
Reversal of ABCG2/BCRP-Mediated Multidrug Resistance by 5,3′,5′-Trihydroxy-3,6,7,4′-Tetramethoxyflavone Isolated from the Australian Desert Plant Eremophila galeata Chinnock

 , , , , ,
, , , , ,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Lines and Culture Conditions
2.3. Extraction and Sample Preparation
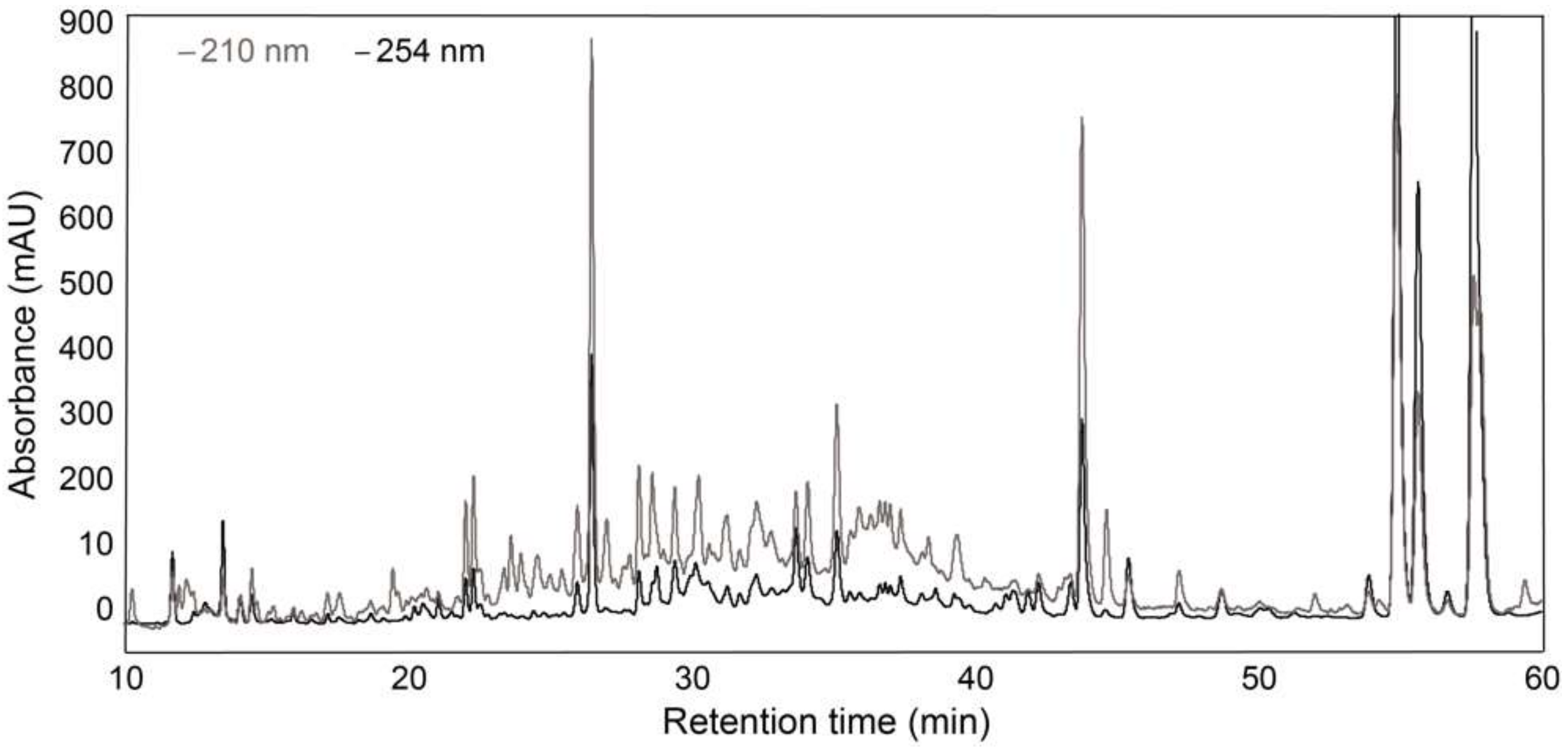
2.4. Analytical-Scale HPLC-PDA-HRMS
2.5. Analytical-Scale HPLC-PDA and Fraction Collection
2.6. NMR Experiments
2.7. MTT Cell Viability Assay
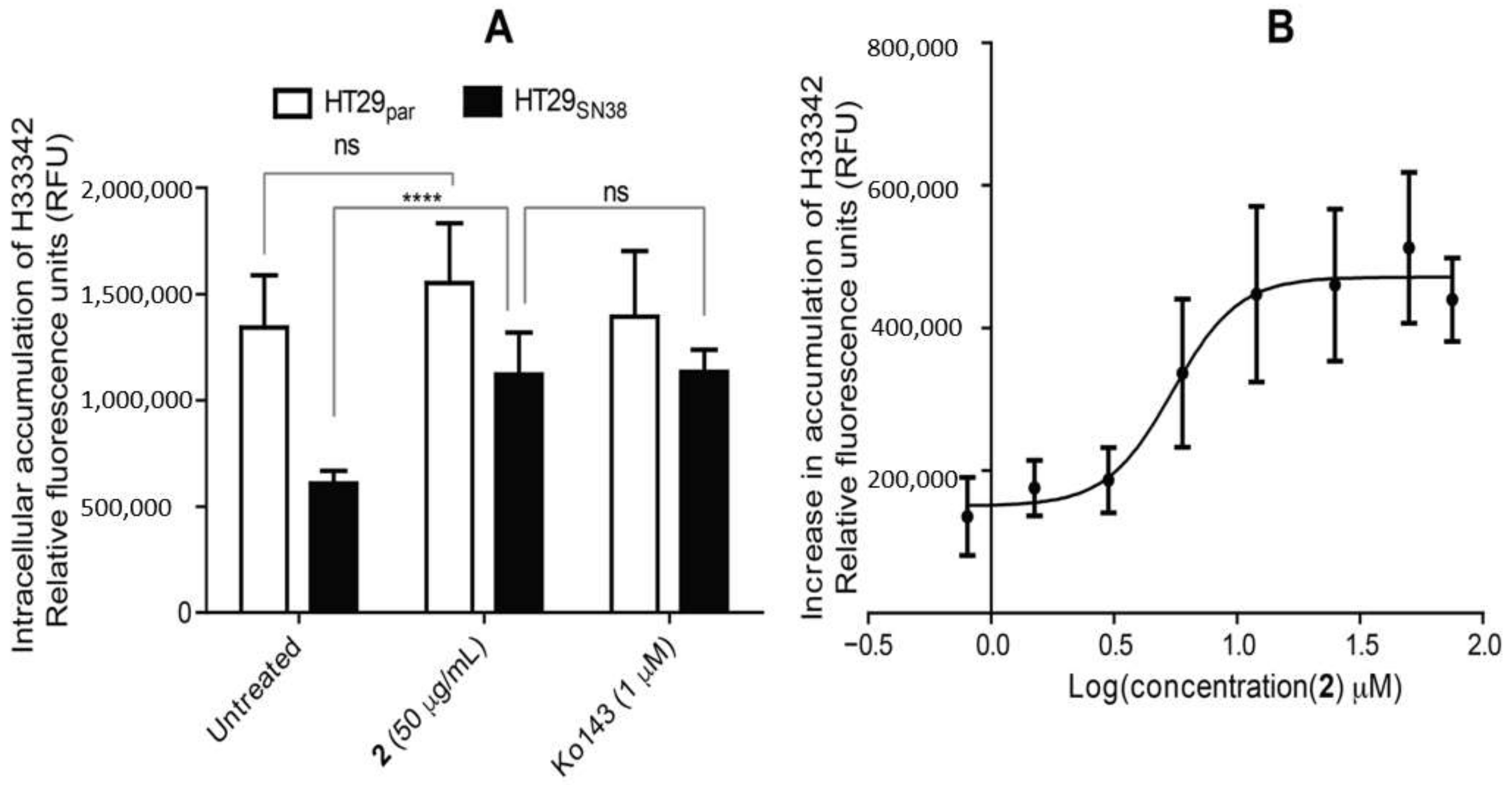
2.8. Dye Accumulation Assay
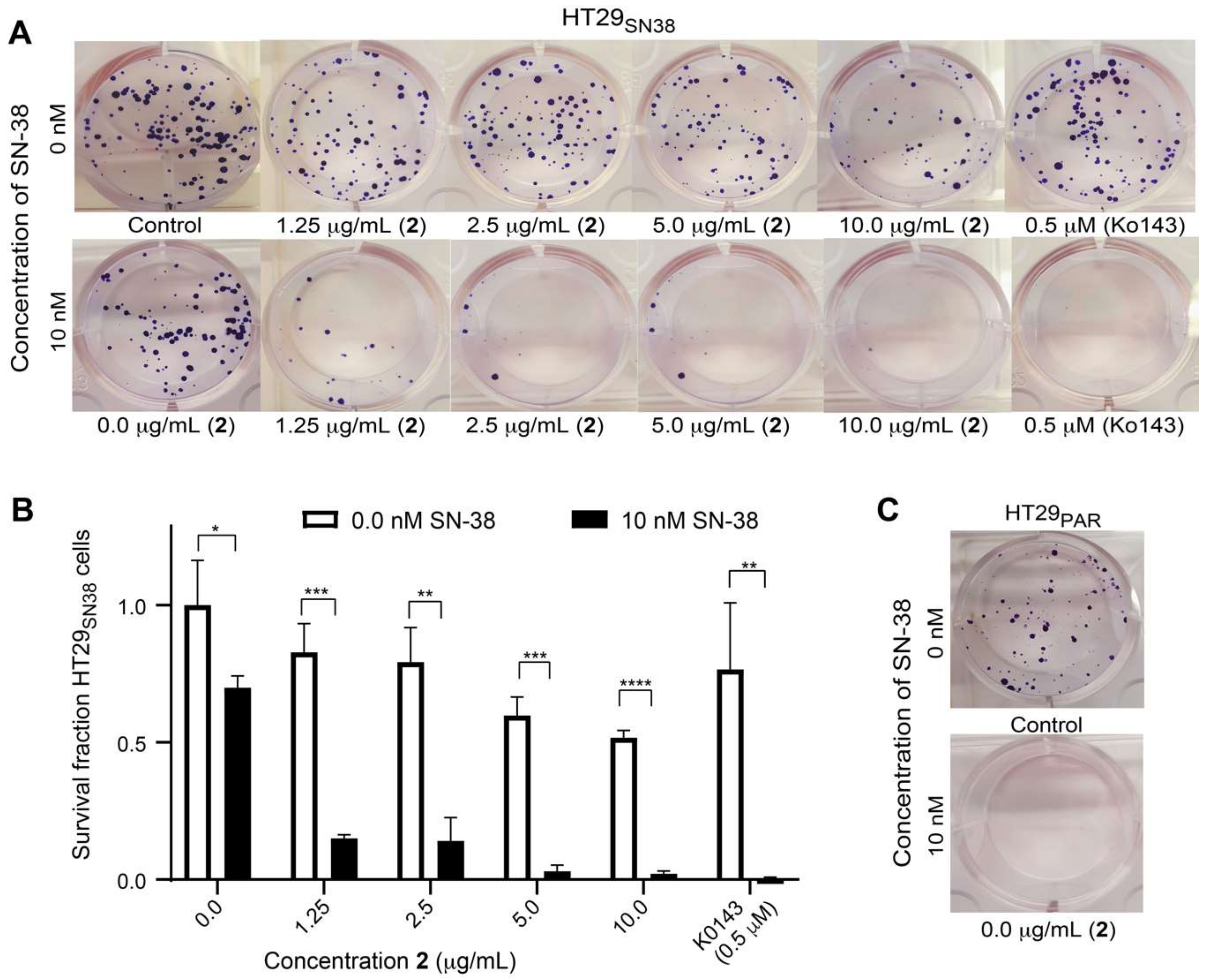
2.9. Colony Formation Assay
2.10. Statistical Analyses
2.11. Molecular Interaction Modelling
3. Results
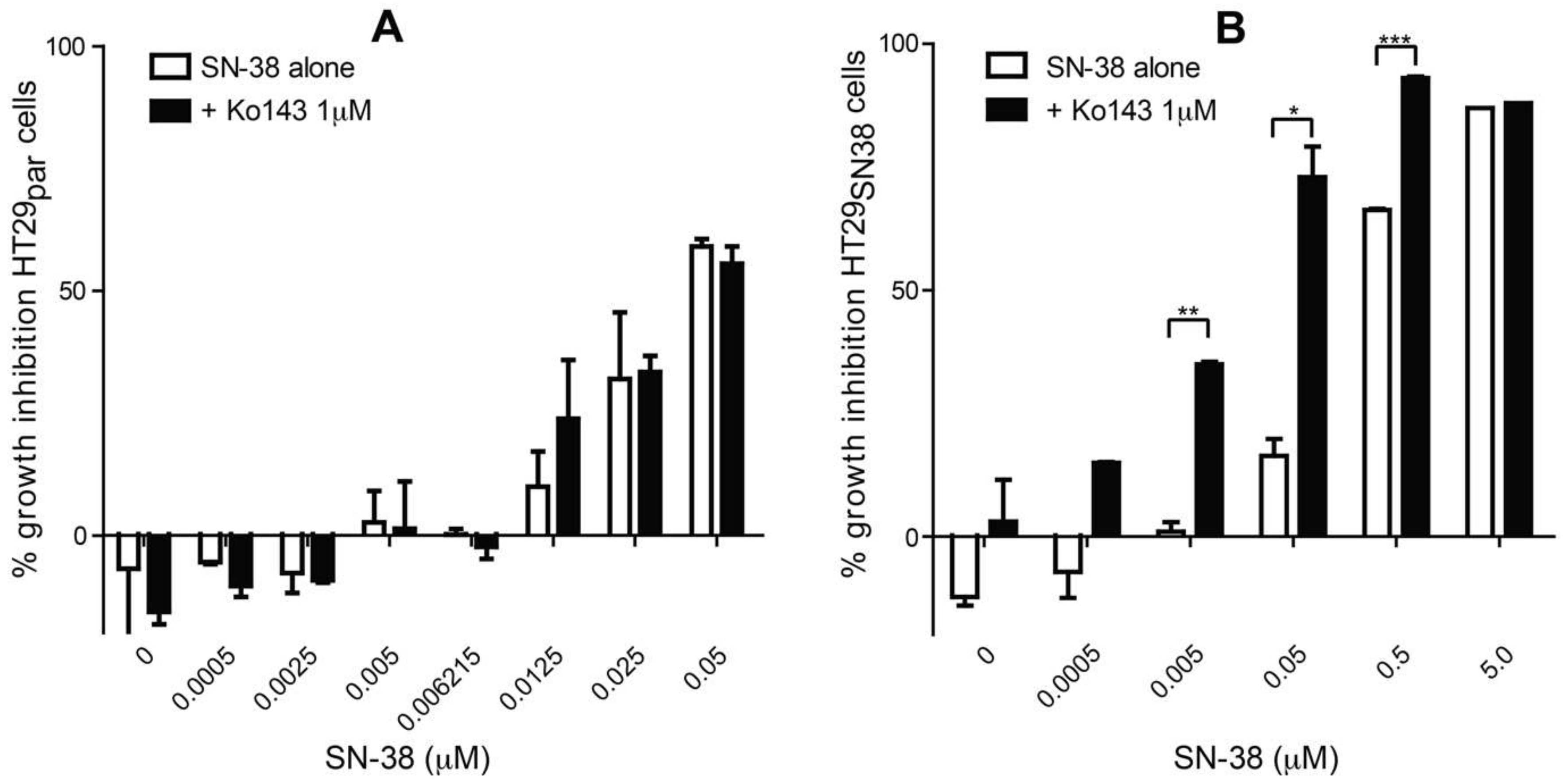
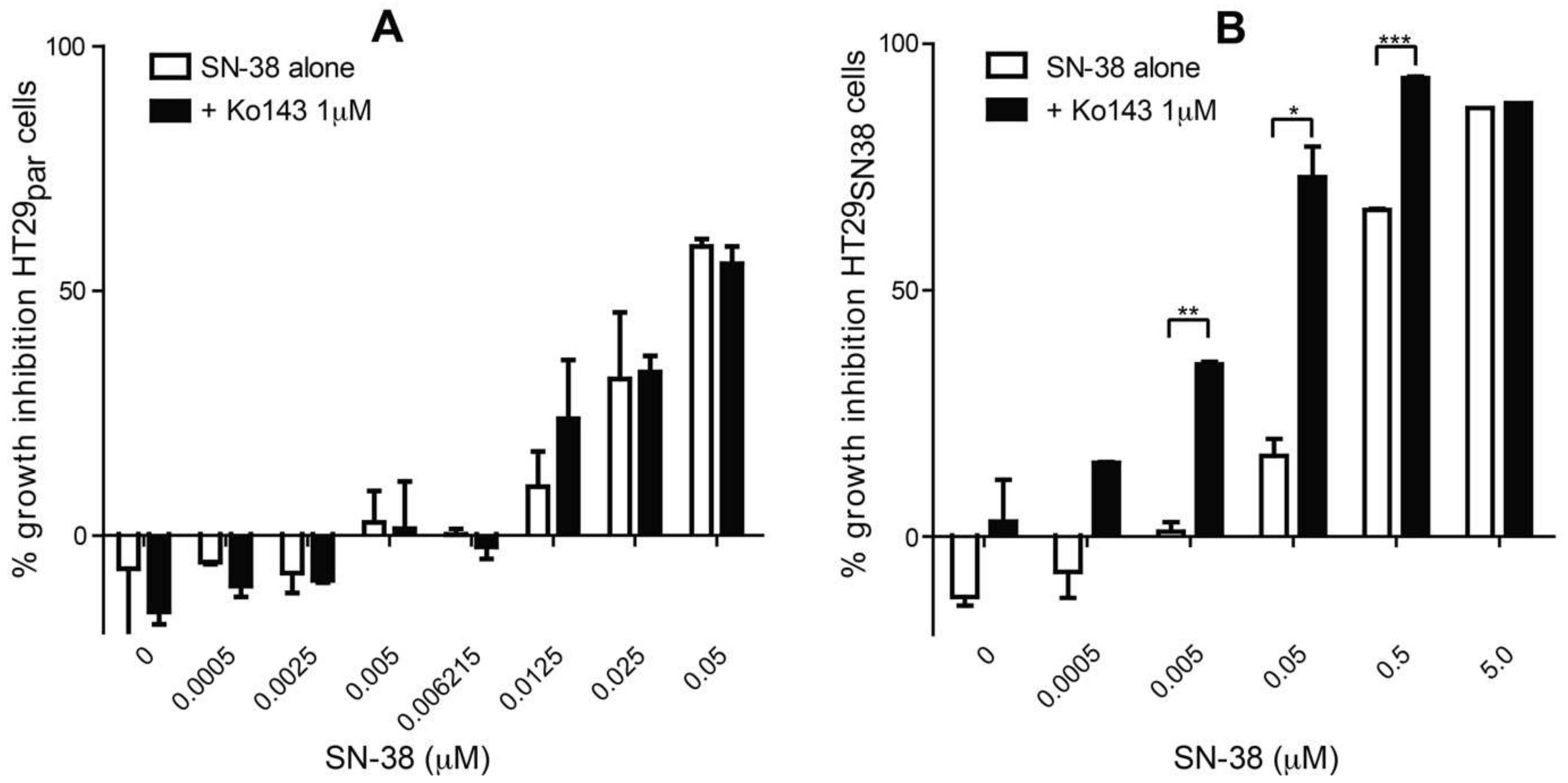
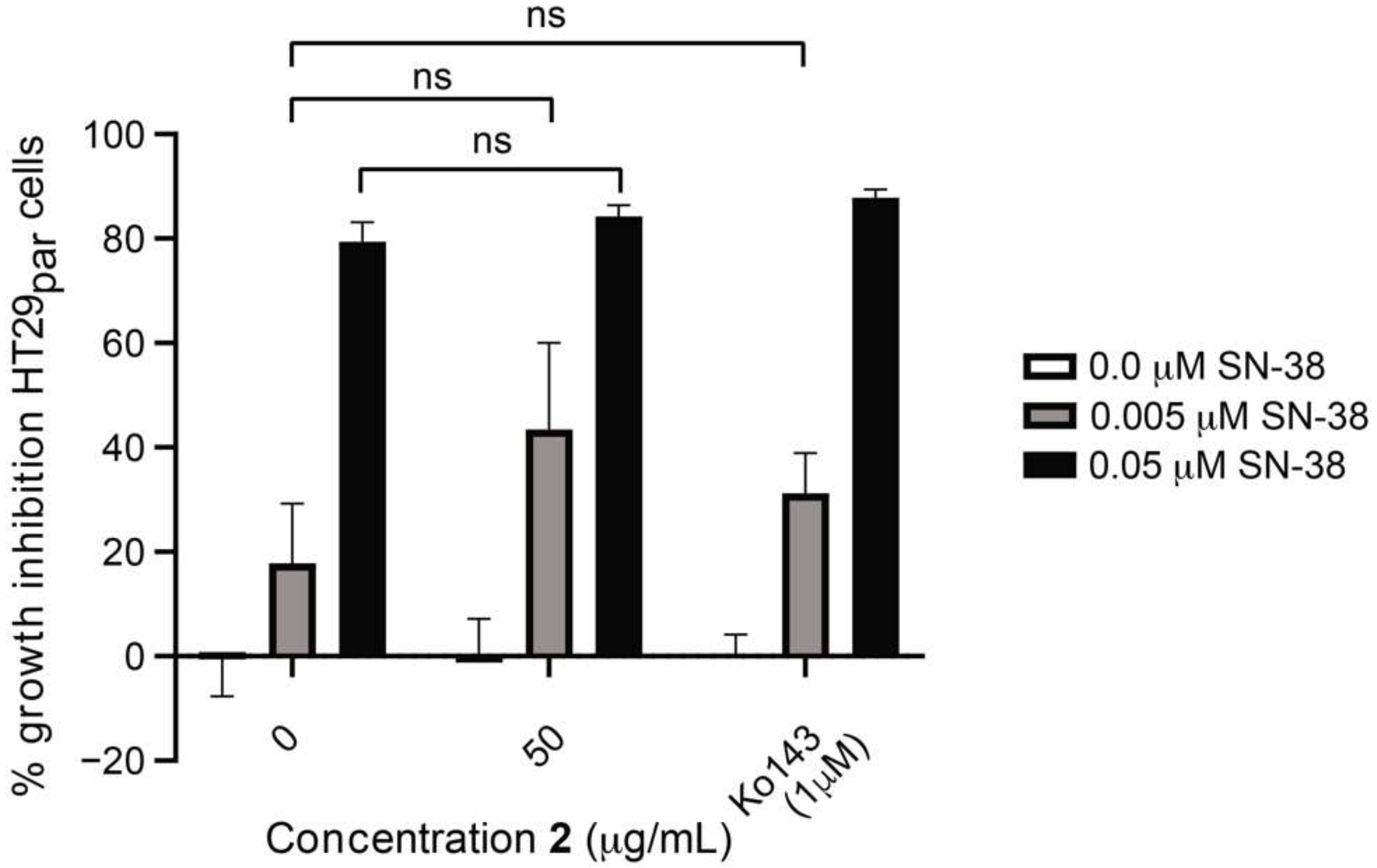
3.1. The Effect of SN-38 Alone and in Combination with BCRP Inhibitor Ko143 on HT29 Colon Cancer Cells
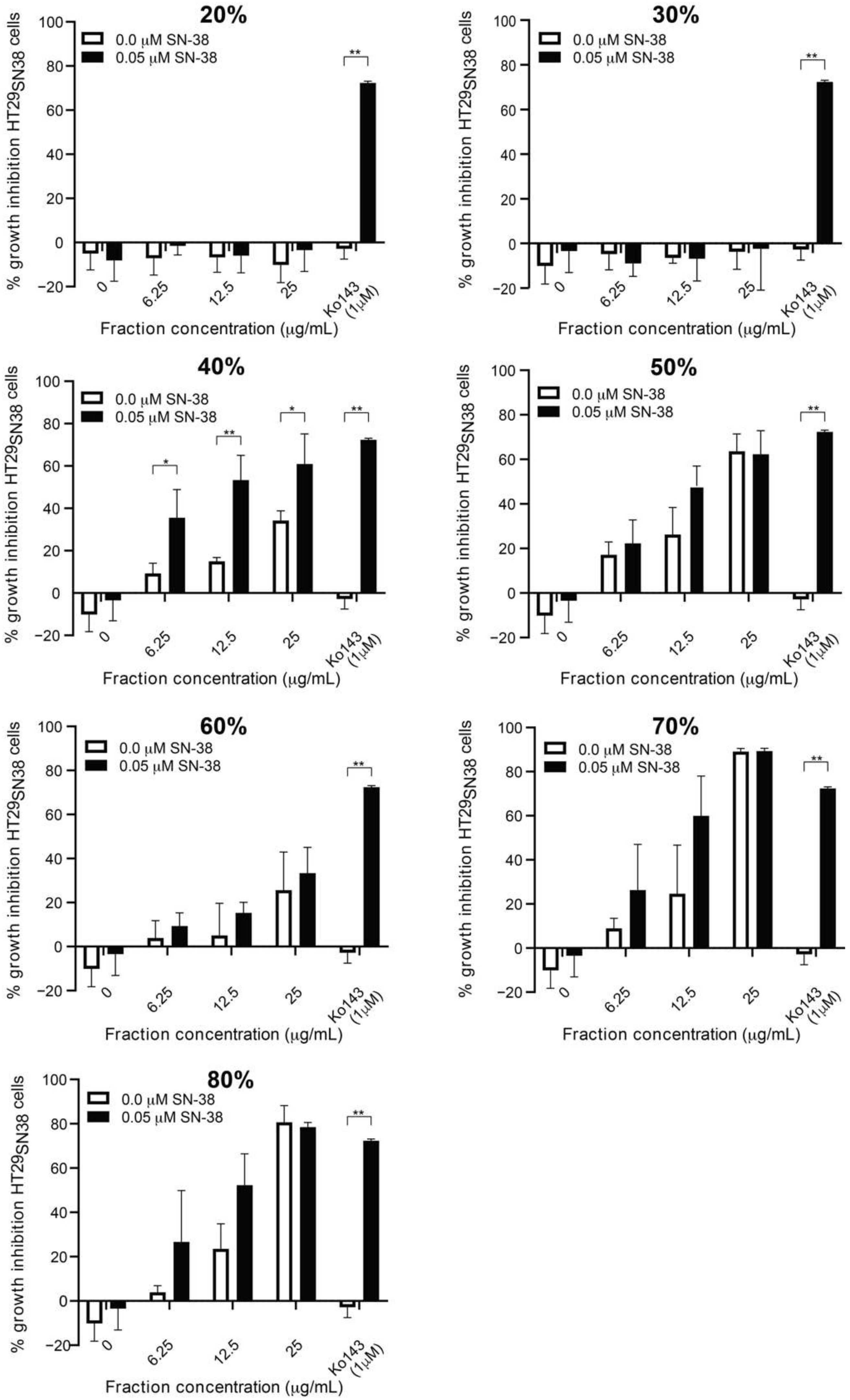
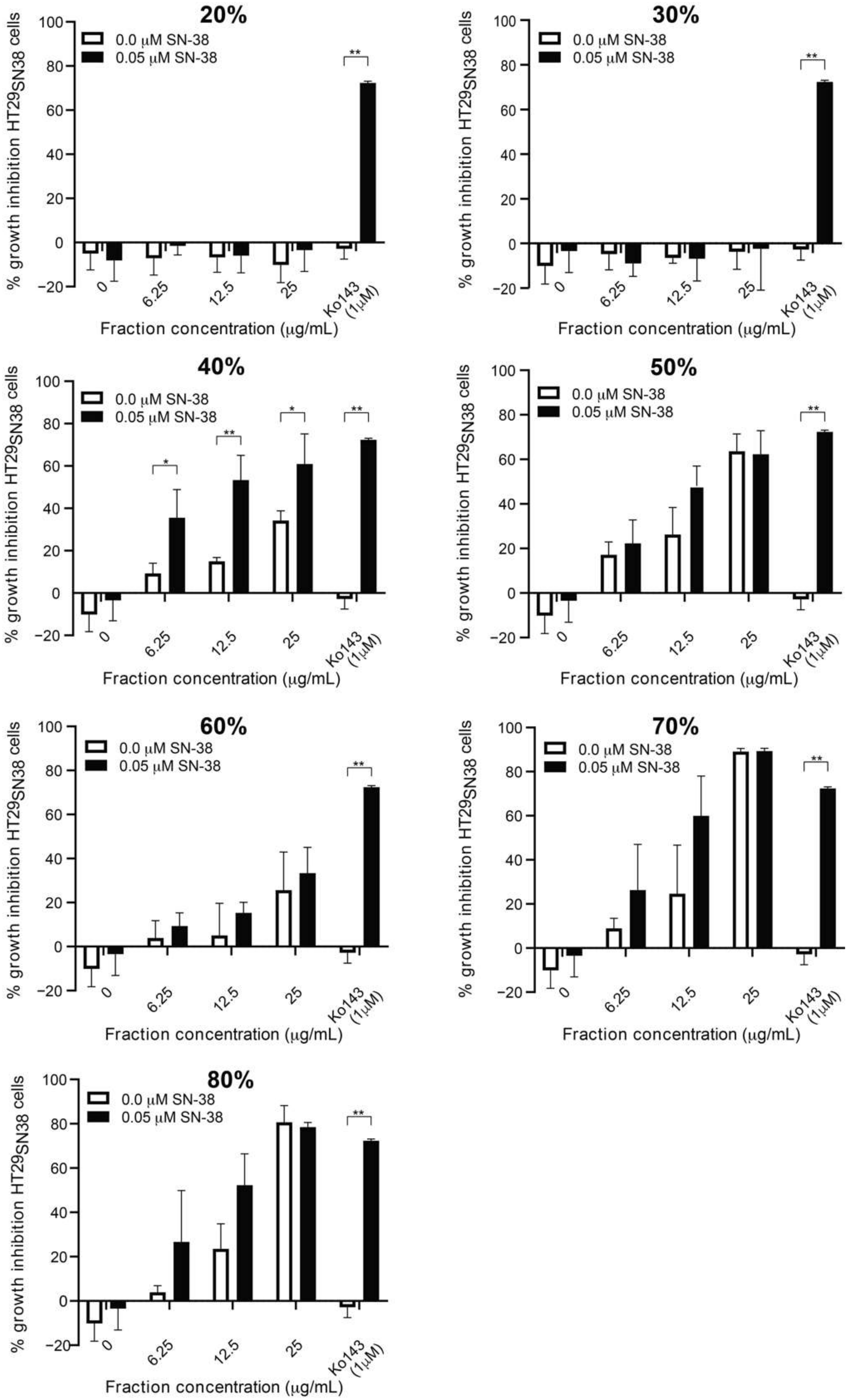
3.2. Screening of SPE Fractions
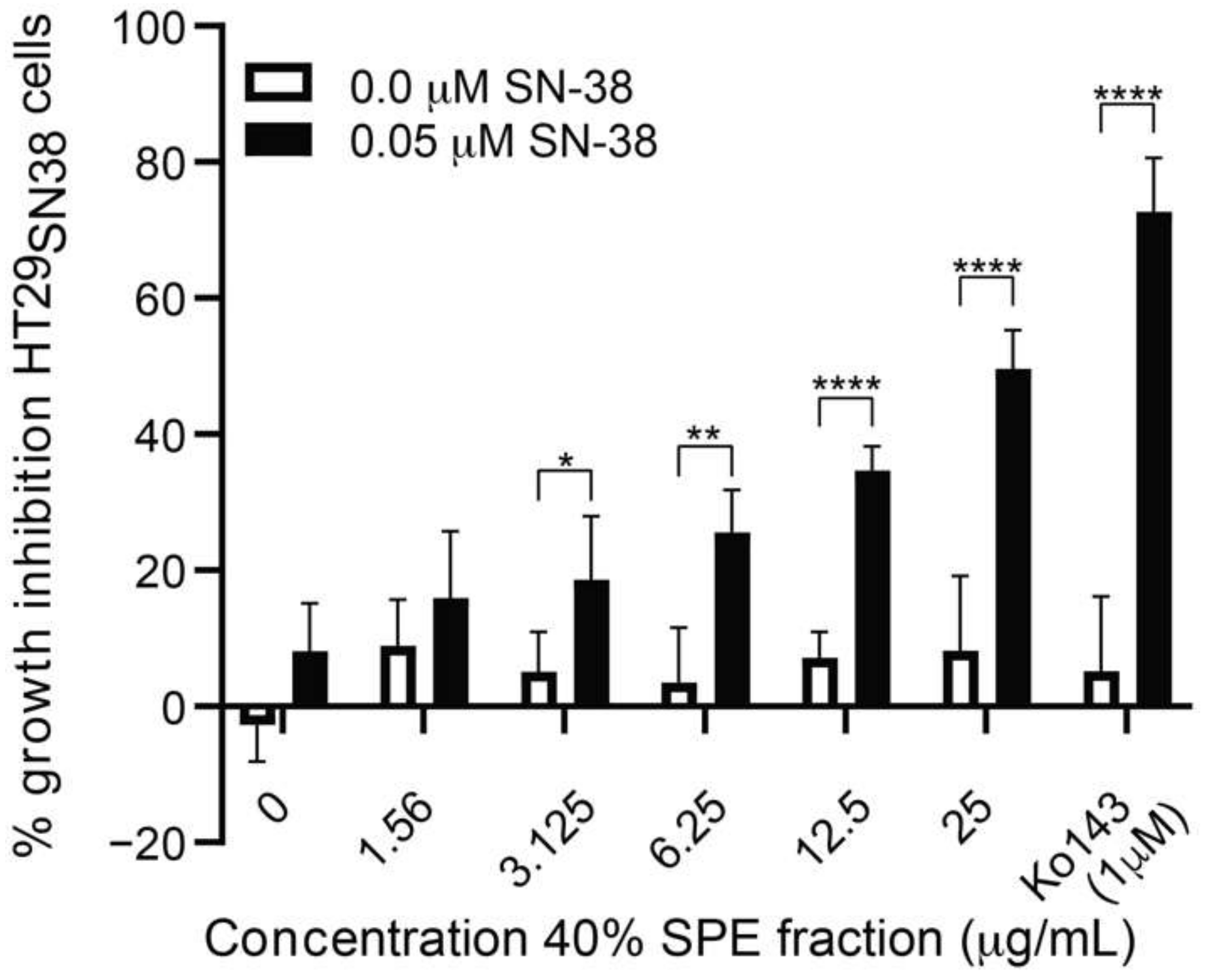
3.3. Further Testing of the Most Active Fraction
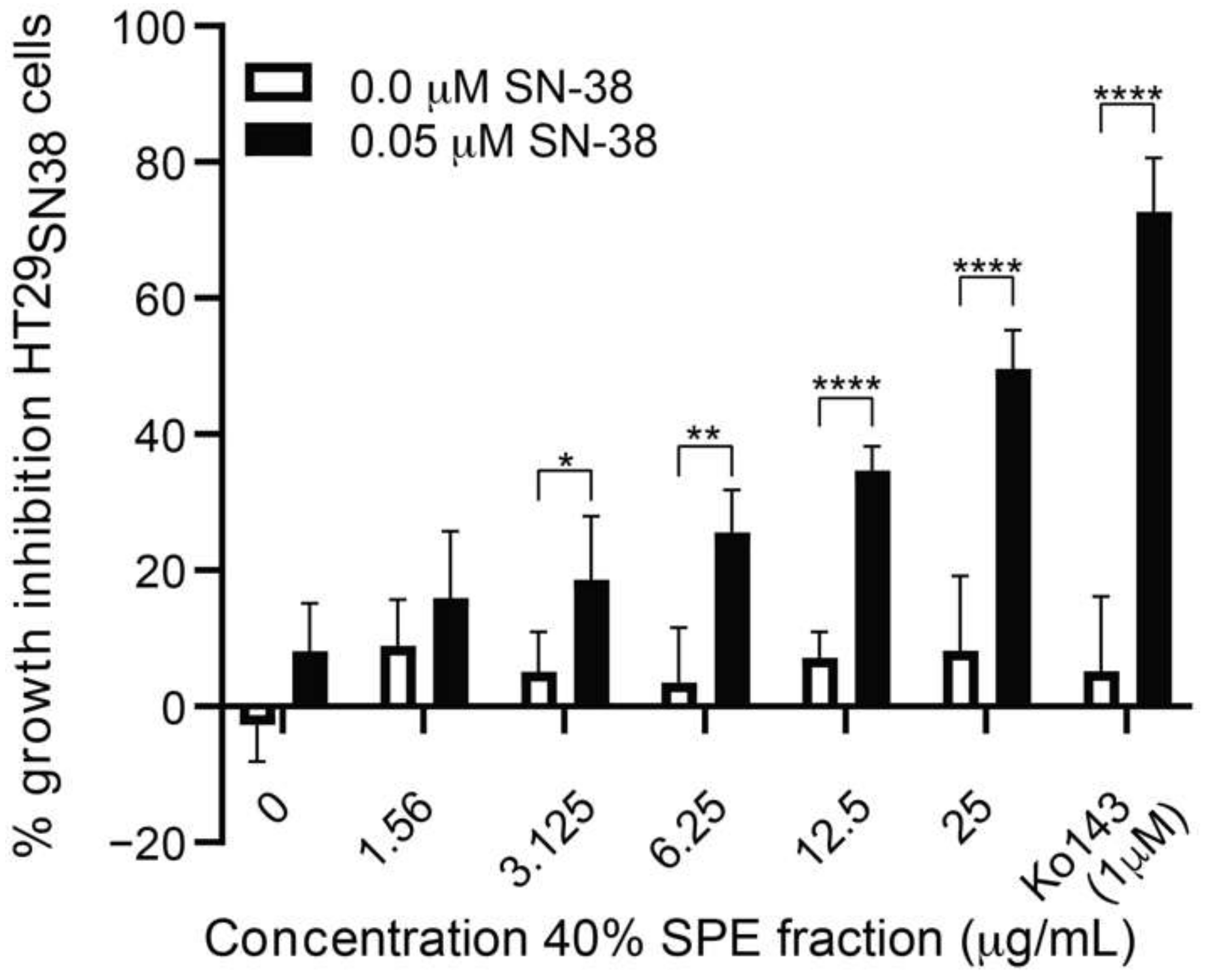
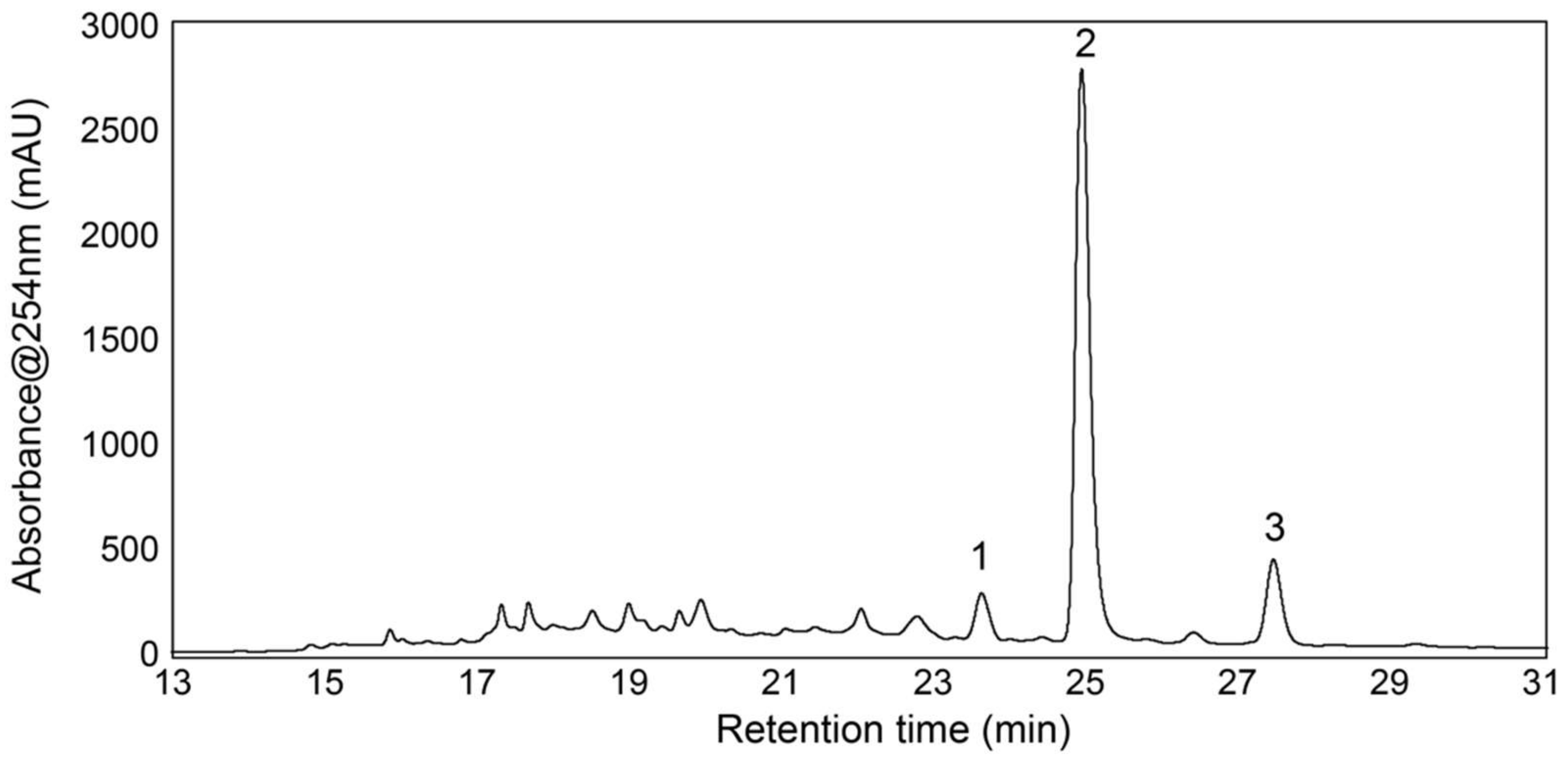
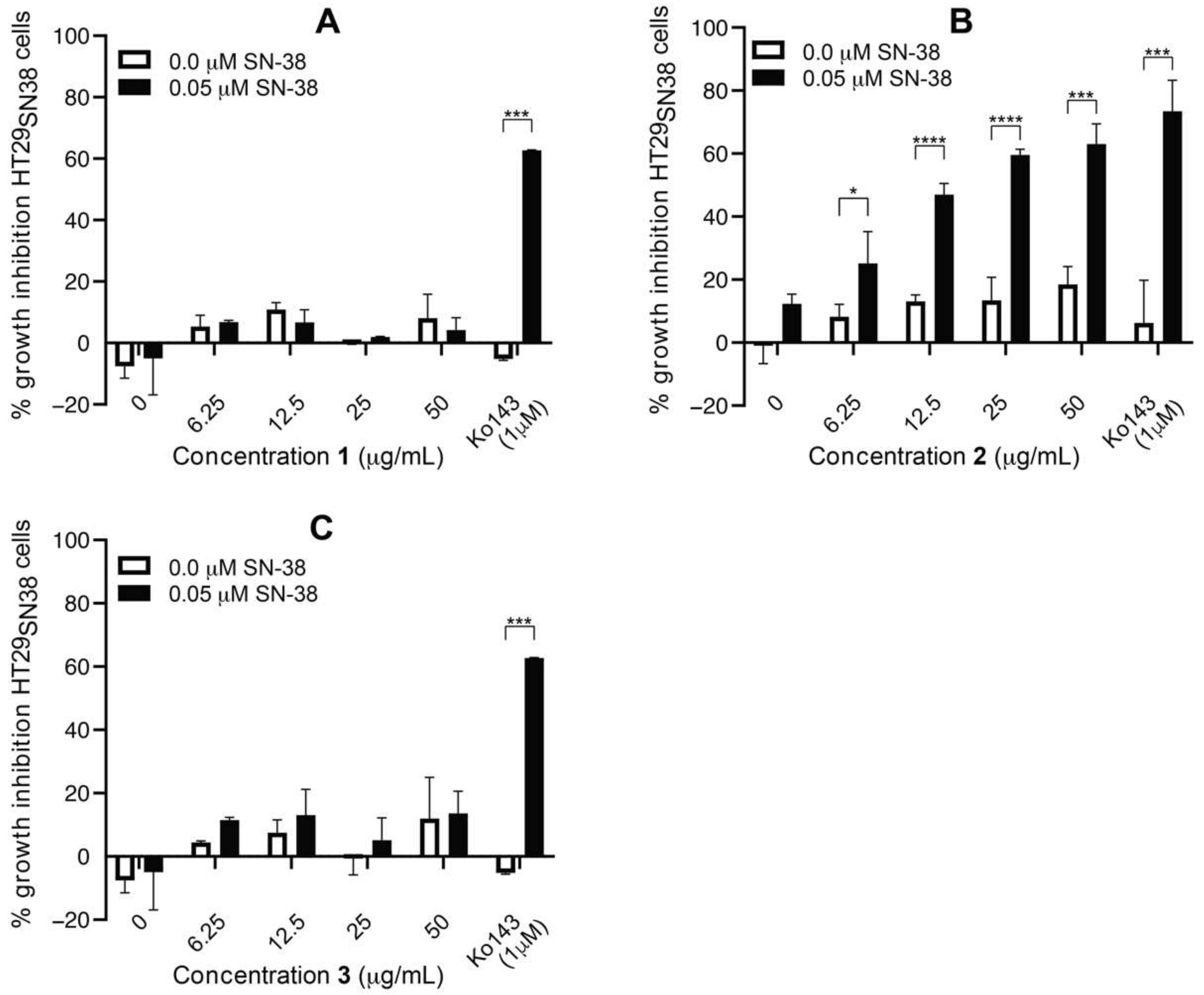
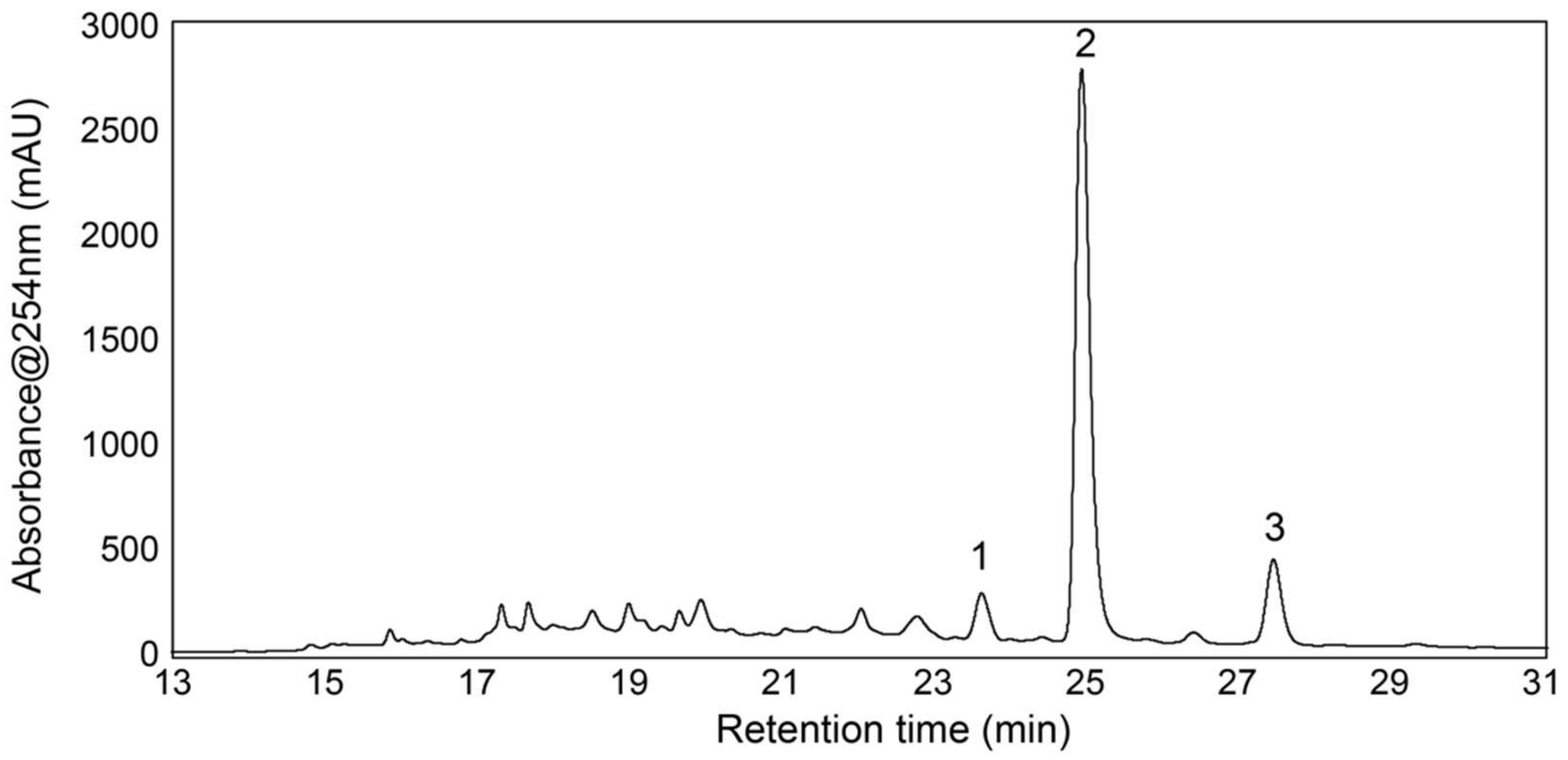
3.4. Isolation and Test of Pure Compounds from the 40% Acetonitrile Fraction
3.5. Structure Elucidation
