
Coffee Bioactive N-Methylpyridinium Attenuates Tumor Necrosis Factor (TNF)-α-Mediated Insulin Resistance and Inflammation in Human Adipocytes

 ,
,  ,
,  ,
,  ,
, 
 , ,
, ,  , , ,
, , ,  ,
,  and
and 
Abstract
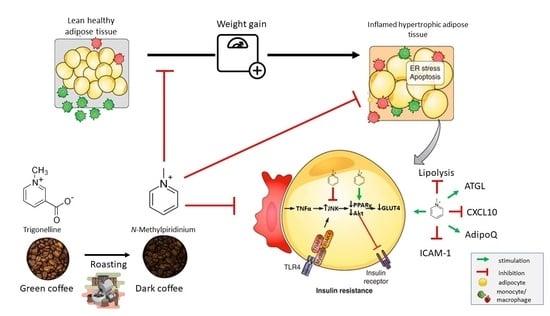
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture and Treatments
2.3. Cell Viability
2.4. Assessment of Cellular Lipid Accumulation
2.5. Determination of Lipolysis
2.6. RNA Isolation and Real-Time Quantitative Polymerase Chain Reaction
2.7. Cell Lysis and Immunoblotting
2.8. 2-NBDG Uptake Assays
2.9. In Vitro THP-1 Adhesion Assay
2.10. In Silico Molecular Docking
2.11. Statistical Analysis
3. Results
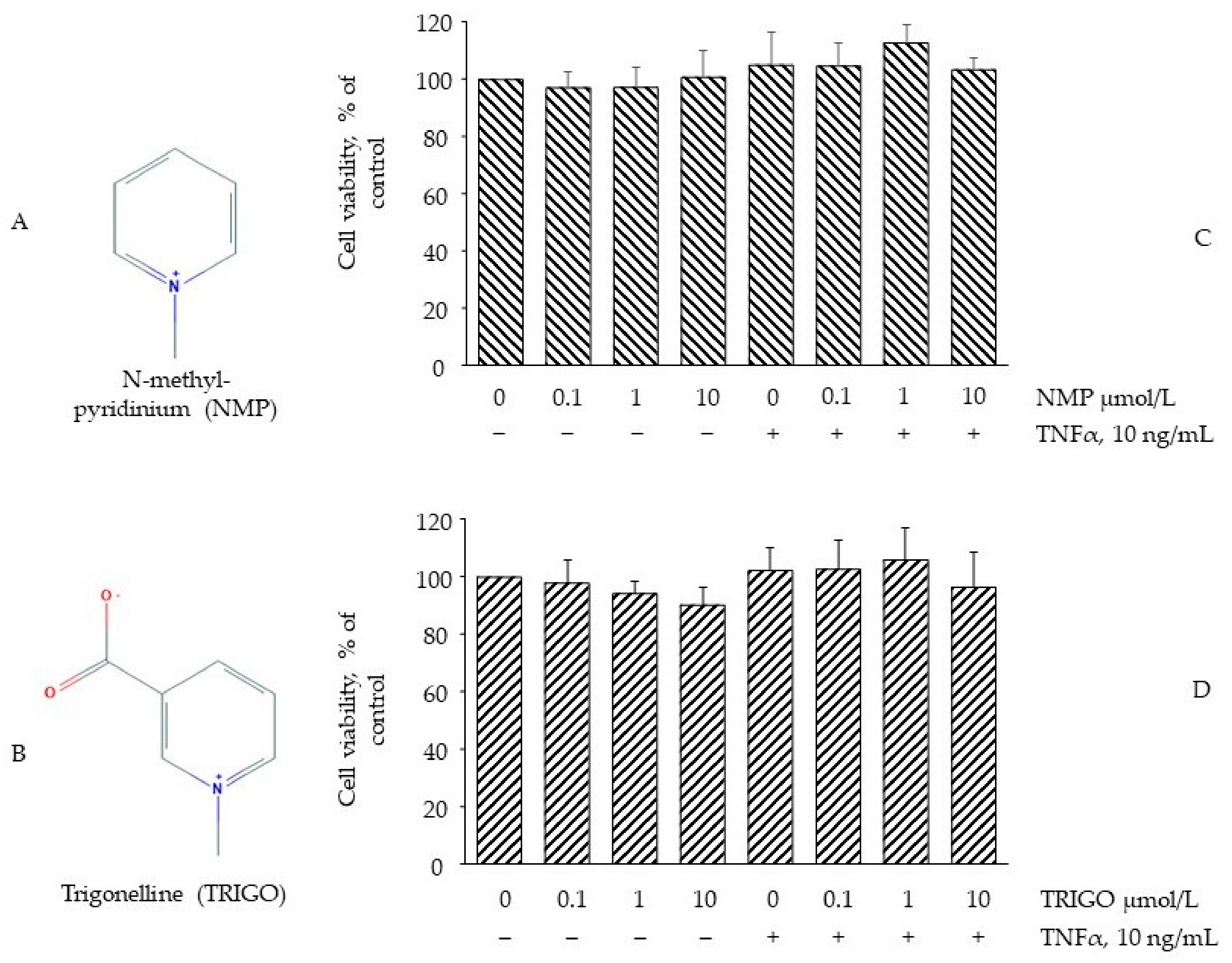
3.1. Effect of NMP on Cell Viability
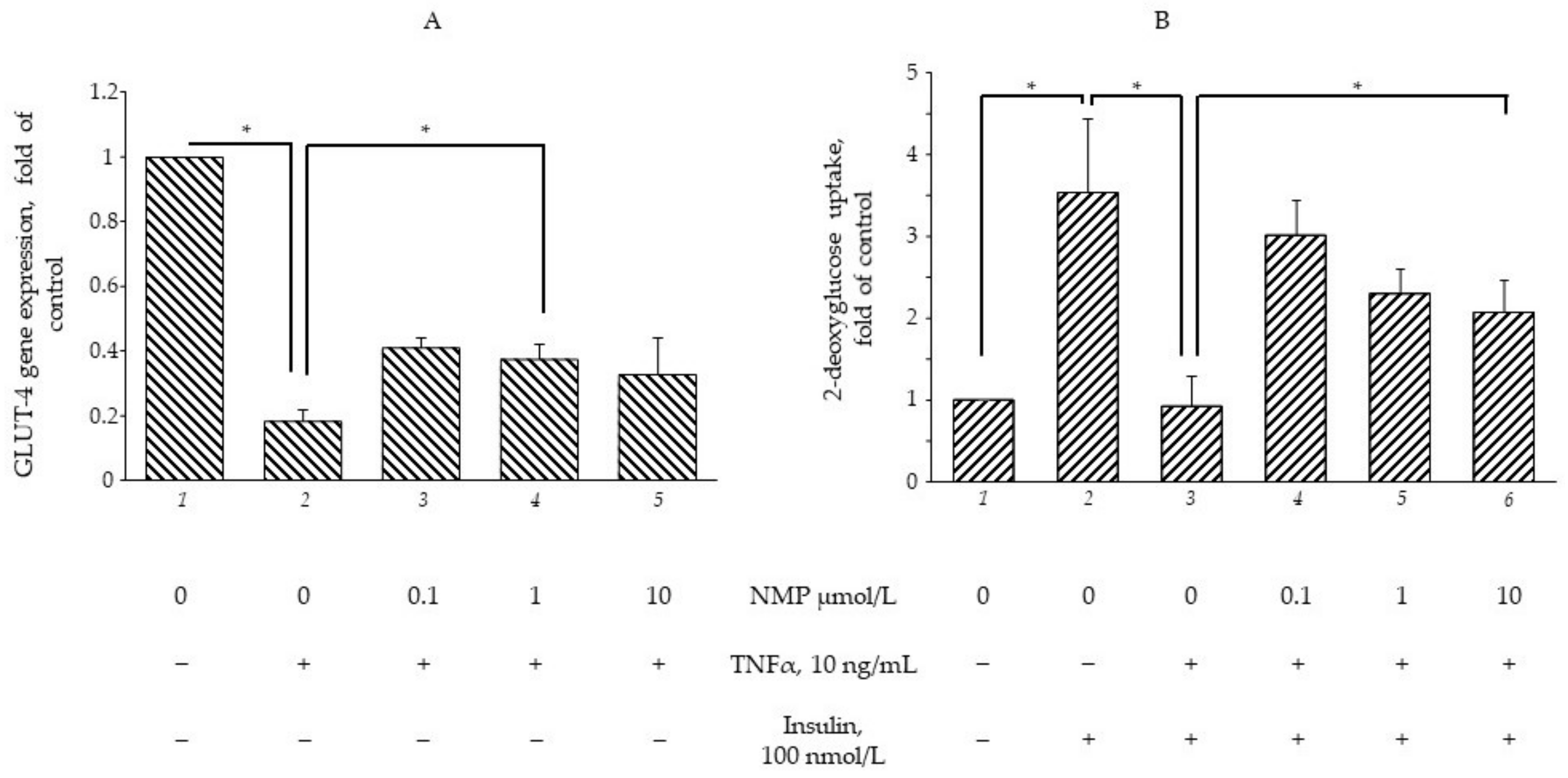
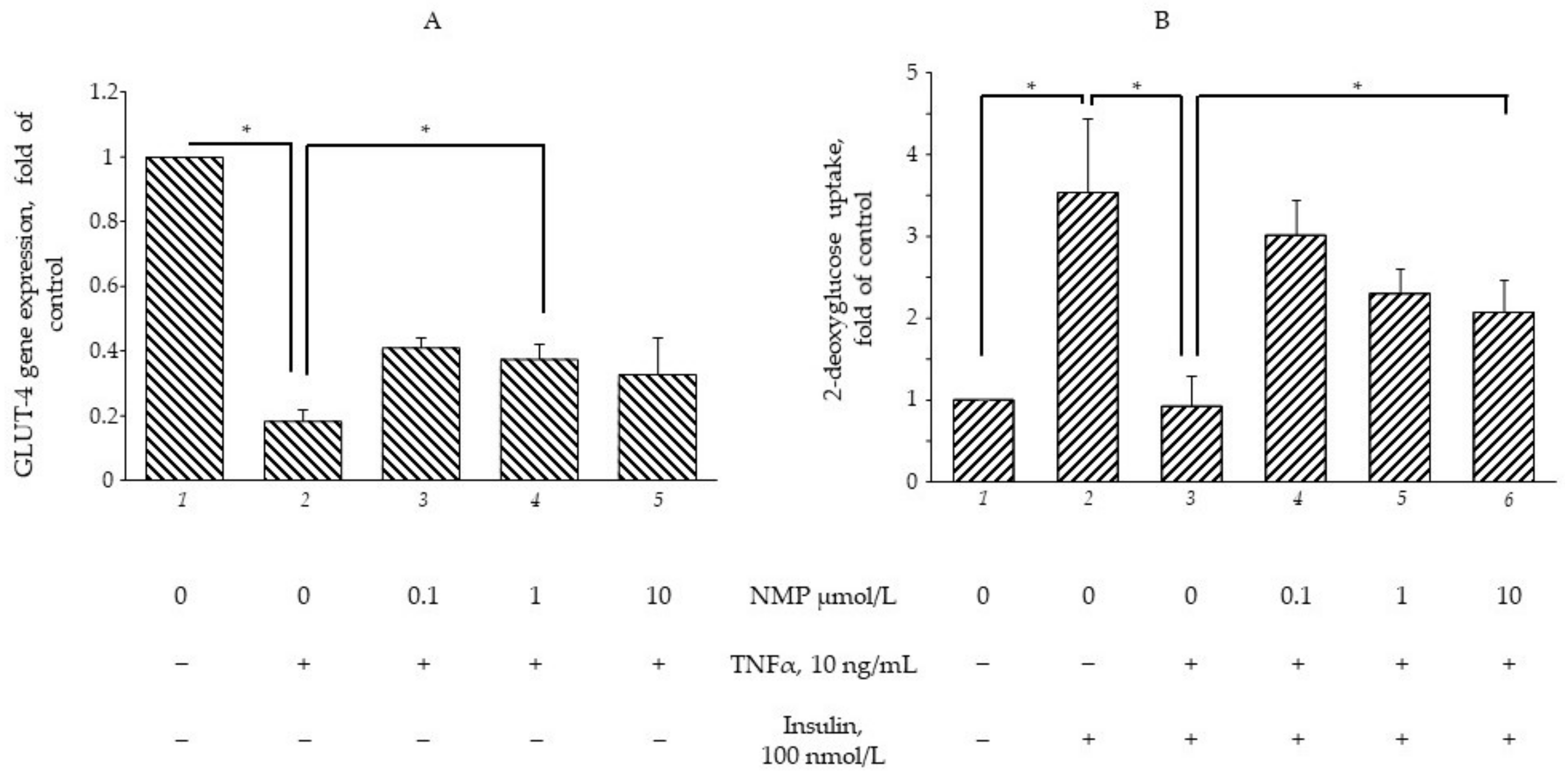
3.2. NMP Restores the TNF-α-Mediated Impairment of GLUT-4 Gene Expression and Insulin-Stimulated Glucose Uptake
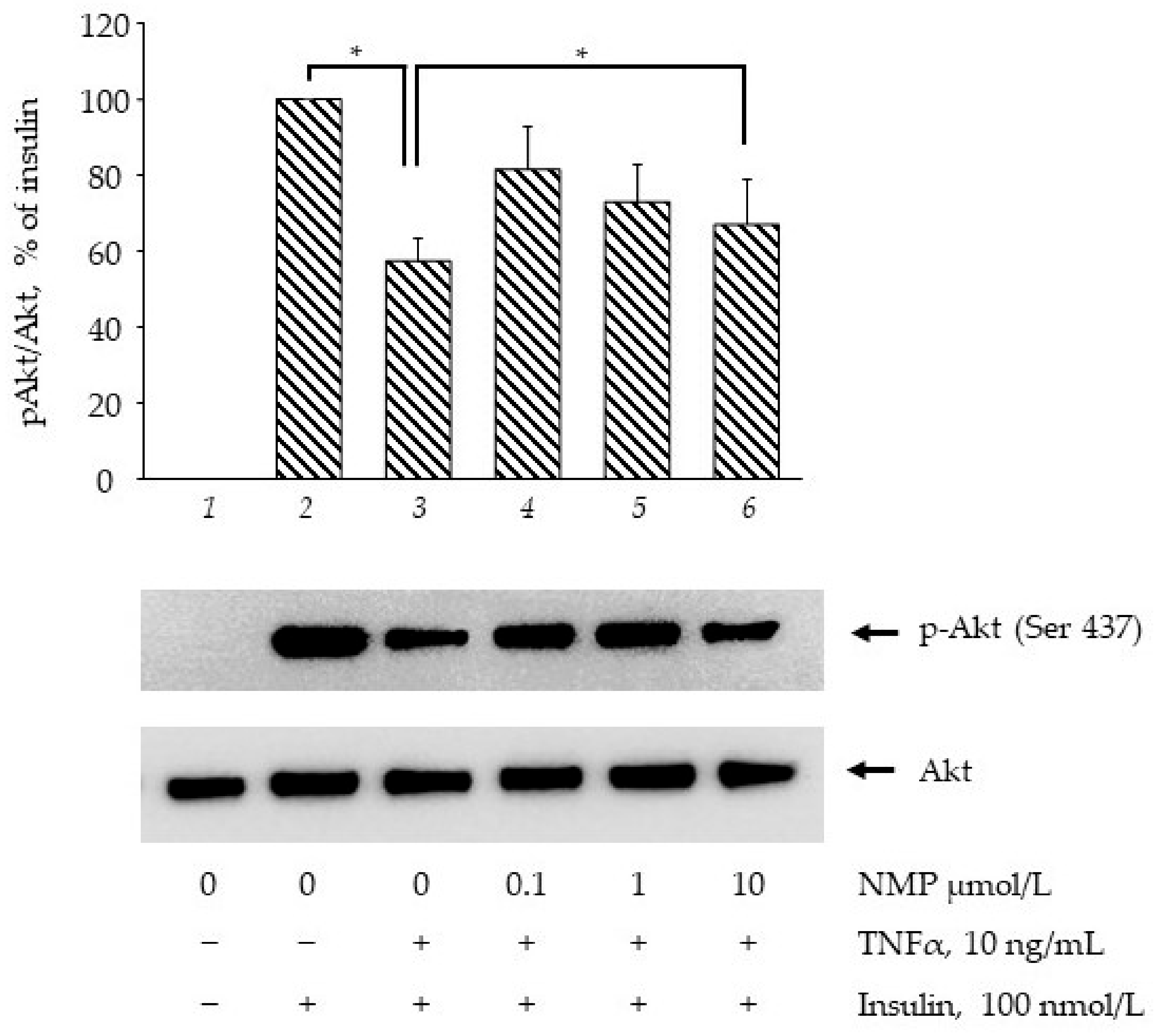
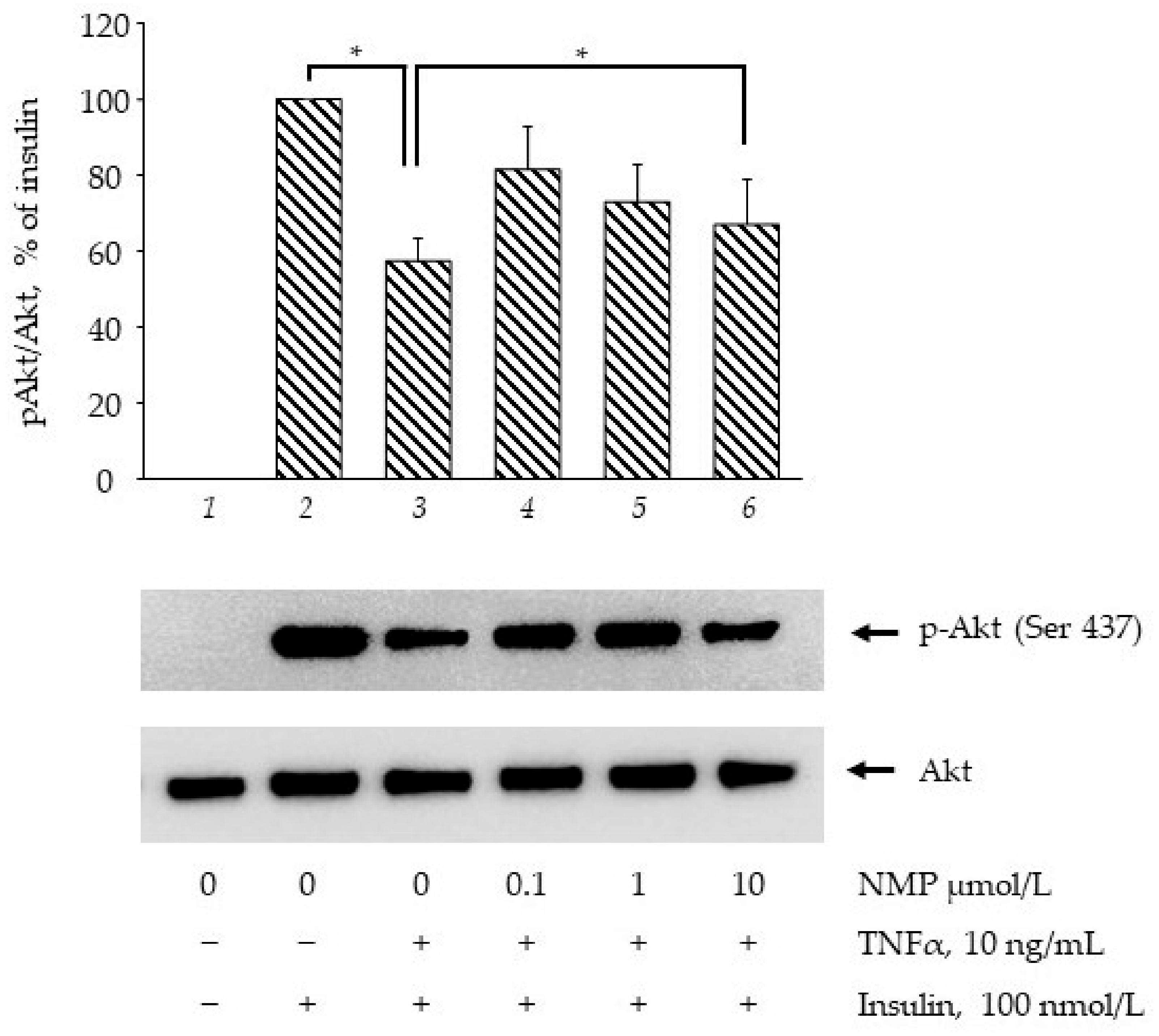
3.3. NMP Improves the Insulin Signaling Impaired by TNF-α
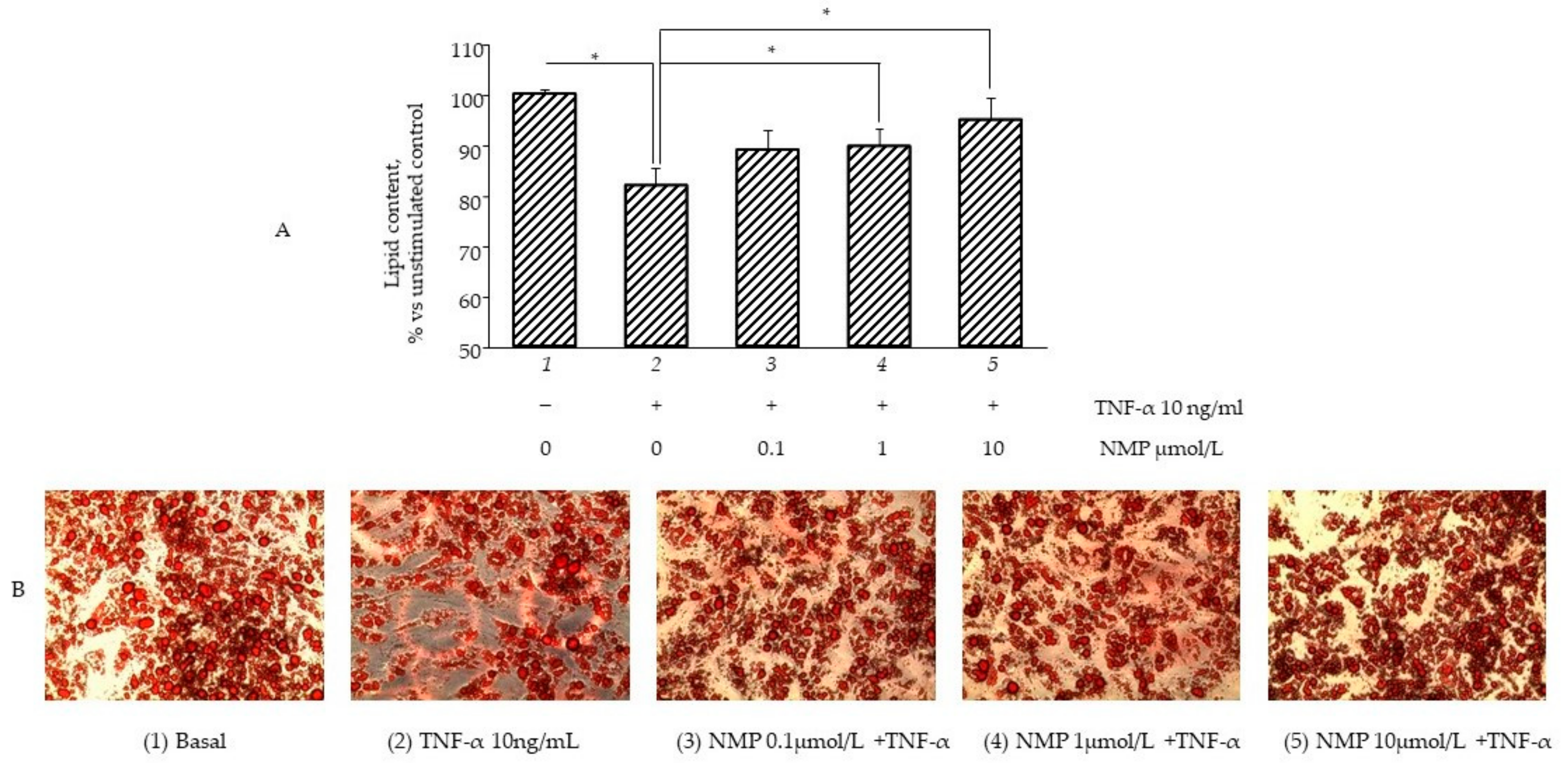
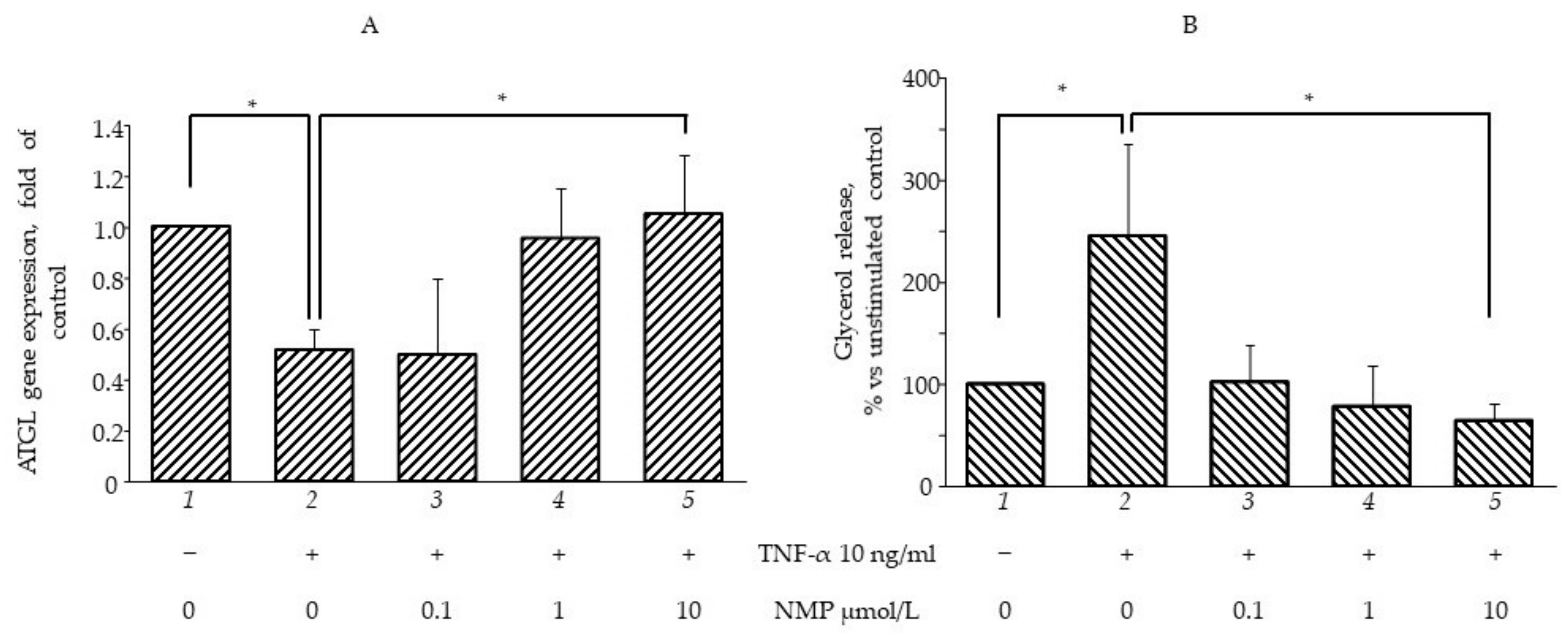
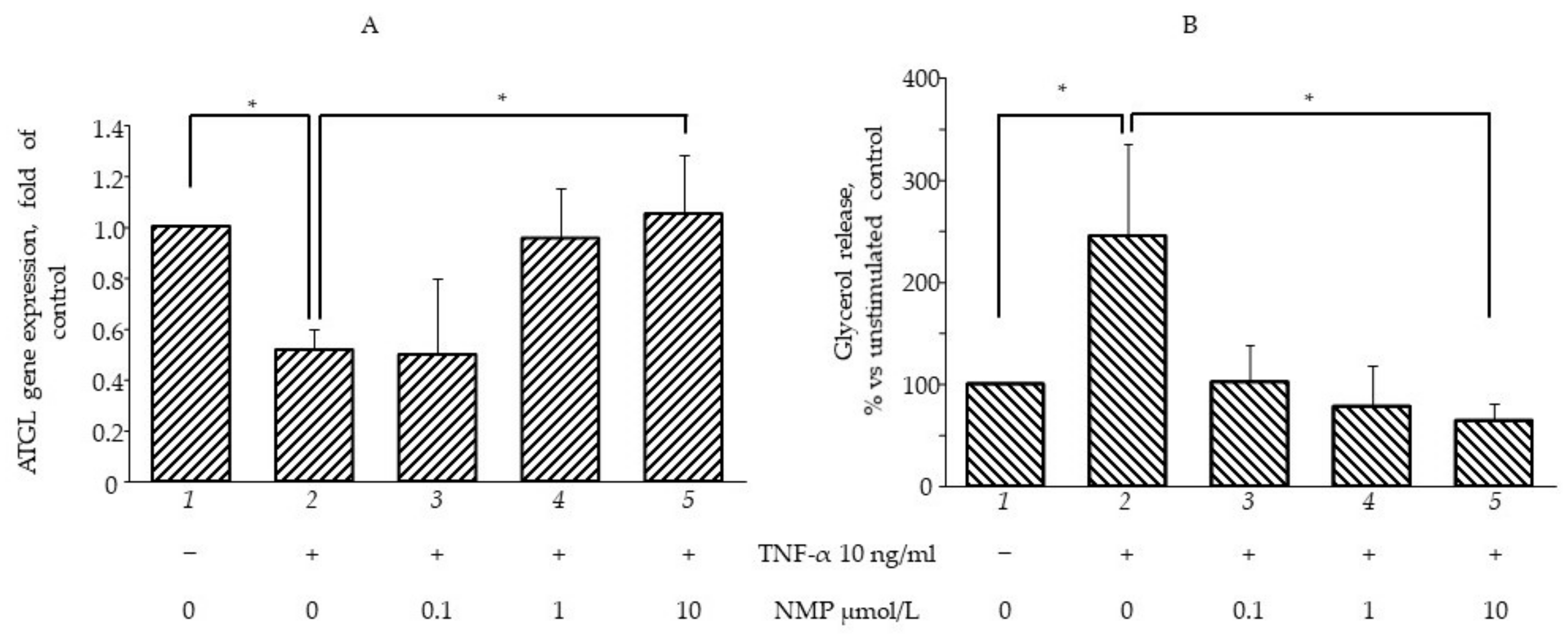
3.4. NMP Modulates Fat Accumulation in Adipocytes Reverting TNF-α Lipolytic Activity
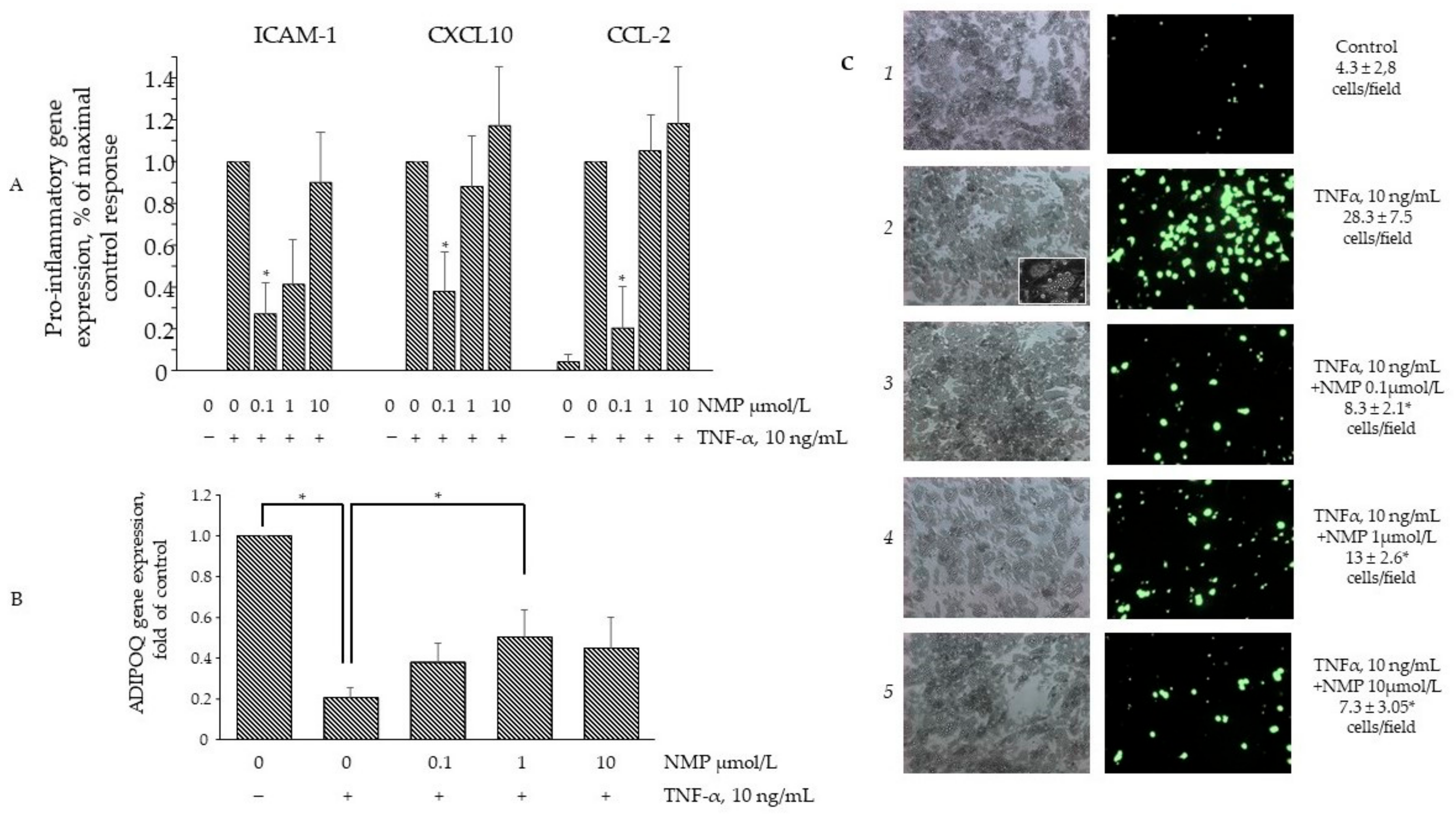
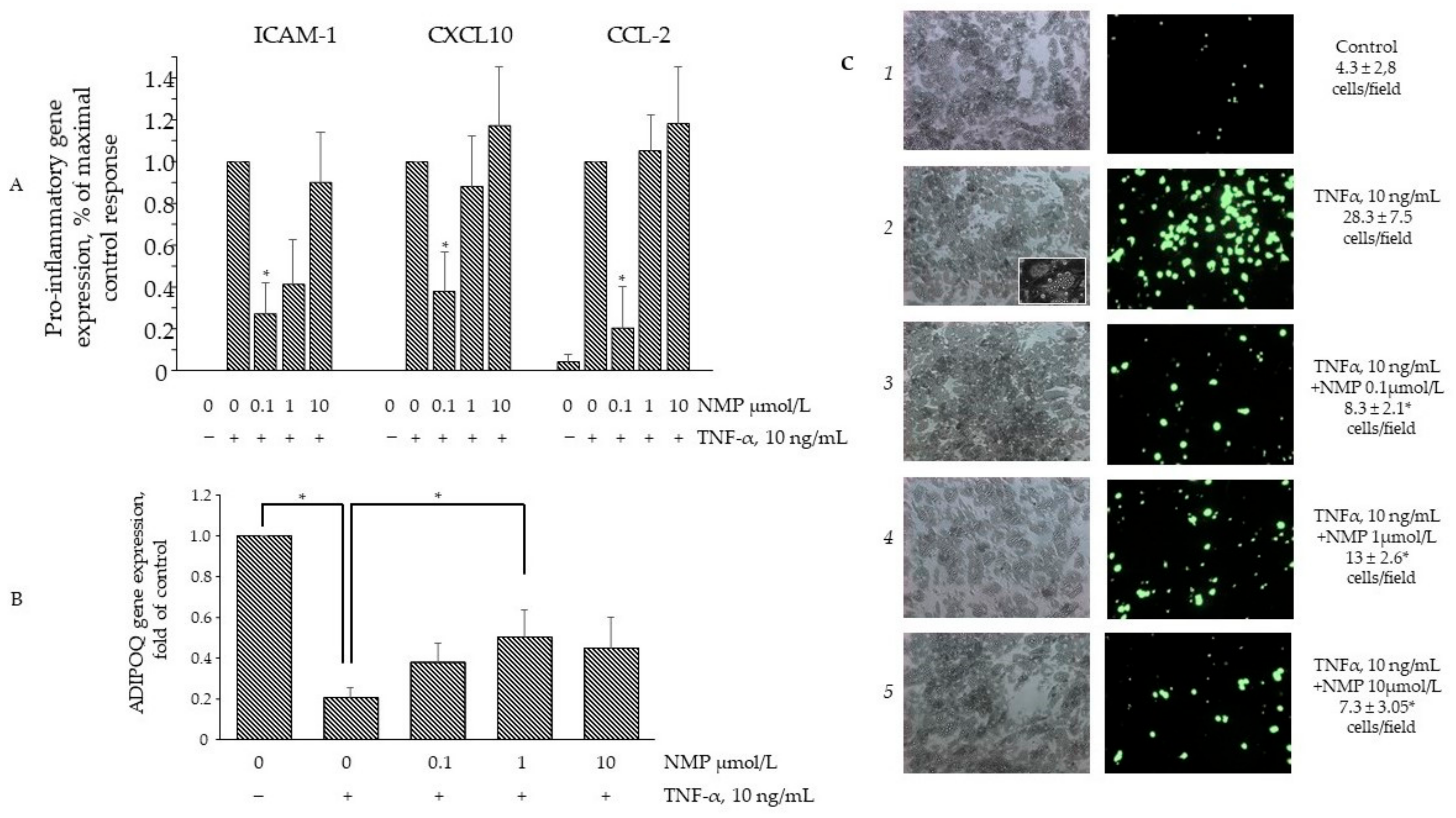
3.5. NMP Inhibits TNF-α Mediated Inflammatory Gene Expression and Reduces Monocyte Adhesion
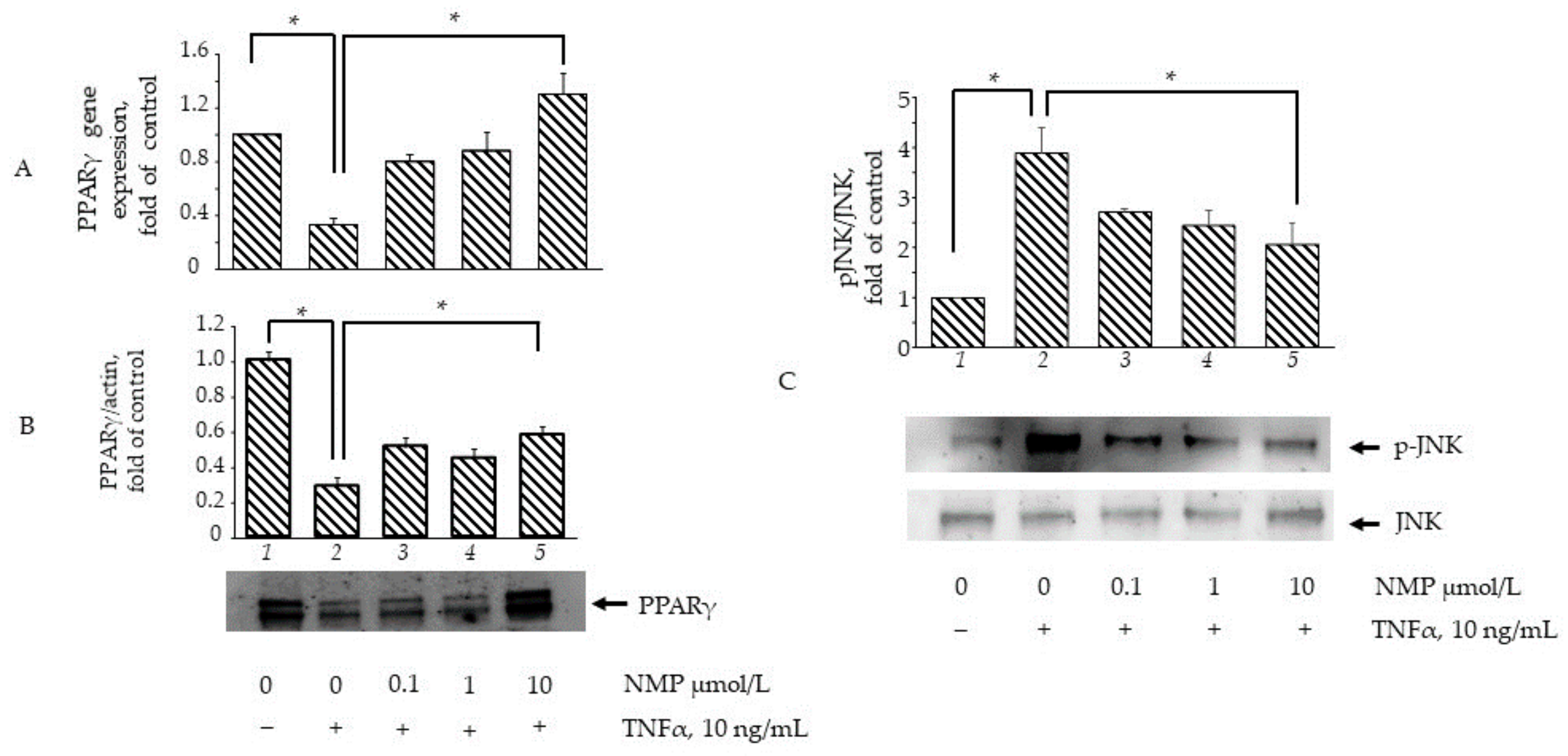
3.6. NMP Restores the Expression and Activity of Adipogenic and Inflammatory Molecular Switches PPARγ and JNK
4. Discussion
4.1. NMP Demonstrated Biological Effects
4.2. Role of TNF in Insulin Signaling and Lipogenesis
4.3. Sites of NMP Interference within TNF-α Signaling
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bluher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.C.T.; Delgado, V.; Borlaug, B.A.; Bax, J.J. Diabesity: The combined burden of obesity and diabetes on heart disease and the role of imaging. Nat. Rev. Cardiol. 2021, 18, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Dal Canto, E.; Ceriello, A.; Ryden, L.; Ferrini, M.; Hansen, T.B.; Schnell, O.; Standl, E.; Beulens, J.W. Diabetes as a cardiovascular risk factor: An overview of global trends of macro and micro vascular complications. Eur. J. Prev. Cardiol. 2019, 26, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillon, N.J.; Loos, R.J.F.; Marshall, S.M.; Zierath, J.R. Metabolic consequences of obesity and type 2 diabetes: Balancing genes and environment for personalized care. Cell 2021, 184, 1530–1544. [Google Scholar] [CrossRef] [PubMed]
- Shimobayashi, M.; Albert, V.; Woelnerhanssen, B.; Frei, I.C.; Weissenberger, D.; Meyer-Gerspach, A.C.; Clement, N.; Moes, S.; Colombi, M.; Meier, J.A.; et al. Insulin resistance causes inflammation in adipose tissue. J. Clin. Investig. 2018, 128, 1538–1550. [Google Scholar] [CrossRef]
- Lee, M.W.; Lee, M.; Oh, K.J. Adipose Tissue-Derived Signatures for Obesity and Type 2 Diabetes: Adipokines, Batokines and MicroRNAs. J. Clin. Med. 2019, 8, 854. [Google Scholar] [CrossRef] [Green Version]
