
Bisphenol A Induces Accelerated Cell Aging in Murine Endothelium

 ,
,  , , ,
, , , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Animal Model
2.3. MTT Cell Viability Assay
2.4. Senescence-Associated β-Galactosidase Assay
2.5. Western Blot
2.6. Protein Oxidation
2.7. qPCR
2.8. Immunohistochemistry
2.9. Confocal Microscopy
2.10. Superoxide Anion Production
2.11. Statistical Analysis
3. Results
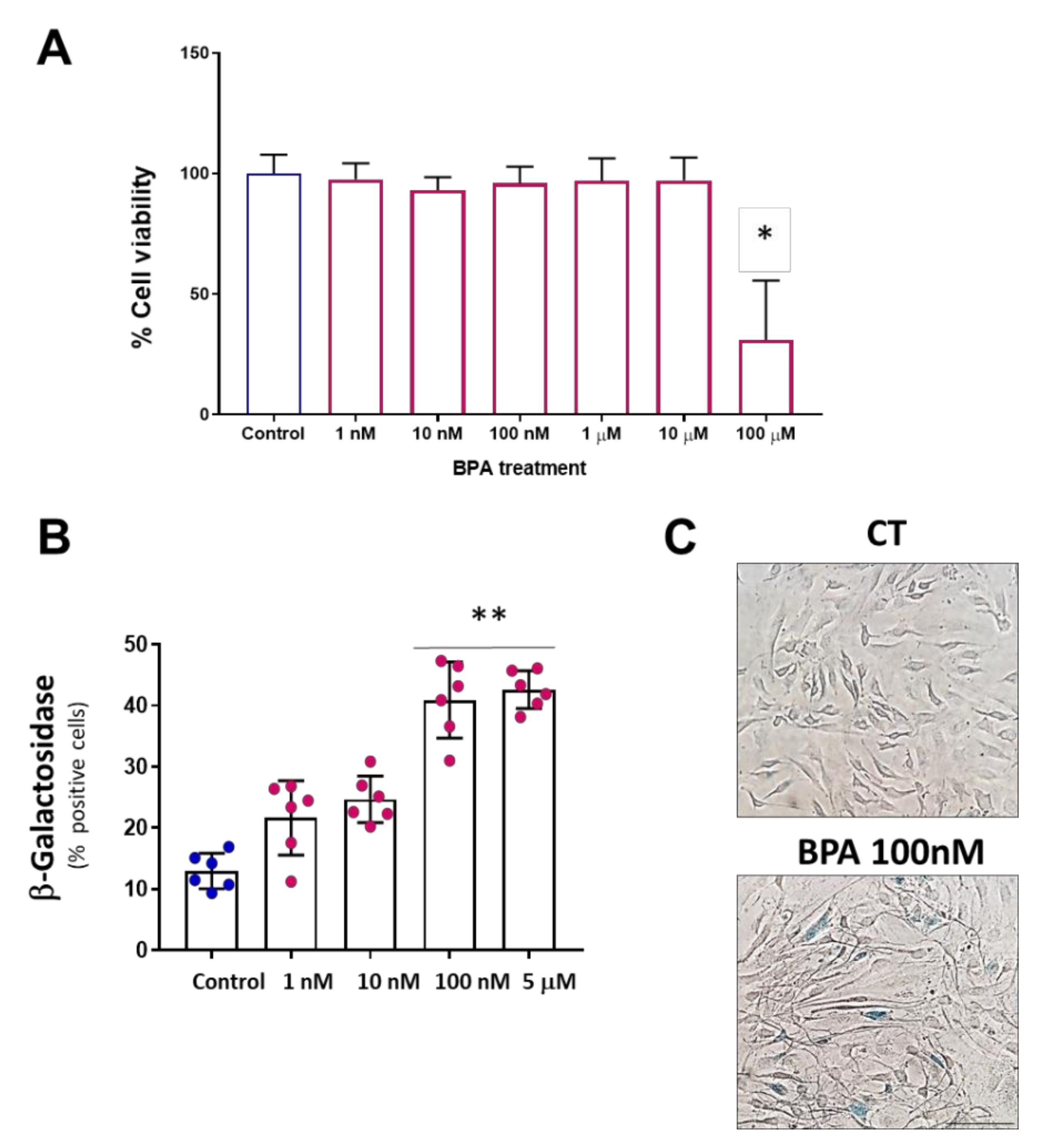
3.1. BPA Increases Cellular Senescence at Low Concentrations
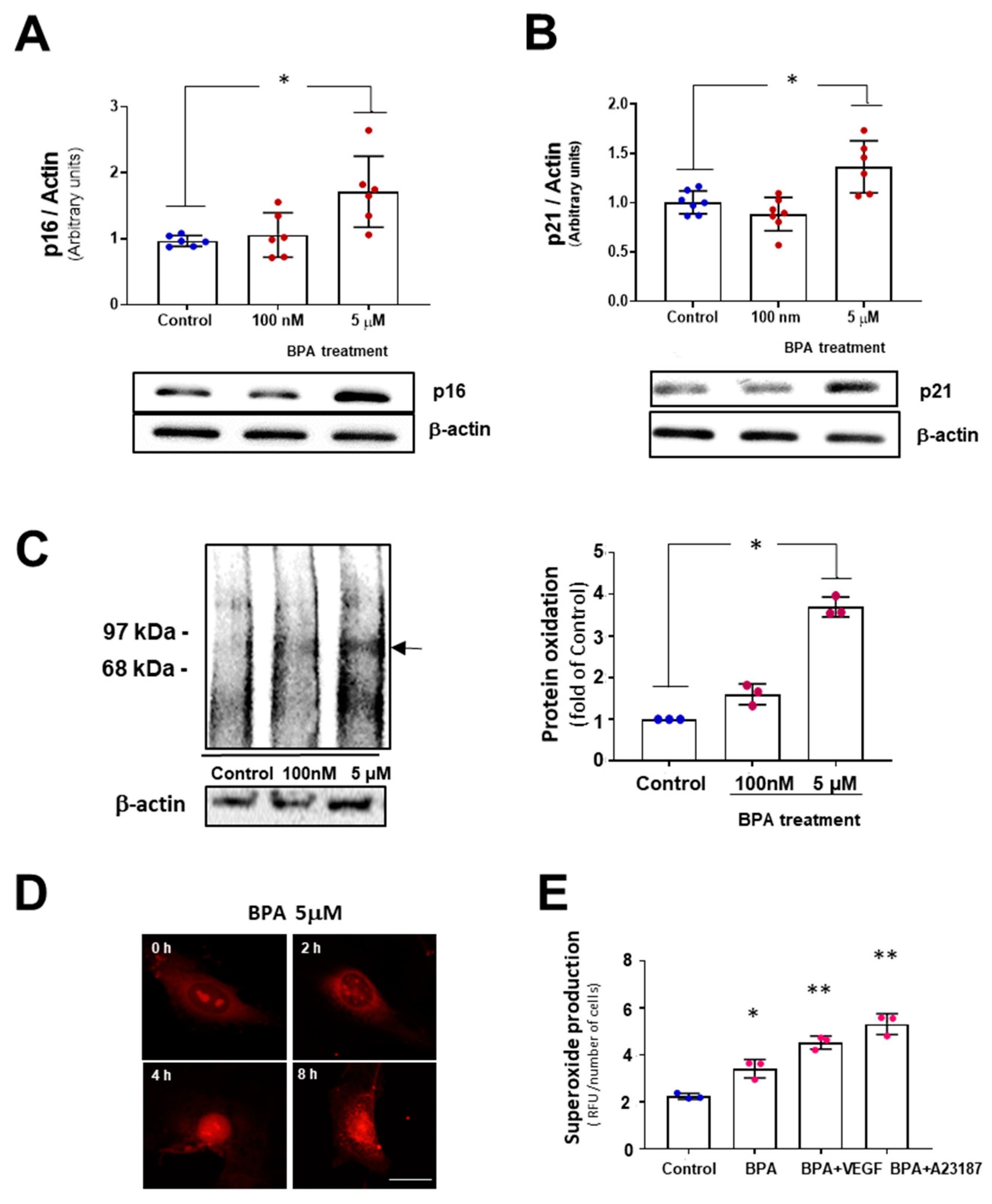
3.2. BPA Modulates the PERK-ATF4-CHOP Pathway after Five Days Treatment
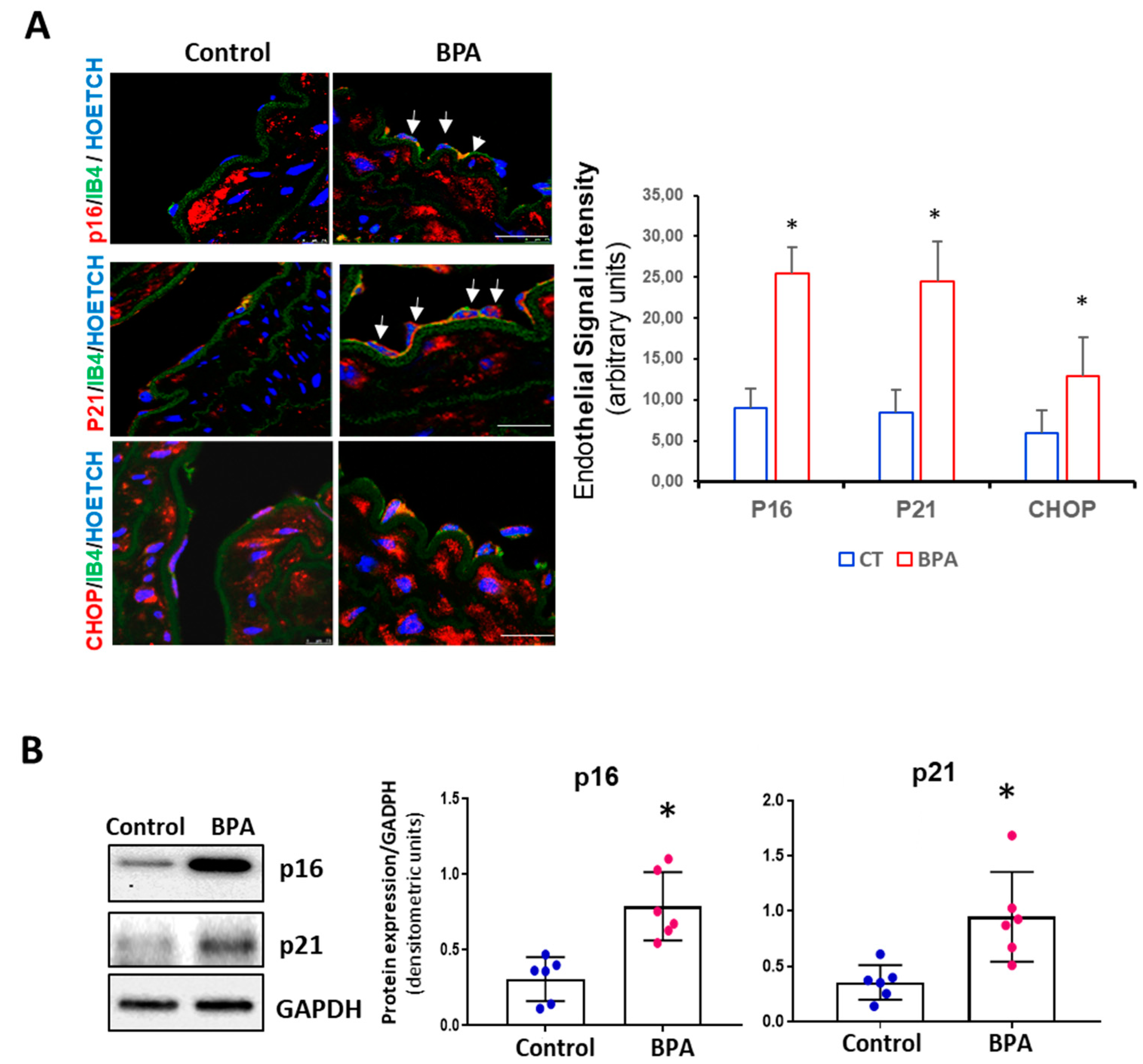
3.3. BPA Induces Senescence and Activation of the PERK-ATF4-CHOP Pathway in Mice
