
Annotation of 1350 Common Genetic Variants of the 19 ALDH Multigene Family from Global Human Genome Aggregation Database (gnomAD)


Abstract
:1. Introduction
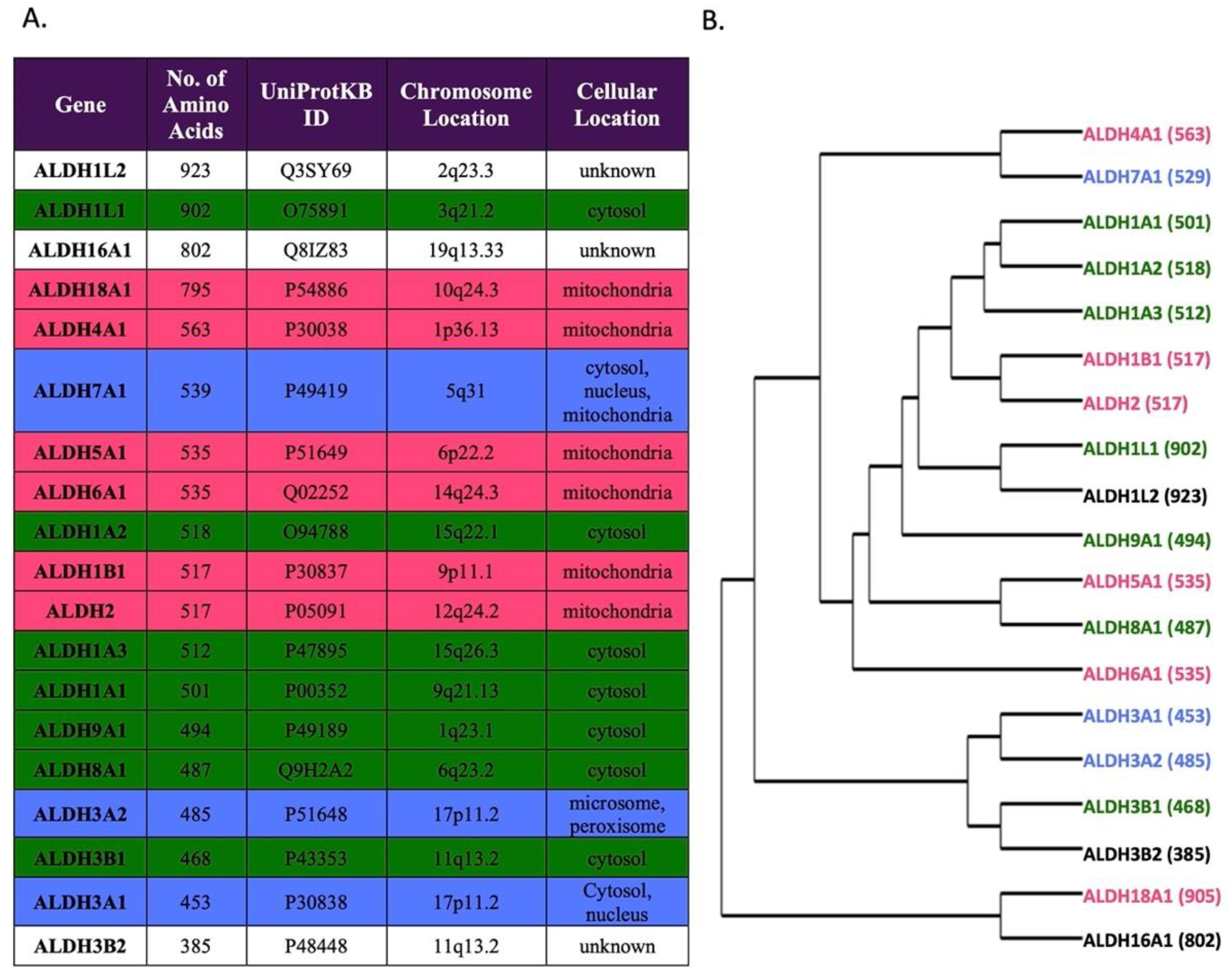
2. Materials and Methods
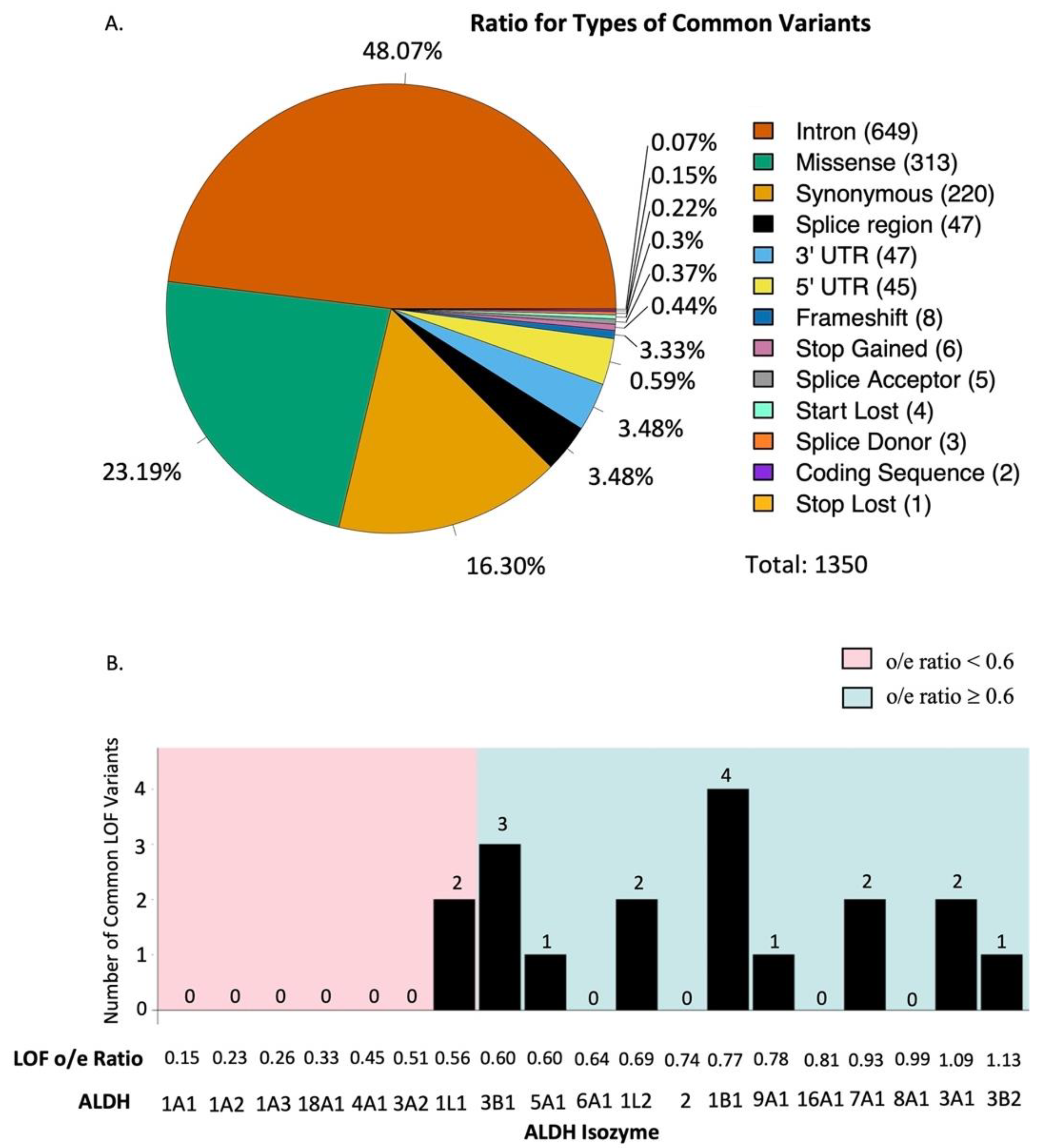
3. Results
3.1. Analysis of Common ALDH Variants
3.2. Gene Constraint Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pray, L. Eukaryotic genome complexity. Nat. Educ. 2008, 1, 96. [Google Scholar]
- Vasiliou, V.; Pappa, A.; Estey, T. Role of human aldehyde dehydrogenases in endobiotic and xenobiotic metabolism. Drug Metab. Rev. 2004, 36, 279–299. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, J.; Nakamura, M. DNA-protein crosslink formation by endogenous aldehydes and AP sites. DNA Repair 2020, 88, 102806. [Google Scholar] [CrossRef] [PubMed]
- Marchitti, S.A.; Brocker, C.; Stagos, D.; Vasiliou, V. Non-P450 aldehyde oxidizing enzymes: The aldehyde dehydrogenase superfamily. Expert Opin. Drug Metab. Toxicol. 2008, 4, 697–720. [Google Scholar] [CrossRef] [Green Version]
- Marchitti, S.A.; Deitrich, R.A.; Vasiliou, V. Neurotoxicity and metabolism of the catecholamine-derived 3,4-dihydroxyphenylacetaldehyde and 3,4-dihydroxyphenylglycolaldehyde: The role of aldehyde dehydrogenase. Pharmacol. Rev. 2007, 59, 125–150. [Google Scholar] [CrossRef] [PubMed]
- Zakhari, S. Overview: How is alcohol metabolized by the body? Alcohol Res. Health 2006, 29, 245–254. [Google Scholar] [PubMed]
- Sapkota, M.; Wyatt, T.A. Alcohol, Aldehydes, Adducts and Airways. Biomolecules 2015, 5, 2987–3008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, R.J.; Ludeman, S.M.; Paikoff, S.J.; Pegg, A.E.; Dolan, M.E. Role of MGMT in protecting against cyclophosphamide-induced toxicity in cells and animals. DNA Repair 2007, 6, 1145–1154. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, A.; Hsu, L.C.; Dave, V. Retinal oxidation activity and biological role of human cytosolic aldehyde dehydrogenase. Enzyme 1992, 46, 239–244. [Google Scholar] [CrossRef]
