
The Interactions of H2TMPyP, Analogues and Its Metal Complexes with DNA G-Quadruplexes—An Overview

 ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
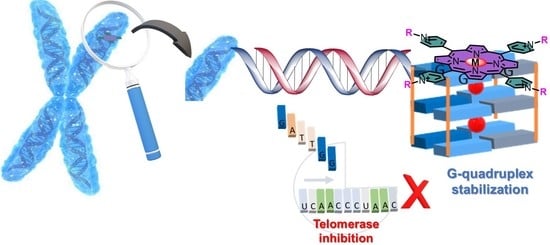
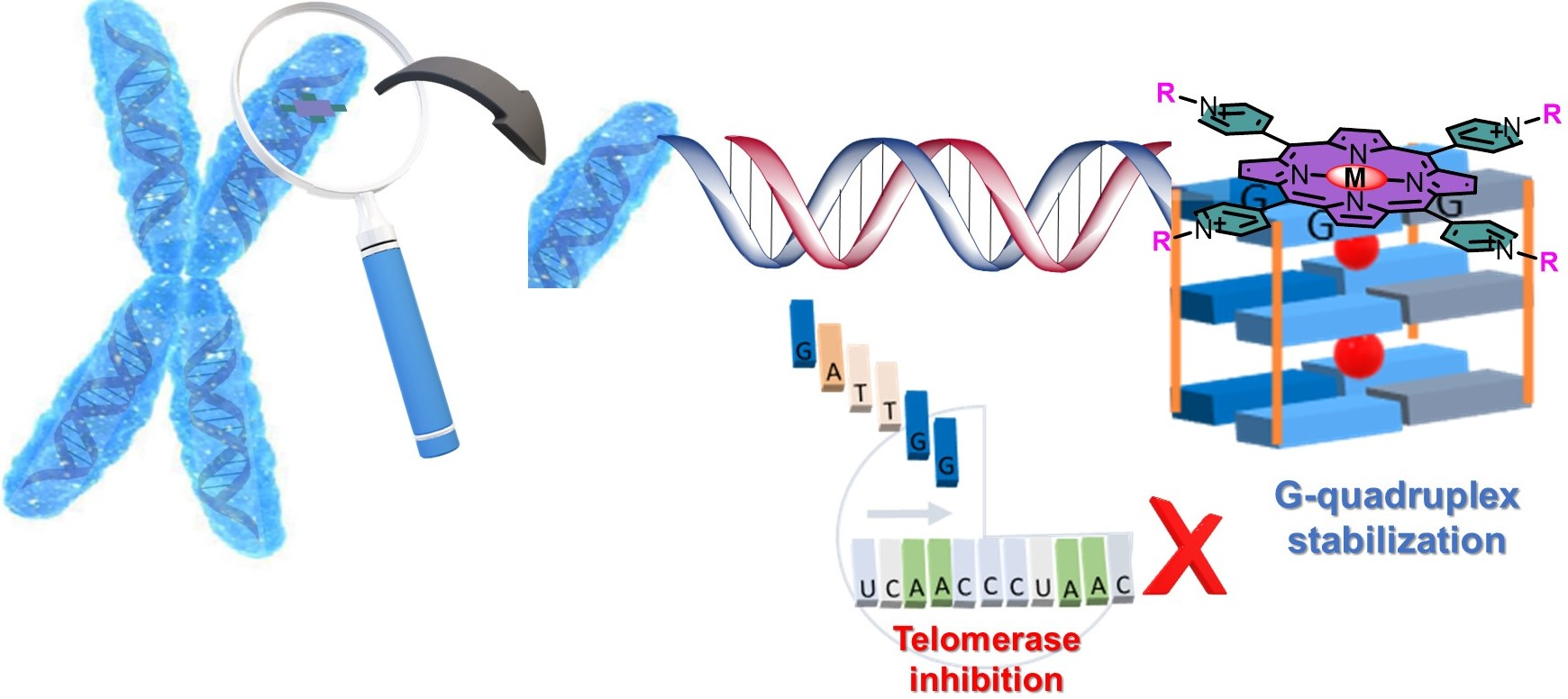
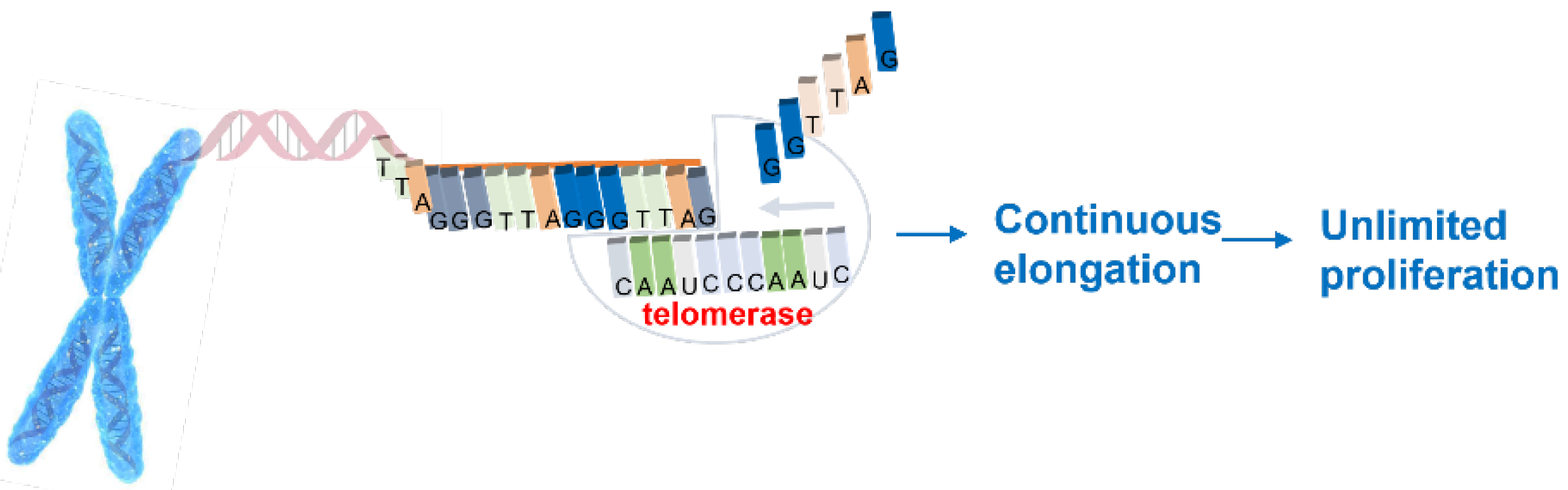
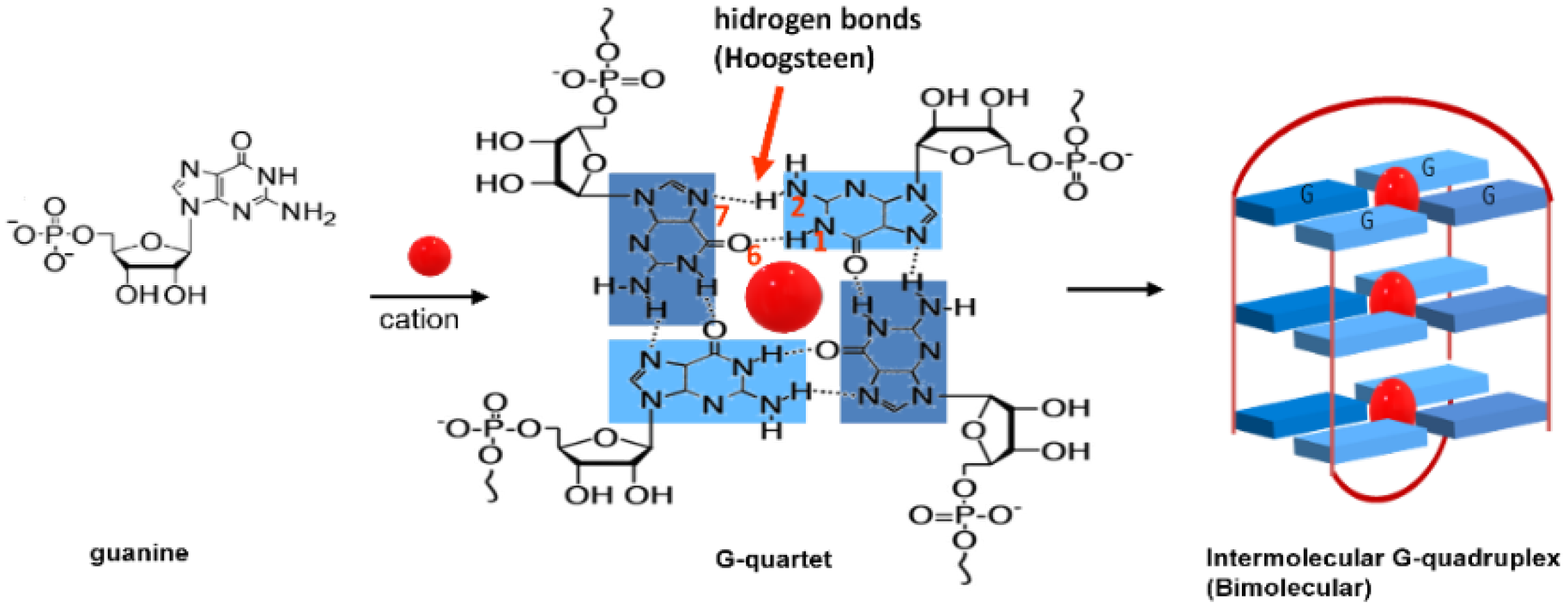
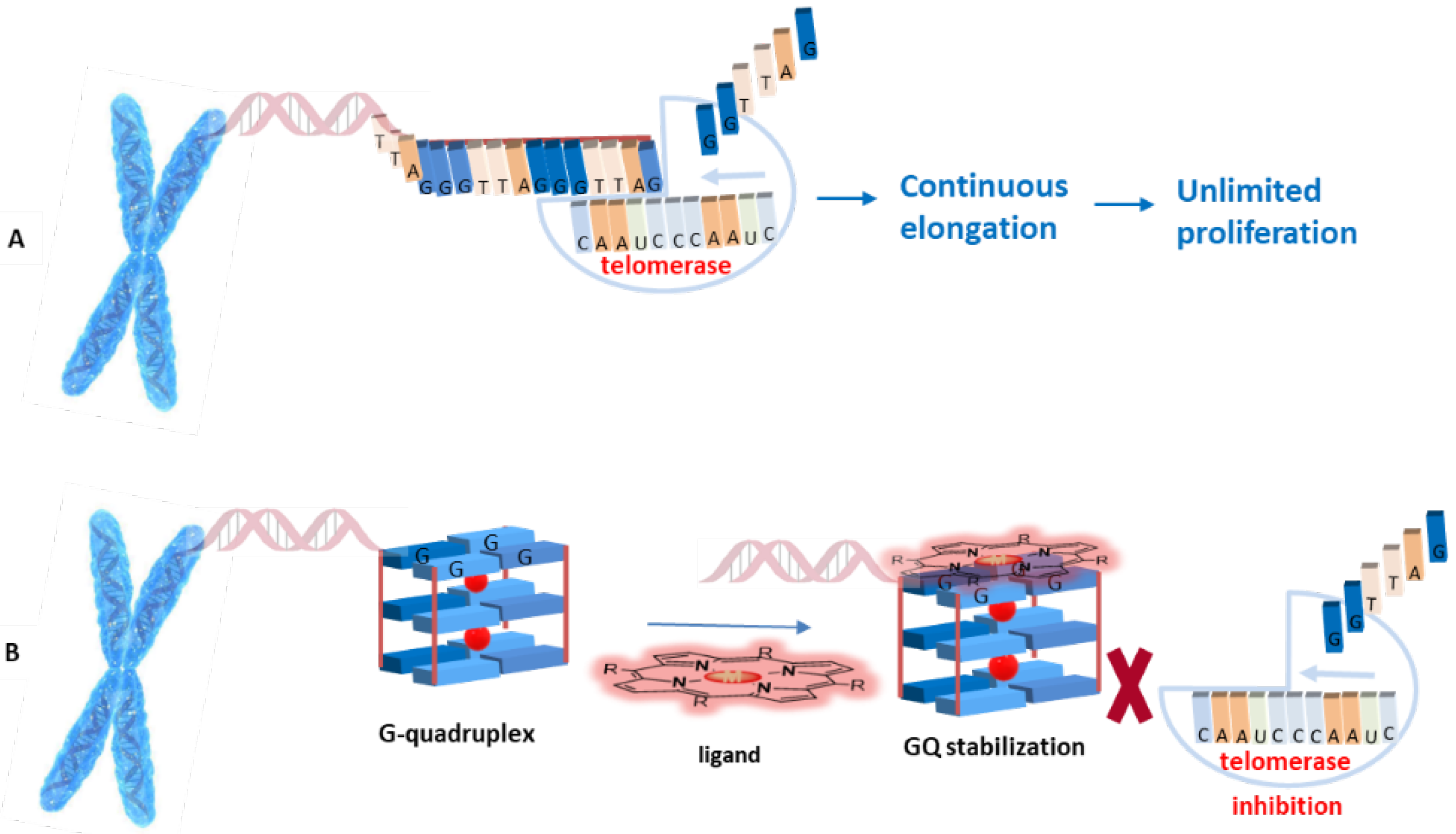
1. Introduction
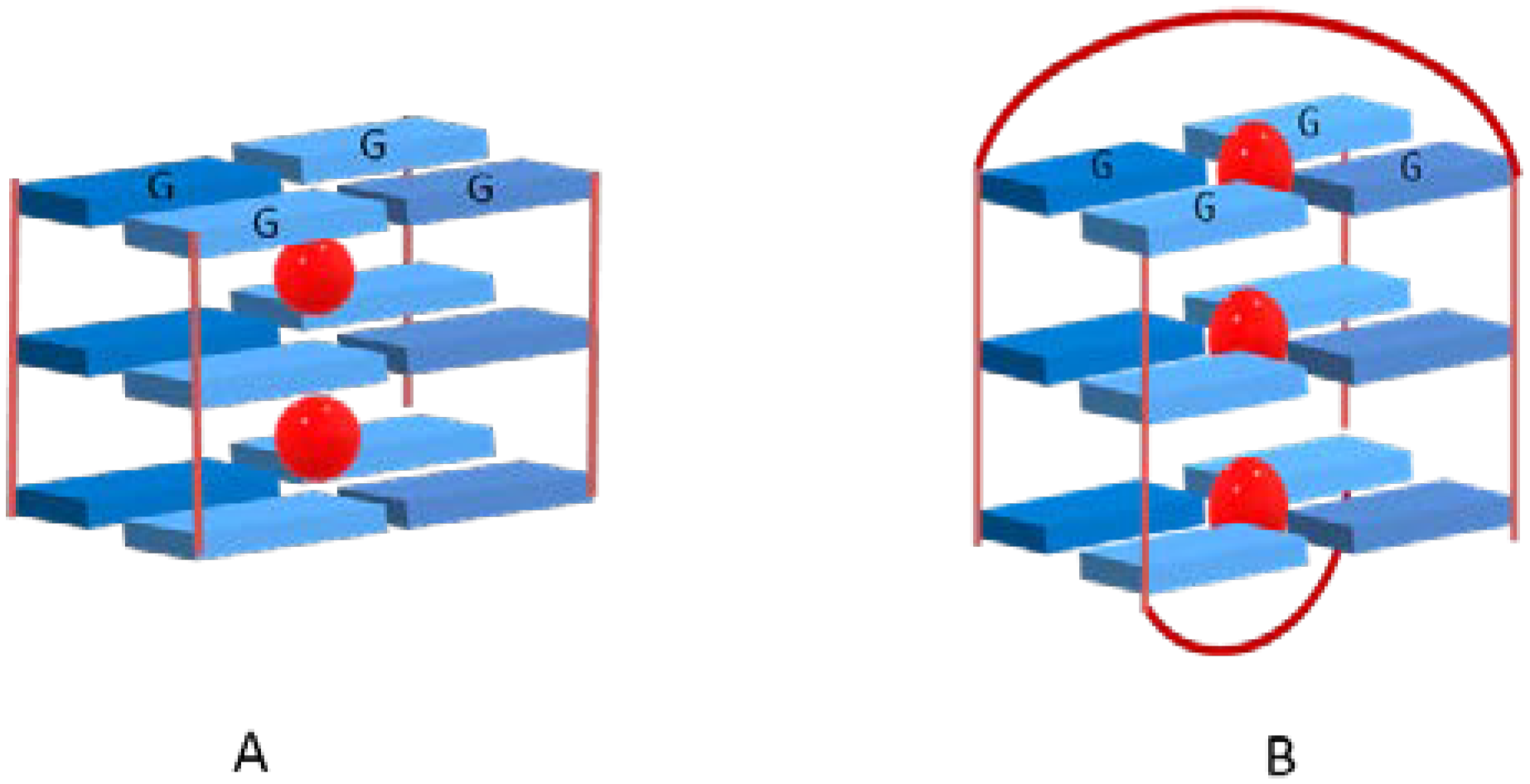
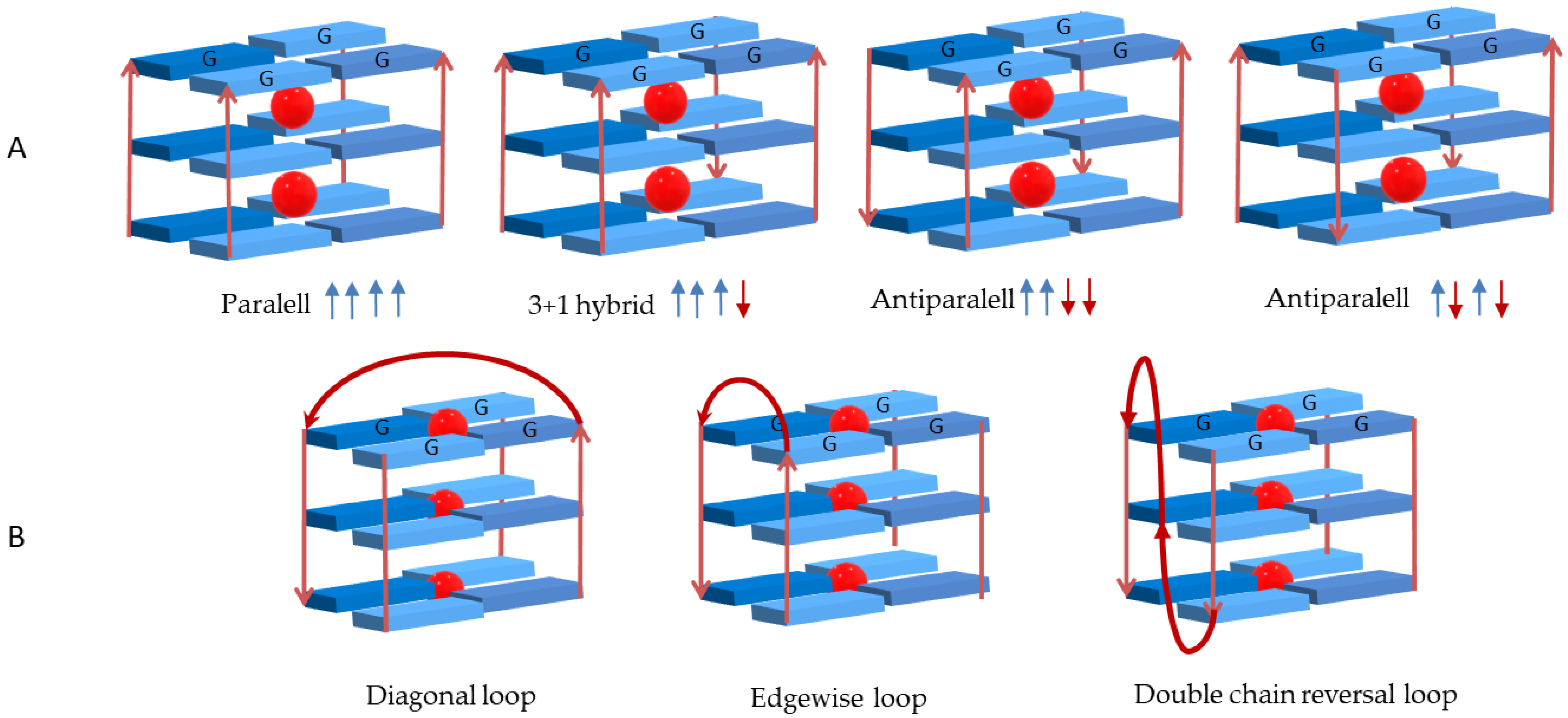
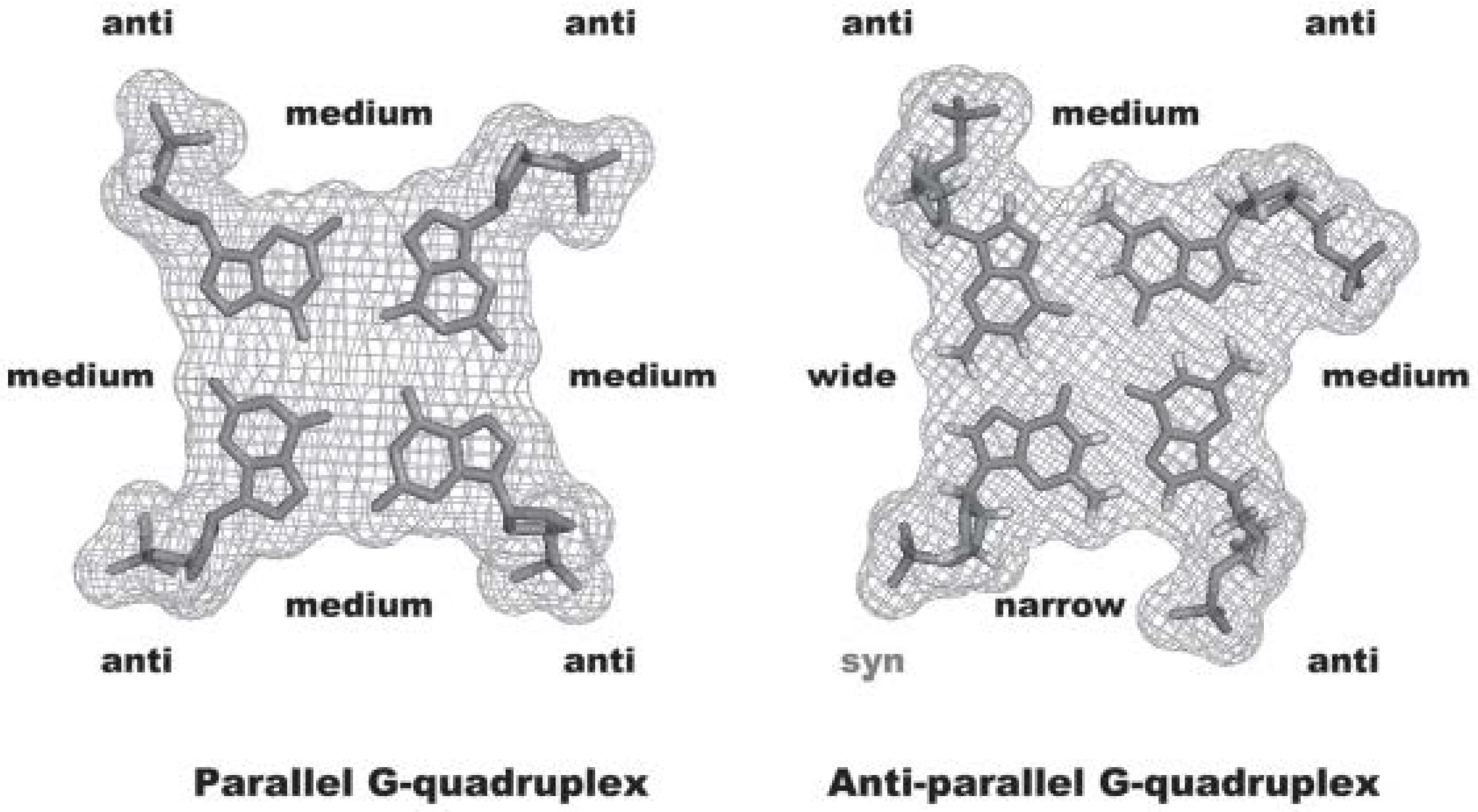
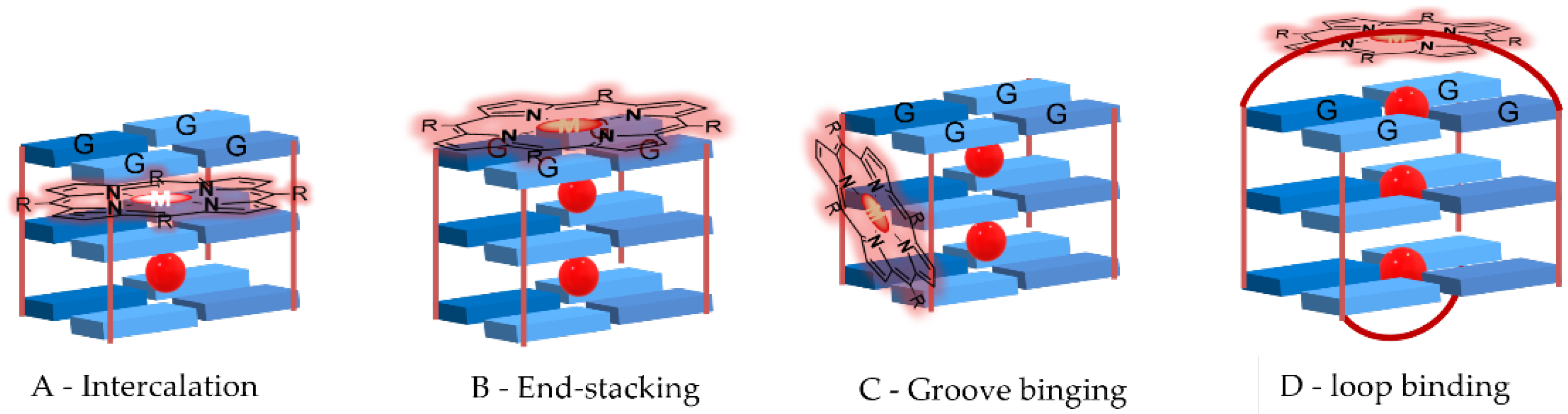
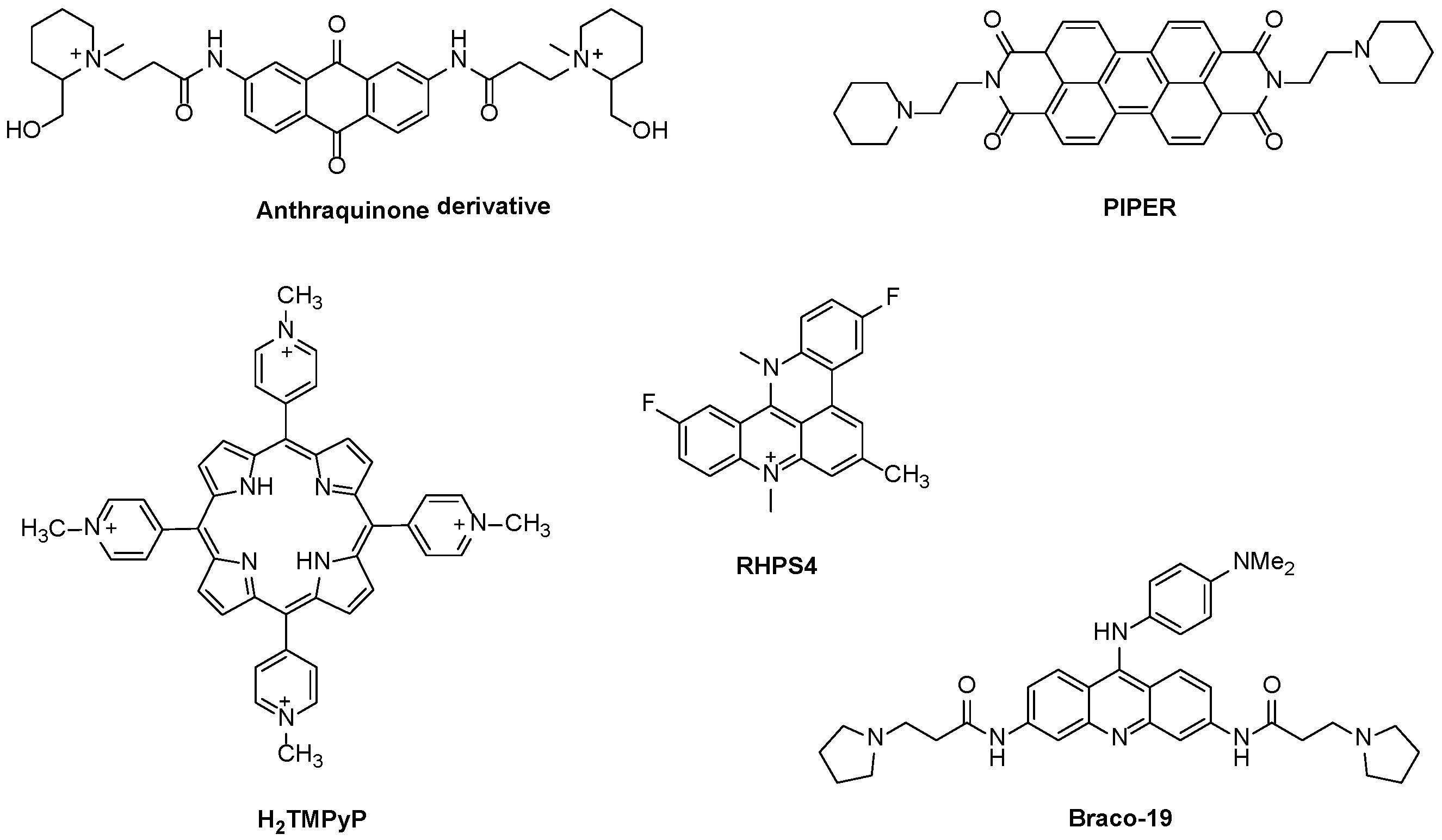
2. Interactive G-Quadruplex Ligands and GQ Recognition Modes
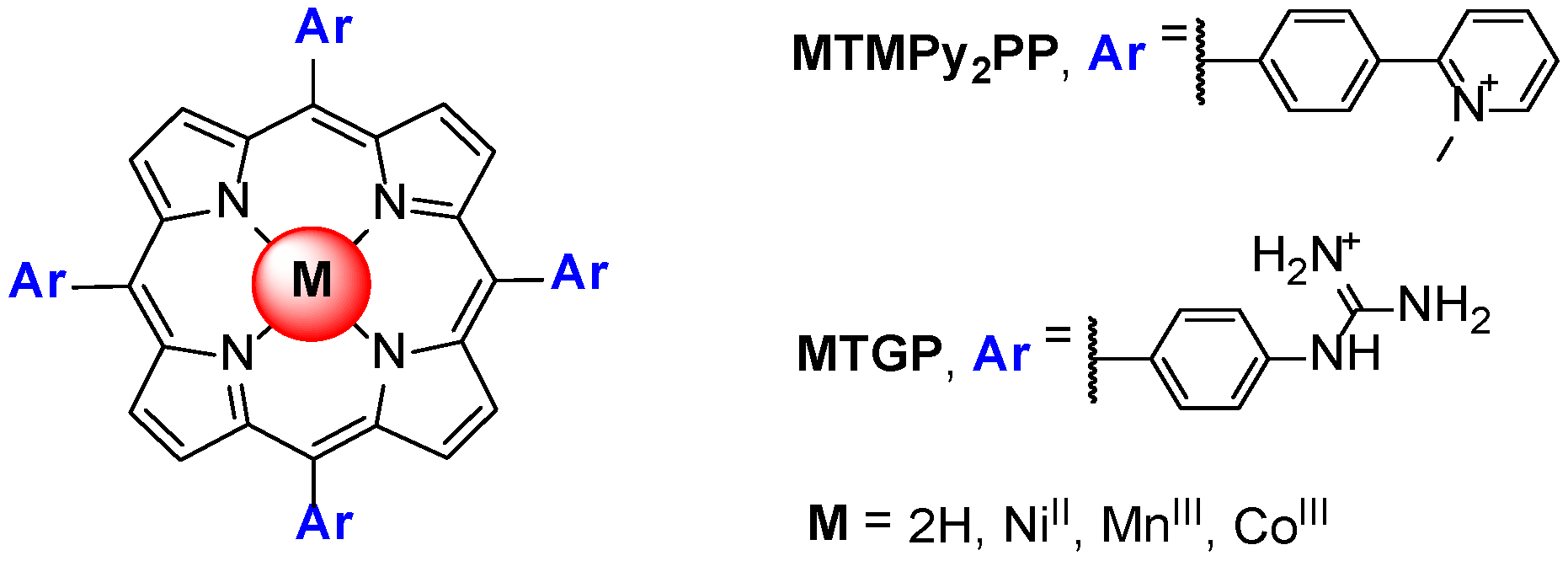
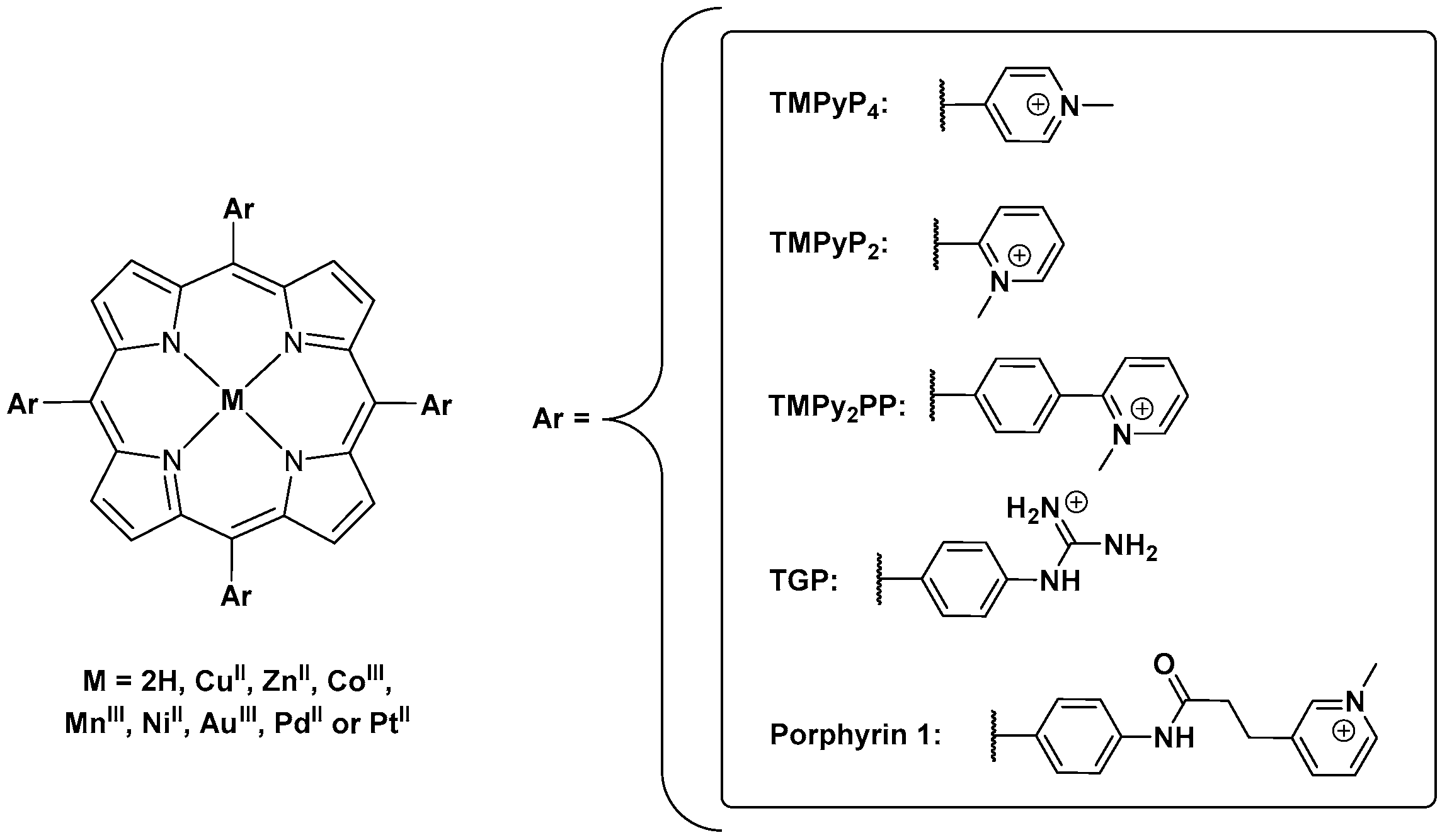
Porphyrins and Metalloporphyrins as Interactive G-Quadruplex Ligands
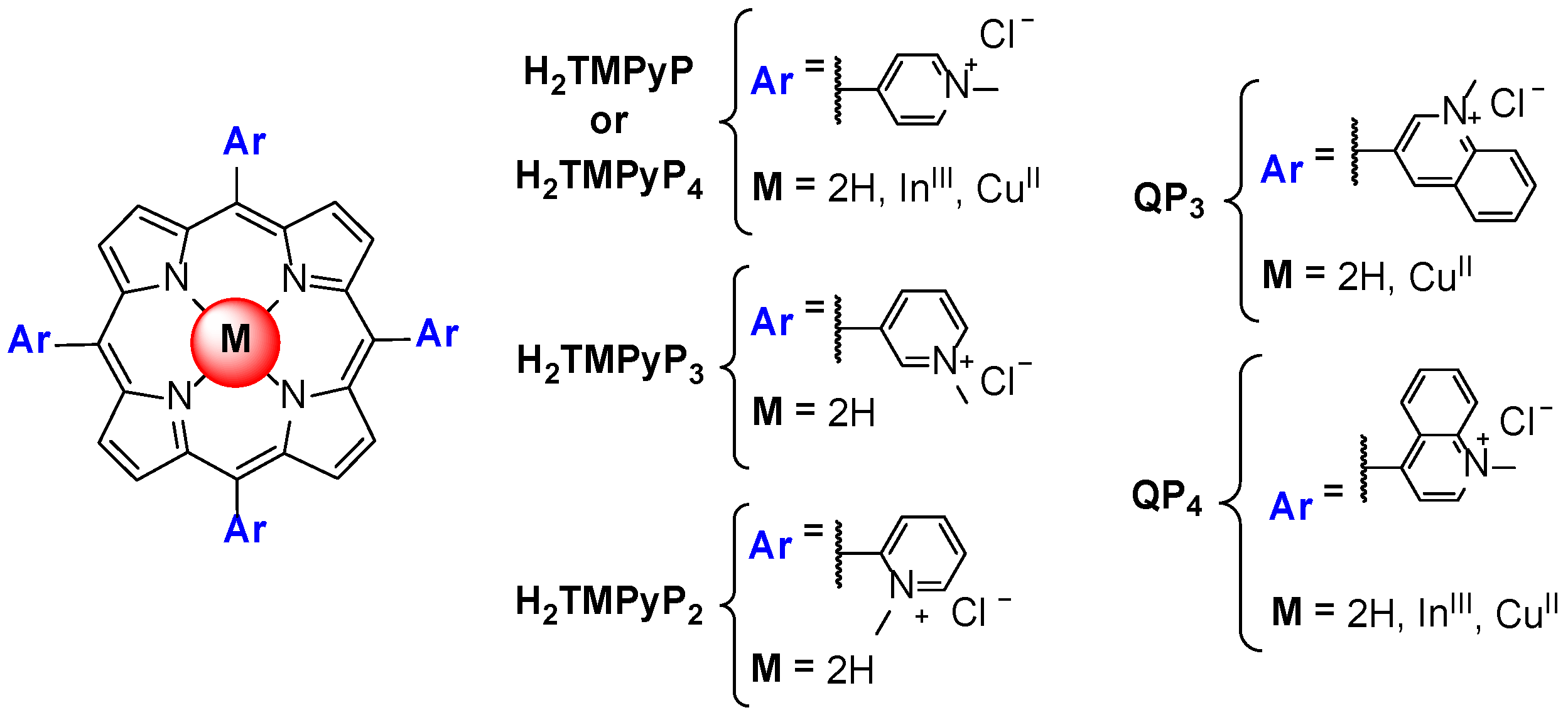
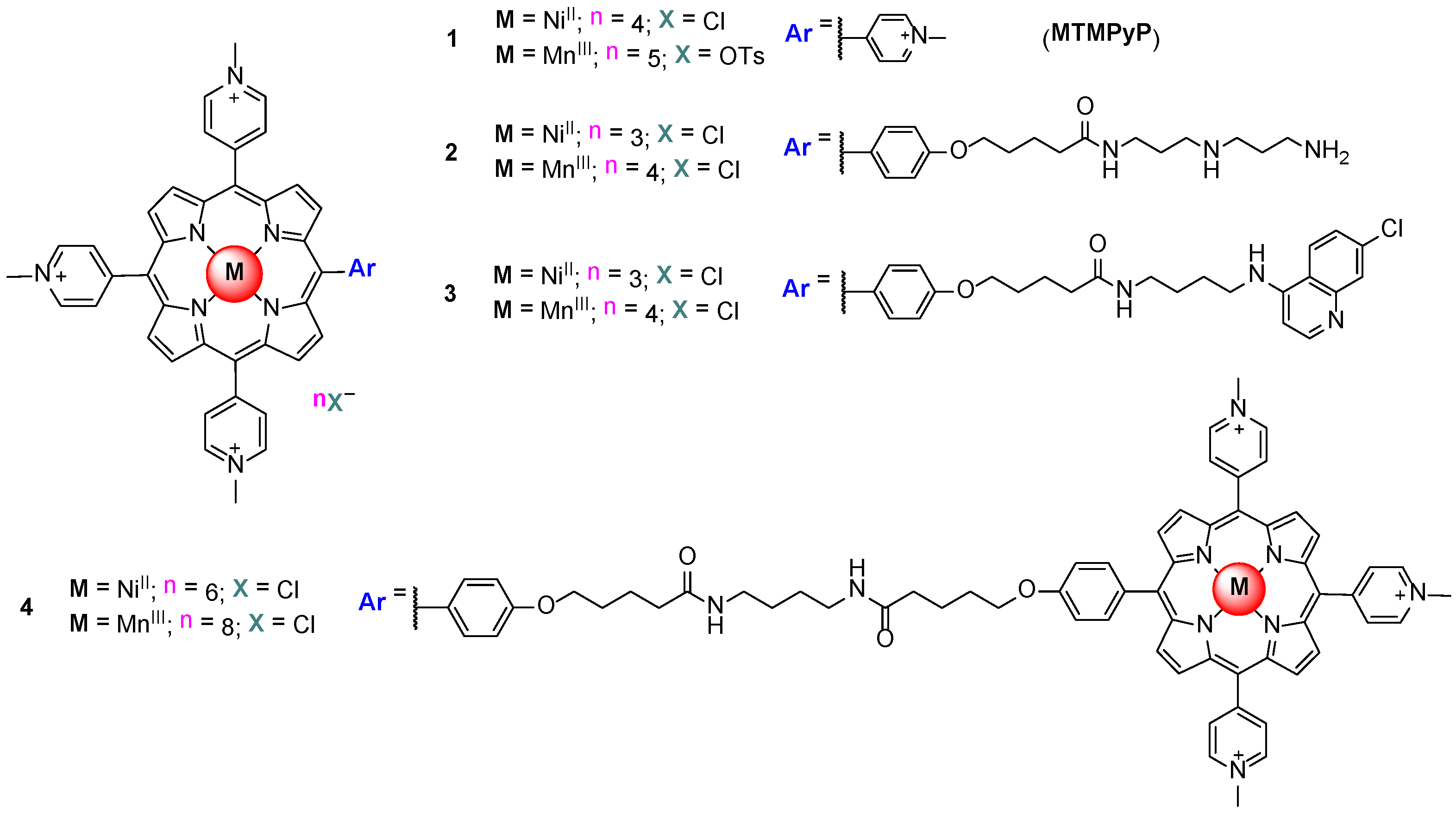
3. Evaluation of the Interactions H2TMPyP and of Its Metal Complexes (M = AgII, ZnII, CoIII, NiII, PdII, MnIII and CuII) with GQ and ds DNA Structures
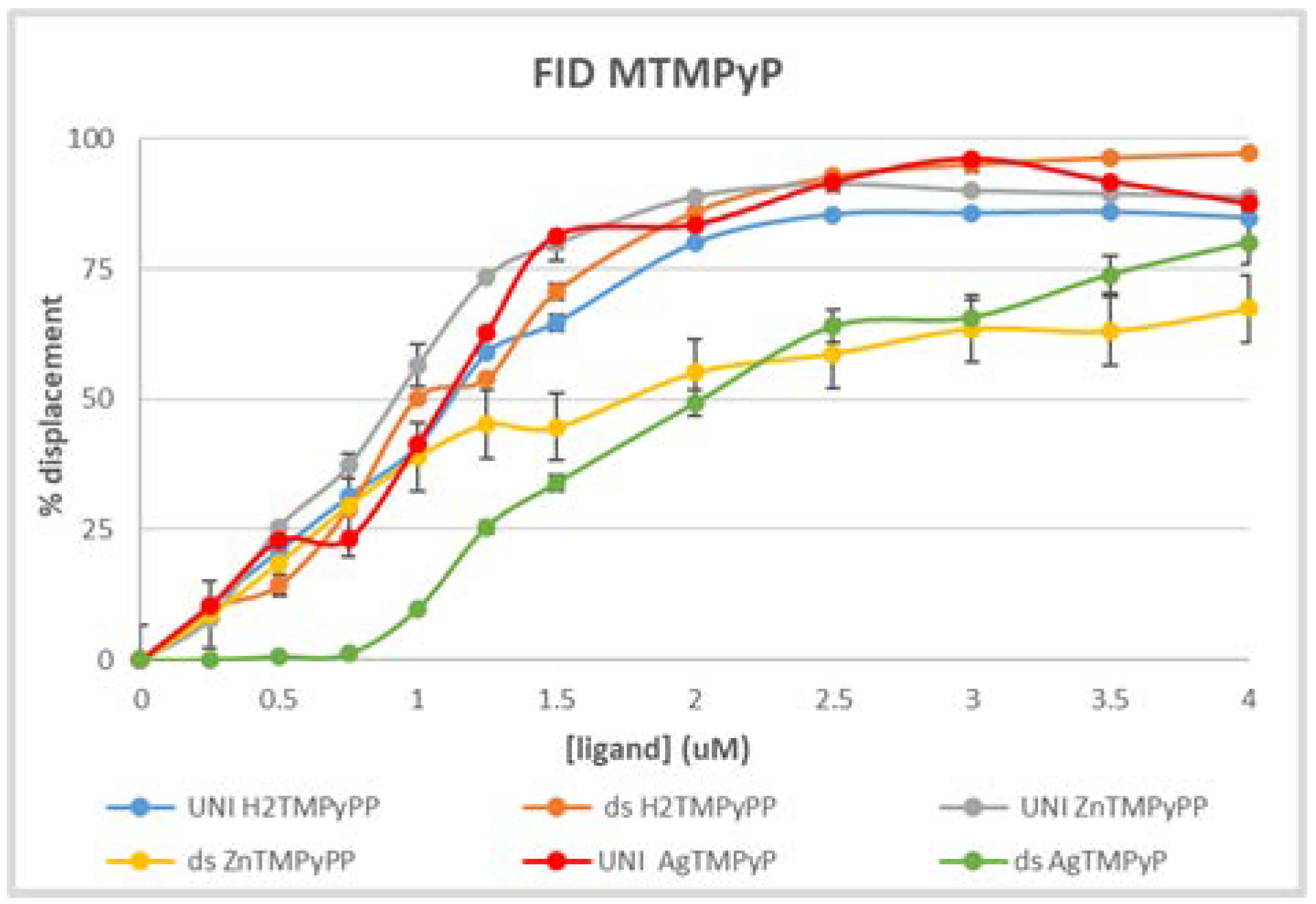
3.1. UV-Vis Spectroscopy
3.2. Fluorescence Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McClintock, B. The stability of broken ends of chromosomes in Zea mays. Genetics 1941, 26, 234. [Google Scholar] [CrossRef]
- Nandakumar, J.; Cech, T.R. Finding the end: Recruitment of telomerase to the telomere. Nat. Rev. Mol. Cell Biol. 2013, 14, 69. [Google Scholar] [CrossRef]
- Blackburn, E.H. A history of telomere biology. In Telomeres, 2nd ed.; de Lange, T., Lundblad, V., Blackburn, E.H., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2006; pp. 1–19. [Google Scholar]
- Shay, J.W.; Zou, Y.; Hiyama, E.; Wright, W.E. Telomerase and cancer. Hum. Mol. Genet. 2001, 10, 677. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, Y. Functional and mechanistic analysis of telomerase: An antitumor drug target. Pharmacol. Ther. 2016, 163, 24. [Google Scholar] [CrossRef]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.C.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific Association of Human Telomerase Activity with Immortal Cells and Cancer. Science 1994, 266, 2011–2016. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Hurley, L.H.; Neidle, S.; Targeting, G. quadruplexes in gene promoters: A novel anticancer strategy? Nat. Rev. Drug Discov. 2011, 10, 261. [Google Scholar] [CrossRef]
- Mishra, S.; Kota, S.; Chaudhary, R.; Misra, H.S. Guanine quadruplexes and their roles in molecular processes. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 482. [Google Scholar] [CrossRef]
- Neidle, S. Quadruplex Nucleic Acids as Novel therapeutic targets. J. Med. Chem. 2016, 16, 5987. [Google Scholar] [CrossRef] [PubMed]
- Collie, G.W.; Parkinson, G.N. The application of DNA and RNA G-quadruplexes to therapeutic medicines. Chem. Soc. Rev. 2011, 40, 5867. [Google Scholar] [CrossRef]
- De Cian, A.; Lacroix, L.; Douarre, C.; Temime-Smaali, N.; Trentesaux, C.; Riou, J.-F.; Mergny, J.-L. Targeting Telomeres and Telomerase. Biochimie 2008, 90, 131. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Okamoto, K. Structural Insights into G-Quadruplexes: Towards New Anticancer Drugs. Future Med. Chem. 2010, 2, 619. [Google Scholar] [CrossRef]
- Hadjiliadis, N.; Sletten, E. Metal Complex-DNA Interactions; Blackwell Publishing Ltd. Wiley: Chippenham, UK, 2009. [Google Scholar]
- Ma, Y.; Iida, K.; Nagasawa, K. Topologies of G-Quadruplexes: Biological Functions and Regulation by Ligands. Biochem. Biophys. Res. Commun. 2020, 531, 3. [Google Scholar] [CrossRef]
- Adrian, M.; Heddi, B.; Phan, A. NMR Spectroscopy of G-Quadruplexes. Methods 2012, 57, 11. [Google Scholar] [CrossRef]
- Largy, E.; Mergny, J.-L.; Gabelica, V. Role of Alkali Metal Ions in G-Quadruplex Nucleic Acid Structure and Stability. The Alkali Metal Ions: Their Role for Life; Springer: Berlin/Heidelberg, Germany, 2016; pp. 203–258. [Google Scholar]