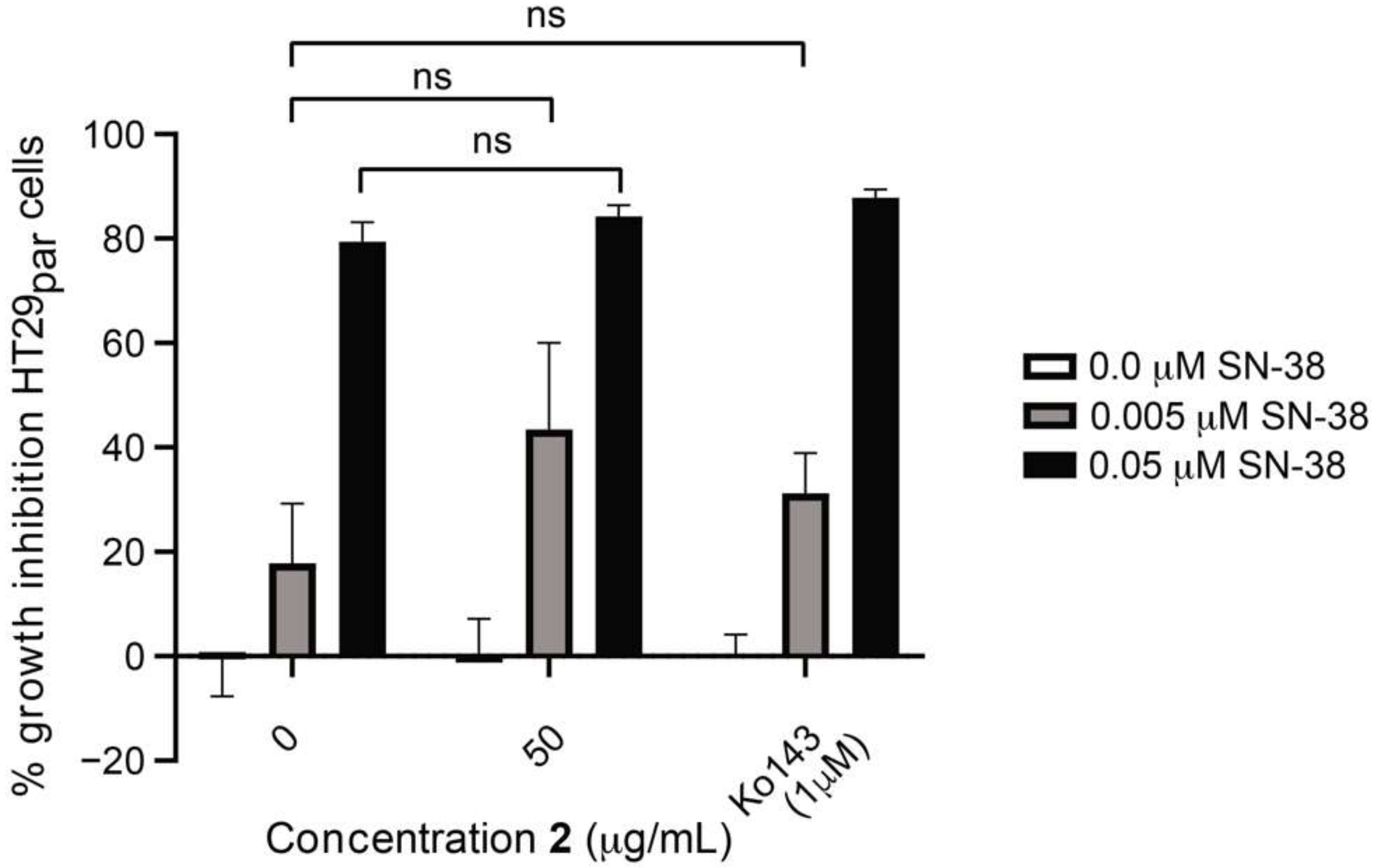
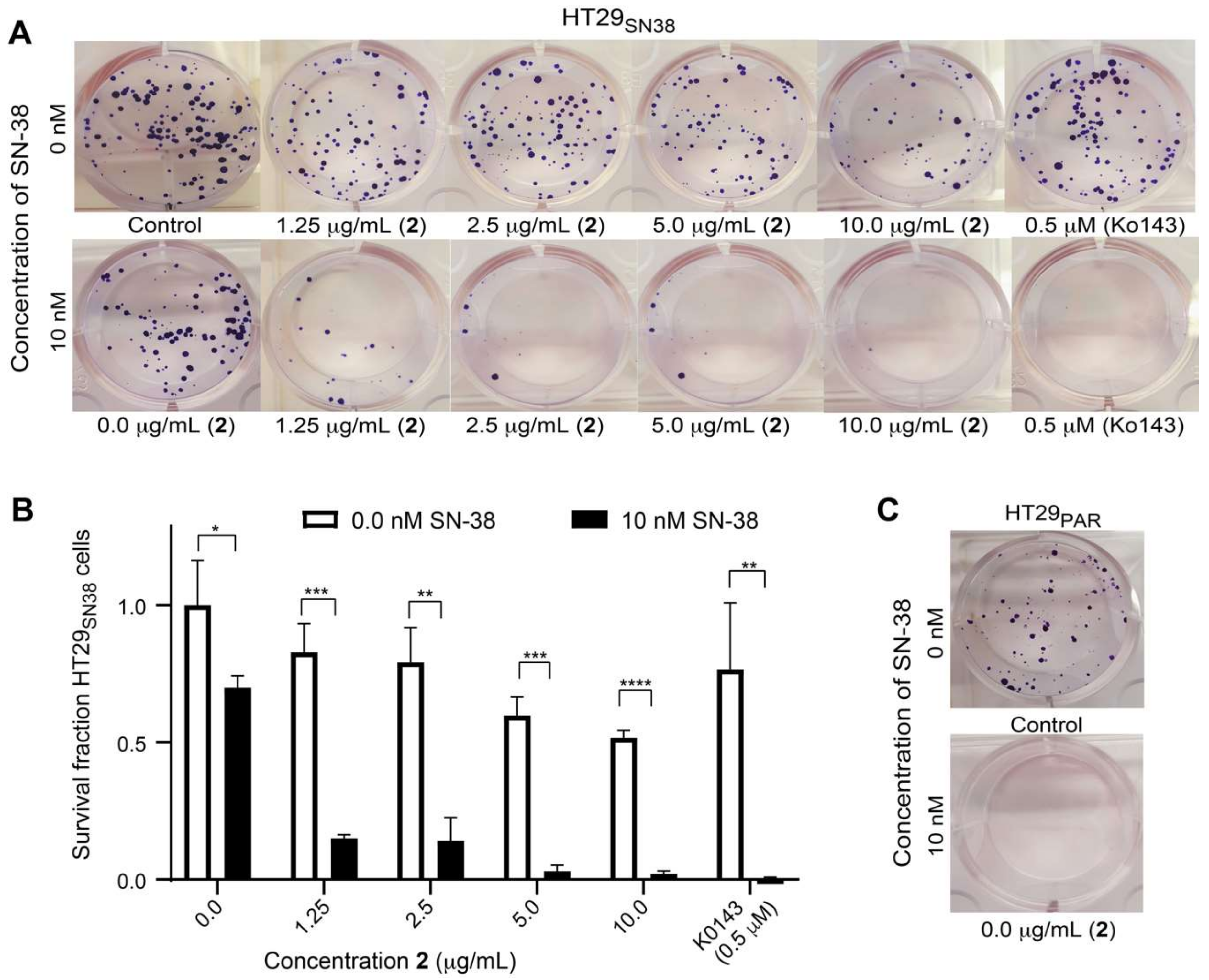
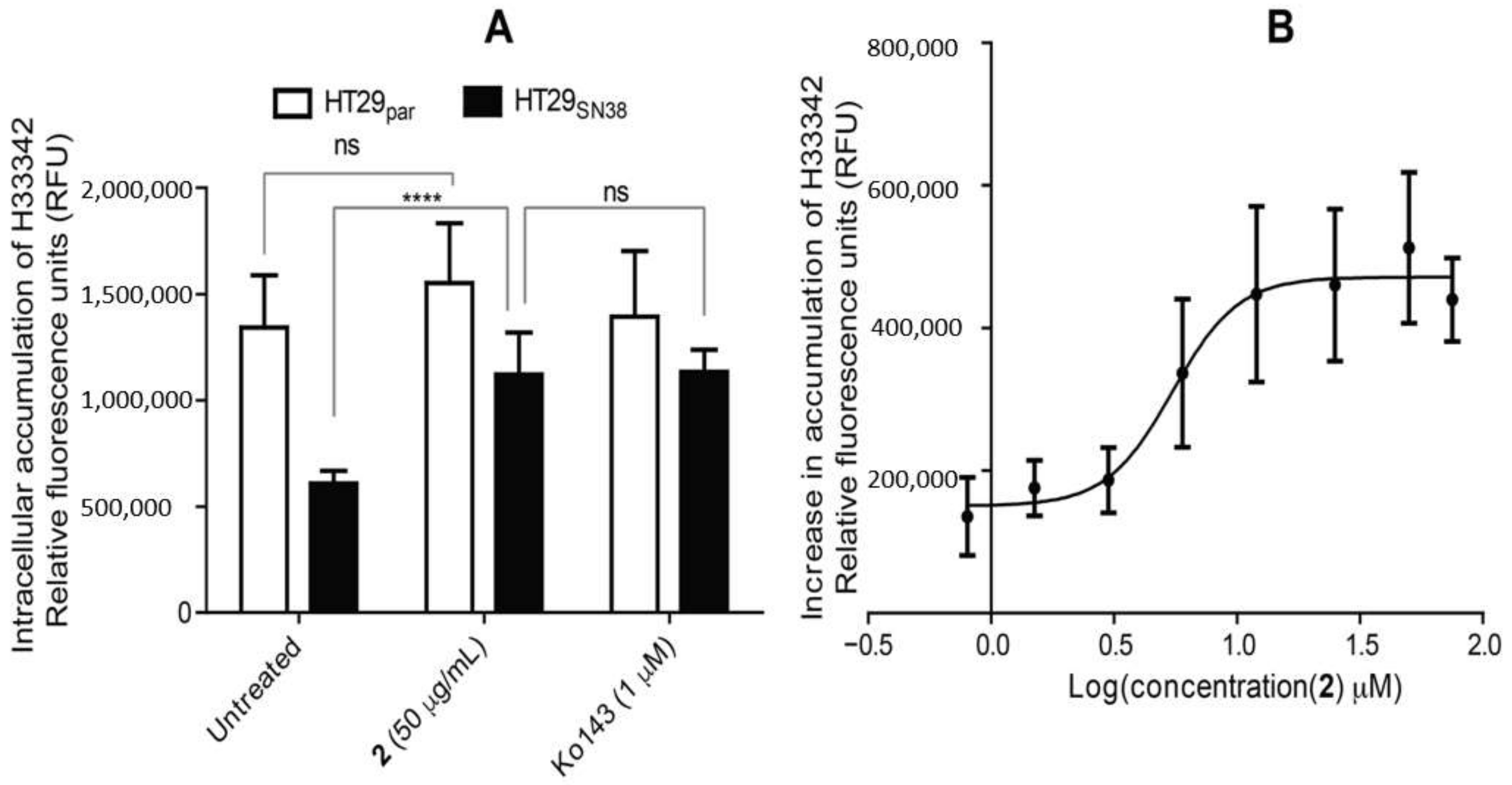
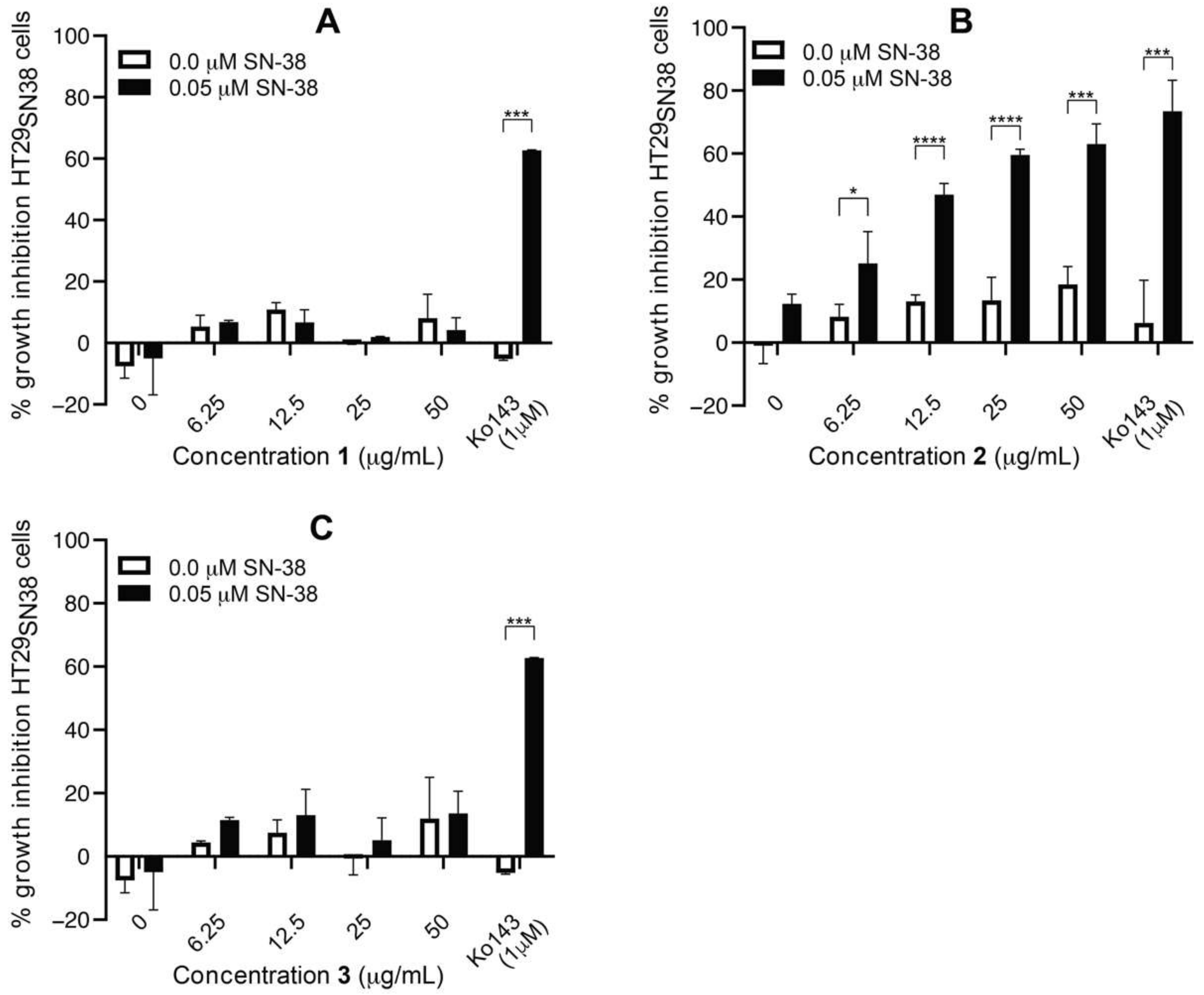
3.6. Efflux-Pump Inhibition Study on Isolated Compound
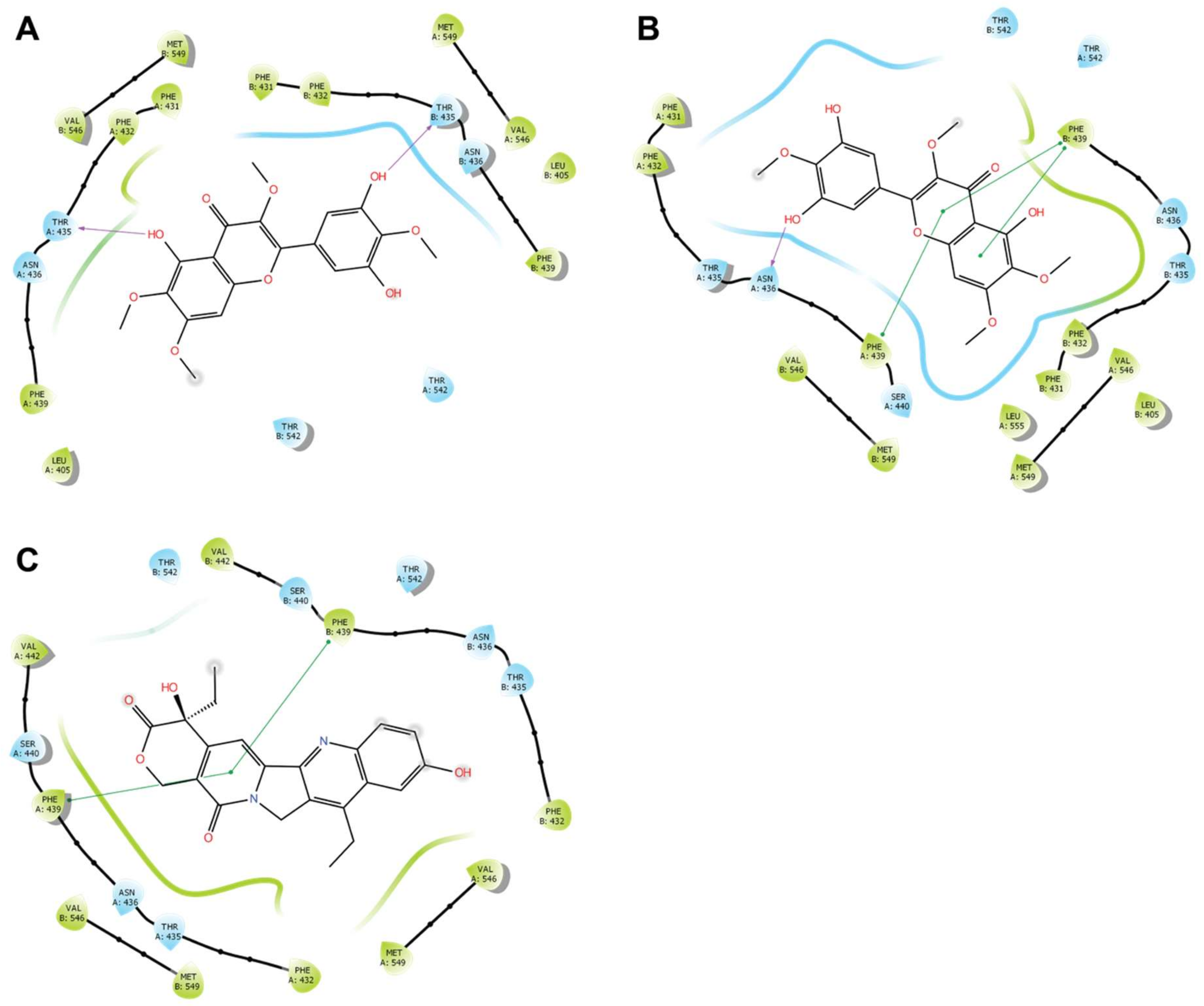
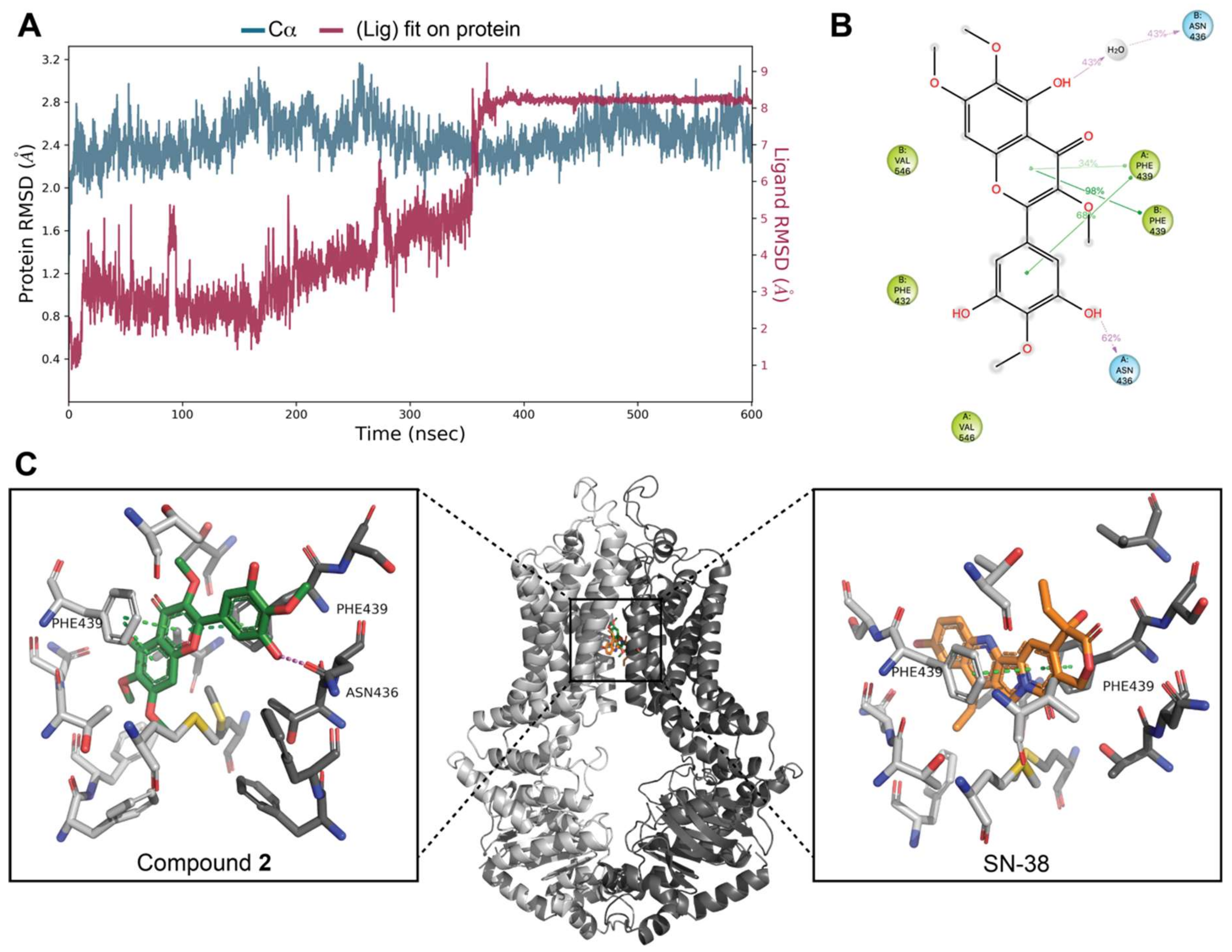
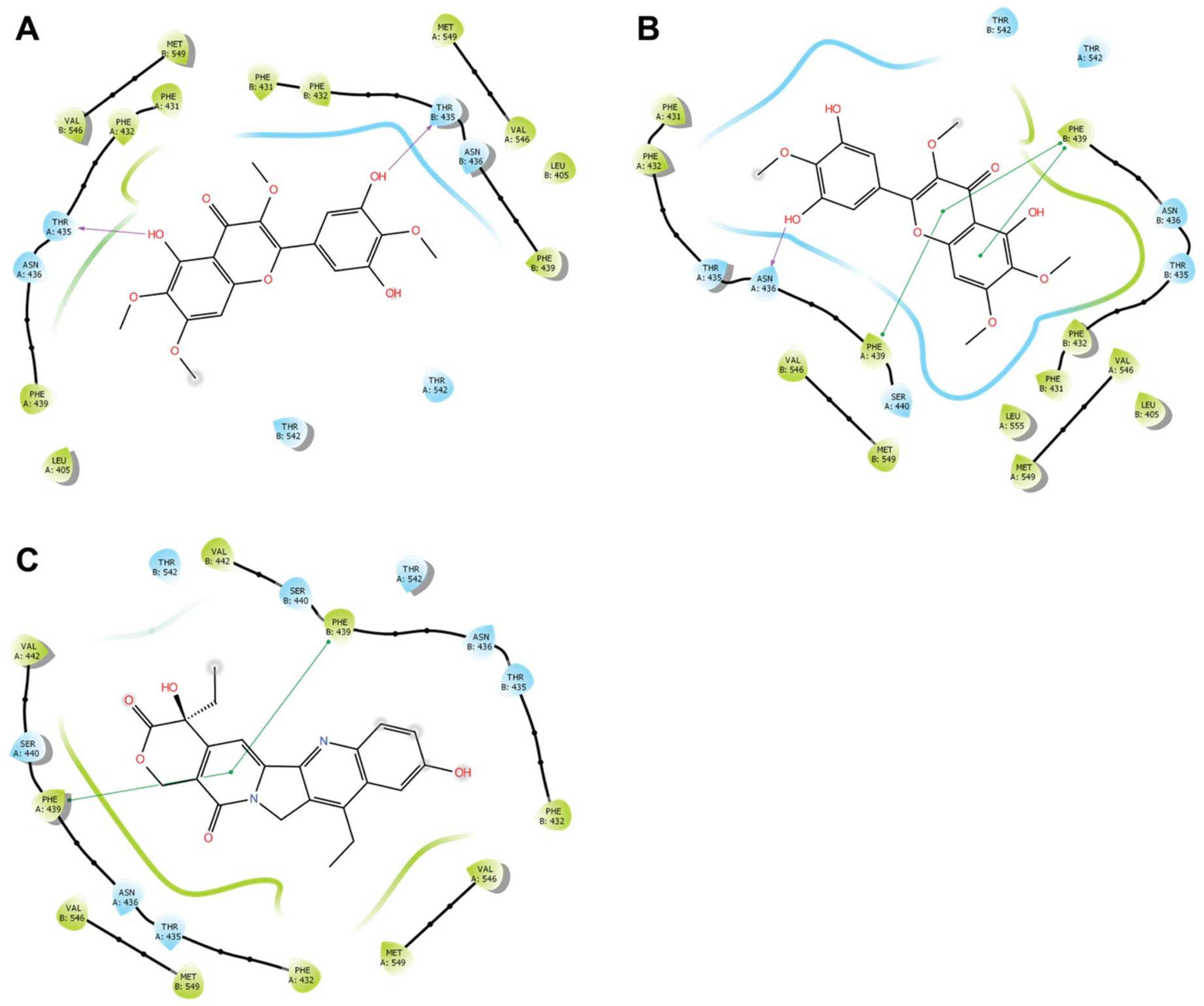
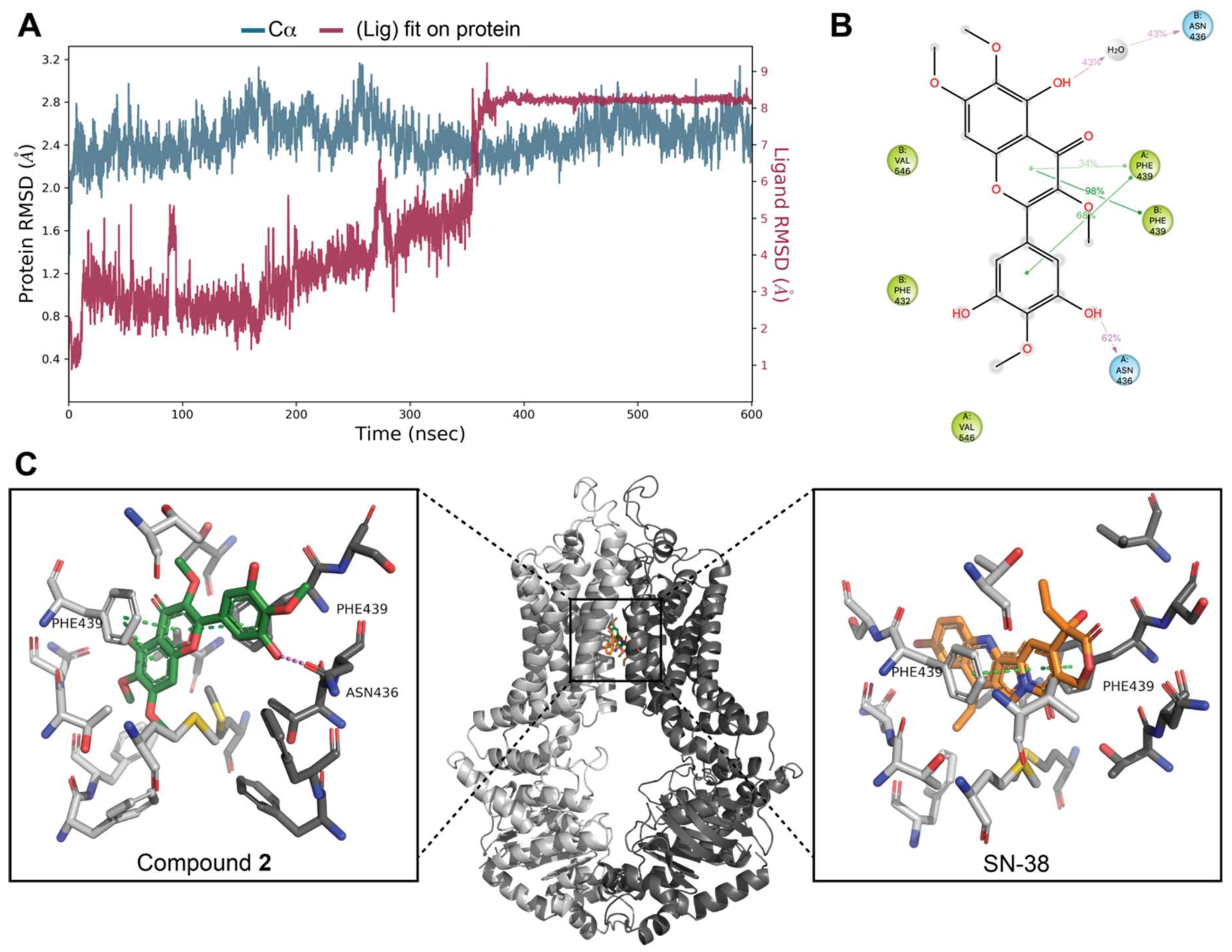
3.7. Docking Studies on BCRP
4. Discussion
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Wu, Q.; Yang, Z.; Nie, Y.; Shi, Y.; Fan, D. Multi-drug resistance in cancer chemotherapeutics: Mechanisms and lab approaches. Cancer Lett. 2014, 347, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Gillet, J.P.; Gottesman, M.M. Mechanisms of multidrug resistance in cancer. Methods Mol. Biol. 2010, 596, 47–76. [Google Scholar] [CrossRef]
- Assaraf, Y.G.; Brozovic, A.; Concalves, A.C.; Jurkovicova, D.; Line, A.; Machuqueiro, M.; Saponara, S.; Sarmento-Ribeiro, A.B.; Xavier, C.P.R.; Vasconcelos, M.H. The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist. Updat. 2019, 46, 100645. [Google Scholar] [CrossRef] [PubMed]
- DeGorter, M.K.; Xia, C.Q.; Yang, J.J.; Kim, R.B. Drug transporters in drug efficacy and toxicity. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 249–273. [Google Scholar] [CrossRef]
- Manolaridis, I.; Jackson, S.M.; Taylor, N.M.I.; Kowal, J.; Stahlberg, H.; Locher, K.P. Cryo-EM structures of a human ABCG2 mutant trapped in ATP-bound and substrate-bound states. Nature 2018, 563, 426–430. [Google Scholar] [CrossRef] [Green Version]