- Grun, F.; Hirose, Y.; Kawauchi, S.; Ogura, T.; Umesono, K. Aldehyde dehydrogenase 6, a cytosolic retinaldehyde dehydrogenase prominently expressed in sensory neuroepithelia during development. J. Biol. Chem. 2000, 275, 41210–41218. [Google Scholar] [CrossRef] [Green Version]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Mills, P.B.; Struys, E.; Jakobs, C.; Plecko, B.; Baxter, P.; Baumgartner, M.; Willemsen, M.A.; Omran, H.; Tacke, U.; Uhlenberg, B.; et al. Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nat. Med. 2006, 12, 307–309. [Google Scholar] [CrossRef] [PubMed]
- Klyosov, A.A. Kinetics and specificity of human liver aldehyde dehydrogenases toward aliphatic, aromatic, and fused polycyclic aldehydes. Biochemistry 1996, 35, 4457–4467. [Google Scholar] [CrossRef] [PubMed]
- Lassen, N.; Pappa, A.; Black, W.J.; Jester, J.V.; Day, B.J.; Min, E.; Vasiliou, V. Antioxidant function of corneal ALDH3A1 in cultured stromal fibroblasts. Free Radic. Biol. Med. 2006, 41, 1459–1469. [Google Scholar] [CrossRef] [PubMed]
- Pappa, A.; Chen, C.; Koutalos, Y.; Townsend, A.J.; Vasiliou, V. Aldh3a1 protects human corneal epithelial cells from ultraviolet- and 4-hydroxy-2-nonenal-induced oxidative damage. Free Radic. Biol. Med. 2003, 34, 1178–1189. [Google Scholar] [CrossRef]
- Estey, T.; Cantore, M.; Weston, P.A.; Carpenter, J.F.; Petrash, J.M.; Vasiliou, V. Mechanisms involved in the protection of UV-induced protein inactivation by the corneal crystallin ALDH3A1. J. Biol. Chem. 2007, 282, 4382–4392. [Google Scholar] [CrossRef] [Green Version]
- Nsg, D.C. Beliefs and practices of antenatal mothers in a rural setting. Nurs. J. India 1995, 86, 4–6. [Google Scholar]
- Uma, L.; Hariharan, J.; Sharma, Y.; Balasubramanian, D. Corneal aldehyde dehydrogenase displays antioxidant properties. Exp. Eye Res. 1996, 63, 117–120. [Google Scholar] [CrossRef]
- Vasiliou, V.; Nebert, D.W. Analysis and update of the human aldehyde dehydrogenase (ALDH) gene family. Hum. Genom. 2005, 2, 138–143. [Google Scholar] [CrossRef] [Green Version]
- Jackson, B.; Brocker, C.; Thompson, D.C.; Black, W.; Vasiliou, K.; Nebert, D.W.; Vasiliou, V. Update on the aldehyde dehydrogenase gene (ALDH) superfamily. Hum. Genom. 2011, 5, 283–303. [Google Scholar] [CrossRef] [Green Version]
- Shortall, K.; Djeghader, A.; Magner, E.; Soulimane, T. Insights into Aldehyde Dehydrogenase Enzymes: A Structural Perspective. Front. Mol. Biosci. 2021, 8, 659550. [Google Scholar] [CrossRef]