- Mohanty, D.; Bansa, M. Conformational Polymorphism in G-Tetraplex Structures: Strand Reversal by Base Flipover or Sugar Flipover. Nucleic Acids Res. 1993, 21, 1767. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huang, Z.-S.; Tan, J.-H.; Ou, T.-M.; Li, D.; Gu, L.-Q. G-Quadruplex DNA and its Ligands in Anticancer Therapy. In Medicinal Chemistry of Nucleic Acids; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; Wiley Chap 5; pp. 206–257. [Google Scholar]
- Tanpure, A.A.; Srivatsan, S.G. Conformation-sensitive nucleoside analogues as topology-specific fluorescence turn-on probes for DNA and RNA G-quadruplexes. Nucleic Acids Res. 2015, 43, e149. [Google Scholar] [CrossRef]
- Schrank, Z.; Khan, N.; Osude, C.; Singh, S.; Miller, R.J.; Merrick, C.; Mabel, A.; Kuckovic, A.; Puri, N. Oligonucleotides targeting telomeres and telomerase in cancer. Molecules 2018, 9, 2267. [Google Scholar] [CrossRef] [PubMed]
- Yaku, H.; Murashima, T.; Miyoshi, D.; Sugimoto, N. Specific binding of anionic porphyrin and phthalocyanine to the G-quadruplex with a variety of in vitro and in vivo applications. Molecules 2012, 17, 10586. [Google Scholar] [CrossRef] [PubMed]
- Bidzinska, J.; Cimino-Reale, G.; Zaffaroni, N.; Folini, M. G-quadruplex structures in the human genome as novel therapeutic targets. Molecules 2013, 18, 12368. [Google Scholar] [CrossRef]
- Moye, A.L.; Porter, K.C.; Cohen, S.B.; Phan, T.; Zyner, K.G.; Sasaki, N.; Lovrecz, G.O.; Beck, J.L.; Bryan, T.M. Telomeric G-quadruplexes are a substrate and site of localization for human telomerase. Nat. Commun. 2015, 6, 7643. [Google Scholar] [CrossRef]
- Hou, J.Q.; Chen, S.-B.; Zan, L.P.; Ou, T.M.; Tan, J.H.; Luyt, L.G.; Huang, Z.S. Identification of a selective G-quadruplex DNA binder using a multistep virtual screening approach. Chem. Commun. 2015, 51, 198. [Google Scholar] [CrossRef]
- Cavaleiro, J.A.S.; Tomé, A.C.; Neves, M.G.P.M.S. Handbook of Porphyrin Science. In Meso-Tetraphenylporphyrin Derivatives: New Synthetic Methodologies; Smith, K.M.K., Kadish, K.M., Guilard, R., Eds.; World Scientific Publishing Company Co.: Singapore, 2010; Volume 2, pp. 193–294. [Google Scholar]
- Moura, N.M.M.; Ramos, C.I.V.; Linhares, I.; Santos, S.M.; Faustino, M.A.F.; Almeida, A.; Cavaleiro, J.A.S.; Amado, F.M.L.; Lodeiro, C.; Neves, M.G.P.M.S. Synthesis, characterization and biological evaluation of cationic porphyrin-terpyridine derivatives. RSC Adv. 2016, 6, 110674. [Google Scholar] [CrossRef]
- Ramos, C.I.V.; Figueira, F.; De Polêto, M.; Amado, F.M.L.; Verli, H.; Tomé, J.P.C.; Neves, M.G.P.M.S. ESI-MS/MS of Expanded Porphyrins: A Look into Their Structure and Aromaticity. J. Mass Spectrom. 2016, 51, 342. [Google Scholar] [CrossRef]
- Ramos, C.I.V.; Moura, N.M.M.; Santos, S.M.F.; Faustino, M.A.F.; Tomé, J.P.C.; Amado, F.M.L.; Santana-Marques, M.G.; De Paula, R.; Simões, M.M.Q.; Neves, M.G.P.M.S. An insight into the gas-phase fragmentations of potential molecular sensors with porphyrin-chalcone structures. Int. J. Mass Spectrom. 2015, 392, 164. [Google Scholar] [CrossRef]
- Eddahmi, M.; Moura, N.M.M.; Ramos, C.I.V.; Bouissane, L.; Faustino, M.A.F.; Cavaleiro, J.A.S.; Rakib, M.; Neves, M.G.P.M.S. An insight into the vicarious nucleophilic substitution reaction of 2-nitro-5,10,15,20-tetraphenylporphyrin with p-chlorophenoxyacetonitrile: Synthesis and gas-phase fragmentation studies. Arab. J. Chem. 2020, 13, 5849. [Google Scholar] [CrossRef]
