Class A G Protein-Coupled Receptor Antagonist Famotidine as a Therapeutic Alternative against SARS-CoV2: An In Silico Analysis


Abstract
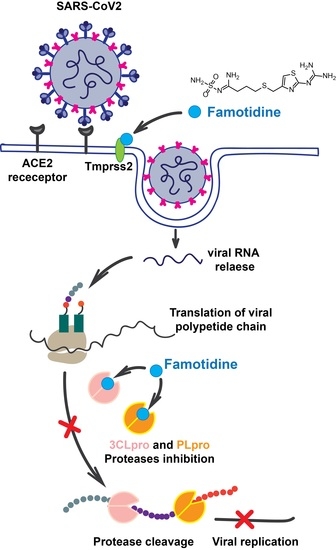
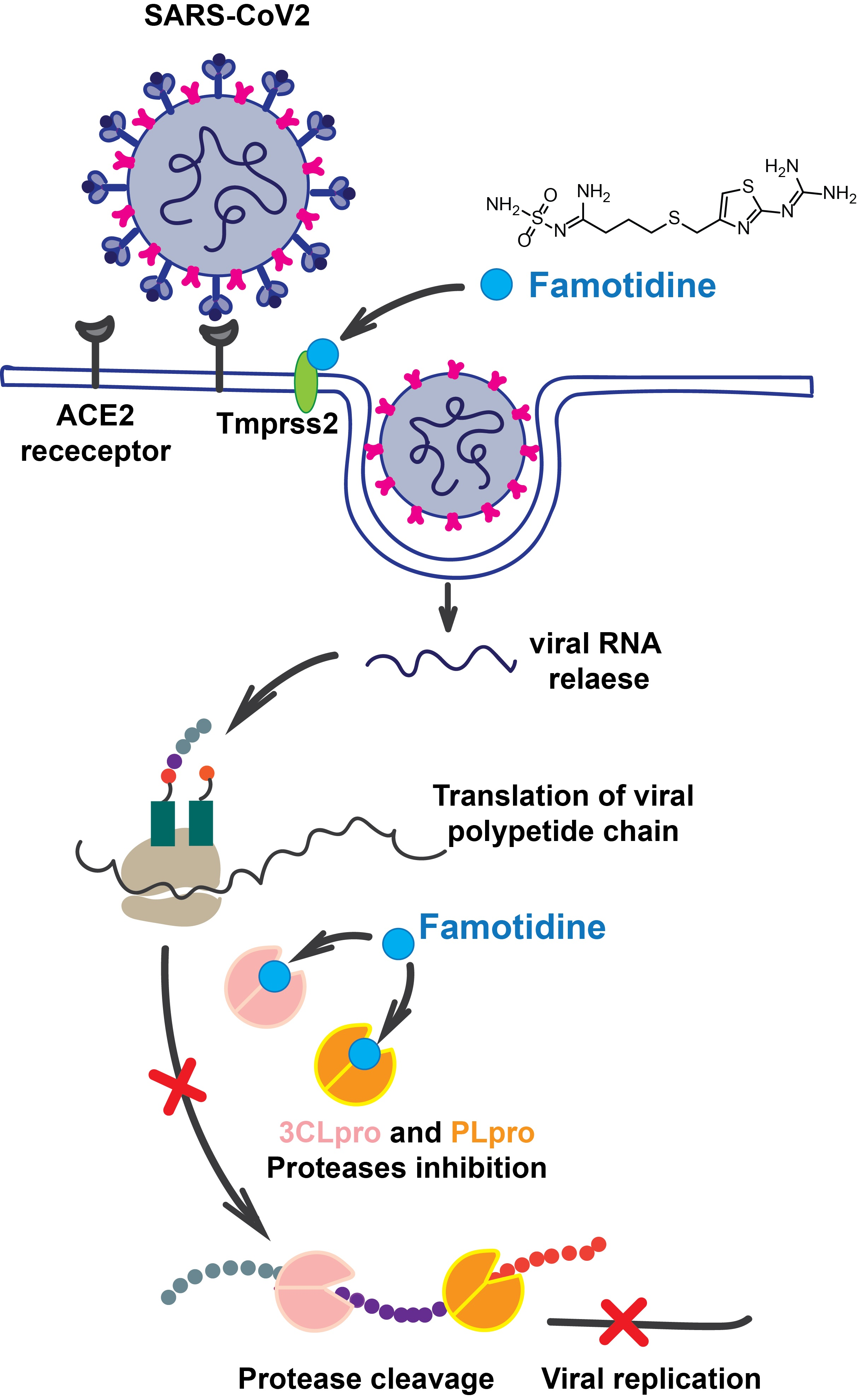
1. Introduction
2. Materials and Methods
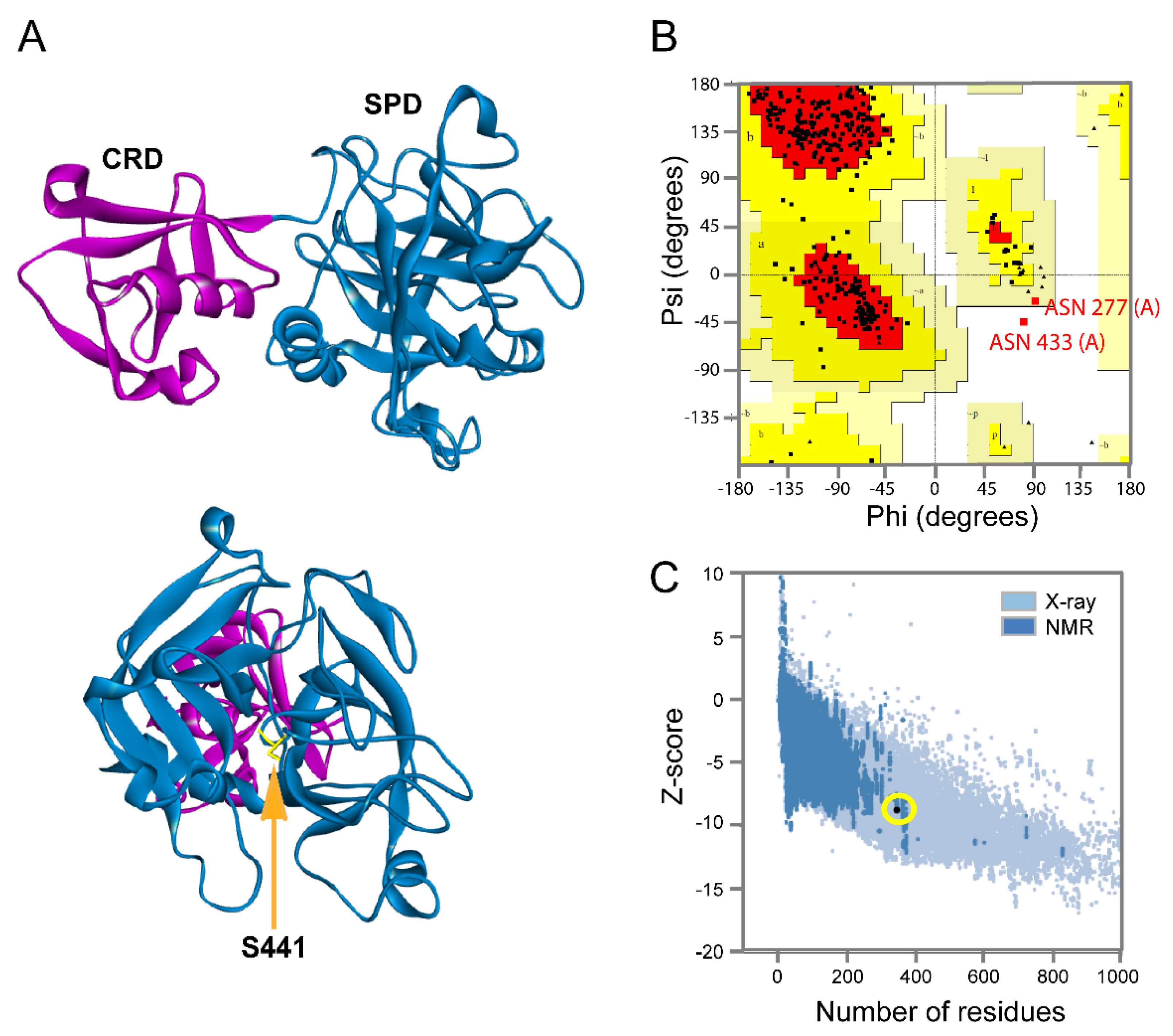
2.1. Protein Modeling
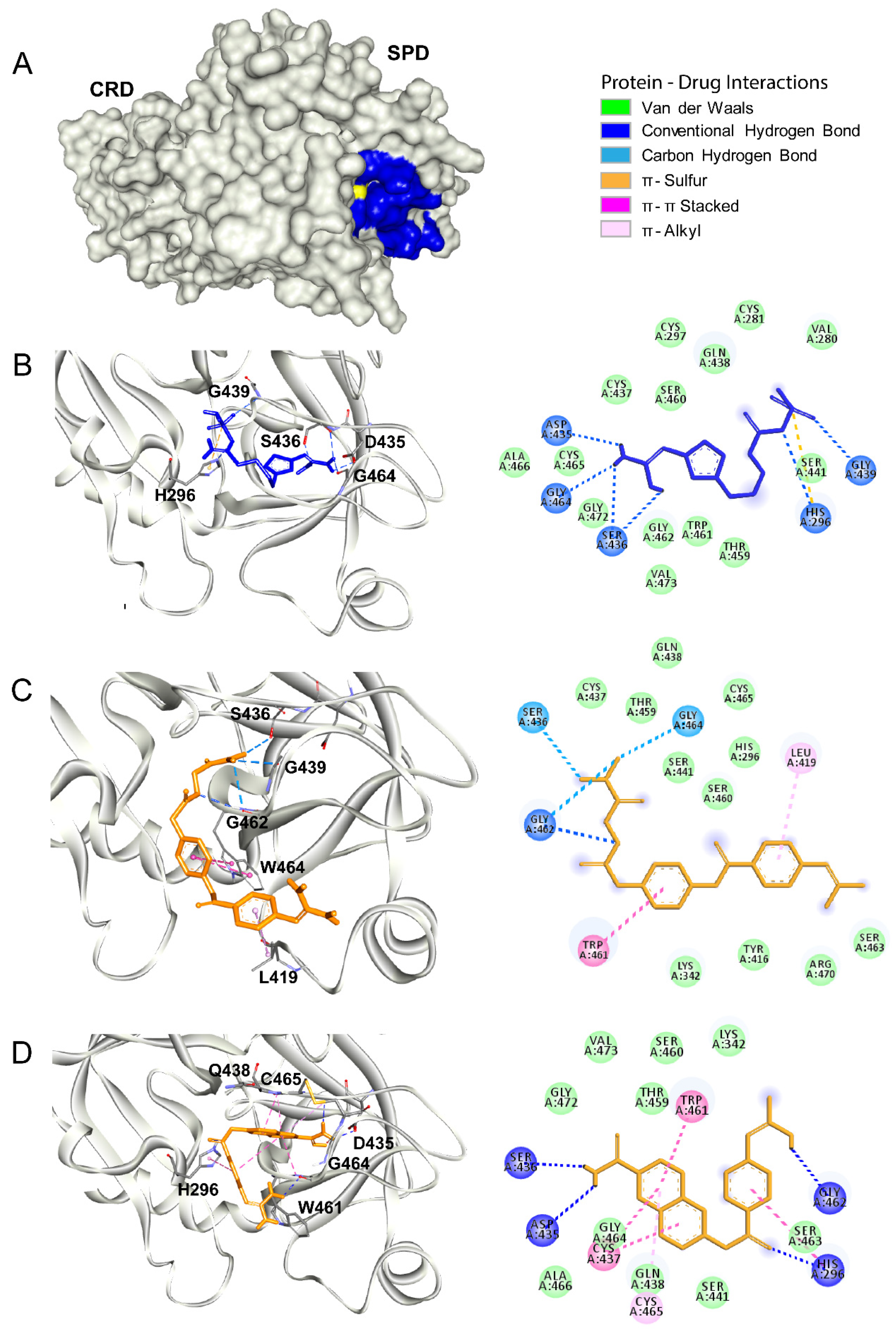