- Stagos, D.; Chen, Y.; Brocker, C.; Donald, E.; Jackson, B.C.; Orlicky, D.J.; Thompson, D.C.; Vasiliou, V. Aldehyde dehydrogenase 1B1: Molecular cloning and characterization of a novel mitochondrial acetaldehyde-metabolizing enzyme. Drug Metab. Dispos. 2010, 38, 1679–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, L.C.; Chang, W.C. Cloning and characterization of a new functional human aldehyde dehydrogenase gene. J. Biol. Chem. 1991, 266, 12257–12265. [Google Scholar] [CrossRef]
- Nene, A.; Chen, C.H.; Disatnik, M.H.; Cruz, L.; Mochly-Rosen, D. Aldehyde dehydrogenase 2 activation and coevolution of its epsilonPKC-mediated phosphorylation sites. J. Biomed. Sci. 2017, 24, 3. [Google Scholar] [CrossRef] [Green Version]
- Abriola, D.P.; Fields, R.; Stein, S.; MacKerell, A.D., Jr.; Pietruszko, R. Active site of human liver aldehyde dehydrogenase. Biochemistry 1987, 26, 5679–5684. [Google Scholar] [CrossRef] [PubMed]
- Hempel, J.; Liu, Z.J.; Perozich, J.; Rose, J.; Lindahl, R.; Wang, B.C. Conserved residues in the aldehyde dehydrogenase family. Locations in the class 3 tertiary structure. Adv. Exp. Med. Biol. 1997, 414, 9–13. [Google Scholar] [CrossRef]
- Liu, Z.J.; Sun, Y.J.; Rose, J.; Chung, Y.J.; Hsiao, C.D.; Chang, W.R.; Kuo, I.; Perozich, J.; Lindahl, R.; Hempel, J.; et al. The first structure of an aldehyde dehydrogenase reveals novel interactions between NAD and the Rossmann fold. Nat. Struct. Biol. 1997, 4, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Vasiliou, V.; Pappa, A. Polymorphisms of human aldehyde dehydrogenases. Consequences for drug metabolism and disease. Pharmacology 2000, 61, 192–198. [Google Scholar] [CrossRef]
- Brooks, P.J.; Enoch, M.A.; Goldman, D.; Li, T.K.; Yokoyama, A. The alcohol flushing response: An unrecognized risk factor for esophageal cancer from alcohol consumption. PLoS Med. 2009, 6, e50. [Google Scholar] [CrossRef]
- Rizzo, W.B.; Carney, G. Sjogren-Larsson syndrome: Diversity of mutations and polymorphisms in the fatty aldehyde dehydrogenase gene (ALDH3A2). Hum. Mutat. 2005, 26, 1–10. [Google Scholar] [CrossRef]