- Monteiro, A.R.; Ramos, C.I.V.; Fateixa, S.; Moura, N.M.M.; Neves, M.G.P.M.S.; Trindade, T. Hybrids based on graphene oxide and porphyrin as a tool for detection and stabilization of DNA G-quadruplexes. ACS Omega 2018, 3, 11184. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.I.V.; Santana-Marques, M.G.; Tomé, J.P.C. Charge and substituent effects on the stability of porphyrin/G-quadruplex adducts. J. Mass Spectrom. 2012, 47, 173. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.I.V.; Almeida, S.P.; Lourenço, L.M.O.; Pereira, P.M.; Fernandes, R.; Faustino, M.A.F.; Tomé, J.P.C.; Carvalho, J.; Cruz, C.; Neves, M.G.P.M.S. Multicharged phthalocyanines as selective ligands for G-Quadruplex DNA structures. Molecules 2019, 24, 733. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Nunes, J.; Carvalho, J.; Figueiredo, J.; Ramos, C.I.V.; Lourenço, L.M.O.; Tomé, J.P.C.; Neves, M.G.P.M.S.; Mergny, J.-L.; Queiroz, J.A.; Salgado, G.F.; et al. Phthalocyanines for G-quadruplex aptamers binding. Bioorg. Chem. 2020, 100, 103920. [Google Scholar] [CrossRef]
- Madureira, J.; Marques, M.; Maia, C.; Sousa, B.; Campino, L.; Santana-Marques, M.G.; Farrell, N. Nonclassic Metallointercalators with Dipyridophenazine: DNA Interaction Studies and Leishmanicidal Activity. Inorg. Chem. 2013, 52, 8881. [Google Scholar] [CrossRef]
- Ramos, C.I.V.; Barros, C.M.; Fernandes, A.M.; Santana-Marques, M.G.; Correia, A.J.F.; Tomé, J.P.C.; Carrilho, M.D.C.T.; Faustino, M.A.F.; Tomé, A.C.; Neves, M.G.P.M.S.; et al. Interactions of cationic porphyrins with double-stranded oligodeoxynucleotides: A study by electrospray ionisation mass spectrometry. J. Mass Spectrom. 2005, 40, 1439. [Google Scholar] [CrossRef]
- Ramos, C.I.V.; Santana-Marques, M.G. Electrospray mass spectrometry for the study of the non-covalent interactions of porphyrins and duplex desoxyribonucleotides. J. Porphyr. Phthalocyanines 2009, 13, 518. [Google Scholar] [CrossRef]
- Silva, E.M.P.; Ramos, C.I.V.; Pereira, P.M.R.; Giuntini, F.; Faustino, M.A.F.; Tomé, J.P.C.; Tomé, A.C.; Silva, A.M.S.; Santana-Marques, M.G.; Neves, M.G.P.M.S.; et al. Cationic-vinyl substituted meso-tetraphenylporphyrins: Synthesis and non-covalent interactions with a short poly(dGdC) duplex. J. Porphyr. Phthalocyanines 2012, 16, 101. [Google Scholar] [CrossRef]
- Sun, D.; Thompson, B.; Cathers, B.E.; Salazar, M.; Kerwin, S.M.; Trent, J.O.; Jenkins, T.C.; Neidle, S.; Hurley, L.H. Inhibition of human telomerase by a G-quadruplex interactive compound. J. Med. Chem. 1997, 40, 2113. [Google Scholar] [CrossRef] [PubMed]
- Ou, T.-M.; Lu, Y.-J.; Tan, J.; Huang, Z.; Wong, K.; Gu, L. G-Quadruplexes: Targets in anticancer drug design. ChemMedChem 2008, 3, 690. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.R.; Cadoni, E.; Ressurreição, A.S.; Moreira, R.; Paulo, A. Design of Modular G-quadruplex Ligands. ChemMedChem 2018, 13, 869. [Google Scholar] [CrossRef]
- Jaumot, J.; Gargallo, R. Experimental Methods for Studying the Interactions between G-Quadruplex Structures and Ligands. Curr. Pharm Design. 2012, 18, 1900. [Google Scholar] [CrossRef]
- Murat, P.; Singh, Y.; Defrancq, E. Methods for investigating G-quadruplex DNA / ligand interactions. Chem. Soc. Rev. 2011, 40, 5293. [Google Scholar] [CrossRef] [PubMed]
- Sirajuddin, M.; Ali, S.; Badshah, A. Drug-DNA interactions and their study by UV-Visible, fluorescence spectroscopies and cyclic voltammetry. J. Photochem. Photobiol. B Biol. 2013, 124, 1–19. [Google Scholar] [CrossRef]
- Carvalho, J.; Quintela, T.; Gueddouda, N.M.; Bourdoncle, A.; Mergny, J.-L.; Salgado, G.F.; Queiroz, J.A.; Cruz, C. Phenanthroline polyazamacrocycles as G-quadruplex DNA binders. Org. Biomol. Chem. 2018, 16, 2776. [Google Scholar] [CrossRef]
- Largy, E.; Hamon, F.; Teulade-Fichou, M.-P. Development of a high-throughput G4-FID assay for screening and evaluation of small molecules binding quadruplex nucleic acid structures. Anal. Bioanal. Chem. 2011, 400, 3419. [Google Scholar] [CrossRef]
