
The Role of the C-Terminal Lysine of S100P in S100P-Induced Cell Migration and Metastasis


 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Site-Directed Mutagenesis and Recombinant Proteins
2.2. Transfection of Mammary Cell Lines
2.3. Cell Migration Assays
2.4. Metastasis Assays In Vivo
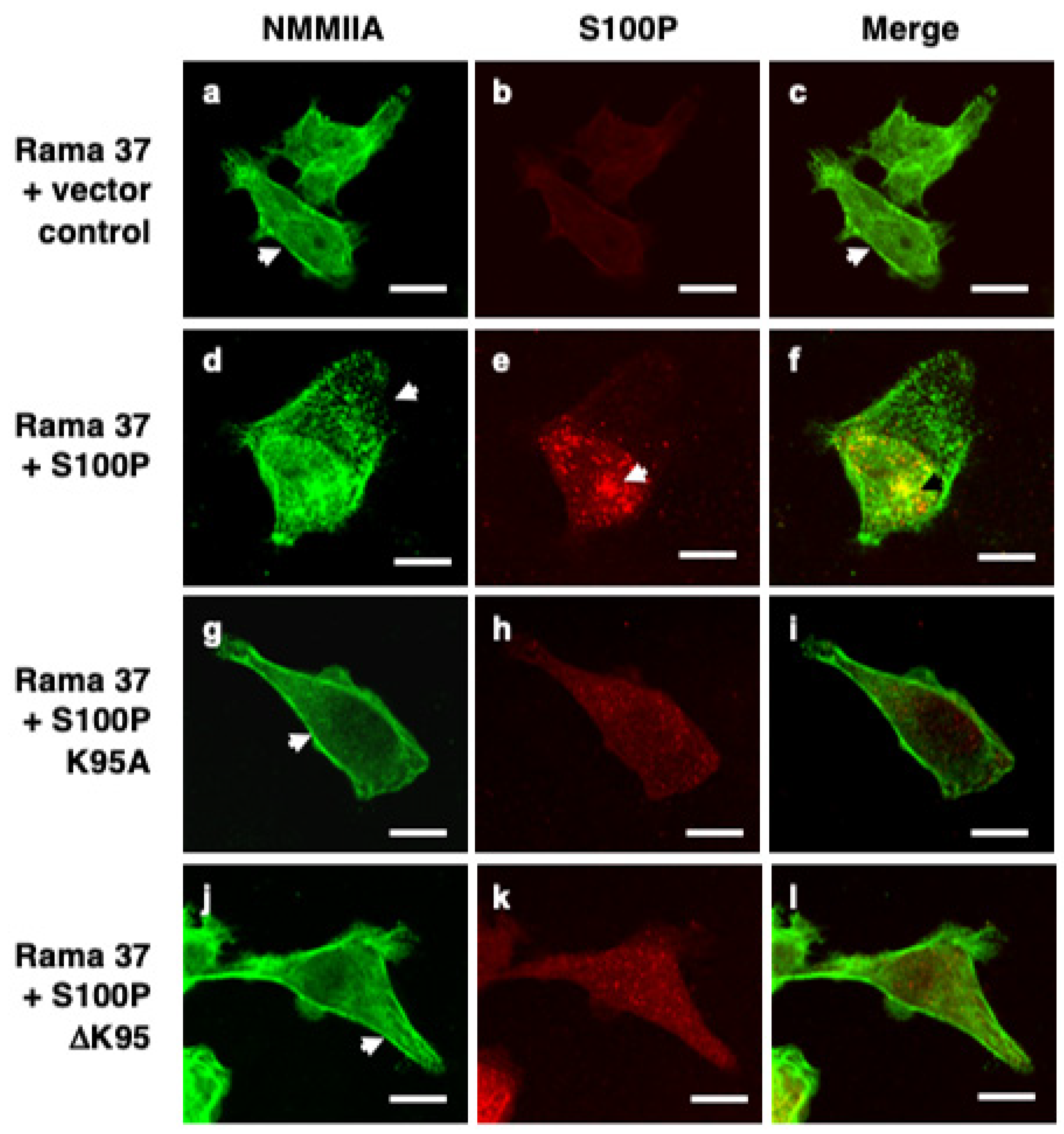
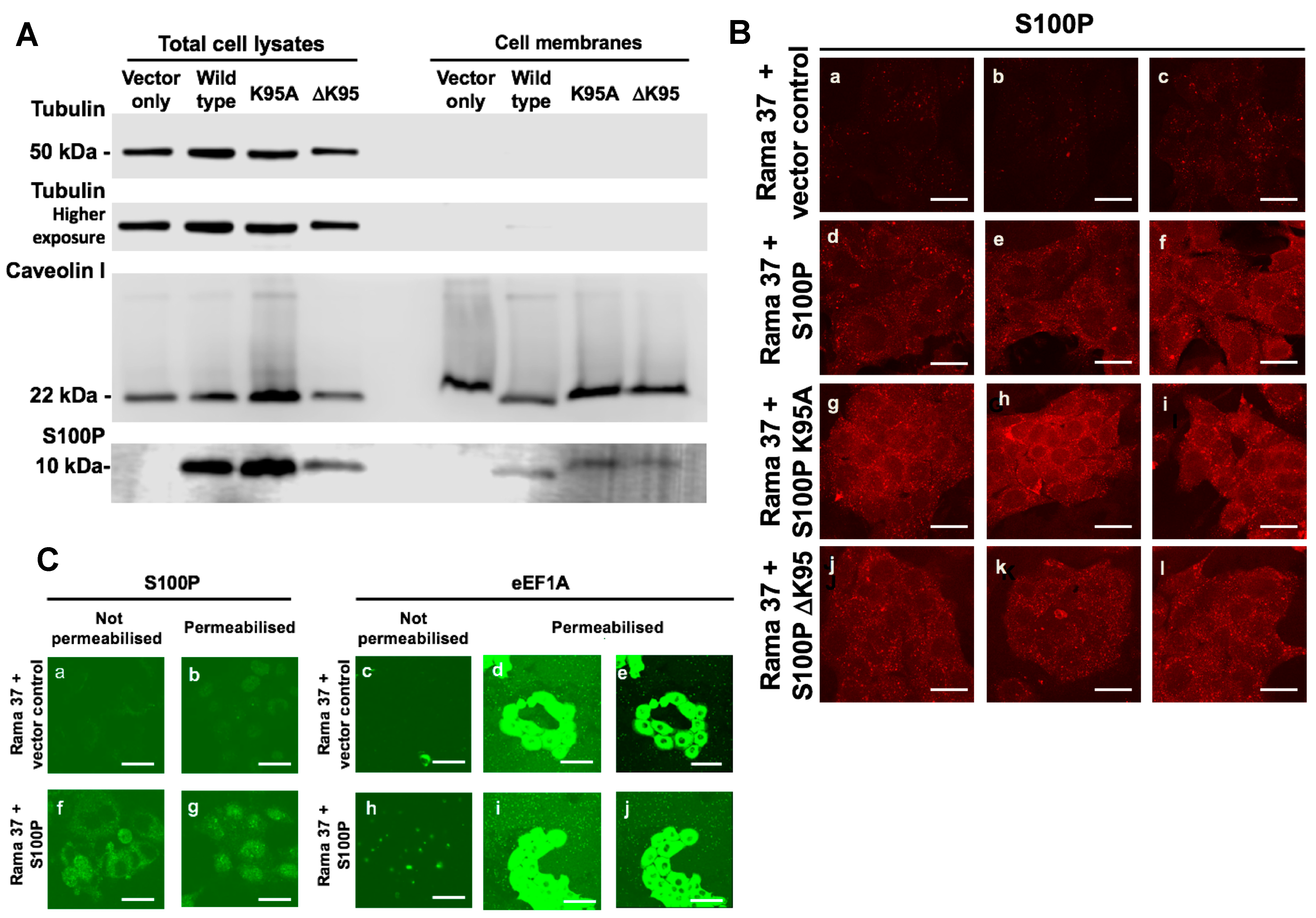
2.5. Immunofluorescence Staining of Cultured Cells
2.6. Isolation of Membrane Fractions
2.7. Interaction between S100P and Recombinant NMMIIA
2.8. Western Blotting
2.9. Statistical Analyses
3. Results
3.1. The Effect of C-Terminal Mutants of S100P on Tumorigenesis and Metastasis
3.2. S100P Mutants and Altered Cytoskeletal Organisation
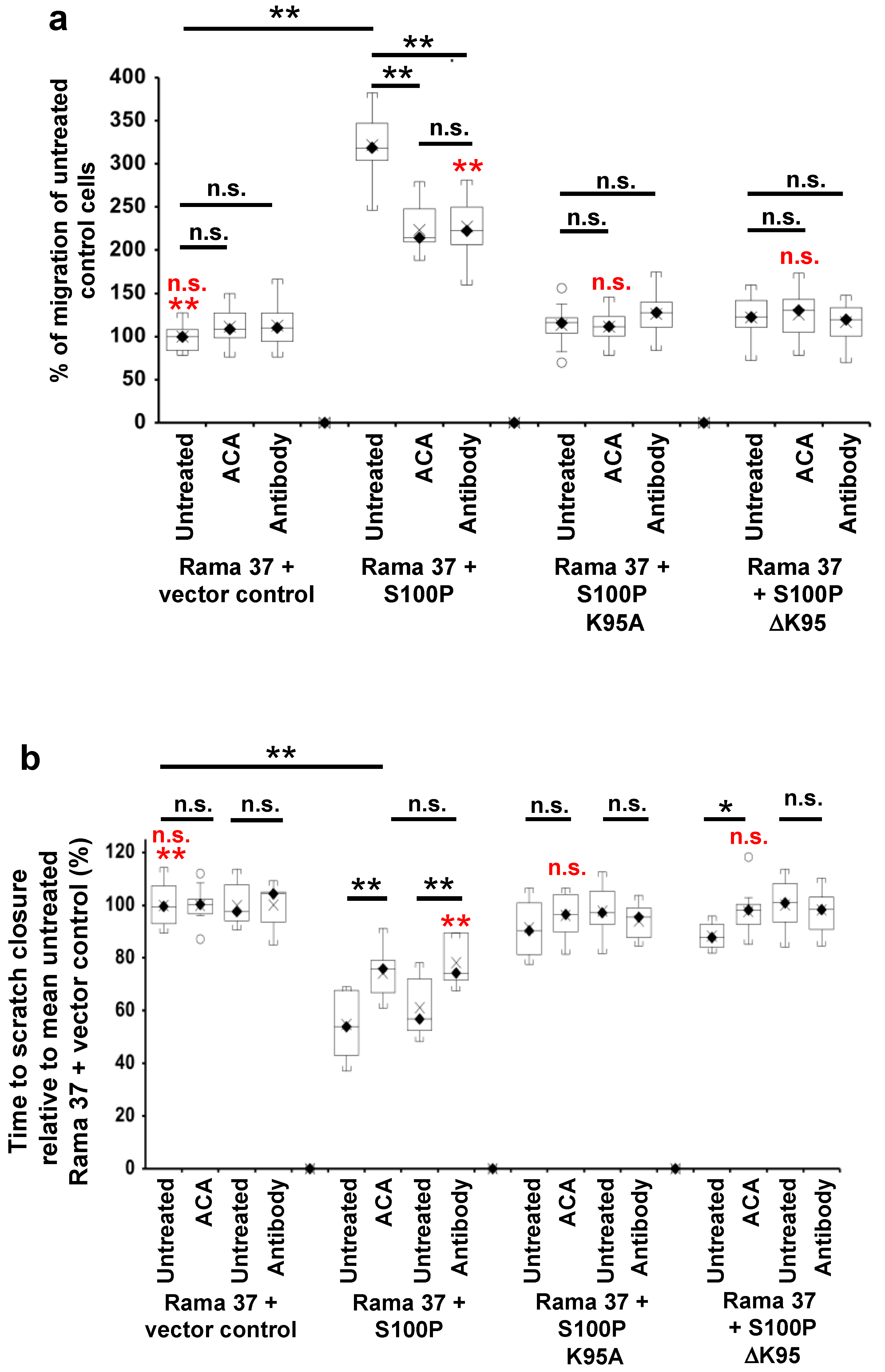
3.3. The Effect of S100P Mutants on Transwell and Scratch-Wound Cell Migration
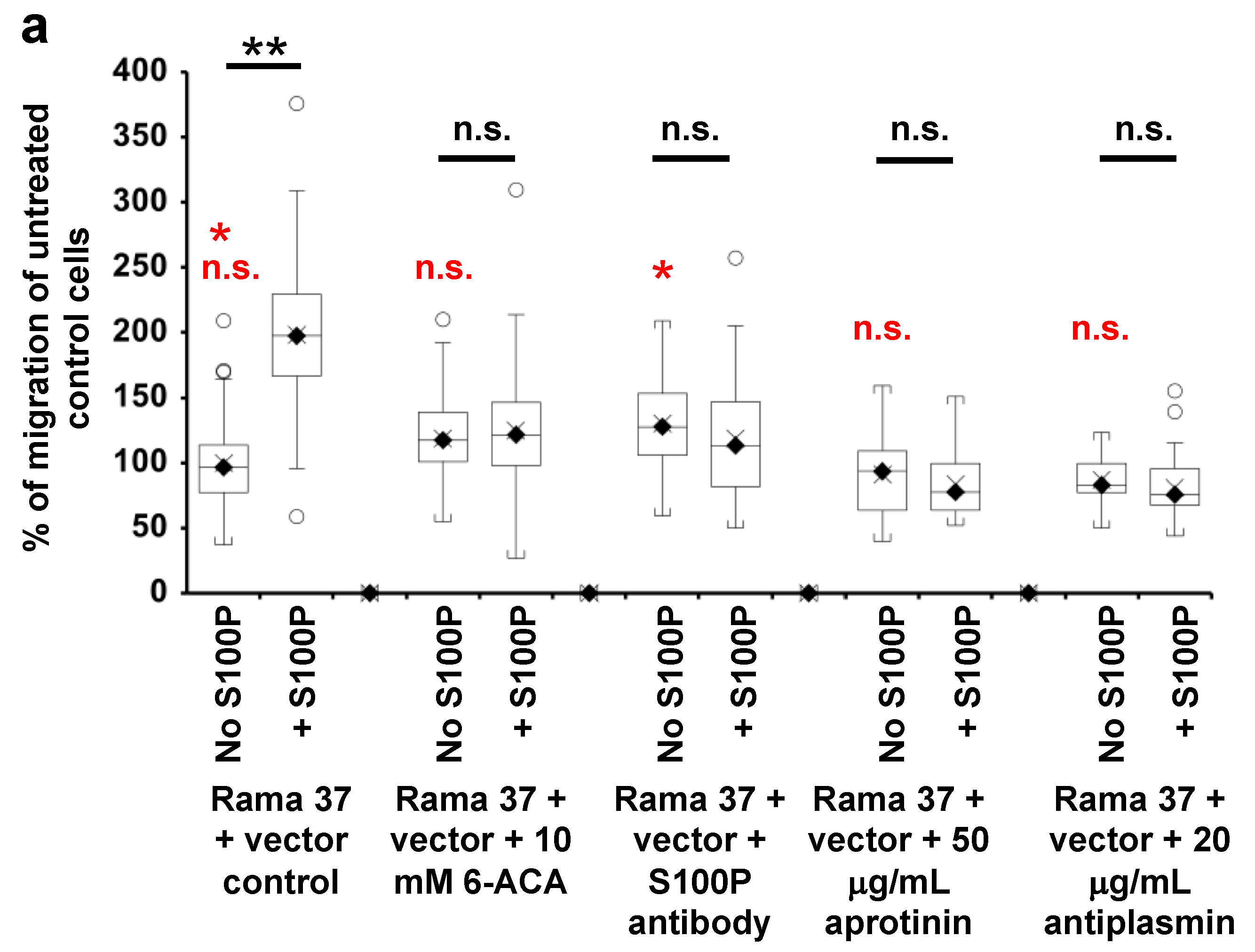
3.4. Effect of 6-Aminocaproic Acid or S100P Antibody on the Migration of Rama 37 Cells Expressing Wild-Type or Mutant S100P Proteins
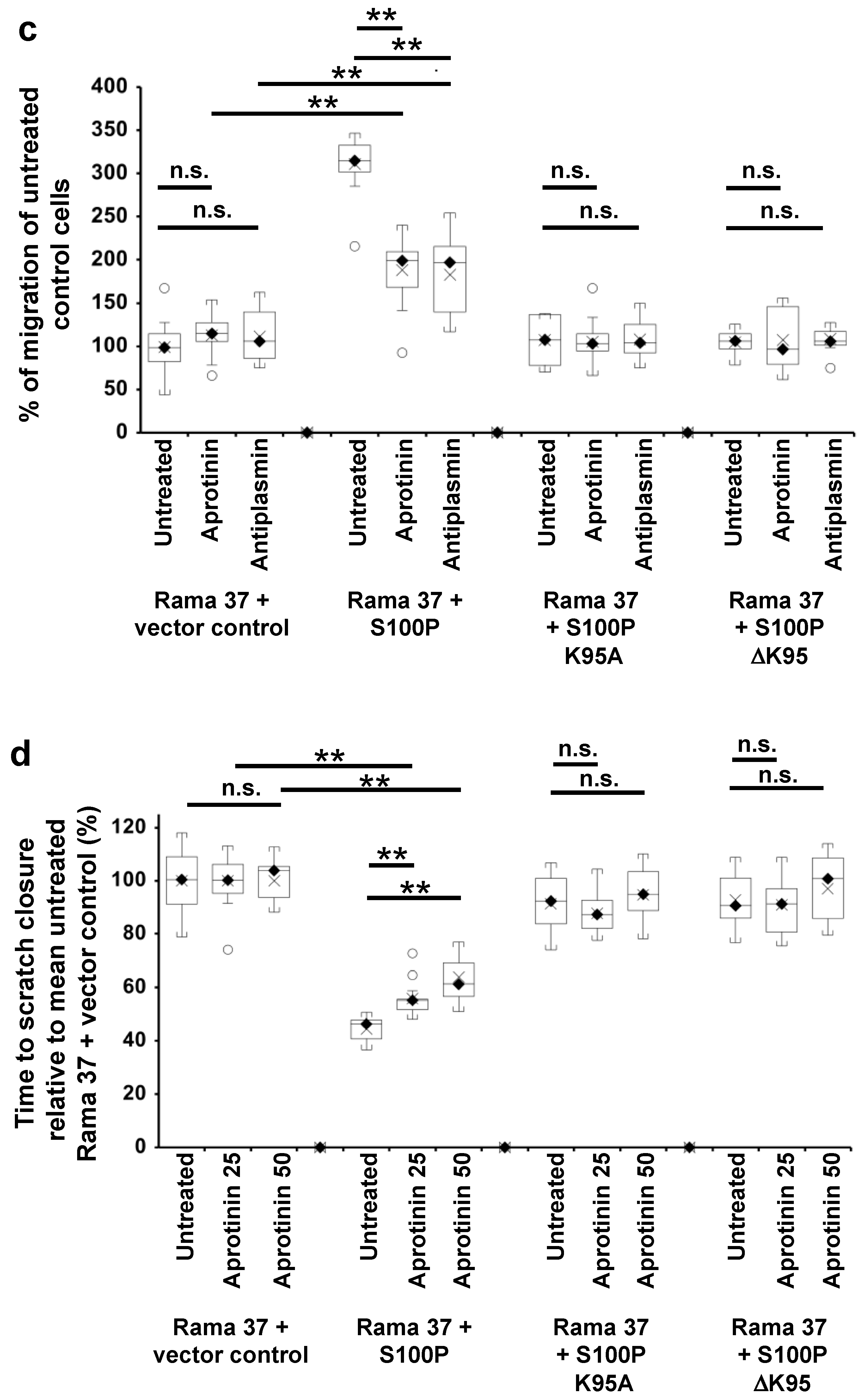
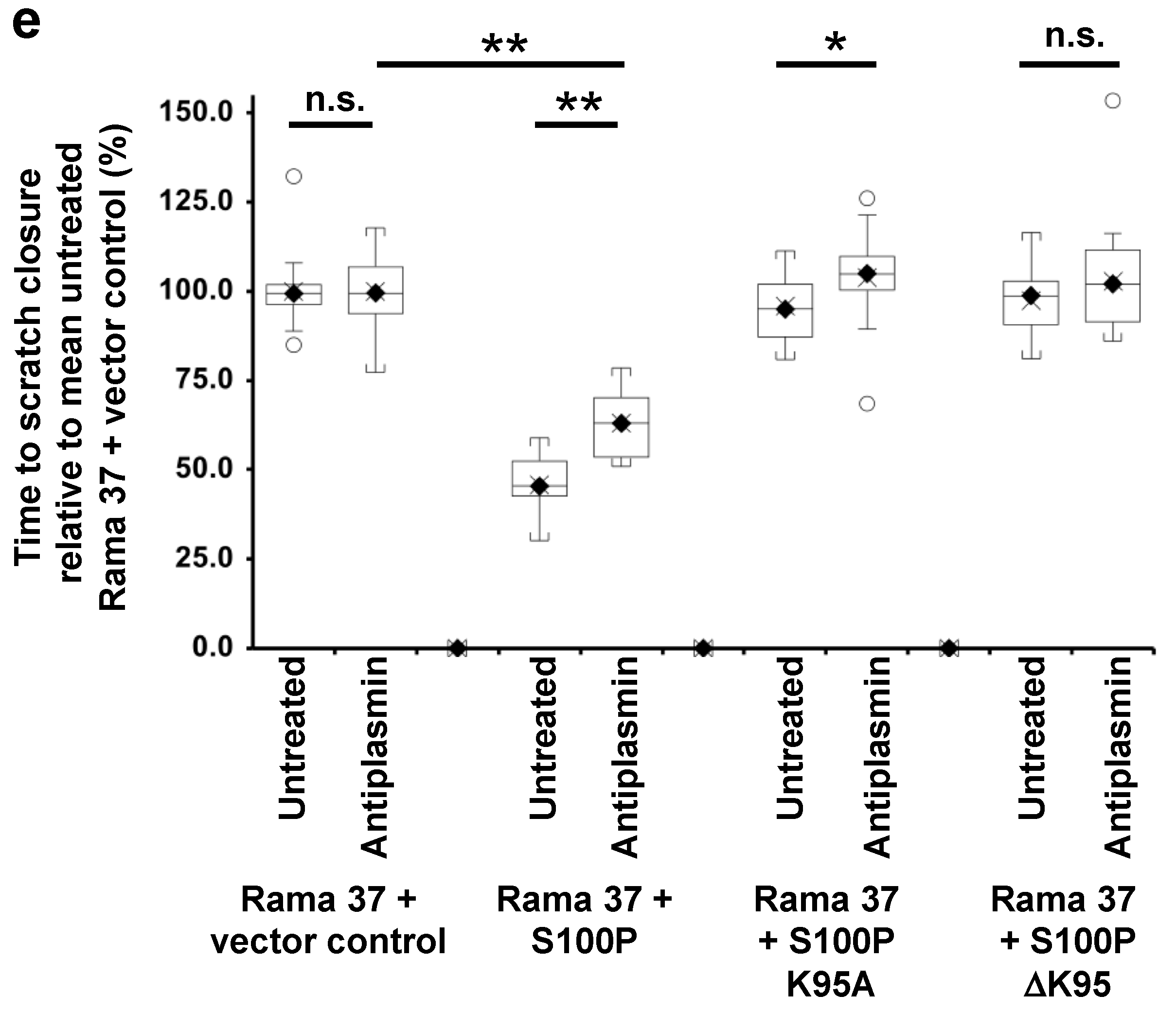
3.5. Effect of Plasmin Inhibitors on the Migration of Rama 37 Cells Expressing Wild-Type and Mutant S100P Proteins
3.6. Membrane-Associated S100P
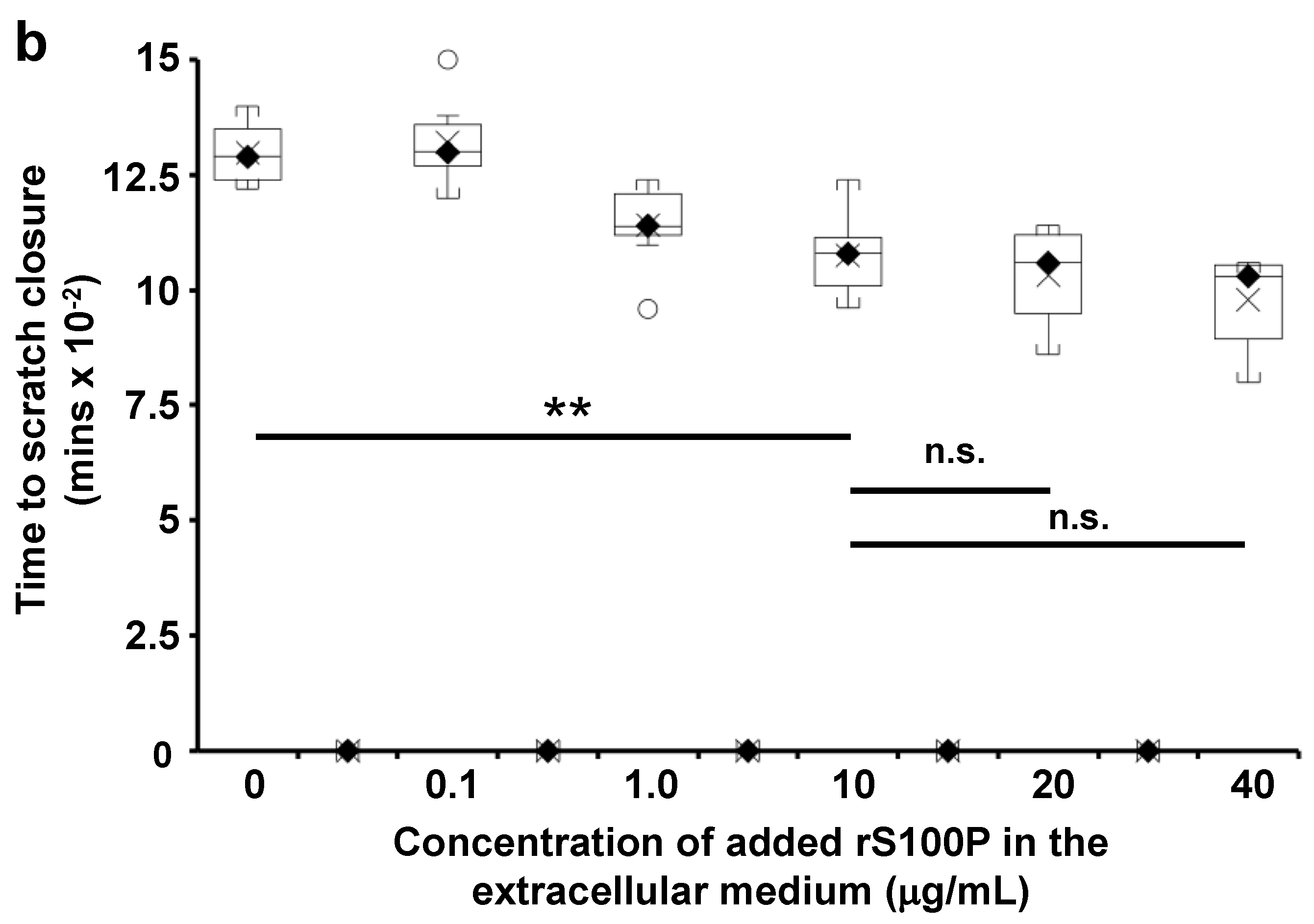
3.7. The Effect of Extracellularly Added S100P on the Migration of S100P-Negative Control Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Salama, I.; Malone, P.S.; Mihaimeed, F.; Jones, J.L. A review of the S100 proteins in cancer. Eur. J. Surg. Oncol. 2007, 34, 357–364. [Google Scholar] [CrossRef]
- Gross, S.R.; Sin, C.G.; Barraclough, R.; Rudland, P.S. Joining S100 proteins and migration: For better or for worse, in sickness and in health. Cell. Mol. Life Sci. CMLS 2014, 71, 1551–1579. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Platt-Higgins, A.; Carroll, J.; de Silva Rudland, S.; Winstanley, J.; Barraclough, R.; Rudland, P.S. Induction of metastasis by S100P in a rat mammary model and its association with poor survival of breast cancer patients. Cancer Res. 2006, 66, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Maciejczyk, A.; Lacko, A.; Ekiert, M.; Jagoda, E.; Wysocka, T.; Matkowski, R.; Halon, A.; Gyorffy, B.; Lage, H.; Surowiak, P. Elevated nuclear S100P expression is associated with poor survival in early breast cancer patients. Histol. Histopathol. 2013, 28, 513–524. [Google Scholar] [PubMed]