- Kaur, R.; Paria, P.; Saini, A.G.; Suthar, R.; Bhatia, V.; Attri, S.V. Metabolic epilepsy in hyperprolinemia type II due to a novel nonsense ALDH4A1 gene variant. Metab. Brain Dis. 2021, 36, 1413–1417. [Google Scholar] [CrossRef] [PubMed]
- Motte, J.; Fisse, A.L.; Grüter, T.; Schneider, R.; Breuer, T.; Lücke, T.; Krueger, S.; Nguyen, H.P.; Gold, R.; Ayzenberg, I.; et al. Novel variants in a patient with late-onset hyperprolinemia type II: Diagnostic key for status epilepticus and lactic acidosis. BMC Neurol. 2019, 19, 345. [Google Scholar] [CrossRef] [PubMed]
- Pearl, P.L.; Wiwattanadittakul, N.; Roullet, J.B.; Gibson, K.M. Succinic Semialdehyde Dehydrogenase Deficiency. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Korasick, D.A.; Tanner, J.J. Impact of missense mutations in the ALDH7A1 gene on enzyme structure and catalytic function. Biochimie 2021, 183, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Koppaka, V.; Thompson, D.C.; Chen, Y.; Ellermann, M.; Nicolaou, K.C.; Juvonen, R.O.; Petersen, D.; Deitrich, R.A.; Hurley, T.D.; Vasiliou, V. Aldehyde dehydrogenase inhibitors: A comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application. Pharmacol. Rev. 2012, 64, 520–539. [Google Scholar] [CrossRef] [Green Version]
- Priyadharshini Christy, J.; George Priya Doss, C. Single amino acid polymorphism in aldehyde dehydrogenase gene superfamily. Front. Biosci. 2015, 20, 335–376. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Collins, R.L.; Brand, H.; Karczewski, K.J.; Zhao, X.; Alföldi, J.; Francioli, L.C.; Khera, A.V.; Lowther, C.; Gauthier, L.D.; Wang, H.; et al. A structural variation reference for medical and population genetics. Nature 2020, 581, 444–451. [Google Scholar] [CrossRef]
- Cummings, B.B.; Karczewski, K.J.; Kosmicki, J.A.; Seaby, E.G.; Watts, N.A.; Singer-Berk, M.; Mudge, J.M.; Karjalainen, J.; Satterstrom, F.K.; O’Donnell-Luria, A.H.; et al. Transcript expression-aware annotation improves rare variant interpretation. Nature 2020, 581, 452–458. [Google Scholar] [CrossRef]
- Minikel, E.V.; Karczewski, K.J.; Martin, H.C.; Cummings, B.B.; Whiffin, N.; Rhodes, D.; Alföldi, J.; Trembath, R.C.; van Heel, D.A.; Daly, M.J.; et al. Evaluating drug targets through human loss-of-function genetic variation. Nature 2020, 581, 459–464. [Google Scholar] [CrossRef]