- Marchand, A.; Strzelecka, D.; Gabelica, V. Selective and Cooperative Ligand Binding to Antiparallel Human Telomeric DNA G-Quadruplexes. Chem. Eur. J. 2016, 22, 9551. [Google Scholar] [CrossRef]
- Casagrande, A.; Alvino, A.; Bianco, G.; Franceschin, O.M. Study of binding affinity and selectivity of perylene and coronene derivatives towards duplex and quadruplex DNA by ESI-MS. J. Mass Spectrom. 2009, 44, 530. [Google Scholar] [CrossRef]
- Lecours, M.J.; Marchand, A.; Anwar, A.; Guetta, C.; Hopkins, W.S.; Gabelica, V. What stoichiometries determined by mass spectrometry reveal about the ligand binding mode to G-quadruplex nucleic acids. Biochim. Biophys. Acta 2017, 1861, 1353. [Google Scholar] [CrossRef]
- Read, M.; Harrison, R.J.; Romagnoli, B.; Tanious, F.A.; Gowan, S.H.; Reszka, A.P.; Wilson, W.D.; Kelland, L.R.; Neidle, S. Structure-based design of selective and potent G-quadruplex-mediated telomerase inhibitors. Proc. Natl. Acad. Sci. USA 2001, 98, 4844. [Google Scholar] [CrossRef]
- Fedoroff, O.Y.; Salazar, M.; Han, H.Y.; Chemeris, V.V.; Kerwin, S.M.; Hurley, L.H. NMR-based model of a telomerase-inhibiting compound bound to G-quadruplex DNA. Biochemistry 1998, 37, 12367. [Google Scholar] [CrossRef] [PubMed]
- Mergny, J.L.; Lacroix, L.; Teulade-Fichou, M.P.; Hounsou, C.; Guittat, L.; Hoarau, M.; Arimondo, P.B.; Vigneron, J.P.; Lehn, J.M.; Riou, J.F.; et al. Telomerase inhibitors based on quadruplex ligands selected by a fluorescence assay. Proc. Natl. Acad. Sci. USA 2001, 98, 3062. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, J.; Mergny, J.-L.; Salgado, G.F.; Queiroz, J.A.; Cruz, C. G-quadruplex, Friend or Foe: The Role of the G-quartet in Anticancer Strategies. Trends Mol. Med. 2020, 26, 848. [Google Scholar] [CrossRef] [PubMed]
- Wheelhouse, R.T.; Sun, D.K.; Han, H.Y.; Han, F.X.G.; Hurley, L.H. Cationic porphyrins as telomerase inhibitors: The interaction of tetra-(N-methyl-4-pyridyl)porphine with quadruplex DNA. J. Am. Chem. Soc. 1998, 120, 3261. [Google Scholar] [CrossRef]
- Boschi, E.; Davis, S.; Taylor, S.; Butterworth, A.; Chirayath, L.A.; Purohit, V.; Siegel, L.K.; Buenaventura, J.; Sheriff, A.H.; Jin, R.; et al. Interaction of a Cationic Porphyrin and Its Metal derivatives with G-Quadruplex DNA. J. Phys. Chem. B 2016, 120, 12807. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-M.; Cui, Y.-X.; Zhu, L.-N.; Chu, J.-Q.; Kong, D.-M. Cationic porphyrins with large side arm substituents as resonance light scattering ratiometric probes for specific recognition of nucleic acid G-quadruplexes. Nucleic Acids Res. 2019, 47, 2727. [Google Scholar] [CrossRef]
- Monchaud, D.; Teulade-Fichou, M.-P. A hitchhiker’s guide to G-quadruplex ligands. Org. Biomol. Chem. 2008, 6, 627. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wu, Y.; Zhang, W. G-quadruplex structures and their interaction diversity with ligands. Chem. Med. Chem. 2014, 9, 899. [Google Scholar] [CrossRef] [PubMed]
- Neidle, S.; Balasubramanian, S. Quadruplex Nucleic Acids; RSC Publishing: Cambridge, UK, 2006. [Google Scholar] [CrossRef]
- Georgiades, S.N.; Karim, N.H.A.; Suntharalingam, K.; Vilar, R. Interaction of Metal Complexes with G-Quadruplex DNA. Angew. Chem. Int. Ed. 2010, 49, 4020. [Google Scholar] [CrossRef] [PubMed]
- Romera, C.; Bombarde, O.; Bonnet, R.; Gomez, D.; Dumy, P.; Calsou, P.; Gwan, J.F.; Lin, J.H.; Defrancq, E.; Pratviel, G. Improvement of porphyrins for G-quadruplex DNA targeting. Biochimie 2011, 93, 1310. [Google Scholar] [CrossRef]
- Milagrom, L.R. The Colours of Life; Oxford University Press: Oxford, UK, 1997. [Google Scholar]
- Vallejo, M.C.S.; Reis, M.J.A.; Pereira, A.M.V.M.; Serra, V.V.; Cavaleiro, J.A.S.; Moura, N.M.M.; Neves, M.G.P.M.S. Merging pyridine(s) with porphyrins and analogues: An overview of synthetic approaches. Dye. Pigment. 2021, 191, 109298. [Google Scholar] [CrossRef]