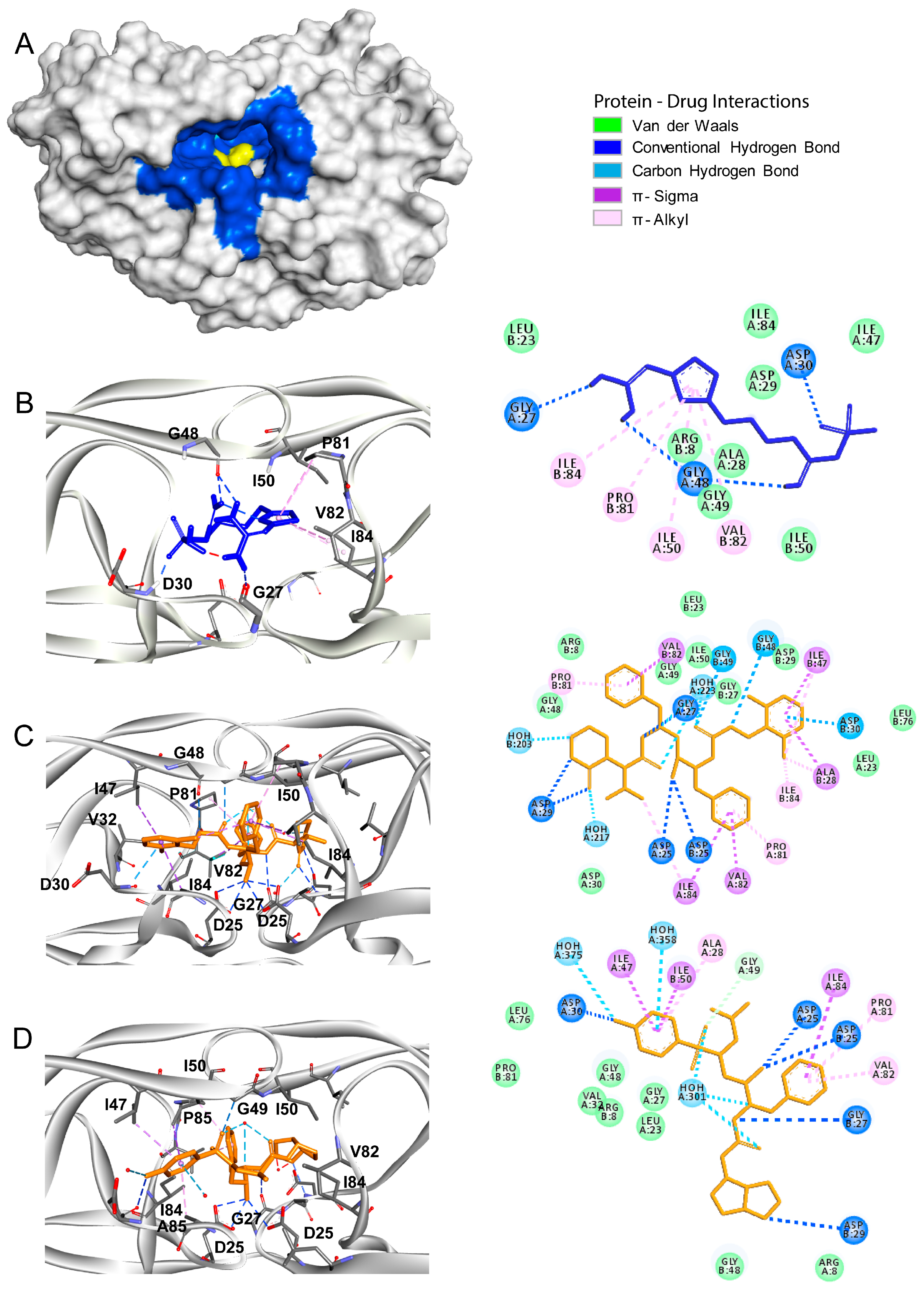
2.2. Molecular Docking
2.3. Analysis of Pharmacokinetic Drug Properties
3. Results
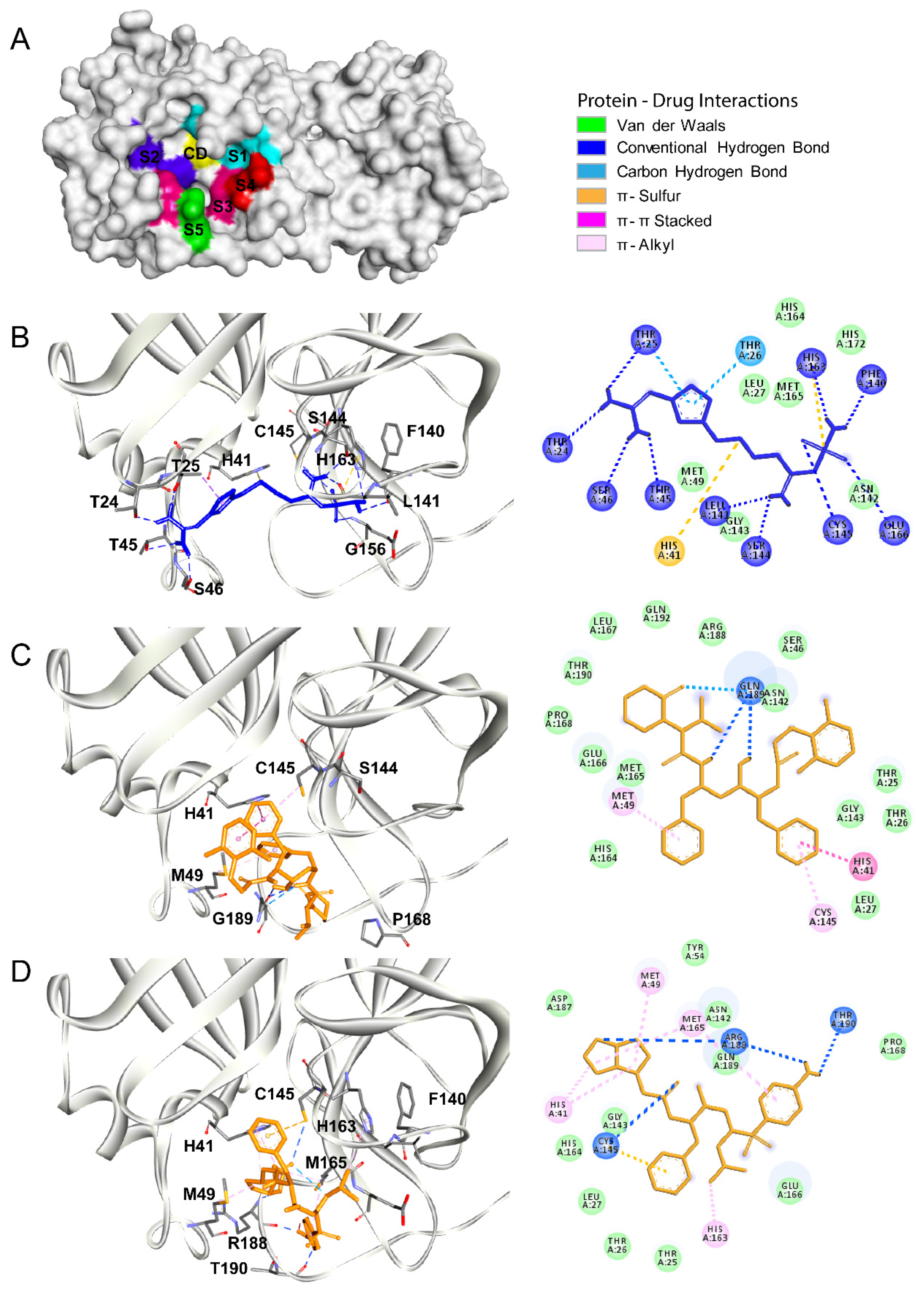
