
RNR-R2 Upregulation by a Short Non-Coding Viral Transcript

 , , , , , and
, , , , , and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Tissue Culture, Treatments and Reagents
2.2. Preparation of HBV
2.3. Preparation of Lentiviral Transducing Particles and Transduction
2.4. Primary Human Hepatocytes
2.5. RNA and Protein Extraction and Analysis
2.6. Extracellular and Intracellular DNA
2.7. HBsAg Chemiluminescence Immunoassay
2.8. Liquid Chromatography/Mass Spectrometry
2.9. RNA-Seq Analysis
2.10. Statistical Analysis
3. Results
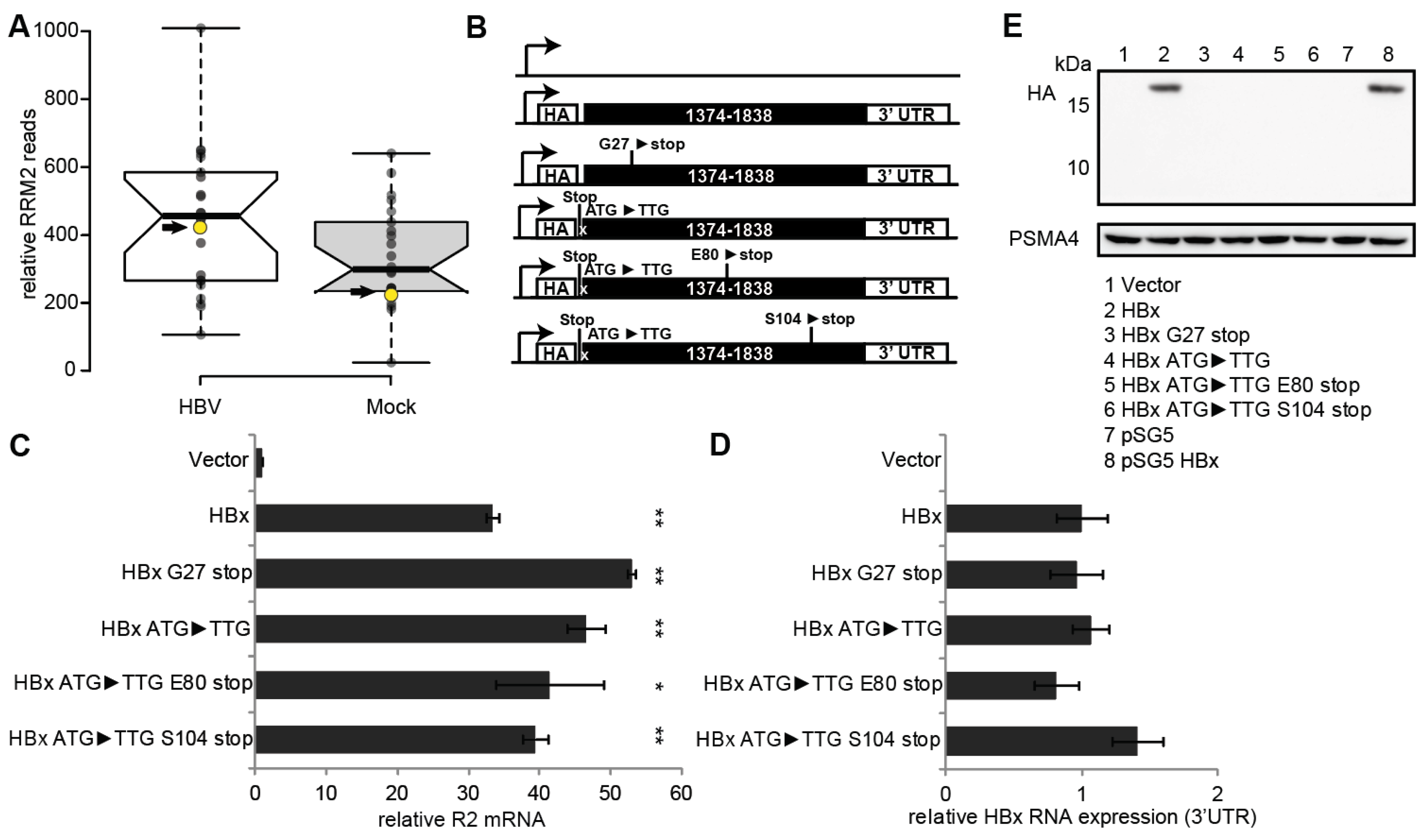
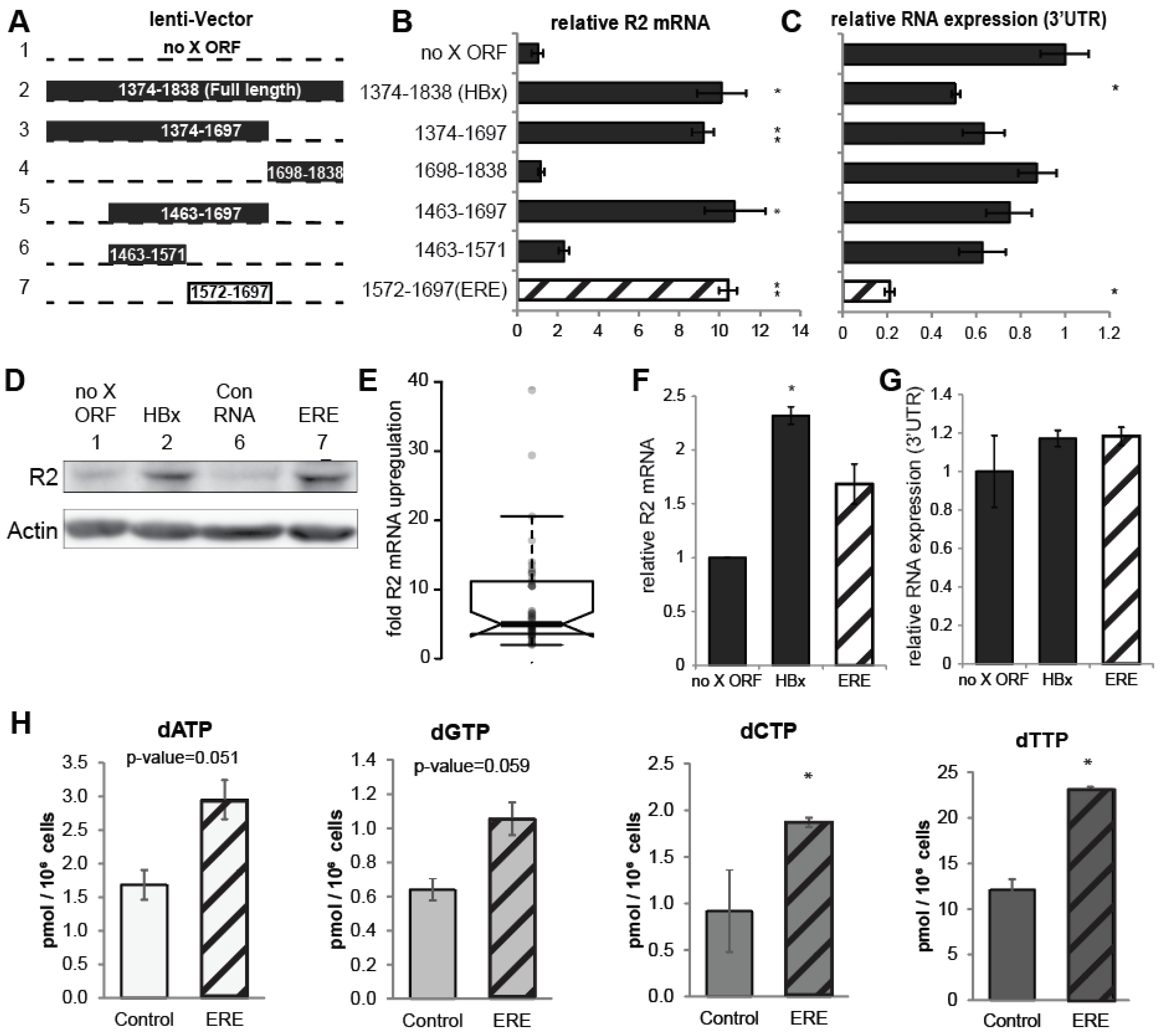
3.1. RNR-R2 Induction by Non-Coding HBx ORF Mutants
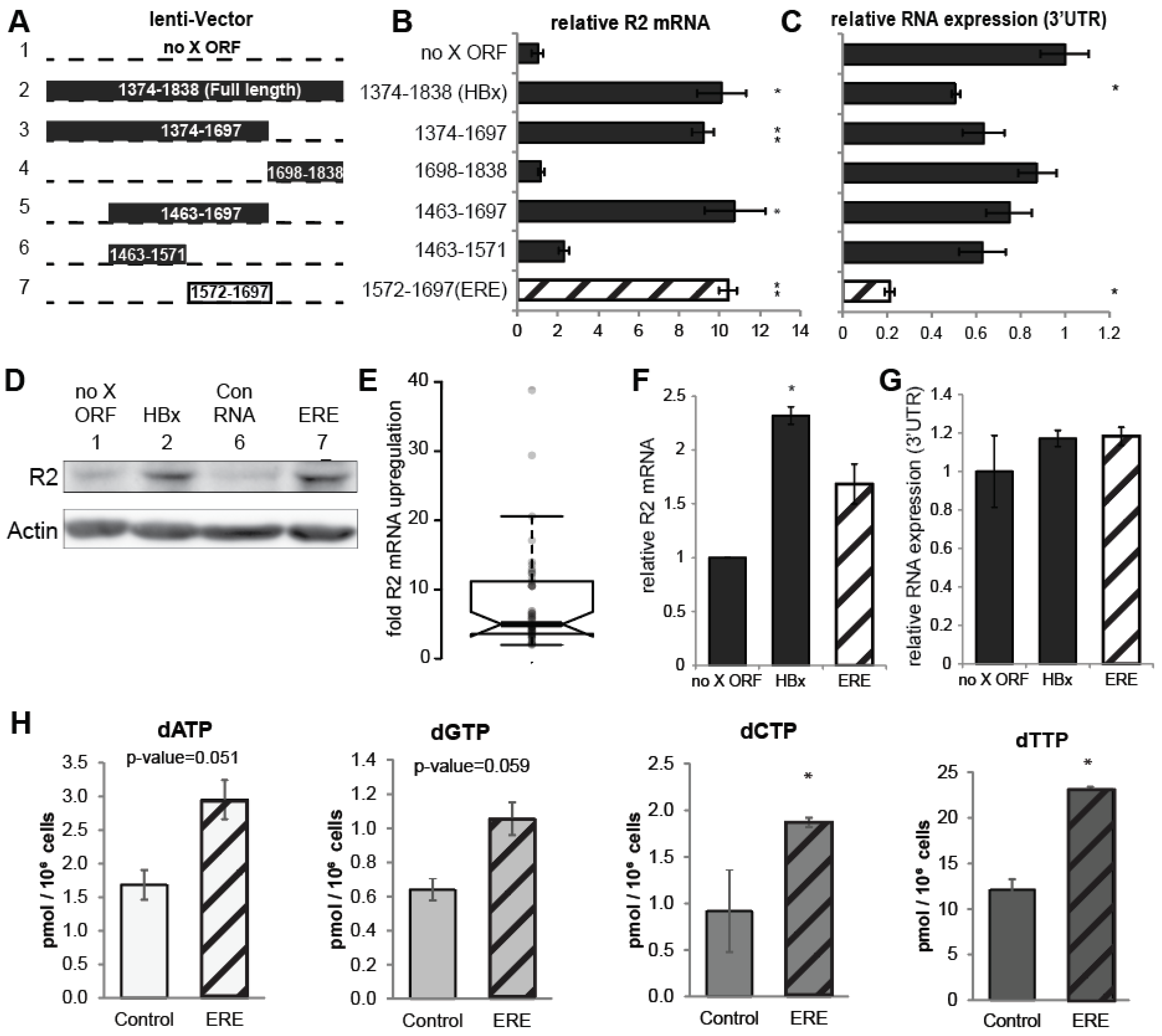
3.2. A Short RNA Region of the HBx ORF Activates RNR-R2
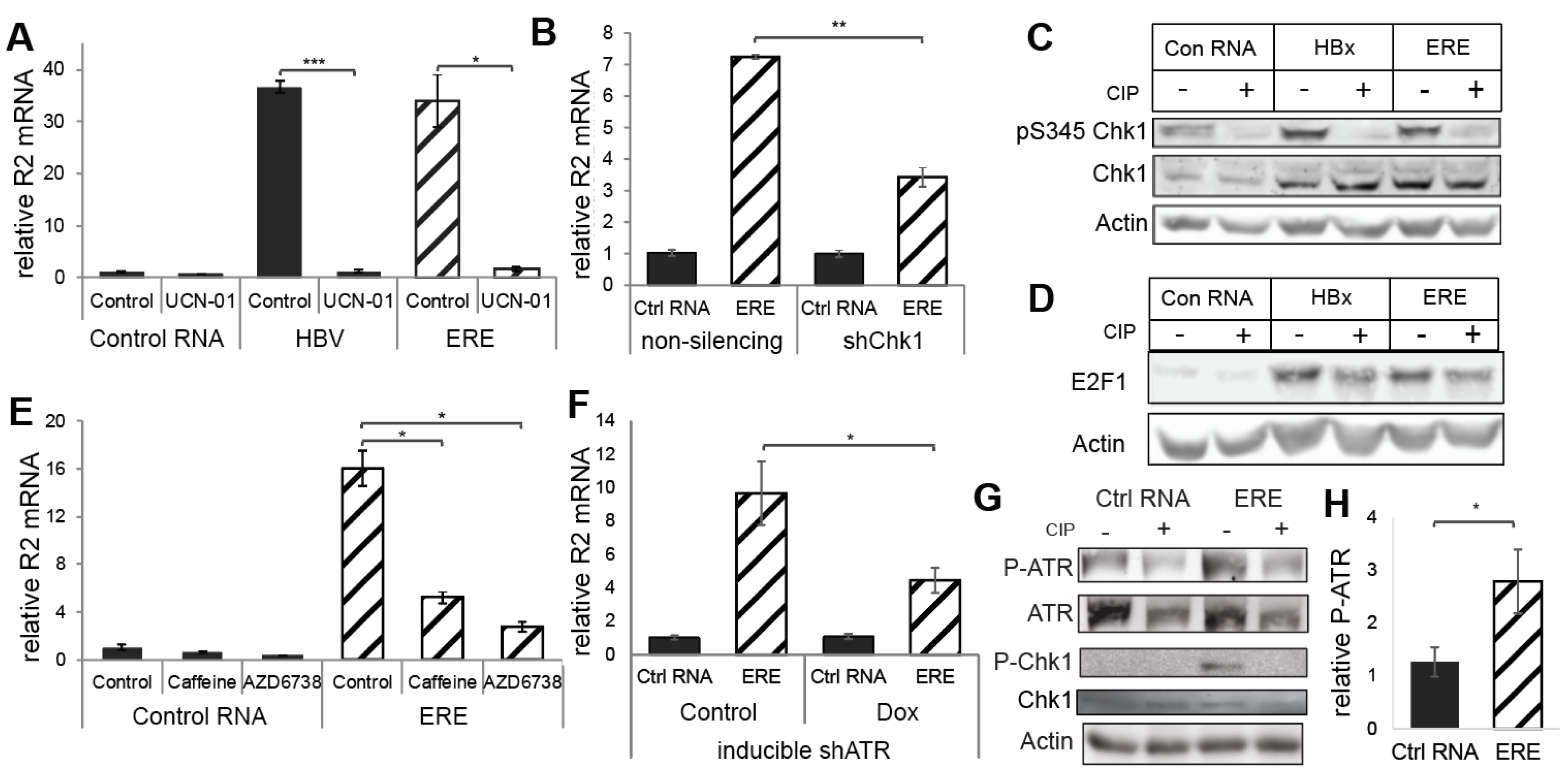
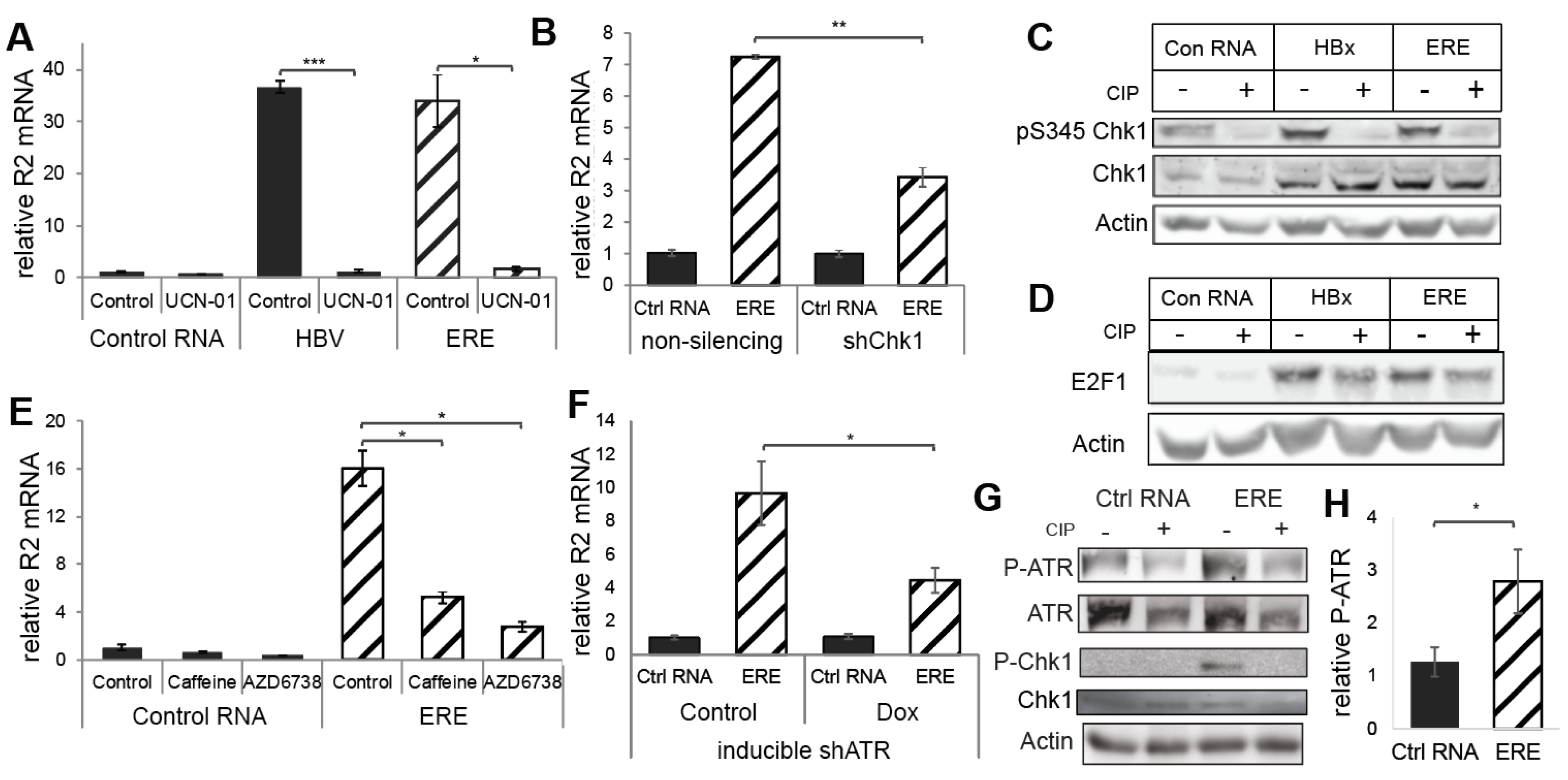
3.3. ERE Induced the ATR-Chk1-E2F1-RNR-R2 Axis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mehta, K.; Laimins, L. Human papillomaviruses preferentially recruit DNA repair factors to viral genomes for rapid repair and amplification. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anacker, D.C.; Aloor, H.L.; Shepard, C.N.; Lenzi, G.M.; Johnson, B.A.; Kim, B.; Moody, C.A. HPV31 utilizes the ATR-Chk1 pathway to maintain elevated RRM2 levels and a replication-competent environment in differentiating Keratinocytes. Virology 2016, 499, 383–396. [Google Scholar] [CrossRef]