- Monchaud, D.; Granzhan, A.; Saettel, N.; Guédin, A.; Mergny, J.L.; Teulade-Fichou, M.P. One ring to bind them all-part I: The efficiency of the macrocyclic scaffold for g-quadruplex DNA recognition. J. Nucleic Acids 2010, 2010, 525862. [Google Scholar] [CrossRef]
- Rowland, G.B.; Barnett, K.; DuPont, J.I.; Akurathi, G.; Le, V.H.; Lewis, E.A. The effect of pyridyl substituents on the thermodynamics of porphyrin binding to G-quadruplex DNA. Bioorg. Med. Chem. 2013, 21, 7515. [Google Scholar] [CrossRef]
- Izbicka, E.; Wheelhouse, R.T.; Raymond, E.; Davidson, K.K.; Lawrence, R.A.; Sun, D.; Windle, B.E.; Hurley, L.H.; Von Hoff, D.D. Effects of Cationic Porphyrins as G-Quadruplex Interactive Agents in Human Tumor Cells. Cancer Res. 1999, 59, 639. [Google Scholar]
- Vialas, C.; Pratviel, G.; Meunier, B. Oxidative Damage Generated by an Oxo-Metalloporphyrin onto the Human Telomeric Sequence. Biochemistry 2000, 39, 9514. [Google Scholar] [CrossRef]
- Shi, D.F.; Wheelhouse, R.T.; Sun, D.; Hurley, L.H. Quadruplex-interactive agents as telomerase inhibitors:Synthesis of porphyrins and structure-activity relationship for the inhibition of telomerase. J. Med. Chem. 2001, 44, 4509. [Google Scholar] [CrossRef]
- Keating, L.R.; Szalai, V.A. Parallel-stranded guanine quadruplex interactions with a copper cationic porphyrin. Biochemistry 2004, 43, 15891. [Google Scholar] [CrossRef]
- Evans, S.E.; Mendez, M.A.; Turner, K.B.; Keating, L.R.; Grimes, R.T.; Melchoir, S.; Szalai, V.A. End-stacking of copper cationic porphyrins on parallel—Stranded guanine quadruplexes. J. Biol. Inorg. Chem. 2007, 12, 1235. [Google Scholar] [CrossRef] [PubMed]
- DuPont, J.I.; Henderson, K.L.; Metz, A.; Le, V.H.; Emerson, J.P.; Lewis, E.A. Calorimetric and spectroscopic investigations of the binding of metallated porphyrins to G-quadruplex DNA. Biochim. Biophys. Acta 2016, 1860, 902. [Google Scholar] [CrossRef] [PubMed]
- Dixon, I.M.; Lopez, F.; Esteve, J.P.; Tejera, A.M.; Blasco, M.A.; Pratviel, G.; Meunier, B. Porphyrin Derivatives for Telomere Binding and Telomerase Inhibition. ChemBioChem 2005, 6, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Dixon, I.M.; Lopez, F.; Tejera, A.M.; Esteve, J.P.; Blasco, M.A.; Pratviel, G.; Meunier, B. A G-quadruplex ligand with 10000-fold selectivity over duplex DNA. J. Am. Chem. Soc. 2007, 129, 1502. [Google Scholar] [CrossRef] [PubMed]
- Sabater, L.; Fang, P.-J.; Chang, C.-F.; Rache, A.; Prado, E.; Dejeu, J.; Garofalo, A.; Lin, J.-H.; Mergny, J.-L.; Defrancq, E.; et al. Cobalt(III)porphyrin to target G-quadruplex DNA. Dalton Trans. 2015, 44, 3701. [Google Scholar] [CrossRef] [PubMed]
- Dejeu, J.; Lavergne, T.; Nora, J.D.; Defrancq, E.; Pratviel, G. Binding of mettaloporphyrins of G-Quadruplex: The role of central metal. Inorg. Chimica Acta 2016, 452, 98. [Google Scholar] [CrossRef]
- Pan, J.; Zhang, S. Interaction between cationic zinc porphyrin and lead ion induced telomeric guanine quadruplexes: Evidence for end-stacking. J. Biol. Inorg. Chem. 2009, 14, 401. [Google Scholar] [CrossRef]
- Bhattacharjee, A.J.; Ahluwalia, K.; Taylor, S.; Jin, O.; Nicoludis, J.M.; Buscaglia, R.; Chaires, J.B.; Kornfilt, D.J.P.; Marquard, D.G.S.; Yatsunyk, L.A. Induction of G-quadruplex DNA structure by Zn(II) 5,10,15,20-tetrakis(N-methyl-4-pyridyl)porphyrin. Biochimie 2011, 93, 1297. [Google Scholar] [CrossRef]
- Sabharwal, N.C.; Mendoza, O.; Nicoludis, J.M.; Ruan, T.; Mergny, J.L.; Yatsunyk, L.A. Investigation of the interactions between Pt(II) and Pd(II) derivatives of 5,10,15,20-tetrakis (N-methyl-4-pyridyl) porphyrin and G-quadruplex DNA. J. Biol. Inorg. Chem. 2016, 21, 227. [Google Scholar] [CrossRef]
- Yao, X.; Song, D.; Qin, T.; Yang, C.; Yu, Z.; Li, X.; Liu, K.; Su, H. Interaction between G-Quadruplex and Zinc Cationic Porphyrin: The Role of the Axial Water. Sci Rep. 2017, 7, 10951. [Google Scholar] [CrossRef] [PubMed]