- Cawthorn, W.P.; Sethi, J.K. TNF-alpha and adipocyte biology. FEBS Lett. 2008, 582, 117–131. [Google Scholar] [CrossRef] [Green Version]
- Chacon, M.R.; Fernandez-Real, J.M.; Richart, C.; Megia, A.; Gomez, J.M.; Miranda, M.; Caubet, E.; Pastor, R.; Masdevall, C.; Vilarrasa, N.; et al. Monocyte chemoattractant protein-1 in obesity and type 2 diabetes. Insulin sensitivity study. Obesity 2007, 15, 664–672. [Google Scholar] [CrossRef]
- Good, M.; Newell, F.M.; Haupt, L.M.; Whitehead, J.P.; Hutley, L.J.; Prins, J.B. TNF and TNF receptor expression and insulin sensitivity in human omental and subcutaneous adipose tissue--influence of BMI and adipose distribution. Diabetes Vasc. Dis. Res. 2006, 3, 26–33. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Arner, P.; Caro, J.F.; Atkinson, R.L.; Spiegelman, B.M. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Investig. 1995, 95, 2409–2415. [Google Scholar] [CrossRef]
- Boden, G. Fatty acid-induced inflammation and insulin resistance in skeletal muscle and liver. Curr. Diabetes Rep. 2006, 6, 177–181. [Google Scholar] [CrossRef]
- Ishizuka, K.; Usui, I.; Kanatani, Y.; Bukhari, A.; He, J.; Fujisaka, S.; Yamazaki, Y.; Suzuki, H.; Hiratani, K.; Ishiki, M.; et al. Chronic Tumor Necrosis Factor-α Treatment Causes Insulin Resistance via Insulin Receptor Substrate-1 Serine Phosphorylation and Suppressor of Cytokine Signaling-3 Induction in 3T3-L1 Adipocytes. Endocrinology 2007, 148, 2994–3003. [Google Scholar] [CrossRef]
- Brake, D.K.; Smith, E.O.; Mersmann, H.; Smith, C.W.; Robker, R.L. ICAM-1 expression in adipose tissue: Effects of diet-induced obesity in mice. Am. J. Physiol. Cell Physiol. 2006, 291, C1232–C1239. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.H.; Lee, Y.R.; Park, H.G.; Lee, W.L. The effects of either resveratrol or exercise on macrophage infiltration and switching from M1 to M2 in high fat diet mice. J. Exerc. Nutr. Biochem. 2015, 19, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Samoggia, A.; Riedel, B. Consumers’ Perceptions of Coffee Health Benefits and Motives for Coffee Consumption and Purchasing. Nutrients 2019, 11, 653. [Google Scholar] [CrossRef] [Green Version]
- Kolb, H.; Martin, S.; Kempf, K. Coffee and Lower Risk of Type 2 Diabetes: Arguments for a Causal Relationship. Nutrients 2021, 13, 1144. [Google Scholar] [CrossRef]
- Lee, A.; Lim, W.; Kim, S.; Khil, H.; Cheon, E.; An, S.; Hong, S.; Lee, D.H.; Kang, S.S.; Oh, H.; et al. Coffee Intake and Obesity: A Meta-Analysis. Nutrients 2019, 11, 1274. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Zhang, D.; Jiang, W. Coffee and caffeine intake and incidence of type 2 diabetes mellitus: A meta-analysis of prospective studies. Eur. J. Nutr. 2014, 53, 25–38. [Google Scholar] [CrossRef]
- Lang, R.; Dieminger, N.; Beusch, A.; Lee, Y.M.; Dunkel, A.; Suess, B.; Skurk, T.; Wahl, A.; Hauner, H.; Hofmann, T. Bioappearance and pharmacokinetics of bioactives upon coffee consumption. Anal. Bioanal. Chem. 2013, 405, 8487–8503. [Google Scholar] [CrossRef]