- Hau, P.M.; Tsao, S.W. Epstein–Barr virus hijacks DNA damage response transducers to orchestrate its life cycle. Viruses 2017, 9, 341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollingworth, R.; Horniblow, R.D.; Forrest, C.; Stewart, G.S.; Grand, R.J. Localisation of double-strand break repair proteins to viral replication compartments following lytic reactivation of KSHV. J. Virol. 2017, 91, e00930-17. [Google Scholar] [CrossRef] [Green Version]
- Cohen, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. Hepatitis B virus activates deoxynucleotide synthesis in nondividing hepatocytes by targeting the R2 gene. Hepatology 2009, 51, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Ricardo-Lax, I.; Ramanan, V.; Michailidis, E.; Shamia, T.; Reuven, N.; Rice, C.M.; Shlomai, A.; Shaul, Y. Hepatitis B virus induces RNR-R2 expression via DNA damage response activation. J. Hepatol. 2015, 63, 789–796. [Google Scholar] [CrossRef]
- Liu, X.; Xu, Z.; Hou, C.; Wang, M.; Chen, X.; Lin, Q.; Song, R.; Lou, M.; Zhu, L.; Qiu, Y.; et al. Inhibition of hepatitis B virus replication by targeting ribonucleotide reductase M2 protein. Biochem. Pharmacol. 2016, 103, 118–128. [Google Scholar] [CrossRef]
- Melegari, M.; Scaglioni, P.P.; Wands, J.R. Hepatitis B virus mutants associated with 3TC and Famciclovir administration are replication defective. Hepatology 1998, 27, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Chouteau, P.; Le Seyec, J.; Dréan, B.S.-L.; Cannie, I.; Brissot, P.; Lescoat, G.; Guguen-Guillouzo, C.; Gripon, P. Inhibition of hepatitis B virus production associated with high levels of intracellular viral DNA intermediates in iron-depleted HepG2.2.15 cells. J. Hepatol. 2001, 34, 108–113. [Google Scholar] [CrossRef]