- Yuan, R.H.; Chang, K.T.; Chen, Y.L.; Hsu, H.C.; Lee, P.H.; Lai, P.L.; Jeng, Y.M. S100P expression is a novel prognostic factor in hepatocellular carcinoma and predicts survival in patients with high tumor stage or early recurrent tumors. PLoS ONE 2013, 8, e65501. [Google Scholar] [CrossRef]
- Diederichs, S.; Bulk, E.; Steffen, B.; Ji, P.; Tickenbrock, L.; Lang, K.; Zanker, K.S.; Metzger, R.; Schneider, P.M.; Gerke, V.; et al. S100 family members and trypsinogens are predictors of distant metastasis and survival in early-stage non-small cell lung cancer. Cancer Res. 2004, 64, 5564–5569. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.Y.; Fang, Y.; Zhen, L.; Zhu, X.J.; Chen, H.; Liu, H.; Jiang, B.; Li, G.X.; Deng, H.J. Analysis of the predictive efficiency of S100P on adverse prognosis and the pathogenesis of S100P-mediated invasion and metastasis of colon adenocarcinoma. Cancer Genet. 2016, 209, 143–153. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.N.; Lin, G.L.; Qiu, H.Z.; Wu, B.; Wu, H.Y.; Zhao, Y.; Chen, Y.J.; Lu, C.M. S100P, a potential novel prognostic marker in colorectal cancer. Oncol. Rep. 2012, 28, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Surowiak, P.; Maciejczyk, A.; Materna, V.; Drag-Zalesinska, M.; Wojnar, A.; Pudelko, M.; Kedzia, W.; Spaczynski, M.; Dietel, M.; Zabel, M.; et al. Unfavourable prognostic significance of S100P expression in ovarian cancers. Histopathology 2007, 51, 125–128. [Google Scholar] [CrossRef]
- Wang, X.; Tian, T.; Li, X.; Zhao, M.; Lou, Y.; Qian, J.; Liu, Z.; Chen, H.; Cui, Z. High expression of S100P is associated with unfavorable prognosis and tumor progression in patients with epithelial ovarian cancer. Am. J. Cancer Res. 2015, 5, 2409–2421. [Google Scholar]
- Zhang, H.; Wang, G.; Ding, Y.; Wang, Z.; Barraclough, R.; Rudland, P.S.; Fernig, D.G.; Rao, Z. The crystal structure at 2Å resolution of the Ca2+ -binding protein S100P. J. Mol. Biol. 2003, 325, 785–794. [Google Scholar] [CrossRef]
