, M.J.; Cambridge, G. Extended B cell phenotype in patients with myalgic encephalomyelitis/chronic fatigue syndrome: A cross-sectional study. Clin. Exp. Immunol. 2016, 184, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Hardcastle, S.L.; Brenu, E.W.; Johnston, S.; Nguyen, T.; Huth, T.; Kaur, M.; Ramos, S.; Salajegheh, A.; Staines, D.; Marshall-Gradisnik, S. Analysis of the relationship between immune dysfunction and symptom severity in patients with Chronic Fatigue Syndrome/Myalgic Encephalomyelitis (CFS/ME). J. Clin. Cell. Immunol. 2014, 5, 2. [Google Scholar] [CrossRef]
- Hardcastle, S.L.; Brenu, E.W.; Johnston, S.; Nguyen, T.; Huth, H.; Ramos, S.; Staines, D.; Marshall-Gradisnik, S. Longitudinal analysis of immune abnormalities in varying severities of Chronic Fatigue Syndrome/Myalgic Encephalomyelitis patients. Transl. Med. 2015, 13, 299. [Google Scholar] [CrossRef] [PubMed]
- Cox, B.S.; Alharshawi, K.; Lafuse, W.P.; Ariza, M.E. Herpesvirus dUTPases contribute to Myalgic Encephalomyelitis/Chronic Fatigue Syndrome by promoting follicular helper T cell differentiation. JCI 2021. Submitted. [Google Scholar]
- Chen, W.J.; ten Dijke, P. Immunoregulation by members of the TGFβ superfamily. Nat. Rev. Immunol. 2016, 16, 723–740. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.L.; Walton, K.L.; Winbanks, C.E.; Murphy, K.T.; Thomson, R.E.; Makanji, Y.; Qian, H.; Lynch, G.S.; Harrison, C.A.; Gregorevic, P. Elevated expression of activins promotes muscle wasting and cachexia. FASEB J. 2014, 28, 1711–1723. [Google Scholar] [CrossRef]
- Chen, J.L.; Walton, K.L.; Hagg, A.; Colgan, T.D.; Johnson, K.; Qian, H.; Gregorevic, P.; Harrison, C.A. Specific targeting of TGF-β family ligands demonstrates distinct role in the regulation of muscle mass in heath and disease. Proc. Natl. Acad. Sci. USA 2017, 114, E5266–E5275. [Google Scholar] [CrossRef]
- Spolski, R.; Leonard, W.J. Interleukin-21: A double-edged sword with therapeutic potential. Nat. Rev. Drug Discov. 2014, 13, 379–395. [Google Scholar] [CrossRef]
- Crotty, S. T follicular helper cell biology: A decade of discovery and diseases. Immunity 2019, 50, 1132–1148. [Google Scholar] [CrossRef]
- Chang, P.P.; Barral, P.; Fitch, J.; Pratama, A.; Ma, C.S.; Kallies, A.; Hogan, J.J.; Cerundolo, V.; Tangye, S.G.; Bittman, R.; et al. Identification of Bcl-6-dependent follicular helper NKT cells that provide cognate help for B cell responses. Nat. Immunol. 2011, 13, 35–43. [Google Scholar] [CrossRef]
- King, I.L.; Fortier, A.; Tighe, M.; Dibble, J.; Watts, G.F.M.; Veerapen, N.; Haberman, A.M.; Besra, G.S.; Mohrs, M.; Brenner, M.B.; et al. Invariant natural killer T cells direct B cell responses to cognate lipid antigen in an IL-21-dependent manner. Nat. Immunol. 2011, 13, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Dellabona, P.; Abrignani, S.; Casorati, G. iNKT-cell help to B cells: A cooperative job between innate and adaptive immune responses. Eur. J. Immunol. 2014, 44, 2230–2237. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Yang, J.Q.; Kim, P.J.; Singh, R.J. Homeostatic regulation of marginal zone B cells by invariant Natural Killer T cells. PLoS ONE 2011, 6, e26536. [Google Scholar] [CrossRef] [PubMed]