- Wing, P.A.; Davenne, T.; Wettengel, J.; Lai, A.G.; Zhuang, X.; Chakraborty, A.; D’Arienzo, V.; Kramer, C.; Ko, C.; Harris, J.; et al. A dual role for SAMHD1 in regulating HBV cccDNA and RT-dependent particle genesis. Life Sci. Alliance 2019, 2, e201900355. [Google Scholar] [CrossRef] [PubMed]
- Sommer, A.F.R.; Rivière, L.; Qu, B.; Schott, K.; Rieß, M.; Ni, Y.; Shepard, C.; Schnellbächer, E.; Finkernagel, M.; Himmelsbach, K.; et al. Restrictive influence of SAMHD1 on Hepatitis B Virus life cycle. Sci. Rep. 2016, 6, 26616. [Google Scholar] [CrossRef] [Green Version]
- Coggins, S.A.; Mahboubi, B.; Schinazi, R.F.; Kim, B. SAMHD1 functions and human diseases. Viruses 2020, 12, 382. [Google Scholar] [CrossRef] [Green Version]
- Elledge, S.J.; Zhou, Z.; Allen, J.B. Ribonucleotide reductase: Regulation, regulation, regulation. Trends Biochem. Sci. 1992, 17, 119–123. [Google Scholar] [CrossRef]
- Guo, H.; Jiang, D.; Zhou, T.; Cuconati, A.; Block, T.M.; Guo, J.-T. Characterization of the Intracellular Deproteinized Relaxed Circular DNA of Hepatitis B Virus: An Intermediate of Covalently Closed Circular DNA Formation. J. Virol. 2007, 81, 12472–12484. [Google Scholar] [CrossRef] [Green Version]