- Boettler, U.; Sommerfeld, K.; Volz, N.; Pahlke, G.; Teller, N.; Somoza, V.; Lang, R.; Hofmann, T.; Marko, D. Coffee constituents as modulators of Nrf2 nuclear translocation and ARE (EpRE)-dependent gene expression. J. Nutr. Biochem. 2011, 22, 426–440. [Google Scholar] [CrossRef]
- Kotyczka, C.; Boettler, U.; Lang, R.; Stiebitz, H.; Bytof, G.; Lantz, I.; Hofmann, T.; Marko, D.; Somoza, V. Dark roast coffee is more effective than light roast coffee in reducing body weight, and in restoring red blood cell vitamin E and glutathione concentrations in healthy volunteers. Mol. Nutr. Food Res. 2011, 55, 1582–1586. [Google Scholar] [CrossRef]
- Riedel, A.; Hochkogler, C.M.; Lang, R.; Bytof, G.; Lantz, I.; Hofmann, T.; Somoza, V. N-methylpyridinium, a degradation product of trigonelline upon coffee roasting, stimulates respiratory activity and promotes glucose utilization in HepG2 cells. Food Funct. 2014, 5, 454–462. [Google Scholar] [CrossRef]
- Massaro, M.; Scoditti, E.; Pellegrino, M.; Carluccio, M.A.; Calabriso, N.; Wabitsch, M.; Storelli, C.; Wright, M.; De Caterina, R. Therapeutic potential of the dual peroxisome proliferator activated receptor (PPAR)alpha/gamma agonist aleglitazar in attenuating TNF-alpha-mediated inflammation and insulin resistance in human adipocytes. Pharmacol. Res. 2016, 107, 125–136. [Google Scholar] [CrossRef]
- Silakari, O.; Singh, P.K. Chapter 6—Molecular docking analysis: Basic technique to predict drug-receptor interactions. In Concepts and Experimental Protocols of Modelling and Informatics in Drug Design; Silakari, O., Singh, P.K., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 131–155. [Google Scholar]
- Lang, R.; Wahl, A.; Skurk, T.; Yagar, E.F.; Schmiech, L.; Eggers, R.; Hauner, H.; Hofmann, T. Development of a hydrophilic liquid interaction chromatography-high-performance liquid chromatography-tandem mass spectrometry based stable isotope dilution analysis and pharmacokinetic studies on bioactive pyridines in human plasma and urine after coffee consumption. Anal. Chem. 2010, 82, 1486–1497. [Google Scholar] [CrossRef]
- Bresciani, L.; Tassotti, M.; Rosi, A.; Martini, D.; Antonini, M.; Dei Cas, A.; Bonadonna, R.; Brighenti, F.; Del Rio, D.; Mena, P. Absorption, Pharmacokinetics, and Urinary Excretion of Pyridines after Consumption of Coffee and Cocoa-Based Products Containing Coffee in a Repeated Dose, Crossover Human Intervention Study. Mol. Nutr. Food Res. 2020, 64, e2000489. [Google Scholar] [CrossRef]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of Cell Viability by the MTT Assay. Cold Spring Harb. Protoc. 2018, 2018, prot095505. [Google Scholar] [CrossRef]
- Ruan, H.; Hacohen, N.; Golub, T.R.; Van Parijs, L.; Lodish, H.F. Tumor necrosis factor-alpha suppresses adipocyte-specific genes and activates expression of preadipocyte genes in 3T3-L1 adipocytes: Nuclear factor-kappaB activation by TNF-alpha is obligatory. Diabetes 2002, 51, 1319–1336. [Google Scholar] [CrossRef] [Green Version]