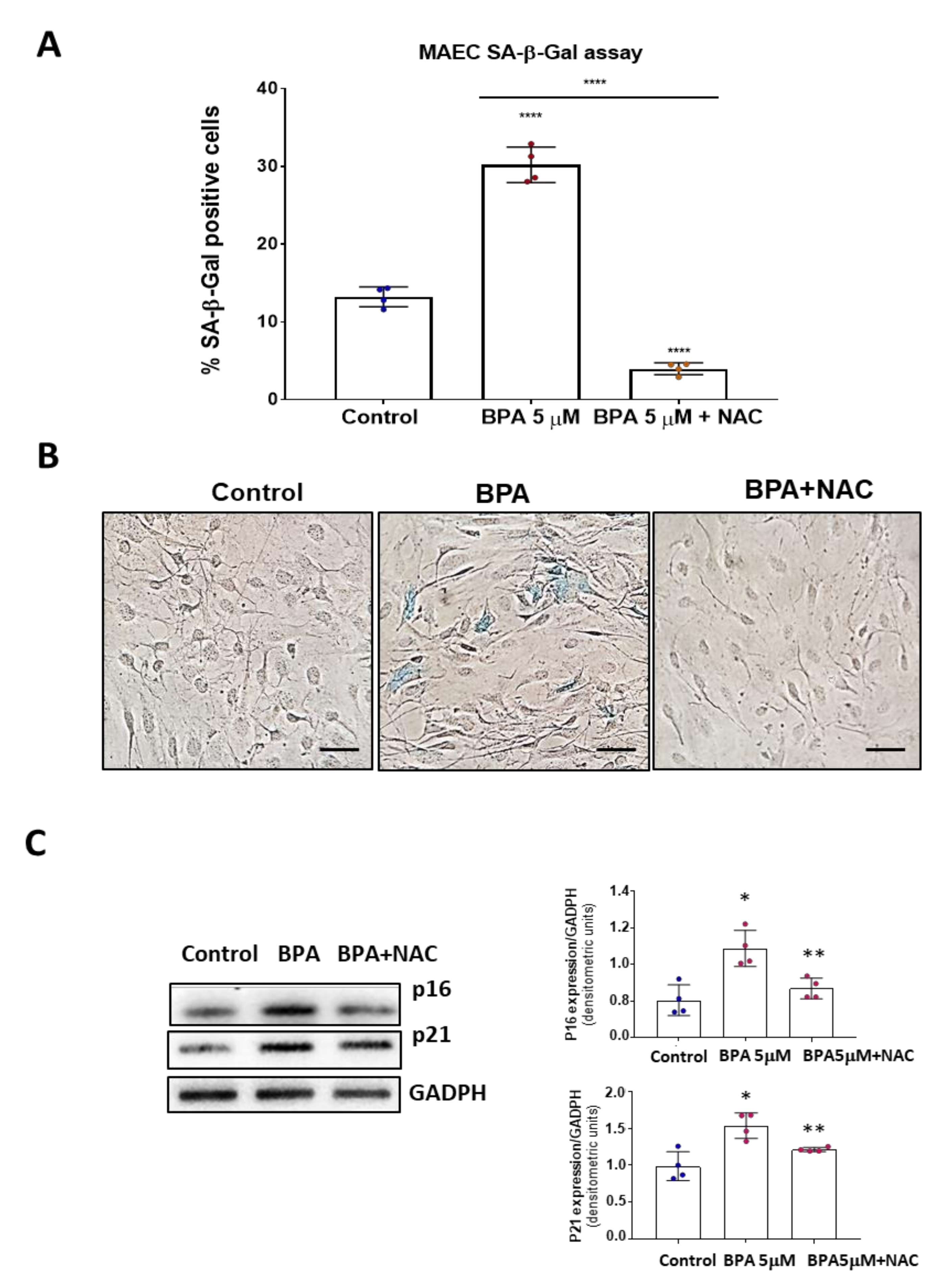
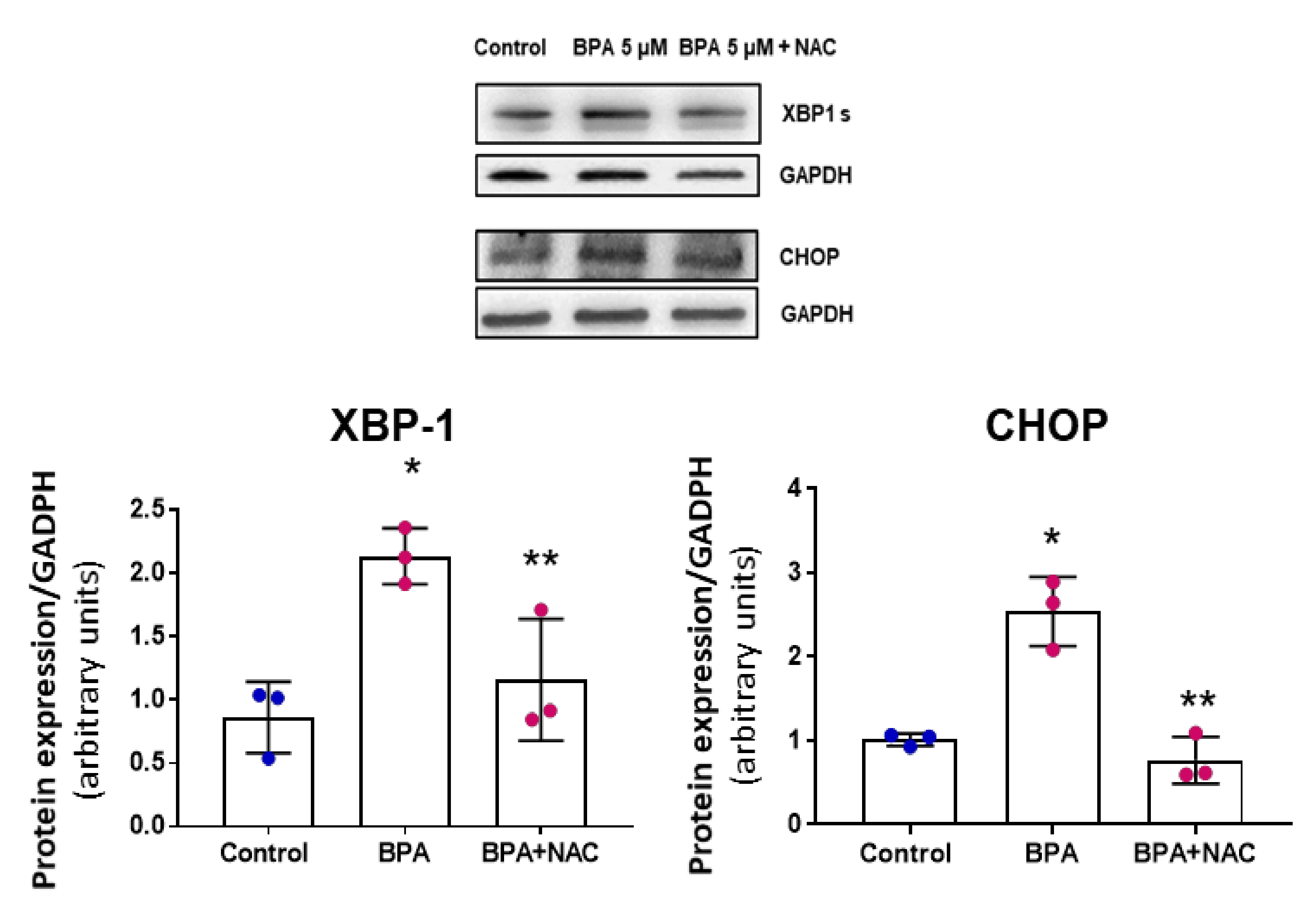
3.4. NAC Reduces Cell Senescence and Attenuates the BPA-Induced Response on the UPR System
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- EFSA. Scientific Opinion on the risks to public health related to the presence of bisphenol A (BPA) in foodstuffs. EFSA J. 2016, 13, 3978. [Google Scholar] [CrossRef]
- Thomas, S.; Visakh, P. (Eds.) Handbook of Engineering and Specialty Thermoplastics, Volume 3: Polyethers and Polyesters; Wiley: Hoboken, NJ, USA, 2011; ISBN 978-0-470-63926-9. [Google Scholar]
- Vandenberg, L.N.; Hauser, R.; Marcus, M.; Olea, N.; Welshons, W.V. Human exposure to bisphenol A (BPA). Reprod. Toxicol. 2007, 24, 139–177. [Google Scholar] [CrossRef]