- Cai, D.; Mills, C.; Yu, W.; Yan, R.; Aldrich, C.E.; Saputelli, J.R.; Mason, W.S.; Xu, X.; Guo, J.-T.; Block, T.M.; et al. Identification of Disubstituted Sulfonamide Compounds as Specific Inhibitors of Hepatitis B Virus Covalently Closed Circular DNA Formation. Antimicrob. Agents Chemother. 2012, 56, 4277–4288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michailidis, E.; Pabon, J.; Xiang, K.; Park, P.; Ramanan, V.; Hoffmann, H.-H.; Schneider, W.M.; Bhatia, S.N.; De Jong, Y.P.; Shlomai, A.; et al. A robust cell culture system supporting the complete life cycle of hepatitis B virus. Sci. Rep. 2017, 7, 16616. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, H.; Takahashi, M.; Gage, F.H.; Verma, I.M. Stable and efficient gene transfer into the retina using an HIV-based lentiviral vector. Proc. Natl. Acad. Sci. USA 1997, 94, 10319–10323. [Google Scholar] [CrossRef] [Green Version]
- Doitsh, G.; Shaul, Y. Enhancer I Predominance in Hepatitis B Virus Gene Expression. Mol. Cell. Biol. 2004, 24, 1799–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michailidis, E.; Vercauteren, K.; Mancio-Silva, L.; Andrus, L.; Jahan, C.; Ricardo-Lax, I.; Zou, C.; Kabbani, M.; Park, P.; Quirk, C.; et al. Expansion, in vivo-ex vivo cycling, and genetic manipulation of primary human hepatocytes. Proc. Natl. Acad. Sci. USA 2020, 117, 1678–1688. [Google Scholar] [CrossRef] [Green Version]