- Taylor, N.M.I.; Manolaridis, I.; Jackson, S.M.; Kowal, J.; Stahlberg, H.; Locher, K.P. Structure of the human multidrug transporter ABCG2. Nature 2017, 546, 504–509. [Google Scholar] [CrossRef]
- Jackson, S.M.; Manolaridis, I.; Kowal, J.; Zechner, M.; Taylor, N.M.I.; Bause, M.; Bartholomaeus, R.; Bernhardt, G.; Koenig, B.; Buschauer, A.; et al. Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nat. Struct. Mol. Biol. 2018, 25, 333–340. [Google Scholar] [CrossRef]
- Orlando, B.J.; Liao, M. ABCG2 transports anticancer drugs via a closed-to-open switch. Nat. Commun. 2020, 11, 2264. [Google Scholar] [CrossRef]
- Mao, Q.; Unadkat, J.D. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport—An update. AAPS J. 2015, 17, 65–82. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Jaitak, V. Natural products as multidrug resistance modulators in cancer. Eur. J. Med. Chem. 2019, 176, 268–291. [Google Scholar] [CrossRef]
- Karthikeyan, S.; Hoti, S.L. Development of fourth generation ABC inhibitors from natural products: A novel approach to overcome cancer multidrug resistance. Anticancer Agents Med. Chem. 2015, 15, 605–615. [Google Scholar] [CrossRef]
- Singab, A.N.; Youssef, F.S.; Ashour, M.L.; Wink, M. The genus Eremophila (Scrophulariaceae): An ethnobotanical, biological and phytochemical review. J. Pharm. Pharmacol. 2013, 65, 1239–1279. [Google Scholar] [CrossRef]