- Wang, Q.; Pierce-Hoffman, E.; Cummings, B.B.; Alfoldi, J.; Francioli, L.C.; Gauthier, L.D.; Hill, A.J.; O’Donnell-Luria, A.H.; Karczewski, K.J.; MacArthur, D.G. Landscape of multi-nucleotide variants in 125,748 human exomes and 15,708 genomes. Nat. Commun. 2020, 11, 2539. [Google Scholar] [CrossRef]
- Whiffin, N.; Armean, I.M.; Kleinman, A.; Marshall, J.L.; Minikel, E.V.; Goodrich, J.K.; Quaife, N.M.; Cole, J.B.; Wang, Q.; Karczewski, K.J.; et al. The effect of LRRK2 loss-of-function variants in humans. Nat. Med. 2020, 26, 869–877. [Google Scholar] [CrossRef]
- Whiffin, N.; Karczewski, K.J.; Zhang, X.; Chothani, S.; Smith, M.J.; Evans, D.G.; Roberts, A.; Quaife, N.M.; Schafer, S.; Rackham, O.; et al. Characterising the loss-of-function impact of 5′ untranslated region variants in 15,708 individuals. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Borinskaya, S.; Yoshimura, K.; Kal’Ina, N.; Marusin, A.; Stepanov, V.; Qin, Z.; Khaliq, S.; Lee, M.-Y.; Yang, Y.; et al. Refined Geographic Distribution of the OrientalALDH2*504Lys(nee487Lys) Variant. Ann. Hum. Genet. 2009, 73, 335–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, A.; Huang, I. Molecular abnormality of an inactive aldehyde dehydrogenase variant commonly found in Orientals. PNAS 1984, 81, 258–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinshilboum, R.; Wang, L. Pharmacogenomics: Bench to bedside. Nat. Rev. Drug Discov. 2004, 3, 739–748. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 229–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaser, R.; Adusumalli, S.; Leng, S.-N.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nat. Methods 2015, 11, 1–9. [Google Scholar] [CrossRef]
- Larson, H.N.; Weiner, H.; Hurley, T.D. Disruption of the coenzyme binding site and dimer interface revealed in the crystal structure of mitochondrial aldehyde dehydrogenase “Asian” variant. J. Biol. Chem. 2005, 280, 30550–30556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sophos, N.A.; Vasiliou, V. Aldehyde dehydrogenase gene superfamily: The 2002 update. Chem. Biol. Interact. 2003, 143, 5–22. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.; Cummings, B.; et al. The ExAC browser: Displaying reference data information from over 60 000 exomes. Nucleic Acids Res. 2017, 45, D840–D845. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-H.; Ferreira, J.C.B.; Joshi, A.U.; Stevens, M.C.; Li, S.-J.; Hsu, J.H.-M.; Maclean, R.; Ferreira, N.D.; Cervantes, P.R.; Martinez, D.D.; et al. Novel and prevalent non-East Asian ALDH2 variants; Implications for global susceptibility to aldehydes’ toxicity. EBioMedicine 2020, 55, 102753. [Google Scholar] [CrossRef]
- Petrović, J.; Pešić, V.; Lauschke, V.M. Frequqencies of clinically important CYP2C19 and CYP2D6 alleles are graded across Europe. Eur. J. Hum. Genet. 2020, 28, 88–94. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Gene | No. of Amino Acid | No. of Variants ≥0.1% | Intron | Missense | Synonymous | Splice Region | 3’ UTR | 5’ UTR | Frameshift | Stop Gained | Splice Acceptor | Start Lost | Splice Donor | Coding Sequence | Stop Lost |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ALDH1A1 | 501 | 49 | 32 | 5 | 5 | 4 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| ALDH1A2 | 518 | 68 | 45 | 6 | 8 | 2 | 1 | 5 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| ALDH1A3 | 512 | 70 | 33 | 10 | 17 | 3 | 5 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| ALDH1B1 | 517 | 36 | 0 | 19 | 10 | 0 | 3 | 0 | 3 | 1 | 0 | 0 | 0 | 0 | 0 |
| ALDH1L1 | 902 | 124 | 63 | 26 | 19 | 7 | 4 | 1 | 0 | 2 | 1 | 0 | 1 | 0 | 0 |
| ALDH1L2 | 923 | 109 | 72 | 19 | 9 | 2 | 3 | 2 | 0 | 2 | 0 | 0 | 0 | 0 | 0 |
| ALDH2 | 517 | 47 | 26 | 9 | 9 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| ALDH3A1 | 453 | 86 | 42 | 24 | 12 | 0 | 4 | 1 | 1 | 0 | 1 | 1 | 0 | 0 | 0 |
| ALDH3A2 | 485 | 34 | 15 | 9 | 6 | 2 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| ALDH3B1 | 468 | 78 | 33 | 14 | 15 | 2 | 8 | 0 | 3 | 0 | 1 | 0 | 0 | 2 | 0 |
| ALDH3B2 | 385 | 84 | 35 | 32 | 8 | 1 | 3 | 4 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| ALDH4A1 | 563 | 102 | 51 | 23 | 17 | 2 | 3 | 6 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| ALDH5A1 | 535 | 59 | 19 | 20 | 9 | 4 | 2 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| ALDH6A1 | 535 | 36 | 17 | 6 | 4 | 6 | 2 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| ALDH7A1 | 539 | 74 | 44 | 13 | 9 | 1 | 1 | 4 | 1 | 0 | 0 | 1 | 0 | 0 | 0 |