- Lee, Y.C.; Volk, D.E.; Thiviyanathan, V.; Kleerekoper, Q.; Gribenko, A.V.; Zhang, S.; Gorenstein, D.G.; Makhatadze, G.I.; Luxon, B.A. NMR structure of the Apo-S100P protein. J. Biomol. NMR 2004, 29, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.R.; Irvine, A.F.; Jung, H.S.; Tozawa, K.; Pastok, M.W.; Picone, R.; Badyal, S.K.; Basran, J.; Rudland, P.S.; Barraclough, R.; et al. Asymmetric mode of Ca(2)(+)-S100A4 interaction with nonmuscle myosin IIA generates nanomolar affinity required for filament remodeling. Structure 2012, 20, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Donato, R. S100: A multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int. J. Biochem. Cell. Biol. 2001, 33, 637–668. [Google Scholar] [CrossRef]
- Penumutchu, S.R.; Chou, R.H.; Yu, C. Structural insights into calcium-bound S100P and the V domain of the RAGE complex. PLoS ONE 2014, 9, e103947. [Google Scholar] [CrossRef]
- Clarke, C.; Gross, S.R.; Ismail, T.M.; Rudland, P.S.; Al-Medhtiy, M.; Santangeli, M.; Barraclough, R. Activation of tissue plasminogen activator by metastasis-inducing S100P protein. Biochem. J. 2017, 474, 3227–3240. [Google Scholar] [CrossRef][Green Version]
- Koltzscher, M.; Neumann, C.; Konig, S.; Gerke, V. Ca2+-dependent binding and activation of dormant ezrin by dimeric S100P. Mol. Biol. Cell 2003, 14, 2372–2384. [Google Scholar] [CrossRef] [PubMed]
- Heil, A.; Nazmi, A.R.; Koltzscher, M.; Poeter, M.; Austermann, J.; Assard, N.; Baudier, J.; Kaibuchi, K.; Gerke, V. S100P is a novel interaction partner and regulator of IQGAP1. J. Biol. Chem. 2011, 286, 7227–7238. [Google Scholar] [CrossRef]
- Du, M.; Wang, G.; Ismail, T.M.; Gross, S.; Fernig, D.G.; Barraclough, R.; Rudland, P.S. S100P dissociates myosin IIA filaments and focal adhesion sites to reduce cell adhesion and enhance cell migration. J. Biol. Chem. 2012, 287, 15330–15344. [Google Scholar] [CrossRef]
- Austermann, J.; Nazmi, A.R.; Muller-Tidow, C.; Gerke, V. Characterization of the Ca2+ -regulated ezrin-S100P interaction and its role in tumor cell migration. J. Biol. Chem. 2008, 283, 29331–29340. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, G.; Liu, D.; Bao, Z.; Fernig, D.G.; Rudland, P.S.; Barraclough, R. The C-terminal region of S100A4 is important for its metastasis-inducing properties. Oncogene 2005, 24, 4401–4411. [Google Scholar] [CrossRef][Green Version]
- Ismail, T.; Fernig, D.; Rudland, P.; Terry, C.; Wang, G.; Barraclough, R. The basic C-terminal amino acids of calcium-binding protein S100A4 promote metastasis. Carcinogenesis 2008, 29, 2259–2266. [Google Scholar] [CrossRef]