- Shlomai, A.; Paran, N.; Shaul, Y. PGC-1alpha controls hepatitis B virus through nutritional signals. Proc. Natl. Acad. Sci. USA 2006, 103, 16003–16008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NNiu, C.; Livingston, C.M.; Li, L.; Beran, R.K.; Daffis, S.; Ramakrishnan, D.; Burdette, D.; Peiser, L.; Salas, E.; Ramos, H.; et al. The Smc5/6 complex restricts HBV when localized to ND10 without inducing an innate immune response and is counteracted by the HBV X protein shortly after infection. PLoS ONE 2017, 12, e0169648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirosh, O.; Cohen, Y.; Shitrit, A.; Shani, O.; Le-Trilling, V.T.K.; Trilling, M.; Friedlander, G.; Tanenbaum, M.; Stern-Ginossar, N. The Transcription and Translation Landscapes during Human Cytomegalovirus Infection Reveal Novel Host-Pathogen Interactions. PLOS Pathog. 2015, 11, e1005288. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Shamay, M.; Barak, O.; Doitsh, G.; Ben-Dor, I.; Shaul, Y. Hepatitis B virus pX interacts with HBXAP, a PHD finger protein to coactivate transcription. J. Biol. Chem. 2002, 277, 9982–9988. [Google Scholar] [CrossRef] [Green Version]
- Maréchal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef] [PubMed]
- Leung-Pineda, V.; Ryan, C.E.; Piwnica-Worms, H. Phosphorylation of Chk1 by ATR is antagonized by a Chk1-regulated protein phosphatase 2A circuit. Mol. Cell. Biol. 2006, 26, 7529–7538. [Google Scholar] [CrossRef] [Green Version]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Kumagai, A.; Wang, S.X.; Dunphy, W.G. Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev. 2000, 14, 2745–2756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138. [Google Scholar] [CrossRef] [Green Version]