- Dursun, E.; Fron-Chabouis, H.H.; Attal, J.-P.; Raskin, A. Bisphenol A Release: Survey of the Composition of Dental Composite Resins. Open Dent. J. 2016, 10, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Iribarne-Durán, L.M.; Artacho-Cordón, F.; Peña-Caballero, M.; Molina-Molina, J.M.; Jiménez-Díaz, I.; Vela-Soria, F.; Serrano, L.; Hurtado, J.A.; Fernández, M.F.; Freire, C.; et al. Presence of Bisphenol A and Parabens in a Neonatal Intensive Care Unit: An Exploratory Study of Potential Sources of Exposure. Environ. Health Perspect. 2019, 127, 117004. [Google Scholar] [CrossRef]
- Duty, S.M.; Mendonca, K.; Hauser, R.; Calafat, A.M.; Ye, X.; Meeker, J.D.; Ackerman, R.; Cullinane, J.; Faller, J.; Ringer, S. Potential Sources of Bisphenol A in the Neonatal Intensive Care Unit. Pediatrics 2013, 131, 483–489. [Google Scholar] [CrossRef] [Green Version]
- Testai, E.; Hartemann, P.; Rodriguez-Farre, E.; Rastogi, S.C.; Bustos, J.; Gundert-Remy, U.; Hensten, A.; Kopperud, H.M.; Olea, N.N.; Piersma, A.; et al. The safety of the use of bisphenol A in medical devices. Regul. Toxicol. Pharmacol. 2016, 79, 106–107. [Google Scholar] [CrossRef] [Green Version]
- Grand View Research Polycarbonate Sheet Market Size & Share|Industry Report, 2018–2025. Available online: https://www.grandviewresearch.com/industry-analysis/bisphenol-a-bpa-market (accessed on 11 September 2020).