- Browne, E.P. Regulation of B-cell responses by Toll-like receptors. Immunology 2012, 136, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Nakatomi, Y.; Mizuno, K.; Ishii, A.; Wada, Y.; Tanaka, M.; Tazawa, S.; Onoe, K.; Fukuda, S.; Kawabe, J.; Takahashi, K.; et al. Neuroinflammation in patients with chronic fatigue syndrome/myalgic encephalomyelitis: An 11C-(R)-PK11195 PET study. J. Nucl. Med. 2014, 55, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.T.; Du, X.M.; Ma, X.J.; Zong, Y.; Chen, J.K.; Yu, C.L.; Liu, Y.G.; Chen, Y.C.; Zhao, Y.C.; Lu, G.C. Activation of the NLRP3 inflammasome in lipopolysaccharide-induced mouse fatigue and its relevance to chronic fatigue syndrome. J. Neuroinflamm. 2016, 13, 71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ma, X.; Xia, Z.; Chen, J.; Liu, Y.; Chen, Y.; Zhu, J.; Li, J.; Yu, H.; Zong, Y.; et al. NLRP3 inflammasome activation mediates fatigue-like behaviors in mice via neuroinflammation. Neuroscience 2017, 358, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.; Lin, J.C.; Sheriff, S.; Maudsley, A.A.; Younger, J.W. Evidence of widespread metabolite abnormalities in Myalgic encephalomyelitis/chronic fatigue syndrome: Assessment with whole-brain magnetic resonance spectroscopy. Brain Imaging Behav. 2019, 14, 562–572. [Google Scholar] [CrossRef]
- Williams, M.V.; Cox, B.; Lafuse, W.P.; Ariza, M.E. Epstein-Barr Virus dUTPase induces neuroinflammatory mediators: Implications for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Clin. Ther. 2019, 41, 848–863. [Google Scholar] [CrossRef]
- Aid, S.; Bosetti, F. Targeting cyclooxygenases-1 and -2 in neuroinflammation: Therapeutic implications. Biochimie 2011, 93, 46–51. [Google Scholar] [CrossRef]
- Guillemin, G.J. Quinolinic acid, the inescapable neurotoxin. FEBS J. 2012, 279, 1356–1365. [Google Scholar] [CrossRef] [PubMed]
- Dobryakova, E.; Genova, H.M.; DeLuca, J.; Wylie, G.R. The dopamine imbalance hypothesis of fatigue in multiple sclerosis and other neurological disorders. Front. Neurol. 2015, 6, 52. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Berk, M.; Galecki, P.; Walder, K.; Maes, M. The neuro-immune pathophysiology of central and peripheral fatigue in systemic immune-inflammatory and neuro-immune diseases. Mol. Neurobiol. 2016, 53, 1195–1219. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Carvalho, A.F.; Anderson, G.; Galecki, P.; Maes, M. The many neuroprogressive actions of tryptophan catabolites (TRYCATS) that may be associated with the pathophysiology of neuro-immune disorders. Curr. Pharm. Des. 2016, 22, 963–977. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ariza, M.E. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: The Human Herpesviruses Are Back! Biomolecules 2021, 11, 185. https://doi.org/10.3390/biom11020185
Ariza ME. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: The Human Herpesviruses Are Back! Biomolecules. 2021; 11(2):185. https://doi.org/10.3390/biom11020185
Chicago/Turabian StyleAriza, Maria Eugenia. 2021. "Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: The Human Herpesviruses Are Back!" Biomolecules 11, no. 2: 185. https://doi.org/10.3390/biom11020185
APA StyleAriza, M. E. (2021). Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: The Human Herpesviruses Are Back! Biomolecules, 11(2), 185. https://doi.org/10.3390/biom11020185