- Pedersen, H.A.; Semple, S.J.; Buirchell, B.; Møller, B.L.; Staerk, D. PTP1B-inhibiting branched-chain fatty acid dimers from Eremophila oppositifolia subsp. angustifolia identified by high-resolution PTP1B inhibition profiling and HPLC-PDA-HRMS-SPE-NMR analysis. J. Nat. Prod. 2020, 83, 1598–1610. [Google Scholar] [CrossRef] [PubMed]
- Gericke, O.; Hansen, N.L.; Pedersen, G.B.; Kjaerulff, L.; Luo, D.; Staerk, D.; Møller, B.L.; Pateraki, I.; Heskes, A.M. Nerylneryl diphosphate is the precursor of serrulatane, viscidane and cembrane-type diterpenoids in Eremophila species. BMC Plant. Biol. 2020, 20, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wubshet, S.G.; Tahtah, Y.; Heskes, A.M.; Kongstad, K.T.; Pateraki, I.; Hamberger, B.; Møller, B.M.; Staerk, D. Identification of PTP1B and α-glucosidase inhibitory serrulatanes from Eremophila spp. by combined use of dual high-resolution PTP1B and α-glucosidase inhibition profiling and HPLC-HRMS-SPE-NMR. J. Nat. Prod. 2016, 79, 1063–1072. [Google Scholar] [CrossRef]
- Tahtah, Y.; Wubshet, S.G.; Kongstad, K.T.; Heskes, A.M.; Pateraki, I.; Møller, B.M.; Jäger, A.K.; Staerk, D. High-resolution PTP1B inhibition profiling combined with high-performance liquid chromatography—High-resolution mass spectrometry—Solid-phase extraction—Nuclear magnetic resonance spectroscopy: Antidiabetic constituents in crude