- Maffini, M.V.; Rubin, B.S.; Sonnenschein, C.; Soto, A.M. Endocrine disruptors and reproductive health: The case of bisphenol-A. Mol. Cell. Endocrinol. 2006, 254–255, 179–186. [Google Scholar] [CrossRef]
- Rochester, J.R. Bisphenol A and human health: A review of the literature. Reprod. Toxicol. 2013, 42, 132–155. [Google Scholar] [CrossRef]
- Tomza-Marciniak, A.; Stępkowska, P.; Kuba, J.; Pilarczyk, B. Effect of bisphenol A on reproductive processes: A review of in vitro, in vivo and epidemiological studies. J. Appl. Toxicol. 2018, 38, 51–80. [Google Scholar] [CrossRef]
- Ziv-Gal, A.; Flaws, J.A. Evidence for bisphenol A-induced female infertility: A review (2007–2016). Fertil. Steril. 2016, 106, 827–856. [Google Scholar] [CrossRef] [Green Version]
- Pergialiotis, V.; Kotrogianni, P.; Christopoulos-Timogiannakis, E.; Koutaki, D.; Daskalakis, G.; Papantoniou, N. Bisphenol A and adverse pregnancy outcomes: A systematic review of the literature. J. Matern. Neonatal Med. 2018, 31, 3320–3327. [Google Scholar] [CrossRef] [PubMed]
- Akash, M.S.H.; Sabir, S.; Rehman, K. Bisphenol A-induced metabolic disorders: From exposure to mechanism of action. Environ. Toxicol. Pharmacol. 2020, 77. [Google Scholar] [CrossRef] [PubMed]
- Provvisiero, D.P.; Pivonello, C.; Muscogiuri, G.; Negri, M.; de Angelis, C.; Simeoli, C.; Pivonello, R.; Colao, A. Influence of Bisphenol A on Type 2 Diabetes Mellitus. Int. J. Environ. Res. Public Health 2016, 13, 989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Li, M.; Liu, A.; Wu, C.; Li, D.; Deng, Q.; Zhang, B.; Du, J.; Gao, X.; Hong, Y. Bisphenol A and the Risk of Obesity a Systematic Review With Meta-Analysis of the Epidemiological Evidence. Dose-Response 2020, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesan, D.; Feighan, K.M.; Antle, M.C.; Kurrasch, D.M. Gestational low-dose BPA exposure impacts suprachiasmatic nucleus neurogenesis and circadian activity with transgenerational effects. Sci. Adv. 2021, 7, eabd1159. [Google Scholar] [CrossRef]
- Saura, M.; Marquez, S.; Reventun, P.; Olea-Herrero, N.; Arenas, M.I.; Moreno-Gómez-Toledano, R.; Gómez-Parrizas, M.; Muñóz-Moreno, C.; González-Santander, M.; Zaragoza, C.; et al. Oral administration of bisphenol A induces high blood pressure through angiotensin II/CaMKII-dependent uncoupling of eNOS. FASEB J. 2014, 28, 4719–4728. [Google Scholar] [CrossRef]
- Reventun, P.; Sanchez-Esteban, S.; Cook, A.; Cuadrado, I.; Roza, C.; Moreno-Gómez-Toledano, R.; Muñoz, C.; Zaragoza, C.; Bosch, R.J.; Saura, M. Bisphenol A induces coronary endothelial cell necroptosis by activating RIP3/CamKII dependent pathway. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Moreno-Gómez-Toledano, R.; Arenas, M.I.; González-Martínez, C.; Olea-Herrero, N.; Reventún, P.; Di Nunzio, M.; Sánchez-Esteban, S.; Arilla-Ferreiro, E.; Saura, M.; Bosch, R.J. Bisphenol A impaired cell adhesion by altering the expression of adhesion and cytoskeleton proteins on human podocytes. Sci. Rep. 2020, 10, 16638. [Google Scholar] [CrossRef]
- Bosch, R.J.; Quiroga, B.; Munoz-Moreno, C.; Olea-Herrero, N.; Arenas, M.I.; Gonzalez-Santander, M.; Reventun, P.; Zaragoza, C.; de Arriba, G.; Saura, M. Bisphenol A: An environmental factor implicated in renal vascular damage. Nefrología 2016, 36, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Shankar, A.; Teppala, S. Urinary bisphenol A and hypertension in a multiethnic sample of US adults. J. Environ. Public Health 2012, 2012, 481641. [Google Scholar] [CrossRef]
- Aekplakorn, W.; Chailurkit, L.-O.O.; Ongphiphadhanakul, B. Association of serum bisphenol a with hypertension in thai population. Int. J. Hypertens. 2015, 2015, 594189. [Google Scholar] [CrossRef]
- Shimizu, I.; Minamino, T. Cellular senescence in cardiac diseases. J. Cardiol. 2019, 74, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 485. [Google Scholar] [CrossRef]
- Khor, E.S.; Wong, P.F. The roles of MTOR and miRNAs in endothelial cell senescence. Biogerontology 2020, 21, 517–530. [Google Scholar] [CrossRef]
- Terzi, M.Y.; Izmirli, M.; Gogebakan, B. The cell fate: Senescence or quiescence. Mol. Biol. Rep. 2016, 43, 1213–1220. [Google Scholar] [CrossRef]
- Mahemuti, L.; Chen, Q.; Coughlan, M.C.; Qiao, C.; Chepelev, N.L.; Florian, M.; Dong, D.; Woodworth, R.G.; Yan, J.; Cao, X.L.; et al. Bisphenol A induces DSB-ATM-p53 signaling leading to cell cycle arrest, senescence, autophagy, stress response, and estrogen release in human fetal lung fibroblasts. Arch. Toxicol. 2018, 92, 1453–1469. [Google Scholar] [CrossRef]