- Davies, B.R.; Davies, M.P.A.; Gibbs, F.E.M.; Barraclough, R.; Rudland, P.S. Induction of the metastatic phenotype by transfection of a benign rat mammary epithelial cell line with the gene for p9Ka, a rat calcium-binding protein but not with the oncogene EJ ras-1. Oncogene 1993, 8, 999–1008. [Google Scholar] [PubMed]
- Oates, A.J.; Barraclough, R.; Rudland, P.S. The identification of osteopontin as a metastasis-related gene product in a rodent mammary tumour model. Oncogene 1996, 13, 97–104. [Google Scholar]
- Liu, D.; Rudland, P.S.; Sibson, D.R.; Platt-Higgins, A.; Barraclough, R. Human homologue of cement gland protein, a novel metastasis inducer associated with breast carcinomas. Cancer Res. 2005, 65, 3796–3805. [Google Scholar] [CrossRef] [PubMed]
- Dunnington, D.J.; Monaghan, P.; Hughes, C.M.; Rudland, P.S. Phenotypic instability of rat mammary tumor epithelial cells. J. Natl. Cancer Inst. 1983, 71, 1227–1240. [Google Scholar]
- Ismail, T.M.; Zhang, S.; Fernig, D.G.; Gross, S.; Martin-Fernandez, M.L.; See, V.; Tozawa, K.; Tynan, C.J.; Wang, G.; Wilkinson, M.C.; et al. Self-association of calcium-binding protein S100A4 and metastasis. J. Biol. Chem. 2010, 285, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, M.E.A.; Lancaster, T.L.; Ismail, T.M.; Georgiadou, A.; Ganguly, A.; Mistry, J.J.; Wang, K.; Rudland, P.S.; Ahmad, S.; Gross, S.R. S100P enhances the motility and invasion of human trophoblast cell lines. Sci. Rep. 2018, 8, 11488. [Google Scholar] [CrossRef] [PubMed]
- Goh, C.; Sin, T.; Hersch, N.; Rudland, P.; Barraclough, R.; Hoffmann, B.; Gross, S. S100A4 downregulates filopodia formation through increased dynamic instability. Cell Adhes. Migr. 2011, 5, 439–447. [Google Scholar]
- Dunnington, D.J.; Kim, U.; Hughes, C.M.; Monaghan, P.; Rudland, P.S. Lack of production of myoepithelial variants by cloned epithelial cell lines derived from the TMT-081 metastasizing rat mammary tumor. Cancer Res. 1984, 44, 5338–5346. [Google Scholar]
- Chen, H.; Fernig, D.G.; Rudland, P.S.; Sparks, A.; Wilkinson, M.C.; Barraclough, R. Binding to intracellular targets of the metastasis-inducing protein, S100A4 (p9Ka). Biochem. Biophys. Res. Commun. 2001, 286, 1212–1217. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, F.E.; Wilkinson, M.C.; Rudland, P.S.; Barraclough, R. Interactions in vitro of p9Ka, the rat S-100-related, metastasis-inducing, calcium-binding protein. J. Biol. Chem. 1994, 269, 18992–18999. [Google Scholar] [CrossRef]