- Pipier, A.; Rache, A.; Modeste, C.; Amrane, S.; Mothes-Martin, E.; Stigliani, J.-L.; Calsou, P.; Stigliani, J.-L.; Pratviel, G.; Gomez, D. G-Quadruplex binding optimization by gold(III) insertion into the center of a porphyrin. Dalton Trans. 2019, 48, 6091. [Google Scholar] [CrossRef]
- Rundstadler, T.; Mothes, E.; Amrane, S.; Stigliani, J.-L.; Verhaeghe, P.; Pratviel, G. Gold(III) porphyrins: Synthesis and interaction with G-quadruplex DNA. J. Inorg. Biochem. 2021, 223, 111551. [Google Scholar] [CrossRef] [PubMed]
- Tovmasyan, A.; Babayan, N.; Poghosyan, D.; Margaryan, K.; Harutyunyan, B.; Grigoryan, R.; Sarkisyan, N.; Spasojevic, I.; Mamyan, S.; Sahakyan, L.; et al. Novel amphiphilic cationic porphyrin and its Ag(II) complex as potential anticancer agents. J. Inorg. Biochem. 2014, 140, 94. [Google Scholar] [CrossRef] [PubMed]
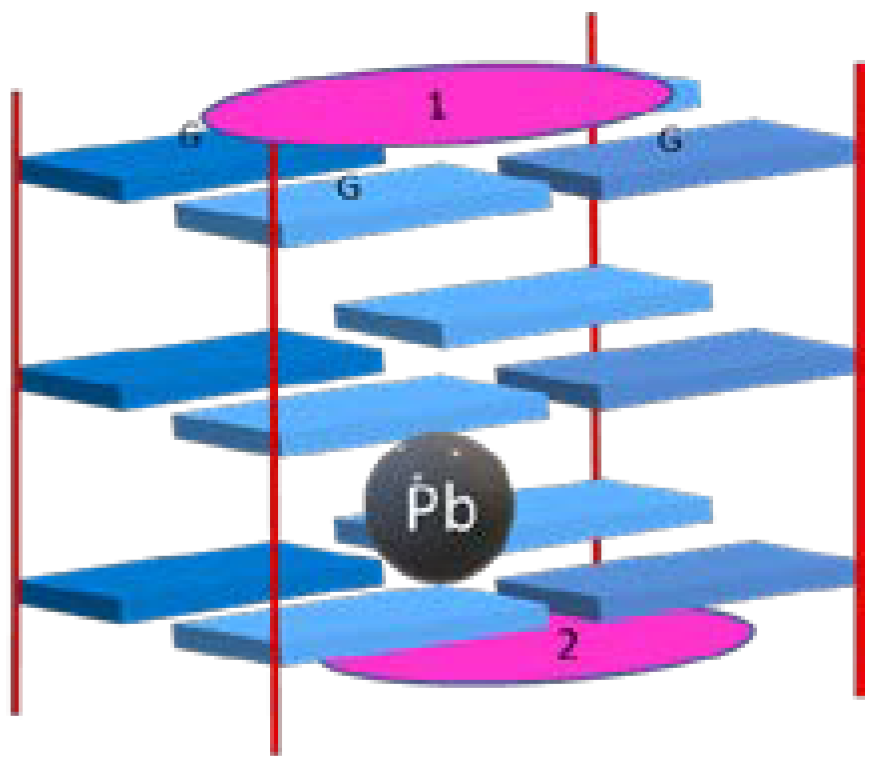
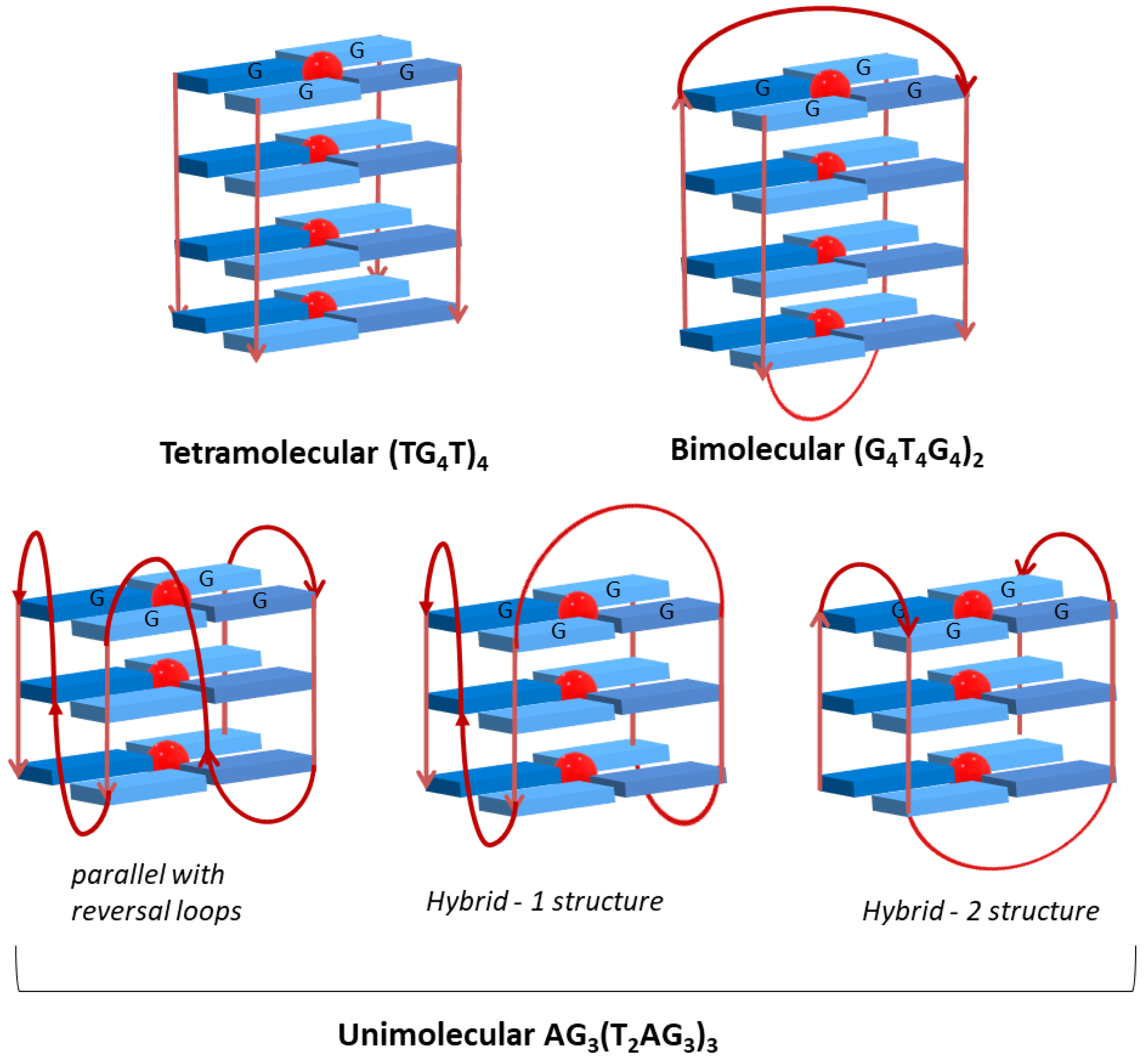
- Schultze, P.; Smith, F.W.; Feigon, J. Refined solution structure of the dimeric quadruplex formed from the Oxytricha telomeric oligonucleotide d(GGGGTTTTGGGG). Structure 1994, 2, 221. [Google Scholar] [CrossRef]
- Wang, Y.; Patel, D.J. Solution structure of the human telomeric repeat d[AG3(T2AG3)3] G-tetraplex. Structure 1993, 1, 263. [Google Scholar] [CrossRef]
- Parkinson, G.N.; Lee, M.P.H.; Neidle, S. Crystal structure of parallel quadruplexes from human telomeric DNA. Nature 2002, 417, 876. [Google Scholar] [CrossRef]
- Wei, C.Y.; Jia, G.Q.; Yuan, J.L.; Feng, Z.C.; Li, C. A spectroscopic study on the interactions of porphyrin with G-quadruplex DNAs. Biochem 2006, 45, 6681. [Google Scholar] [CrossRef]
- Sun, Y.; Ji, F.; Liu, R.; Lin, J.; Xu, Q.; Gao, C. Interaction mechanism of 2-aminobenzothiazole with herring sperm DNA. J. Lumin. 2012, 132, 507. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Sengupta, P.K.; Bhowmik, S. Exploring the preferential interaction of quercetin with VEGF promoter G-quadruplex DNA and construction of a pH-dependent DNA-based logic gate. RSC Adv. 2017, 7, 37230. [Google Scholar] [CrossRef]
- Haq, I.; Trent, J.O.; Chowdhry, B.Z.; Jenkins, T.C. Intercalative G-tetraplex stabilization of telomeric DNA by a cationic porphyrin. J. Am. Chem. Soc. 1999, 121, 1768. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Porphyrin * | Metal | DNA Sequence (5′-3′) | Major Findings | Buffer | Relevant Techniques | |||
|---|---|---|---|---|---|---|---|---|---|
| Ka (M−1) or KD | Binding Mode | Porphyrin: GQ Stoichiometry | Other Parameters | ||||||
| Boschi et al. [54] | TMPyP4 | Free-base | (T4G4)4 | Ka = (3.7 ± 0.9) × 106 | End-stacking and possibly intercalation | 2:1 | T1/2 = 312.6 ± 0.4 K; %H = 35 ± 1; Δλ = 13.4 ± 0.7 nm | KPI | UV-Vis, fluorescence, CD, melting |
| (T4G4T)4 | Ka = (3.0 ± 1.5) × 106 | T1/2 = 317.7 ± 1.0 K; %H = 36 ± 1; Δλ = 15.4 ± 0.3 nm | |||||||
| Cu(II) | (T4G4)4 | Ka = (1.4 ± 0.6) × 106 | End-stacking and possibly intercalation | 1:1 | T1/2 = 310.1 ± 0.4 K; %H = 22 ± 1; Δλ = 7.1 ± 0.7 nm | ||||
| (T4G4T)4 | Ka = (1.1 ± 0.7) × 106 | T1/2 = 315.7 ± 1.3 K; %H = 26 ± 2; Δλ = 8.3 ± 0.1 nm | |||||||
| Zn(II) | (T4G4)4 | Ka = (16 ± 6.0) × 106 | End-stacking and groove- or outside-binding | 2:1 | T1/2 = 324.6 ± 0.6 K; %H = 33 ± 3; Δλ = 11.5 ± 0.2 nm | ||||
| (T4G4T)4 | Ka = (11 ± 2.0) × 106 | T1/2 = 336.2 ± 0.6 K; %H = 32 ± 1; Δλ = 14.2 ± 0.3 nm | |||||||
| TMPyP2 | Free-base | (T4G4)4 | Ka = (2.0 ± 1.0) × 106 | 1:1 | T1/2 = 320.9 ± 0.3 K; %H = 23 ± 4%; Δλ = 5.7 ± 0.6 nm | ||||
| (T4G4T)4 | Ka = (6.3 ± 0.6) × 106 | End-stacking and groove- or outside-binding | 1:1 | T1/2 = 329.8 ± 0.3 K; %H = 22 ± 2%; Δλ = 6.3 ± 0.6 nm | |||||