- Schultze, S.M.; Hemmings, B.A.; Niessen, M.; Tschopp, O. PI3K/AKT, MAPK and AMPK signalling: Protein kinases in glucose homeostasis. Expert Rev. Mol. Med. 2012, 14, e1. [Google Scholar] [CrossRef] [Green Version]
- Capurso, C.; Capurso, A. From excess adiposity to insulin resistance: The role of free fatty acids. Vascul. Pharmacol. 2012, 57, 91–97. [Google Scholar] [CrossRef]
- Han, C.Y.; Umemoto, T.; Omer, M.; Den Hartigh, L.J.; Chiba, T.; LeBoeuf, R.; Buller, C.L.; Sweet, I.R.; Pennathur, S.; Abel, E.D.; et al. NADPH Oxidase-derived Reactive Oxygen Species Increases Expression of Monocyte Chemotactic Factor Genes in Cultured Adipocytes. J. Biol. Chem. 2012, 287, 10379–10393. [Google Scholar] [CrossRef] [Green Version]
- Solinas, G.; Becattini, B. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol. Metab. 2017, 6, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Viedma, V.; Amor, M.; Sarabi, A.; Bilban, M.; Staffler, G.; Zeyda, M.; Stulnig, T.M. Common dysregulated pathways in obese adipose tissue and atherosclerosis. Cardiovasc. Diabetol. 2016, 15, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curat, C.A.; Wegner, V.; Sengenès, C.; Miranville, A.; Tonus, C.; Busse, R.; Bouloumié, A. Macrophages in human visceral adipose tissue: Increased accumulation in obesity and a source of resistin and visfatin. Diabetologia 2006, 49, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Kurylowicz, A.; Kozniewski, K. Anti-Inflammatory Strategies Targeting Metaflammation in Type 2 Diabetes. Molecules 2020, 25, 2224. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef] [Green Version]
- Mesas, A.E.; Leon-Muñoz, L.M.; Rodriguez-Artalejo, F.; Lopez-Garcia, E. The effect of coffee on blood pressure and cardiovascular disease in hypertensive individuals: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2011, 94, 1113–1126. [Google Scholar] [CrossRef]
- Shahinfar, H.; Jayedi, A.; Khan, T.A.; Shab-Bidar, S. Coffee consumption and cardiovascular diseases and mortality in patients with type 2 diabetes: A systematic review and dose-response meta-analysis of cohort studies. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 2526–2538. [Google Scholar] [CrossRef]
- Saeed, M.; Naveed, M.; BiBi, J.; Ali Kamboh, A.; Phil, L.; Chao, S. Potential nutraceutical and food additive properties and risks of coffee: A comprehensive overview. Crit. Rev. Food Sci. Nutr. 2019, 59, 3293–3319. [Google Scholar] [CrossRef]
- Semen, S.; Mercan, S.; Yayla, M.; Acikkol, M. Elemental composition of green coffee and its contribution to dietary intake. Food Chem. 2017, 215, 92–100. [Google Scholar] [CrossRef]
- Wei, F.; Tanokura, M. Chapter 17—Organic Compounds in Green Coffee Beans. In Coffee in Health and Disease Prevention; Preedy, V.R., Ed.; Academic Press: San Diego, CA, USA, 2015; pp. 149–162. [Google Scholar]
- Vieira Porto, A.C.; Farah, A. CHAPTER 18 Potential Effects of Trigonelline and Derivatives on Health. In Coffee: Consumption and Health Implications; The Royal Society of Chemistry: London, UK, 2019; pp. 432–455. [Google Scholar]