- Lloyd, B.H.; Platt-Higgins, A.; Rudland, P.S.; Barraclough, R. Human S100A4 (p9Ka) induces the metastatic phenotype upon benign tumour cells. Oncogene 1998, 17, 465–473. [Google Scholar] [CrossRef] [PubMed][Green Version]
- El-Tanani, M.K.; Barraclough, R.; Wilkinson, M.C.; Rudland, P.S. Regulatory region of metastasis-inducing DNA is the binding site for T cell factor-4. Oncogene 2001, 20, 1793–1797. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rudland, P.S.; Platt-Higgins, A.; Renshaw, C.; West, C.R.; Winstanley, J.H.R.; Robertson, L.; Barraclough, R. Prognostic significance of the metastasis-inducing protein S100A4 (p9Ka) in human breast cancer. Cancer Res. 2000, 60, 1595–1603. [Google Scholar] [PubMed]
- Rudland, P.S.; Platt-Higgins, A.; El-Tanani, M.; De Silva Rudland, S.; Barraclough, R.; Winstanley, J.H.R.; Howitt, R.; West, C.R. Prognostic significance of the metastasis-associated protein osteopontin in human breast cancer. Cancer Res. 2002, 62, 3417–3427. [Google Scholar]
- Barraclough, D.L.; Platt-Higgins, A.; de Silva Rudland, S.; Barraclough, R.; Winstanley, J.; West, C.R.; Rudland, P.S. The metastasis-associated anterior gradient 2 protein is correlated with poor survival of breast cancer patients. Am. J. Pathol. 2009, 175, 1848–1857. [Google Scholar] [CrossRef]
- Jenkinson, S.R.; Barraclough, R.; West, C.R.; Rudland, P.S. S100A4 regulates cell motility and invasion in an in vitro model for breast cancer metastasis. Br. J. Cancer 2004, 90, 253–262. [Google Scholar] [CrossRef]
- Chen, H.C. Boyden chamber assay. Methods Mol. Biol. 2005, 294, 15–22. [Google Scholar]
- Cory, G. Scratch-wound assay. Methods Mol. Biol. 2011, 769, 25–30. [Google Scholar] [CrossRef]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Markus, G.; DePasquale, J.L.; Wissler, F.C. Quantitative determination of the binding of epsilon-aminocaproic acid to native plasminogen. J. Biol. Chem. 1978, 253, 727–732. [Google Scholar] [CrossRef]
- McNicol, G.P.; Fletcher, A.P.; Alkjaersig, N.; Sherry, S. The absorption, distribution, and excretion of ϵ-amino-caproic acid following oral or intravenous administration to man. J. Lab. Clin. Med. 1962, 59, 15–24. [Google Scholar]