extract of Eremophila lucida. Fitoterapia 2016, 110, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Kjærulff, L.; Kongstad, K.T.; Heskes, A.M.; Møller, B.L.; Staerk, D. 2(5H)-Furanosesquiterpenes from Eremophila bignoniiflora: High-resolution inhibition profiling and PTP1B inhibitory activity. Phytochemistry 2019, 166, 112054. [Google Scholar] [CrossRef]
- Semple, S.J.; Reynolds, G.D.; O’Leary, M.C.; Flower, R.L.P. Screening of Australian medicinal plants for antiviral activity. J. Ethnopharmacol. 1998, 60, 163–172. [Google Scholar] [CrossRef]
- Ndi, C.P.; Semple, S.J.; Griesser, H.J.; Barton, M.D. Antimicrobial activity of some Australian plant species from the genus Eremophila. J. Basic. Microbiol. 2007, 47, 158–164. [Google Scholar] [CrossRef]
- Beattie, K.D.; Watermann, P.G.; Forster, P.I.; Thompson, D.R.; Leach, D.N. Chemical composition and cytotoxicity of oils and eremophilanes derived from various parts of Eremophila mitchellii Benth. (Myoporaceae). Phytochemistry 2011, 72, 400–408. [Google Scholar] [CrossRef] [Green Version]
- Mon, H.H.; Christo, S.N.; Ndi, C.P.; Jasieniak, M.; Rickard, H.; Hayball, J.D.; Griesser, H.J.; Semple, S.J. Serrulatane diterpenoid from Eremophila neglecta exhibits bacterial biofilm dispersion and inhibits release of pro-inflammatory cytokines from activated macrophages. J. Nat. Prod. 2015, 78, 3031–3040. [Google Scholar] [CrossRef] [PubMed]