- Hamden, K.; Mnafgui, K.; Amri, Z.; Aloulou, A.; Elfeki, A. Inhibition of key digestive enzymes related to diabetes and hyperlipidemia and protection of liver-kidney functions by trigonelline in diabetic rats. Sci. Pharm. 2013, 81, 233–246. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, Q.; Wang, C.; Lou, Z.; Li, Q. Trigonelline reduced diabetic nephropathy and insulin resistance in type 2 diabetic rats through peroxisome proliferator-activated receptor-γ. Exp. Ther. Med. 2019, 18, 1331–1337. [Google Scholar] [CrossRef] [Green Version]
- Stadler, R.H.; Varga, N.; Hau, J.; Vera, F.A.; Welti, D.H. Alkylpyridiniums. 1. Formation in model systems via thermal degradation of trigonelline. J. Agric. Food Chem. 2002, 50, 1192–1199. [Google Scholar] [CrossRef]
- Somoza, V.; Lindenmeier, M.; Wenzel, E.; Frank, O.; Erbersdobler, H.F.; Hofmann, T. Activity-guided identification of a chemopreventive compound in coffee beverage using in vitro and in vivo techniques. J. Agric. Food Chem. 2003, 51, 6861–6869. [Google Scholar] [CrossRef]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef] [Green Version]
- Tzanavari, T.; Giannogonas, P.; Karalis, K.P. TNF-alpha and obesity. Curr. Dir. Autoimmun. 2010, 11, 145–156. [Google Scholar] [CrossRef]
- Gao, P.; Hu, Y.; Wang, J.; Ni, Y.; Zhu, Z.; Wang, H.; Yang, J.; Huang, L.; Fang, L. Underlying Mechanism of Insulin Resistance: A Bioinformatics Analysis Based on Validated Related-Genes from Public Disease Databases. Med. Sci. Monit. 2020, 26, e924334. [Google Scholar] [CrossRef]
- Allott, E.H.; Oliver, E.; Lysaght, J.; Gray, S.G.; Reynolds, J.V.; Roche, H.M.; Pidgeon, G.P. The SGBS cell strain as a model for the in vitro study of obesity and cancer. Clin. Transl. Oncol. 2012, 14, 774–782. [Google Scholar] [CrossRef]
- Rebollo-Hernanz, M.; Zhang, Q.; Aguilera, Y.; Martín-Cabrejas, M.A.; Gonzalez de Mejia, E. Relationship of the Phytochemicals from Coffee and Cocoa By-Products with their Potential to Modulate Biomarkers of Metabolic Syndrome In Vitro. Antioxidants 2019, 8, 279. [Google Scholar] [CrossRef] [Green Version]
- Vasileva, L.V.; Savova, M.S.; Amirova, K.M.; Balcheva-Sivenova, Z.; Ferrante, C.; Orlando, G.; Wabitsch, M.; Georgiev, M.I. Caffeic and Chlorogenic Acids Synergistically Activate Browning Program in Human Adipocytes: Implications of AMPK- and PPAR-Mediated Pathways. Int. J. Mol. Sci. 2020, 21, 9740. [Google Scholar] [CrossRef]
- Baek, J.H.; Kim, N.J.; Song, J.K.; Chun, K.H. Kahweol inhibits lipid accumulation and induces Glucose-uptake through activation of AMP-activated protein kinase (AMPK). BMB Rep. 2017, 50, 566–571. [Google Scholar] [CrossRef]
- Rebollo-Hernanz, M.; Zhang, Q.; Aguilera, Y.; Martín-Cabrejas, M.A.; Gonzalez de Mejia, E. Phenolic compounds from coffee by-products modulate adipogenesis-related inflammation, mitochondrial dysfunction, and insulin resistance in adipocytes, via insulin/PI3K/AKT signaling pathways. Food Chem. Toxicol. 2019, 132, 110672. [Google Scholar] [CrossRef]