- Andersson, L.; Nilsoon, I.M.; Colleen, S.; Granstrand, B.; Melander, B. Role of urokinase and tissue activator in sustaining bleeding and the management thereof with EACA and AMCA. Ann. N. Y. Acad. Sci. 1968, 146, 642–658. [Google Scholar] [CrossRef]
- Ueshima, S.; Okada, K.; Matsuo, O. Stabilization of plasmin by lysine derivatives. Clin. Chim. Acta 1996, 245, 7–18. [Google Scholar] [CrossRef]
- Mahdy, A.M.; Webster, N.R. Perioperative systemic haemostatic agents. Br. J. Anaesth. 2004, 93, 842–858. [Google Scholar] [CrossRef]
- Shieh, B.H.; Travis, J. The reactive site of human alpha 2-antiplasmin. J. Biol. Chem. 1987, 262, 6055–6059. [Google Scholar] [CrossRef]
- Smith, R.T. Characterising the Effect of S100P Overexpression in Cell Adhesion and Cancer Metastasis. Ph.D. Thesis, University of Liverpool, Liverpool, UK, 2015. [Google Scholar]
- Arumugam, T.; Ramachandran, V.; Logsdon, C.D. Effect of cromolyn on S100P interactions with RAGE and pancreatic cancer growth and invasion in mouse models. J. Natl. Cancer Inst. 2006, 98, 1806–1818. [Google Scholar] [CrossRef]
- Arumugam, T.; Ramachandran, V.; Gomez, S.B.; Schmidt, A.M.; Logsdon, C.D. S100P-derived RAGE antagonistic peptide reduces tumor growth and metastasis. Clin. Cancer Res. 2012, 18, 4356–4364. [Google Scholar] [CrossRef]
- Tan, B.S.; Yang, M.C.; Singh, S.; Chou, Y.C.; Chen, H.Y.; Wang, M.Y.; Wang, Y.C.; Chen, R.H. LncRNA NORAD is repressed by the YAP pathway and suppresses lung and breast cancer metastasis by sequestering S100P. Oncogene 2019, 38, 5612–5626. [Google Scholar] [CrossRef]
- Donato, R. Functional roles of S100 proteins, calcium-binding proteins of the EF-hand type. Biochim. Biophys. Acta 1999, 1450, 191–231. [Google Scholar] [CrossRef]
- Baudier, J.; Deloulme, J.C.; Shaw, G.S. The Zn(2+) and Ca(2+) -binding S100B and S100A1 proteins: Beyond the myths. Biol. Rev. Camb. Philos. Soc. 2020, 95, 738–758. [Google Scholar] [CrossRef]
- Duelli, A.; Kiss, B.; Lundholm, I.; Bodor, A.; Petoukhov, M.V.; Svergun, D.I.; Nyitray, L.; Katona, G. The C-terminal random coil region tunes the Ca(2)(+)-binding affinity of S100A4 through conformational activation. PLoS ONE 2014, 9, e97654. [Google Scholar] [CrossRef]
- Ramagopal, U.A.; Dulyaninova, N.G.; Varney, K.M.; Wilder, P.T.; Nallamsetty, S.; Brenowitz, M.; Weber, D.J.; Almo, S.C.; Bresnick, A.R. Structure of the S100A4/myosin-IIA complex. BMC Struct. Biol. 2013, 13, 31. [Google Scholar] [CrossRef]