- Jensen, N.F.; Stenvang, J.; Beck, M.K.; Hanáková, B.; Belling, K.C.; Do, K.N.; Viuff, B.; Nygård, S.B.; Gupta, R.; Rasmussen, M.H.; et al. Establishment and characterization of models of chemotherapy resistance in colorectal cancer: Towards a predictive signature of chemoresistance. Mol. Oncol. 2015, 9, 1169–1185. [Google Scholar] [CrossRef] [PubMed]
- Franken, N.A.P.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shaw, D.E.; Shelley, M.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aid. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Ambjørner, S.E.B.; Wiese, M.; Köhler, S.C.; Svindt, J.; Lund, X.L.; Gajhede, M.; Saaby, L.; Brodin, B.; Rump, S.; Weigt, H.; et al. The pyrazolo[3,4-d]pyrimidine derivative, SCO-201, reverses multidrug resistance mediated by ABCG2/BCRP. Cells 2020, 9, 613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar] [CrossRef] [Green Version]
- Reutrakul, R.; Krachangchaeng, C.; Pohmakotr, M.; Jaiptech, T.; Yoosook, C.; Kasisit, J.; Sophsan, S.; Sujarit, K.; Santisuk, T. Cytotoxic and anti-HIV-1 constituents from leaves and twigs of Gard. Tubifera. Tetrahedon 2004, 60, 1517–1523. [Google Scholar] [CrossRef]
- Chang, R.; Wang, C.; Zeng, Q.; Guan, B.; Zhang, W.; Jin, H. Chemical constituents of the stems of Celastrus rugosus. Arch. Pharm. Res. 2013, 36, 1291–1301. [Google Scholar] [CrossRef]