- Ribeiro-Varandas, E.; Sofia Pereira, H.; Monteiro, S.; Neves, E.; Brito, L.; Ferreira, R.B.; Viegas, W.; Delgado, M. Bisphenol A Disrupts Transcription and Decreases Viability in Aging Vascular Endothelial Cells. Int. J. Mol. Sci. 2014, 15, 15791–15805. [Google Scholar] [CrossRef] [Green Version]
- Soundararajan, A.; Yoganantharajah, P.; Raghavan, S.; Mohan, V.; Balasubramanyam, M.; Gibert, Y. Bisphenol A exposure under metabolic stress induces accelerated cellular senescence in vivo in a p53 independent manner. Sci. Total Environ. 2019, 689, 1201–1211. [Google Scholar] [CrossRef]
- Soundararajan, A.; Prabu, P.; Mohan, V.; Gibert, Y.; Balasubramanyam, M. Novel insights of elevated systemic levels of bisphenol-A (BPA) linked to poor glycemic control, accelerated cellular senescence and insulin resistance in patients with type 2 diabetes. Mol. Cell. Biochem. 2019, 458, 171–183. [Google Scholar] [CrossRef]
- Kunieda, T.; Minamino, T.; Nishi, J.I.; Tateno, K.; Oyama, T.; Katsuno, T.; Miyauchi, H.; Orimo, M.; Okada, S.; Takamura, M.; et al. Angiotensin II induces premature senescence of vascular smooth muscle cells and accelerates the development of atherosclerosis via a p21-dependent pathway. Circulation 2006, 114, 953–960. [Google Scholar] [CrossRef]
- Li, P.; Guo, X.; Lei, P.; Shi, S.; Luo, S.; Cheng, X. PI3K/Akt/uncoupling protein 2 signaling pathway may be involved in cell senescence and apoptosis induced by angiotensin II in human vascular endothelial cells. Mol. Biol. Rep. 2014, 41, 6931–6937. [Google Scholar] [CrossRef]
- Donato, A.J.; Eskurza, I.; Silver, A.E.; Levy, A.S.; Pierce, G.L.; Gates, P.E.; Seals, D.R. Direct evidence of endothelial oxidative stress with aging in humans: Relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-κB. Circ. Res. 2007, 100, 1659–1666. [Google Scholar] [CrossRef] [Green Version]
- Durrant, J.R.; Seals, D.R.; Connell, M.L.; Russell, M.J.; Lawson, B.R.; Folian, B.J.; Donato, A.J.; Lesniewski, L.A. Voluntary wheel running restores endothelial function in conduit arteries of old mice: Direct evidence for reduced oxidative stress, increased superoxide dismutase activity and down-regulation of NADPH oxidase. J. Physiol. 2009, 587, 3271–3285. [Google Scholar] [CrossRef]
- Miller, S.J.; Watson, W.C.; Kerr, K.A.; Labarrere, C.A.; Chen, N.X.; Deeg, M.A.; Unthank, J.L. Development of progressive aortic vasculopathy in a rat model of aging. Am. J. Physiol. Heart Circ. Physiol. 2007, 293. [Google Scholar] [CrossRef]
- Imanishi, T.; Hano, T.; Nishio, I. Angiotensin II accelarates endothelial progenitor cell senescence through induction of oxidative stress. J. Hypertens. 2005, 23, 97–104. [Google Scholar] [CrossRef]
- Endtmann, C.; Ebrahimian, T.; Czech, T.; Arfa, O.; Laufs, U.; Fritz, M.; Wassmann, K.; Werner, N.; Petoumenos, V.; Nickenig, G.; et al. Angiotensin II impairs endothelial progenitor cell number and function in vitro and in vivo: Implications for vascular regeneration. Hypertension 2011, 58, 394–403. [Google Scholar] [CrossRef]
- Van Der Loo, B.; Labugger, R.; Skepper, J.N.; Bachschmid, M.; Kilo, J.; Powell, J.M.; Palacios-Callender, M.; Erusalimsky, J.D.; Quaschning, T.; Malinski, T.; et al. Enhanced peroxynitrite formation is associated with vascular aging. J. Exp. Med. 2000, 192, 1731–1743. [Google Scholar] [CrossRef] [Green Version]
- Pierce, G.L.; Lesniewski, L.A.; Lawson, B.R.; Beske, S.D.; Seals, D.R. Nuclear factor-κB activation contributes to vascular endothelial dysfunction via oxidative stress in overweight/obese middle-aged and older humans. Circulation 2009, 119, 1284–1292. [Google Scholar] [CrossRef] [Green Version]
- Pluquet, O.; Pourtier, A.; Abbadie, C. The unfolded protein response and cellular senescence. A review in the theme: Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am. J. Physiol. Cell Physiol. 2015, 308, 415–425. [Google Scholar] [CrossRef] [Green Version]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubra, K.T.; Akhter, M.S.; Uddin, M.A.; Barabutis, N. Unfolded protein response in cardiovascular disease. Cell. Signal. 2020, 73, 109699. [Google Scholar] [CrossRef]
- He, L.; Yuan, J.; Xu, Q.; Chen, R.; Chen, L.; Fang, M. miRNA-1283 Regulates the PERK/ATF4 Pathway in Vascular Injury by Targeting ATF4. PLoS ONE 2016, 11, e0159171. [Google Scholar] [CrossRef]
- Vanhoutte, D.; Schips, T.G.; Vo, A.; Grimes, K.M.; Baldwin, T.A.; Brody, M.J.; Accornero, F.; Sargent, M.A.; Molkentin, J.D. Thbs1 induces lethal cardiac atrophy through PERK-ATF4 regulated autophagy. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Xin, Y.; Wu, W.; Qu, J.; Wang, X.; Lei, S.; Yuan, L.; Liu, X. Inhibition of mitofusin-2 promotes cardiac fibroblast activation via the PERK/ATF4 pathway and reactive oxygen species. Oxid. Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Masuda, M.; Miyazaki-Anzai, S.; Levi, M.; Ting, T.C.; Miyazaki, M. PERK-eIF2α-ATF4-CHOP signaling contributes to TNFα-induced vascular calcification. J. Am. Heart Assoc. 2013, 2. [Google Scholar] [CrossRef] [Green Version]