- Nam, E.A.; Zhao, R.; Glick, G.G.; Bansbach, C.E.; Friedman, D.B.; Cortez, D. Thr-1989 phosphorylation is a marker of active ataxia telangiectasia- mutated and Rad3-related (ATR) kinase. J. Biol. Chem. 2011, 286, 28707–28714. [Google Scholar] [CrossRef] [Green Version]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamontagne, R.J.; Mell, J.C.; Bouchard, M.J. Transcriptome-Wide Analysis of Hepatitis B Virus-Mediated Changes to Normal Hepatocyte Gene Expression. PLOS Pathog. 2016, 12, e1005438. [Google Scholar] [CrossRef] [Green Version]
- Beck, J.; Nassal, M. Hepatitis B virus replication. World J. Gastroenterol. 2007, 13, 48–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, T.J. National Institue of Health-Hepatitis B: The Virus and Disease. Hepatology 2010, 49, 1–17. [Google Scholar] [CrossRef]
- Raney, A.K.; Eggers, C.M.; Kline, E.F.; Guidotti, L.G.; Pontoglio, M.; Yaniv, M.; McLachlan, A.; Raney, A.K.; Eggers, C.M.; Kline, E.F.; et al. Nuclear Covalently Closed Circular Viral Genomic DNA in the Liver of Hepatocyte Nuclear Factor 1 -Null Hepatitis B Virus Transgenic Mice. J. Virol. 2002, 75, 2900–2911. [Google Scholar] [CrossRef] [Green Version]
- Altinel, K.; Hashimoto, K.; Wei, Y.; Neuveut, C.; Gupta, I.; Suzuki, A.M.; Dos Santos, A.; Moreau, P.; Xia, T.; Kojima, S.; et al. Single-Nucleotide Resolution Mapping of Hepatitis B Virus Promoters in Infected Human Livers and Hepatocellular Carcinoma. J. Virol. 