- Szabo, I.; Simon, M., Jr.; Hunyadi, J. Plasmin promotes keratinocyte migration and phagocytic-killing accompanied by suppression of cell proliferation which may facilitate re-epithelialization of wound beds. Clin. Dev. Immunol. 2004, 11, 233–240. [Google Scholar] [CrossRef]
- Legrand, C.; Polette, M.; Tournier, J.M.; de Bentzmann, S.; Huet, E.; Monteau, M.; Birembaut, P. uPA/plasmin system-mediated MMP-9 activation is implicated in bronchial epithelial cell migration. Exp. Cell Res. 2001, 264, 326–336. [Google Scholar] [CrossRef]
- Tarui, T.; Majumdar, M.; Miles, L.A.; Ruf, W.; Takada, Y. Plasmin-induced migration of endothelial cells. A potential target for the anti-angiogenic action of angiostatin. J. Biol. Chem. 2002, 277, 33564–33570. [Google Scholar] [CrossRef]
- Majumdar, M.; Tarui, T.; Shi, B.; Akakura, N.; Ruf, W.; Takada, Y. Plasmin-induced migration requires signaling through protease-activated receptor 1 and integrin alpha(9)beta(1). J. Biol. Chem. 2004, 279, 37528–37534. [Google Scholar] [CrossRef] [PubMed]
- Pendurthi, U.R.; Tran, T.T.; Post, M.; Rao, L.V. Proteolysis of CCN1 by plasmin: Functional implications. Cancer Res. 2005, 65, 9705–9711. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lorenz, N.; Loef, E.J.; Kelch, I.D.; Verdon, D.J.; Black, M.M.; Middleditch, M.J.; Greenwood, D.R.; Graham, E.S.; Brooks, A.E.; Dunbar, P.R.; et al. Plasmin and regulators of plasmin activity control the migratory capacity and adhesion of human T cells and dendritic cells by regulating cleavage of the chemokine CCL21. Immunol. Cell Biol. 2016, 94, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Zhao, Y.; Brown, T.L.; Morgan, A.C.; Kohler, H. TransMabs: Cell-penetrating antibodies, the next generation. Expert Opin. Biol. Ther. 2005, 5, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.; MacLeod, T.J.; Zhang, Y.; Waisman, D.M. S100A10, annexin A2, and annexin a2 heterotetramer as candidate plasminogen receptors. Front. Biosci. 2005, 10, 300–325. [Google Scholar] [CrossRef] [PubMed]
- Semov, A.; Moreno, M.J.; Onichtchenko, A.; Abulrob, A.; Ball, M.; Ekiel, I.; Pietrzynski, G.; Stanimirovic, D.; Alakhov, V. Metastasis-associated protein S100A4 induces angiogenesis through interaction with Annexin II and accelerated plasmin formation. J. Biol. Chem. 2005, 280, 20833–20841. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transfected DNA (Designation of Cell Line) a | Incidence of Mammary Tumours (%) b | p-Value c | Incidence of Metastasis (%) d | p-Value e |