- Toth, A.; Nickson, P.; Mandl, A.; Bannister, M.; Toth, K.; Erhardt, P. Endoplasmic Reticulum Stress as a Novel Therapeutic Target in Heart Diseases. Cardiovasc. Hematol. Disord. Targets 2008, 7, 205–218. [Google Scholar] [CrossRef]
- Nishitoh, H. CHOP is a multifunctional transcription factor in the ER stress response. J. Biochem. 2012, 151, 217–219. [Google Scholar] [CrossRef] [Green Version]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Asahi, J.; Kamo, H.; Baba, R.; Doi, Y.; Yamashita, A.; Murakami, D.; Hanada, A.; Hirano, T. Bisphenol A induces endoplasmic reticulum stress-associated apoptosis in mouse non-parenchymal hepatocytes. Life Sci. 2010, 87, 431–438. [Google Scholar] [CrossRef]
- Yin, L.; Dai, Y.; Cui, Z.; Jiang, X.; Liu, W.; Han, F.; Lin, A.; Cao, J.; Liu, J. The regulation of cellular apoptosis by the ROS-triggered PERK/EIF2α/chop pathway plays a vital role in bisphenol A-induced male reproductive toxicity. Toxicol. Appl. Pharmacol. 2017, 314, 98–108. [Google Scholar] [CrossRef]
- Abbadie, C.; Pluquet, O. Unfolded Protein Response (UPR) Controls Major Senescence Hallmarks. Trends Biochem. Sci. 2020, 45, 371–374. [Google Scholar] [CrossRef]
- López-Rivera, E.; Lizarbe, T.R.; Martínez-Moreno, M.; López-Novoa, J.M.; Rodríguez-Barbero, A.; Rodrigo, J.; Fernández, A.P.; Álvarez-Barrientos, A.; Lamas, S.; Zaragoza, C. Matrix metalloproteinase 13 mediates nitric oxide activation of endothelial cell migration. Proc. Natl. Acad. Sci. USA 2005, 102, 3685–3690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, J.; Wang, H.; Zhou, L.; Fan, D.; Shi, L.; Ji, G.; Gu, A. Oxidative stress in bisphenol AF-induced cardiotoxicity in zebrafish and the protective role of N-acetyl N-cysteine. Sci. Total Environ. 2020, 731. [Google Scholar] [CrossRef] [PubMed]
- Lei, B.; Sun, S.; Xu, J.; Feng, C.; Yu, Y.; Xu, G.; Wu, M.; Peng, W. Low-concentration BPAF- and BPF-induced cell biological effects are mediated by ROS in MCF-7 breast cancer cells. Environ. Sci. Pollut. Res. 2018, 25, 3200–3208. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wang, H.-S. Impact of bisphenol a on the cardiovascular system—Epidemiological and experimental evidence and molecular mechanisms. Int. J. Environ. Res. Public Health 2014, 11, 8399–8413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho, L.; Lewis, S.M.; Vanlandingham, M.M.; Olson, G.R.; Davis, K.J.; Patton, R.E.; Twaddle, N.C.; Doerge, D.R.; Churchwell, M.I.; Bryant, M.S.; et al. A two-year toxicology study of bisphenol A (BPA) in Sprague-Dawley rats: CLARITY-BPA core study results. Food Chem. Toxicol. 2019, 132, 110728. [Google Scholar] [CrossRef]
- Prins, G.S.; Patisaul, H.B.; Belcher, S.M.; Vandenberg, L.N. CLARITY-BPA academic laboratory studies identify consistent low-dose Bisphenol A effects on multiple organ systems. Basic Clin. Pharmacol. Toxicol. 2019, 125, 14–31. [Google Scholar] [CrossRef]
- Olea-Herrero, N.; Arenas, M.I.; Muñόz-Moreno, C.; Moreno-Gόmez-Toledano, R.; González-Santander, M.; Arribas, I.; Bosch, R.J. Bisphenol-A induces podocytopathy with proteinuria in mice. J. Cell. Physiol. 2014, 229, 2057–2066. [Google Scholar] [CrossRef]
- Herranz, B.; Marquez, S.; Guijarro, B.; Aracil, E.; Aicart-Ramos, C.; Rodriguez-Crespo, I.; Rodríguez-Puyol, M.; Zaragoza, C.; Saura, M. Integrin-linked kinase regulates vasomotor function by preventing endothelial nitric oxide synthase uncoupling: Role in atherosclerosis. Circ. Res. 2012, 110, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, I.; Piedras, M.J.G.M.; Herruzo, I.; del Carmen Turpin, M.; Castejón, B.; Reventun, P.; Martin, A.; Saura, M.; Zamorano, J.L.; Zaragoza, C. EMMPRIN-Targeted magnetic nanoparticles for in vivo visualization and regression of acute myocardial infarction. Theranostics 2016, 6, 545–557. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Bredhult, C.; Bäcklin, B.M.; Olovsson, M. Effects of some endocrine disruptors on the proliferation and viability of human endometrial endothelial cells in vitro. Reprod. Toxicol. 2007, 23, 550–559. [Google Scholar] [CrossRef]
- Bosch-Panadero, E.; Mas, S.; Civantos, E.; Abaigar, P.; Camarero, V.; Ruiz-Priego, A.; Ortiz, A.; Egido, J.; González-Parra, E.; Gonzalez-Parra, E. Bisphenol A is an exogenous toxin that promotes mitochondrial injury and death in tubular cells. Environ. Toxicol. 2018, 33, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Mohsenzadeh, M.S.; Razavi, B.M.; Imenshahidi, M.; Mohajeri, S.A.; Rameshrad, M.; Hosseinzadeh, H. Evaluation of green tea extract and epigallocatechin gallate effects on bisphenol A-induced vascular toxicity in isolated rat aorta and cytotoxicity in human umbilical vein endothelial cells. Phyther. Res. 2021, 35, 996–1009. [Google Scholar] [CrossRef]