| ALDH8A1 | 487 | 35 | 6 | 10 | 10 | 4 | 1 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| ALDH9A1 | 494 | 62 | 22 | 22 | 7 | 2 | 1 | 6 | 0 | 0 | 0 | 1 | 1 | 0 | 0 |
| ALDH16A1 | 802 | 136 | 63 | 35 | 33 | 2 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| ALDH18A1 | 795 | 61 | 31 | 11 | 13 | 3 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Total | 1350 | 649 | 313 | 220 | 47 | 47 | 45 | 8 | 6 | 5 | 4 | 3 | 2 | 1 |
| Synonymous Variants | Missense Variants | Loss of Function Variants | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | UniProtKB ID | No. of Amino Acid | Exp. SNVs | Obs. SNVs | Constraint Metrics (o/e Ratio) | Exp. SNVs | Obs. SNVs | Constraint Metrics (o/e Ratio) | Exp. SNVs | Obs. SNVs | Constraint Metrics (o/e Ratio) |
| ALDH1A1 | P00352 | 501 | 101.6 | 106.0 | 1.04 (0.89–1.23) | 276.4 | 199.0 | 0.72 (0.64–0.81) | 27.3 | 4.0 | 0.15 (0.07–0.34) |
| ALDH1A2 | O94788 | 518 | 105.2 | 128.0 | 1.22 (1.05–1.41) | 283.1 | 215.0 | 0.76 (0.68–0.85) | 26.5 | 6.0 | 0.23 (0.12–0.45) |
| ALDH1A3 | P47895 | 512 | 123.0 | 122.0 | 0.99 (0.85–1.15) | 301.2 | 192.0 | 0.64 (0.57–0.72) | 23.2 | 6.0 | 0.26 (0.14–0.51) |
| ALDH18A1 | P54886 | 795 | 165.8 | 158.0 | 0.95 (0.84–1.09) | 441.2 | 325.0 | 0.74 (0.67–0.81) | 39.8 | 13.0 | 0.33 (0.21–0.52) |
| ALDH4A1 | P30038 | 563 | 156.0 | 150.0 | 0.96 (0.84–1.10) | 354.5 | 357.0 | 1.01 (0.92–1.10) | 28.9 | 13.0 | 0.45 (0.29–0.71) |
| ALDH3A2 | P51648 | 485 | 104.9 | 85.0 | 0.81 (0.68–0.97) | 277.0 | 245.0 | 0.88 (0.80–0.98) | 23.6 | 12.0 | 0.51 (0.33–0.82) |
| ALDH1L1 | O75891 | 902 | 235.3 | 241.0 | 1.02 (0.92–1.14) | 552.0 | 512.0 | 0.93 (0.86–1.00) | 45.0 | 25.0 | 0.56 (0.40–0.78) |
| ALDH3B1 | P43353 | 468 | 128.5 | 130.0 | 1.01 (0.88–1.17) | 306.2 | 259.0 | 0.85 (0.76–0.94) | 18.3 | 11.0 | 0.60 (0.38–0.99) |
| ALDH5A1 | P51649 | 535 | 119.5 | 112.0 | 0.94 (0.80–1.10) | 289.1 | 254.0 | 0.88 (0.79–0.97) | 23.4 | 14.0 | 0.60 (0.40–0.94) |
| ALDH6A1 | Q02252 | 535 | 103.6 | 87.0 | 0.84 (0.70–1.00) | 292.3 | 242.0 | 0.83 (0.74–0.92) | 29.5 | 19.0 | 0.64 (0.45–0.94) |