- Geenen, S.R.; Presser, L.; Hölzel, T.; Ganter, C.; Müller, T.J.J. Electronic finetuning of 8-methoxy psoralens by palladium-catalyzed coupling: Acidochromicity and solvatochromicity. Chem. Eur. J. 2020, 26, 8064–8075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.; Bai, J.; Zhao, S.; Hu, M.; Sun, Y.; Wang, B.; Ji, M.; Jin, J.; Wang, X.; Hu, J.; et al. Evaluation of inhibitory effects of flavonoids on breast cancer resistance protein (BCRP): From library screening to biological evaluation to structure-activity relationship. Toxicol. Vitr. 2019, 61, 104642. [Google Scholar] [CrossRef] [PubMed]
- Pick, A.; Müller, H.; Mayer, R.; Haenisch, B.; Pajeva, I.K.; Weight, M.; Bönisch, H.; Müller, C.E.; Wiese, M. Structure-activity relationships of flavonoids as inhibitors of breast cancer resistance protein (BCRP). Bioorg. Med. Chem. 2011, 19, 2090–2102. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | 1H NMR δ (nH, m) | 13C NMR δ |
|---|---|---|
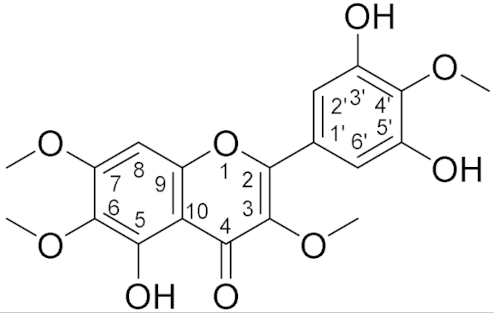 | 12.47 (1H, s, 5-OH), 7.31 (2H, s, H2′/H6′), 6.50 (1H, s, H8), 4.0 (3H, s, 4′-OMe), 3.96 (3H, s, 7-OMe), 3.92 (3H, s, 6-OMe), 3.88 (3H, s, 3-OMe) | 154 (C-2), 139 (C-3), 178 (C-4), 152 (C5), 132 (C-6), 158 (C-7), 90.5 (C-8), 152.5 (C-9), 106.6 (C-10), 126.4 (C-1′), 108.9 (C-2′), 148 (C-3′), 136 (C-4′), 148 (C-5′), 108(C-6′), 61.4 (4′-OMe), 56.4 (7-OMe), 61.0 (6-OMe), 60.5 (3-OMe) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petersen, M.J.; Lund, X.L.; Semple, S.J.; Buirchell, B.; Franzyk, H.; Gajhede, M.; Kongstad, K.T.; Stenvang, J.; Staerk, D. Reversal