- Suganami, T.; Nishida, J.; Ogawa, Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: Role of free fatty acids and tumor necrosis factor alpha. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2062–2068. [Google Scholar] [CrossRef] [Green Version]
- Bozulic, L.; Hemmings, B.A. PIKKing on PKB: Regulation of PKB activity by phosphorylation. Curr. Opin. Cell Biol. 2009, 21, 256–261. [Google Scholar] [CrossRef]
- Czech, M.P. Mechanisms of insulin resistance related to white, beige, and brown adipocytes. Mol. Metab. 2020, 34, 27–42. [Google Scholar] [CrossRef]
- Lee, Y.H.; Giraud, J.; Davis, R.J.; White, M.F. c-Jun N-terminal kinase (JNK) mediates feedback inhibition of the insulin signaling cascade. J. Biol. Chem. 2003, 278, 2896–2902. [Google Scholar] [CrossRef] [Green Version]
- Nandipati, K.C.; Subramanian, S.; Agrawal, D.K. Protein kinases: Mechanisms and downstream targets in inflammation-mediated obesity and insulin resistance. Mol. Cell Biochem. 2017, 426, 27–45. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, G.R.; Michell, B.J.; van Denderen, B.J.; Watt, M.J.; Carey, A.L.; Fam, B.C.; Andrikopoulos, S.; Proietto, J.; Gorgun, C.Z.; Carling, D.; et al. Tumor necrosis factor alpha-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab. 2006, 4, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Kain, V.; Kapadia, B.; Viswakarma, N.; Seshadri, S.; Prajapati, B.; Jena, P.K.; Teja Meda, C.L.; Subramanian, M.; Kaimal Suraj, S.; Kumar, S.T.; et al. Co-activator binding protein PIMT mediates TNF-alpha induced insulin resistance in skeletal muscle via the transcriptional down-regulation of MEF2A and GLUT4. Sci. Rep. 2015, 5, 15197. [Google Scholar] [CrossRef] [Green Version]
- Berger, J.; Moller, D.E. The mechanisms of action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef] [Green Version]
- Filip-Ciubotaru, F.; Foia, L.; Manciuc, C.; Grigore, C. PPARs: Structure, mechanisms of action and control. Note I. Rev. Med. Chir. Soc. Med. Nat. Iasi 2011, 115, 477–484. [Google Scholar]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kershaw, E.E.; Schupp, M.; Guan, H.P.; Gardner, N.P.; Lazar, M.A.; Flier, J.S. PPARgamma regulates adipose triglyceride lipase in adipocytes in vitro and in vivo. Am. J. Physiol. Endocrinol. Metab. 2007, 293, 1736–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.Y.; Tillison, K.; Lee, J.H.; Rearick, D.A.; Smas, C.M. The adipose tissue triglyceride lipase ATGL/PNPLA2 is downregulated by insulin and TNF-alpha in 3T3-L1 adipocytes and is a target for transactivation by PPARgamma. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E115–E127. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-Y.; Kim, J.K.; Jeon, J.H.; Yoon, S.R.; Choi, I.; Yang, Y. c-Jun N-terminal kinase is involved in the suppression of adiponectin expression by TNF-α in 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 2005, 327, 460–467. [Google Scholar] [CrossRef]
- Jin, D.; Sun, J.; Huang, J.; He, Y.; Yu, A.; Yu, X.; Yang, Z. TNF-α reduces g0s2 expression and stimulates lipolysis through PPAR-γ inhibition in 3T3-L1 adipocytes. Cytokine 2014, 69, 196–205. [Google Scholar] [CrossRef]
- Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptors: Nuclear control of metabolism. Endocr. Rev. 1999, 20, 649–688. [Google Scholar] [CrossRef] [Green Version]
- Tamori, Y.; Masugi, J.; Nishino, N.; Kasuga, M. Role of peroxisome proliferator-activated receptor-gamma in maintenance of the characteristics of mature 3T3-L1 adipocytes. Diabetes 2002, 51, 2045–2055. [Google Scholar] [CrossRef] [Green Version]
- Polvani, S.; Tarocchi, M.; Tempesti, S.; Bencini, L.; Galli, A. Peroxisome proliferator activated receptors at the crossroad of obesity, diabetes, and pancreatic cancer. World J. Gastroenterol. 2016, 22, 2441–2459. [Google Scholar] [CrossRef]
- Lettieri Barbato, D.; Aquilano, K.; Baldelli, S.; Cannata, S.M.; Bernardini, S.; Rotilio, G.; Ciriolo, M.R. Proline oxidase-adipose triglyceride lipase pathway restrains adipose cell death and tissue inflammation. Cell Death Differ. 2014, 21, 113–123. [Google Scholar] [CrossRef]
- Haemmerle, G.; Moustafa, T.; Woelkart, G.; Buttner, S.; Schmidt, A.; van de Weijer, T.; Hesselink, M.; Jaeger, D.; Kienesberger, P.C.; Zierler, K.; et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-alpha and PGC-1. Nat. Med. 2011, 17, 1076–1085. [Google Scholar] [CrossRef] [Green Version]
- Lettieri Barbato, D.; Tatulli, G.; Aquilano, K.; Ciriolo, M.R. Inhibition of age-related cytokines production by ATGL: A mechanism linked to the anti-inflammatory effect of resveratrol. Mediat. Inflamm. 2014, 2014, 917698. [Google Scholar] [CrossRef]
- Feng, J.; Lu, S.; Ou, B.; Liu, Q.; Dai, J.; Ji, C.; Zhou, H.; Huang, H.; Ma, Y. The Role of JNk Signaling Pathway in Obesity-Driven Insulin Resistance. Diabetes Metab. Syndr. Obes. 2020, 13, 1399–1406. [Google Scholar] [CrossRef]
- Boden, G. Obesity, insulin resistance and free fatty acids. Curr. Opin. Endocrinol. Diabetes Obes. 2011, 18, 139–143. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Kim, J.W.; Osborne, O.; Oh, D.Y.; Sasik, R.; Schenk, S.; Chen, A.; Chung, H.; Murphy, A.; Watkins, S.M.; et al. Increased adipocyte O2 consumption triggers HIF-1alpha, causing inflammation and insulin resistance in obesity. Cell 2014, 157, 1339–1352. [Google Scholar] [CrossRef] [Green Version]
- Ye, J. Emerging role of adipose tissue hypoxia in obesity and insulin resistance. Int. J. Obes. 2009, 33, 54–66. [Google Scholar] [CrossRef] [Green Version]
- Hurrle, S.; Hsu, W.H. The etiology of oxidative stress in insulin resistance. Biomed. J. 2017, 40, 257–262. [Google Scholar] [CrossRef]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
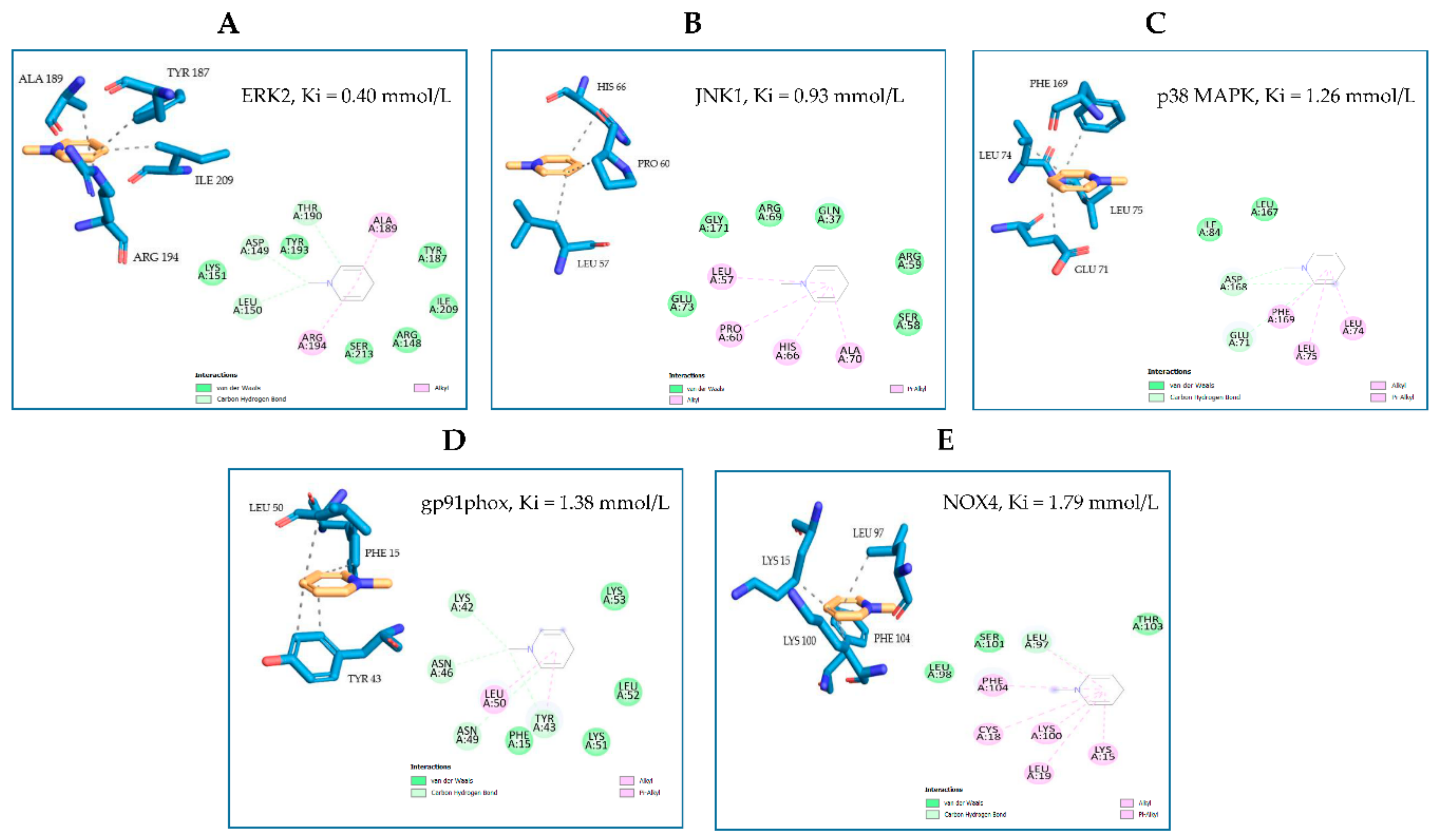{kind=link}
{kind=link}
| Gene Symbol | Full Name | Forward Primer (5′-3′) | Revers Primer (3′-5′) | Accession Number |
|---|---|---|---|---|
| MCP-1/CCL-2 | monocyte chemoattractant protein-1/C-C Motif chemokine ligand 2 | CCCCAGTCACCTGCTGTTAT | TCCTGAACCCACTTCTGCTT | NM_002982.3 |
| CXC-L10 | C-X-C Motif chemokine ligand 10 | CAAGGATGGACCACACAGAG | GCAGGGTCAGAACATCCACT | NM_001565.2 |