2016, 90, 10811–10822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadelmayer, B.; Diederichs, A.; Chapus, F.; Rivoire, M.; Neveu, G.; Alam, A.; Fraisse, L.; Carter, K.; Testoni, B.; Zoulim, F. Full-length 5′RACE identifies all major HBV transcripts in HBV-infected hepatocytes and patient serum. J. Hepatol. 2020, 73, 40–51. [Google Scholar] [CrossRef]
- Knaus, T.; Nassal, M. The encapsidation signal on the hepatitis B virus RNA pregenome forms a stem-loop structure that is critical for its function. Nucleic Acids Res. 1993, 21, 3967–3975. [Google Scholar] [CrossRef]
- Gajer, M.; Dörnbrack, K.; Rösler, C.; Schmid, B.; Beck, J.; Nassal, M. Few basepairing-independent motifs in the apical half of the avian HBV ϵ RNA stem-loop determine site-specific initiation of protein-priming. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.M.; Yen, T.S. Role of the hepatitis B virus posttranscriptional regulatory element in export of intronless transcripts. Mol. Cell. Biol. 1995, 15, 3864–3869. [Google Scholar] [CrossRef] [Green Version]
- Lim, C.S.; Brown, C.M. Hepatitis B virus nuclear export elements: RNA stem-loop α and β, key parts of the HBV post-transcriptional regulatory element. RNA Biol. 2016, 6286, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Liang, T.J. A novel hepatitis B virus (HBV) genetic element with Rev response element-like properties that is essential for expression of HBV gene products. Mol. Cell. Biol. 1993, 13, 