| ALDH1L2 | Q3SY69 | 923 | 183.0 | 167.0 | 0.91 (0.80–1.04) | 500.3 | 414.0 | 0.83 (0.76–0.90) | 49.1 | 34.0 | 0.69 (0.53–0.92) |
| ALDH2 | P05091 | 517 | 134.9 | 143.0 | 1.06 (0.92–1.22) | 316.1 | 250.0 | 0.79 (0.71–0.88) | 24.4 | 18.0 | 0.74 (0.51–1.09) |
| ALDH1B1 | P30837 | 517 | 140.5 | 133.0 | 0.95 (0.82–1.09) | 321.5 | 333.0 | 1.04 (0.95–1.13) | 13.0 | 10.0 | 0.77 (0.47–1.30) |
| ALDH9A1 | P49189 | 494 | 107.7 | 119.0 | 1.10 (0.95–1.29) | 292.0 | 312.0 | 1.07 (0.97–1.17) | 25.5 | 20.0 | 0.78 (0.55–1.14) |
| ALDH16A1 | Q8IZ83 | 802 | 228.6 | 228.0 | 1.00 (0.89–1.11) | 503.3 | 530.0 | 1.05 (0.98–1.13) | 43.1 | 35.0 | 0.81 (0.62–1.08) |
| ALDH7A1 | P49419 | 539 | 105.6 | 108.0 | 1.02 (0.87–1.20) | 295.8 | 278.0 | 0.94 (0.85–1.04) | 37.4 | 35.0 | 0.93 (0.71–1.24) |
| ALDH8A1 | Q9H2A2 | 487 | 122.0 | 115.0 | 0.94 (0.81–1.10) | 291.8 | 268.0 | 0.92 (0.83–1.02) | 18.2 | 18.0 | 0.99 (0.68–1.46) |
| ALDH3A1 | P30838 | 453 | 128.6 | 137.0 | 1.07 (0.93–1.23) | 287.1 | 273.0 | 0.95 (0.86–1.05) | 22.0 | 24.0 | 1.09 (0.79–1.53) |
| ALDH3B2 | P48448 | 385 | 114.9 | 123.0 | 1.07 (0.92–1.24) | 250.2 | 256.0 | 1.02 (0.92–1.14) | 16.0 | 18.0 | 1.13 (0.78–1.66) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-H.; Kraemer, B.R.; Lee, L.; Mochly-Rosen, D. Annotation of 1350 Common Genetic Variants of the 19 ALDH Multigene Family from Global Human Genome Aggregation Database (gnomAD). Biomolecules 2021, 11, 1423. https://doi.org/10.3390/biom11101423
Chen C-H, Kraemer BR, Lee L, Mochly-Rosen D. Annotation of 1350 Common Genetic Variants of the 19 ALDH Multigene Family from Global Human Genome Aggregation Database (gnomAD). Biomolecules. 2021; 11(10):1423. https://doi.org/10.3390/biom11101423
Chicago/Turabian StyleChen, Che-Hong, Benjamin R. Kraemer, Lucia Lee, and Daria Mochly-Rosen. 2021. "Annotation of 1350 Common Genetic Variants of the 19 ALDH Multigene Family from Global Human Genome Aggregation Database (gnomAD)" Biomolecules 11, no. 10: 1423. https://doi.org/10.3390/biom11101423
APA StyleChen, C.-H., Kraemer, B. R., Lee, L., & Mochly-Rosen, D. (2021). Annotation of 1350 Common Genetic Variants of the 19 ALDH Multigene Family from Global Human Genome Aggregation Database (gnomAD). Biomolecules, 11(10), 1423. https://doi.org/10.3390/biom11101423