| Dejeu et al. [74] | TMPyP4 | Free-base | (T2AG3T) | KD = 62 nM | End-stacking | not available | not available | HEPES | SPR, FRET |
| (G3T2A)3G3T2 | KD = 345 nM | ||||||||
| (CG)4T4(CG)4 (duplex) | KD = 345 nM | ||||||||
| Co(II) | (T2AG3T) | KD = 6600 nM | |||||||
| (G3T2A)3G3T2 | KD = 5500 nM | ||||||||
| (CG)4T4(CG)4 (duplex) | n.b. | ||||||||
| Mn(III) | (T2AG3T) | KD = 9000 nM | |||||||
| (G3T2A)3G3T2 | KD = 10,000 nM | ||||||||
| (CG)4T4(CG)4 (duplex) | n.b. | n.b. | |||||||
| Ni(II) | (T2AG3T) | KD = 59 nM | End-stacking | not available | |||||
| (G3T2A)3G3T2 | KD = 240 nM | ||||||||
| (CG)4T4(CG)4 (duplex) | KD = 200 nM | ||||||||
| TMPyP2 | Free-base | (T2AG3T) | KD = 52 nM | ||||||
| (G3T2A)3G3T2 | KD = 114 nM | ||||||||
| (CG)4T4(CG)4 (duplex) | KD = 404 nM | ||||||||
| Co(II) | (T2AG3T) | KD = 3.4 nM | |||||||
| (G3T2A)3G3T2 | KD = 15 nM | ||||||||
| (CG)4T4(CG)4 (duplex) | KD = 417 nM | ||||||||
| Mn(III) | (T2AG3T) | KD = 4 nM | not available | ||||||
| (G3T2A)3G3T2 | KD = 29 nM | ||||||||
| (CG)4T4(CG)4 (duplex) | n.b. | n.b. | |||||||
| Ni(II) | (T2AG3T) | KD = 29 nM | End-stacking | ||||||
| (G3T2A)3G3T2 | KD = 5.4 nM | ||||||||
| (CG)4T4(CG)4 (duplex) | KD = 185 nM | ||||||||
| TGP | Free-base | (T2AG3T) | KD = 83 nM | ||||||
| (G3T2A)3G3T2 | KD = 290 nM | ||||||||
| (CG)4T4(CG)4 (duplex) | KD = 16,000 nM | ||||||||
| Mn(III) | (T2AG3T) | KD = 20 nM | |||||||
| (G3T2A)3G3T2 | KD = 24 nM | ||||||||
| (CG)4T4(CG)4 (duplex) | n.b. | n.b. | |||||||
| Ni(II) | (T2AG3T) | KD = 18 nM | End-stacking | not available | |||||
| (G3T2A)3G3T2 | KD = 53 nM | ||||||||
| (CG)4T4(CG)4 (duplex) | KD = 1030 nM | ||||||||
| DuPont et al. [70] | TMPyP4 | Co(III) | AG3(T2AG3)3 | Ka = (1.2 ± 0.7) = × 105 | End-stacking | 2:1 | ΔH1 = −2.8 ± 0.1 kcal/mol; −TΔS1 = −4.1 ± 0.1 kcal/mol | K+ BPES | ICT, CD, ESI/MS |
| Ni(II) | AG3(T2AG3)3 | Ka = 7.4 ± 0.8) × 107 | End-stacking and intercalation | 4:1 | ΔH1 = −3.2 ± 0.3 kcal/mol; −TΔS1 = −7.6 ± 0.4 kcal/mol | ||||
| Cu(II) | AG3(T2AG3)3 | Ka = (1.7 ± 1.1) × 1010 | ΔH1 = −4.2 ± 0.1 kcal/mol; −TΔS1 = −9.2 ± 0.6 kcal/mol | ||||||
| Zn(II) | AG3(T2AG3)3 | Ka = (7.6 ± 0.6) × 105 | End-stacking | 2:1 | ΔH1 = −4.6 ± 0.4 kcal/mol; −TΔS1 = −3.4 ± 0.4 kcal/mol | ||||
| Keating et al. [68] | TMPyP4 | Cu(II) | (T4G4T4)4 | Ka = 5.6 × 106 | End-stacking | 2:1 | %H = 50; Δλ = 9 nm | KPi | UV-Vis, fluorescence, CD, EPR |
| (T4G8T4)4 | Ka = 5.2 × 107 | End-stacking and intercalation | 3:1 | %H = 58; Δλ = 12 nm | |||||
| Romera et al. [60] | TMPyP4 | Free-base | (T2AG3T) | Ka = 1.6 × 107 | End-stacking | not available | not available | HEPES | SPR, FRET, CD, TRAP assay |
| (G3T2A)3G3T2 | Ka = 2.9 × 106 | ||||||||
| (CG)4T4(CG)4 (duplex) | Ka = 2.9 × 106 | ||||||||
| Au(III) | (T2AG3T) | Ka = 2.2 × 106 | |||||||
| (G3T2A)3G3T2 | Ka = 1.2 × 106 | ||||||||
| (CG)4T4(CG)4 (duplex) | Ka = 4.3 × 106 | ||||||||
| Co(III) | (T2AG3T) | Ka = 1.5 × 105 | End-stacking | not available | not available | ||||
| (G3T2A)3G3T2 | Ka = 1.8 × 105 | ||||||||
| Mn(III) | (T2AG3T) | Ka = 1.1 × 105 | End-stacking | not available | not available | HEPES | |||
| (G3T2A)3G3T2 | Ka = 1.0 × 105 | ||||||||
| Ni(II) | (T2AG3T) | Ka = 1.7 × 107 | |||||||
| (G3T2A)3G3T2 | Ka = 4.2 × 106 | ||||||||
| (CG)4T4(CG)4 (duplex) | Ka = 5.0 × 106 | ||||||||
| Porphyrin 1 | Mn(II) | (T2AG3T) | Ka = 1.7 × 107 | ||||||
| (G3T2A)3G3T2 | Ka = 1.8 × 107 | ||||||||
| (CG)4T4(CG)4 (duplex) | n.b. | n.b. | |||||||
| Sabater et al. [73] | MA | Co(III) | (T2AG3T) | KD = (17 ± 0.4) nM | End-stacking | not available | not available | HEPES | FRET melting assay, SPR, CD, NMR |
| (G3T2A)3G3T2 | KD = (60.2 ± 1.9) nM | ||||||||
| (CG)4T4(CG)4 (duplex) | KD = (3660 ± 13.4) nM | ||||||||
| Sabharwal et al. [77] | TMPyP4 | Pd(II) | AG3(T2AG3)3 | Ka = (1.0 ± 0.3) × 107 | End-stacking | 6.5:1 | ΔT = 30.9 ± 0.4 °C; | KPi | UV-Vis, fluorescence and CD spectroscopies, FRET melting assays, and resonance light scattering |