- Rossman, M.J.; Kaplon, R.E.; Hill, S.D.; McNamara, M.N.; Santos-Parker, J.R.; Pierce, G.L.; Seals, D.R.; Donato, A.J. Endothelial cell senescence with aging in healthy humans: Prevention by habitual exercise and relation to vascular endothelial function. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H890–H895. [Google Scholar] [CrossRef] [PubMed]
- Engin, A.B.; Engin, A. The effect of environmental Bisphenol A exposure on breast cancer associated with obesity. Environ. Toxicol. Pharmacol. 2021, 81, 103544. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Li, H.; Van Deursen, J.M. Senescent cells: A therapeutic target for cardiovascular disease. J. Clin. Investig. 2018, 128, 1217–1228. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Matos, L.; Gouveia, A.M.; Almeida, H. ER stress response in human cellular models of senescence. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2015, 70, 924–935. [Google Scholar] [CrossRef] [Green Version]
- Bae, S.; Hong, Y.-C.C. Exposure to bisphenol a from drinking canned beverages increases blood pressure: Randomized crossover trial. Hypertension 2015, 65, 313–319. [Google Scholar] [CrossRef]
- Moreno-Gómez-Toledano, R.; Arenas, M.I.; Sánchez-Esteban, S.; Cook, A.; Saura, M.; Bosch, R.J. Critical Analysis of Human Exposure to Bisphenol a and its Novel Implications on Renal, Cardiovascular and Hypertensive Diseases. In Hot Topics in Endocrinology and Metabolism [Working Title]; IntechOpen: London, UK, 2021; pp. 1–20. [Google Scholar]
- Song, X.X.; Miao, M.H.; Zhou, X.Y.; Li, D.K.; Tian, Y.P.; Liang, H.; Li, R.S.; Yuan, W. Bisphenol A Exposure and Sperm ACHE Hydroxymethylation in Men. Int. J. Environ. Res. Public Health 2019, 16, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.P.; Zhou, X.Y.; Miao, M.H.; Li, D.-K.K.; Wang, Z.L.; Li, R.S.; Liang, H.; Yuan, W. Association of Bisphenol A Exposure with LINE-1 Hydroxymethylation in Human Semen. Int. J. Environ. Res. Public Health 2018, 15, 1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hines, C.J.; Jackson, M.V.; Deddens, J.A.; Clark, J.C.; Ye, X.Y.; Christianson, A.L.; Meadows, J.W.; Calafat, A.M. Urinary Bisphenol A (BPA) Concentrations among Workers in Industries that Manufacture and Use BPA in the USA. Ann. Work Expo. Health 2017, 61, 164–182. [Google Scholar] [CrossRef]
- He, Y.; Miao, M.; Wu, C.; Yuan, W.; Gao, E.; Zhou, Z.; Li, D.-K. Occupational Exposure Levels of Bisphenol A among Chinese Workers. J. Occup. Health 2009, 51, 432–436. [Google Scholar] [CrossRef] [Green Version]
- Mas, S.; Bosch-Panadero, E.; Abaigar, P.; Camarero, V.; Mahillo, I.; Civantos, E.; Sanchez-Ospina, D.; Ruiz-Priego, A.; Egido, J.; Ortiz, A.; et al. Influence of dialysis membrane composition on plasma bisphenol A levels during online hemodiafiltration. PLoS ONE 2018, 13, e0193288. [Google Scholar] [CrossRef] [PubMed]
- Huygh, J.; Clotman, K.; Malarvannan, G.; Covaci, A.; Schepens, T.; Verbrugghe, W.; Dirinck, E.; Van Gaal, L.; Jorens, P.G. Considerable exposure to the endocrine disrupting chemicals phthalates and bisphenol-A in intensive care unit (ICU) patients. Environ. Int. 2015, 81, 64–72. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno-Gómez-Toledano, R.; Sánchez-Esteban, S.; Cook, A.; Mínguez-Moratinos, M.; Ramírez-Carracedo, R.; Reventún, P.; Delgado-Marín, M.; Bosch, R.J.; Saura, M. Bisphenol A Induces Accelerated Cell Aging in Murine Endothelium. Biomolecules 2021, 11, 1429. https://doi.org/10.3390/biom11101429
Moreno-Gómez-Toledano R, Sánchez-Esteban S, Cook A, Mínguez-Moratinos M, Ramírez-Carracedo R, Reventún P, Delgado-Marín M, Bosch RJ, Saura M. Bisphenol A Induces Accelerated Cell Aging in Murine Endothelium. Biomolecules. 2021; 11(10):1429. https://doi.org/10.3390/biom11101429
Chicago/Turabian StyleMoreno-Gómez-Toledano, Rafael, Sandra Sánchez-Esteban, Alberto Cook, Marta Mínguez-Moratinos, Rafael Ramírez-Carracedo, Paula Reventún, María Delgado-Marín, Ricardo J. Bosch, and Marta Saura. 2021. "Bisphenol A Induces Accelerated Cell Aging in Murine Endothelium" Biomolecules 11, no. 10: 1429. https://doi.org/10.3390/biom11101429
APA StyleMoreno-Gómez-Toledano, R., Sánchez-Esteban, S., Cook, A., Mínguez-Moratinos, M., Ramírez-Carracedo, R., Reventún, P., Delgado-Marín, M., Bosch, R. J., & Saura, M. (2021). Bisphenol A Induces Accelerated Cell Aging in Murine Endothelium. Biomolecules, 11(10), 1429. https://doi.org/10.3390/biom11101429