of ABCG2/BCRP-Mediated Multidrug Resistance by 5,3′,5′-Trihydroxy-3,6,7,4′-Tetramethoxyflavone Isolated from the Australian Desert Plant Eremophila galeata Chinnock. Biomolecules 2021, 11, 1534. https://doi.org/10.3390/biom11101534
Petersen MJ, Lund XL, Semple SJ, Buirchell B, Franzyk H, Gajhede M, Kongstad KT, Stenvang J, Staerk D. Reversal of ABCG2/BCRP-Mediated Multidrug Resistance by 5,3′,5′-Trihydroxy-3,6,7,4′-Tetramethoxyflavone Isolated from the Australian Desert Plant Eremophila galeata Chinnock. Biomolecules. 2021; 11(10):1534. https://doi.org/10.3390/biom11101534
Chicago/Turabian StylePetersen, Malene J., Xamuel L. Lund, Susan J. Semple, Bevan Buirchell, Henrik Franzyk, Michael Gajhede, Kenneth T. Kongstad, Jan Stenvang, and Dan Staerk. 2021. "Reversal of ABCG2/BCRP-Mediated Multidrug Resistance by 5,3′,5′-Trihydroxy-3,6,7,4′-Tetramethoxyflavone Isolated from the Australian Desert Plant Eremophila galeata Chinnock" Biomolecules 11, no. 10: 1534. https://doi.org/10.3390/biom11101534
APA StylePetersen, M. J., Lund, X. L., Semple, S. J., Buirchell, B., Franzyk, H., Gajhede, M., Kongstad, K. T., Stenvang, J., & Staerk, D. (2021). Reversal of ABCG2/BCRP-Mediated Multidrug Resistance by 5,3′,5′-Trihydroxy-3,6,7,4′-Tetramethoxyflavone Isolated from the Australian Desert Plant Eremophila galeata Chinnock. Biomolecules, 11(10), 1534. https://doi.org/10.3390/biom11101534