| Pt(II) | AG3(T2AG3)3 | Ka = (5.8 ± 0.8) × 106 | 7:1 | ΔT = 30.7 ± 06 °C | |||||
| Oligonucleotide Sequence | Topology | Abbreviation |
|---|---|---|
| 5′-TGGGGT-3′ (Tetrahymena telomeric repeat) | Tetramolecular G-Quadruplex | (TG4T)4 |
| 5′-GGG GTT TT GGG G-3′ (Oxytricha repeat oligonucleotide) | Bimolecular G-Quadruplex | (G4T4G4)2 |
| 5′-AGG GTT AGG GTTAGG GTT AGGG-3′ (human telomeric repeat) | Unimolecular G-Quadruplex | AG3(T2AG3)3 |
| long single strand | Double-strand DNA | Salmon-sperm DNA |
| G-Quadruplexes (GQ) | Double-Stranded (ds) | |||||
|---|---|---|---|---|---|---|
| Entry | (TG4T)4 | (G4T4G4)2 | AG3 (T2AG3)3 | Salmon Sperm | Ligand (L) | |
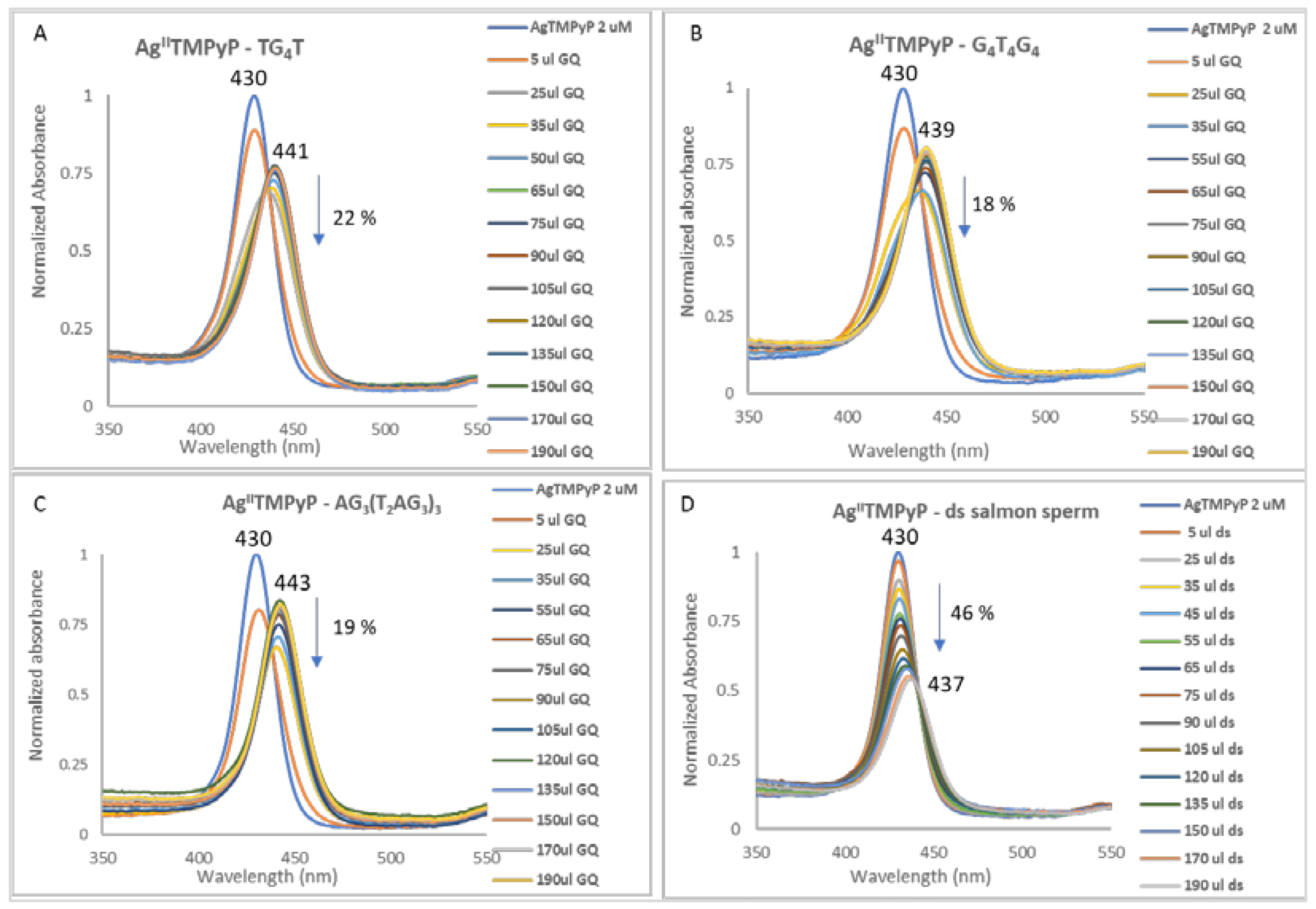
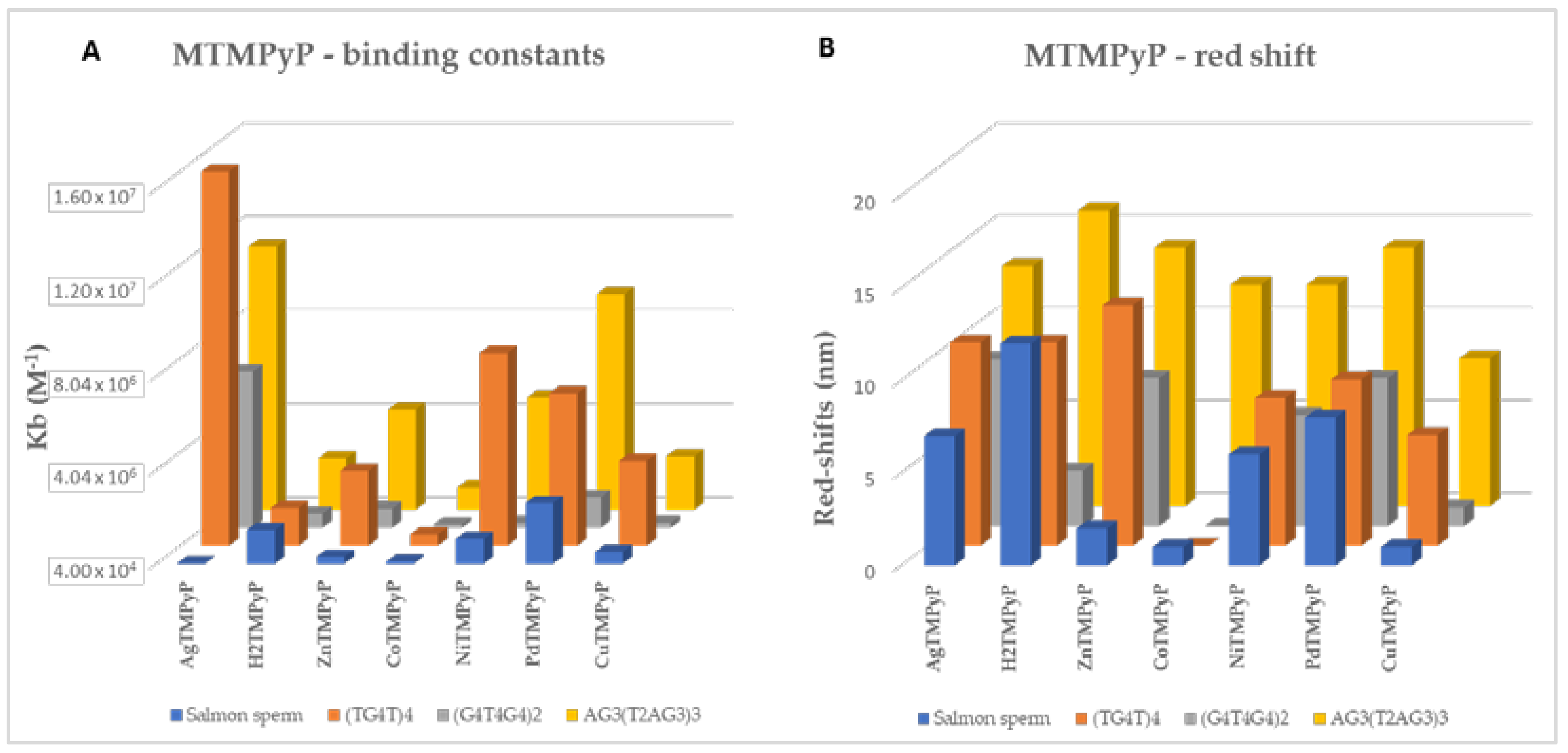
| Red shift (nm) | 11 | 9 | 13 | 7 | AgIITMPyP | |
| (1) | % Hypochromism | 22 | 18 | 19 | 46 | |
| L:DNA Stoichiometry | 3:1 | 3:1 | 4:1 | n.a. | ||
| Kb (M−1) | (1.63 ± 0.31) × 106 | (6.72 ± 0.41) × 106 | (1.13 ± 0.41) × 107 | (6.97 ± 0.37) × 104 | ||
| Red shift (nm) | 11 | 3 | 16 | 12 | H2TMPyP | |
| (2) | % Hypochromism | 23 | 41 | 28 | 36 | |
| L:DNA Stoichiometry | 3:1 | 3:2 | 4:1 | 1:1 | ||
| Kb (M−1) | (1.66 ± 0.41) × 106 | (6.43 ± 0.44) × 105 | (2.57 ± 0.47) × 106 | (1.49 ± 0.32) × 106 | ||
| Red shift (nm) | 13 | 8 | 14 | 2 | ZnIITMPyP | |
| (3) | % Hypochromism | 17 | 74.6 | 27 | 5 | |
| L:DNA Stoichiometry | 3:1 | 3:1 | 4:1 | 2:1 | ||
| Kb (M−1) | (3.25 ± 0.38) × 106 | (8.57 ± 0.76) × 105 | (4.33 ± 0.43) × 106 | (3.52 ± 0.74) × 105 | ||
| Red shift (nm) | 0 | −3 | 12 | 1 | CoIIITMPyP | |
| (4) | % Hypochromism | 26 | 23 | 32 | 49 | |
| L:DNA Stoichiometry | 3:1 | 3:1 | 4:1 | 1:1 | ||
| Kb (M−1) | (5.30 ± 0.89) × 105 | (1.86 ± 0.58) × 105 | (1.00 ± 0.39) × 106 | (1.36 ± 0.40) × 105 | ||
| Red shift (nm) | 8 | 6 | 12 | 6 | NiIITMPyP | |
| (5) | % Hypochromism | 40 | 42 | 35 | 24 | |
| L:DNA Stoichiometry | 3:1 | 3:1 | 3:1 | 1:1 | ||
| Kb (M−1) | (8.28 ± 0.39) × 106 | (2.56 ± 0.41) × 106 | (4.84 ± 0.44) × 106 | (1.11 ± 0.39) × 106 | ||
| Red shift (nm) | 9 | 8 | 14 | 8 | PdIITMPyP | |
| (6) | % Hypochromism | 5 | 12 | 14 | 24 | |
| L:DNA Stoichiometry | 4:1 | 3:1 | 4:1 | 2:1 | ||
| Kb (M−1) | (6.55 ± 0.38) × 106 | (1.35 ± 0.41) × 106 | (9.26 ± 0.36) × 106 | (2.64 ± 0.44) × 106 | ||
| Red shift (nm) | 6 | 1 | 8 | 1 | CuIITMPyP | |
| (7) | % Hypochromism | 28 | 15 | 28 | 3 | |
| L:DNA Stoichiometry | 4:1 | 3:1 | 3:1 | 1:1 | ||
| Kb (M−1) | (3.67 ± 0.42) × 106 | (2.49 ± 0.41) × 105 | (2.33 ± 0.44) × 106 | (5.58 ± 0.79) × 105 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramos, C.I.V.; Monteiro, A.R.; Moura, N.M.M.; Faustino, M.A.F.; Trindade, T.; Neves, M.G.P.M.S. The Interactions of H2TMPyP, Analogues and Its Metal Complexes with DNA G-Quadruplexes—An Overview. Biomolecules 2021, 11, 1404. https://doi.org/10.3390/biom11101404
Ramos CIV, Monteiro AR, Moura NMM, Faustino MAF, Trindade T, Neves MGPMS. The Interactions of H2TMPyP, Analogues and Its Metal Complexes with DNA G-Quadruplexes—An Overview. Biomolecules. 2021; 11(10):1404. https://doi.org/10.3390/biom11101404
Chicago/Turabian StyleRamos, Catarina I. V., Ana R. Monteiro, Nuno M. M. Moura, Maria Amparo F. Faustino, Tito Trindade, and Maria Graça P. M. S. Neves. 2021. "The Interactions of H2TMPyP, Analogues and Its Metal Complexes with DNA G-Quadruplexes—An Overview" Biomolecules 11, no. 10: 1404. https://doi.org/10.3390/biom11101404
APA StyleRamos, C. I. V., Monteiro, A. R., Moura, N. M. M., Faustino, M. A. F., Trindade, T., & Neves, M. G. P. M. S. (2021). The Interactions of H2TMPyP, Analogues and Its Metal Complexes with DNA G-Quadruplexes—An Overview. Biomolecules, 11(10), 1404. https://doi.org/10.3390/biom11101404