|---|---|---|---|---|
| None. Untransfected f | 18/20 (90) | 0/18 | ||
| Rama 37 Vector only f | ||||
| Clone | 19/20 (95) | 0/19 (0) | ||
| Pool | 23/23 (100) | 2/23 (9) | ||
| S100P wild-type | ||||
| Clone | 27/27 (100) | 19/27 (70) | ||
| Pool | 20/20 (100) | 15/20 (75) | ||
| K95A S100P | ||||
| Clone | 21/21 (100) | p > 0.9999 | 7/21 (33) | p = 0.019 |
| Pool | 19/19 (100) | p > 0.9999 | 9/19 (47) | p = 0.105 |
| ΔK95 S100P | ||||
| Clone | 19/19 (100) | p > 0.9999 | 3/19 (16) | p = 0.0003 |
| Pool | 19/21 (90) | p > 0.9999 | 2/19 (11) | p < 0.0001 |
| Cell Clone | Focal Vinculin a | Focal Paxillin a | ||||
|---|---|---|---|---|---|---|
| No of Cells Counted | Mean Focal Adhesions/ Cell ± SD b | Mean Focal Adhesions as % of Vector Control | No of Cells Counted | Mean Focal Adhesions/ Cell ± SD b | Mean Focal Adhesions as % of Vector Control | |
| Vector control | 52 | 16.8 ± 4.0 | 100 | 54 | 15.2 ± 5.4 | 100 |
| Wild-type S100P | 51 | 3.9 ± 2.9 * | 23.2 | 53 | 4.4 ± 3.1 ** | 28.9 |
| K95A-mutant S100P | 52 | 8.1 ± 4.7 ¶ | 48.2 | 51 | 8.0 ± 4.2 ¶¶ | 52.6 |
| ΔK95-mutant S100P | 51 | 19.8 ± 6.2 § | 117.9 | 50 | 20.0 ± 5.2 §§ | 131.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ismail, T.M.; Gross, S.R.; Lancaster, T.; Rudland, P.S.; Barraclough, R. The Role of the C-Terminal Lysine of S100P in S100P-Induced Cell Migration and Metastasis. Biomolecules 2021, 11, 1471. https://doi.org/10.3390/biom11101471
Ismail TM, Gross SR, Lancaster T, Rudland PS, Barraclough R. The Role of the C-Terminal Lysine of S100P in S100P-Induced Cell Migration and Metastasis. Biomolecules. 2021; 11(10):1471. https://doi.org/10.3390/biom11101471
Chicago/Turabian StyleIsmail, Thamir M., Stephane R. Gross, Tara Lancaster, Philip S. Rudland, and Roger Barraclough. 2021. "The Role of the C-Terminal Lysine of S100P in S100P-Induced Cell Migration and Metastasis" Biomolecules 11, no. 10: 1471. https://doi.org/10.3390/biom11101471
APA StyleIsmail, T. M., Gross, S. R., Lancaster, T., Rudland, P. S., & Barraclough, R. (2021). The Role of the C-Terminal Lysine of S100P in S100P-Induced Cell Migration and Metastasis. Biomolecules, 11(10), 1471. https://doi.org/10.3390/biom11101471