| PTGS2/COX-2 | prostaglandin G/H synthase 2/cyclooxygenase-2 | TGCTGTGGAGCTGTATCCTG | GAAACCCACTTCTCCACCA | NM_000963.2 |
| IL-1β | interleukin 1 β | CTGTCCTGCGTGTTGAAAGA | AGTTATATCCTGGCCGCCTT | NM_000576.2 |
| CSF1/M-CSF | colony stimulating factor 1/macrophage-colony stimulating factor | TGGACGCACAGAACAGTCTC | CCTCCAGGGCTCACAATAAA | NM_000757.4 |
| ADIPOQ | adiponectin | AGTCTCACATCTGGTTGGGG | CTCTCTGTGCCTCTGGTTCC | NM_001177800.1 |
| Slc2a4/GLUT-4 | solute carrier family 2 member 4/glucose transporter type 4 | TGGTCCATGTACCCCTCATT | AGAGCCTGTGTGGCAAGAGT | NM_009204.2 |
| PPARγ | peroxisome proliferator-activated receptor γ | TGCAGGTGATCAAGAAGACG | AGTGCAACTGGAAGAAGGGA | NM_005037.5 |
| ATGL/PNPLA2 | adipose triglyceride lipase/patatin-like phospholipase domain-containing protein 2 | CTGACCACCCTCTCCAACAT | TCACCAGGTACTGGCAGATG | NM_020376.4 |
| LIPE/HSL | lipase E, hormone sensitive type/hormone-sensitive lipase | TTCCTCCGGGAGTATGTCAC | TGTGATCCGCTCAAACTCAG | NM_005357 |
| CGI-58 | comparative gene identification-58 | GCACCCTGACATTCCAGTTT | CAGTCCACAGTGTCGCAGAT | AF151816.1 |
| G0S2 | G0/G1 switch gene 2 | GGAAGATGGTGAAGCTGTACG | CTTGCTTCTGGAGAGCCTGT | NM_015714 |
| PLIN1 | perilipin 1 | TCTCGATACACCGTGCAGAC | TGGTCCTCATGATCCTCCTC | NM_002666.5 |
| ICAM-1 | intercellular adhesion molecule-1 | AGACATAGCCCCACCATGAG | CAAGGGTTGGGGTCAGTAGA | NM_000201.2 |
| 18S | 18 ribosomal RNA | AAACGGCTACCACATCCAAG | CCTCCAATGGATCCTCGTTA | NR_003286.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quarta, S.; Scoditti, E.; Carluccio, M.A.; Calabriso, N.; Santarpino, G.; Damiano, F.; Siculella, L.; Wabitsch, M.; Verri, T.; Favari, C.; et al. Coffee Bioactive N-Methylpyridinium Attenuates Tumor Necrosis Factor (TNF)-α-Mediated Insulin Resistance and Inflammation in Human Adipocytes. Biomolecules 2021, 11, 1545. https://doi.org/10.3390/biom11101545
Quarta S, Scoditti E, Carluccio MA, Calabriso N, Santarpino G, Damiano F, Siculella L, Wabitsch M, Verri T, Favari C, et al. Coffee Bioactive N-Methylpyridinium Attenuates Tumor Necrosis Factor (TNF)-α-Mediated Insulin Resistance and Inflammation in Human Adipocytes. Biomolecules. 2021; 11(10):1545. https://doi.org/10.3390/biom11101545
Chicago/Turabian StyleQuarta, Stefano, Egeria Scoditti, Maria Annunziata Carluccio, Nadia Calabriso, Giuseppe Santarpino, Fabrizio Damiano, Luisa Siculella, Martin Wabitsch, Tiziano Verri, Claudia Favari, and et al. 2021. "Coffee Bioactive N-Methylpyridinium Attenuates Tumor Necrosis Factor (TNF)-α-Mediated Insulin Resistance and Inflammation in Human Adipocytes" Biomolecules 11, no. 10: 1545. https://doi.org/10.3390/biom11101545
APA StyleQuarta, S., Scoditti, E., Carluccio, M. A., Calabriso, N., Santarpino, G., Damiano, F., Siculella, L., Wabitsch, M., Verri, T., Favari, C., Del Rio, D., Mena, P., De Caterina, R., & Massaro, M. (2021). Coffee Bioactive N-Methylpyridinium Attenuates Tumor Necrosis Factor (TNF)-α-Mediated Insulin Resistance and Inflammation in Human Adipocytes. Biomolecules, 11(10), 1545. https://doi.org/10.3390/biom11101545