7476–7486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsui, L.V.; Guidotti, L.G.; Ishikawa, T.; Chisari, F.V. Posttranscriptional clearance of hepatitis B virus RNA by cytotoxic T lymphocyte-activated hepatocytes. Proc. Natl. Acad. Sci. USA 1995, 92, 12398–12402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heise, T.; Guidotti, L.G.; Cavanaugh, V.J.; Chisari, F.V. Hepatitis B virus RNA-binding proteins associated with cytokine-induced clearance of viral RNA from the liver of transgenic mice. J. Virol. 1999, 73, 474–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Broennimann, K.; Ricardo-Lax, I.; Adler, J.; Michailidis, E.; de Jong, Y.P.; Reuven, N.; Shaul, Y. RNR-R2 Upregulation by a Short Non-Coding Viral Transcript. Biomolecules 2021, 11, 1822. https://doi.org/10.3390/biom11121822
Broennimann K, Ricardo-Lax I, Adler J, Michailidis E, de Jong YP, Reuven N, Shaul Y. RNR-R2 Upregulation by a Short Non-Coding Viral Transcript. Biomolecules. 2021; 11(12):1822. https://doi.org/10.3390/biom11121822
Chicago/Turabian StyleBroennimann, Karin, Inna Ricardo-Lax, Julia Adler, Eleftherios Michailidis, Ype P. de Jong, Nina Reuven, and Yosef Shaul. 2021. "RNR-R2 Upregulation by a Short Non-Coding Viral Transcript" Biomolecules 11, no. 12: 1822. https://doi.org/10.3390/biom11121822
APA StyleBroennimann, K., Ricardo-Lax, I., Adler, J., Michailidis, E., de Jong, Y. P., Reuven, N., & Shaul, Y. (2021). RNR-R2 Upregulation by a Short Non-Coding Viral Transcript. Biomolecules, 11(12), 1822. https://doi.org/10.3390/biom11121822