3.1. Main Protease
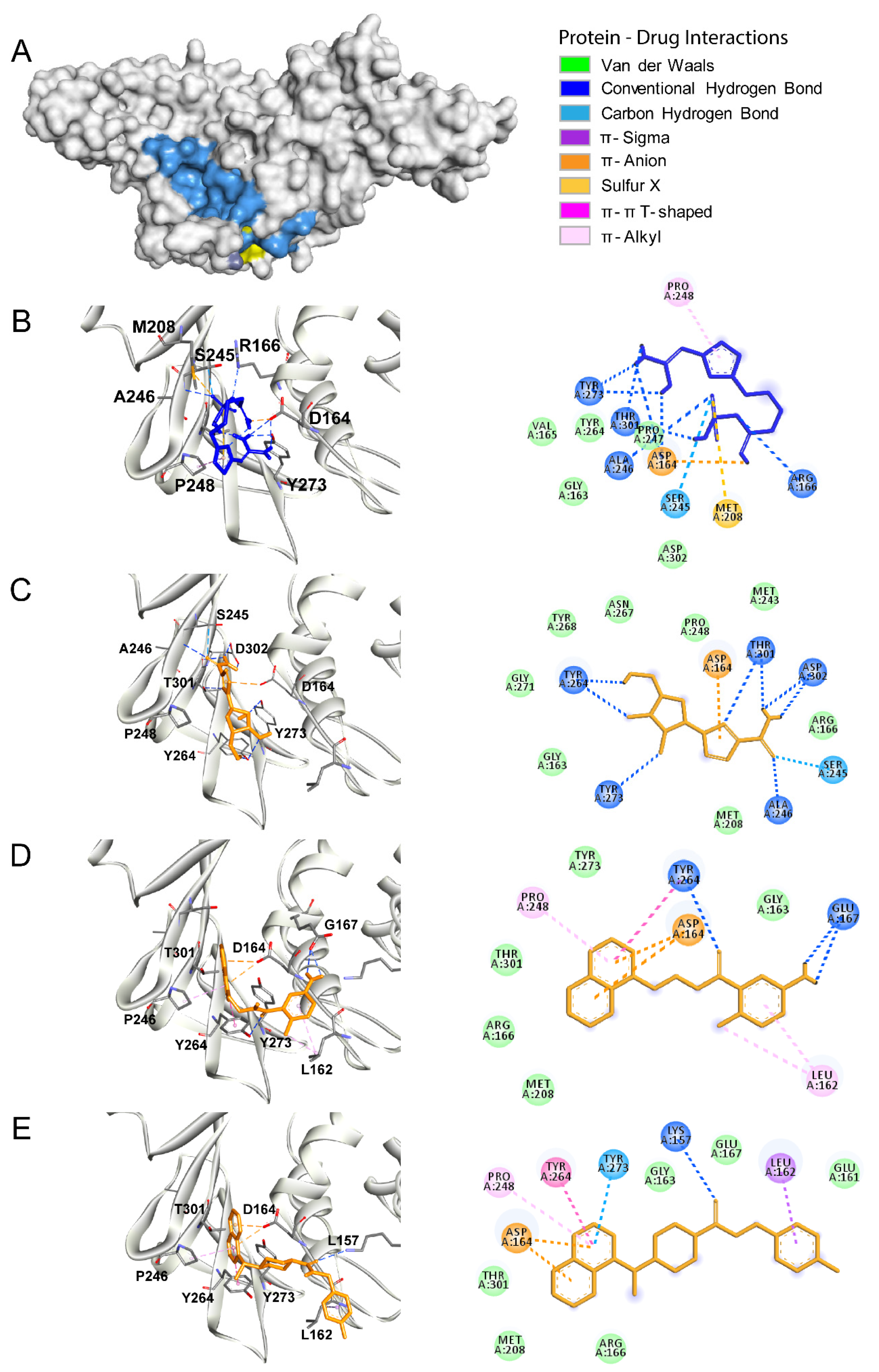
3.2. Papain Like Protease
3.3. Tmprss2
3.4. HIV Protease
3.5. Pharmacokinetics versus Pharmacodynamics in Protease Inhibition
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 3CLpro | SARS-CoV2 main protease |
| ACE2 | angiotensin-converting enzyme 2 |
| ADME | administration, distribution, metabolism, and elimination |
| COVID-19 | Coronavirus infectious disease |
| CRD | cysteine-rich domain |
| FDA | Food and Drug Administration |
| FLEX | flexibility |
| GPCR | G protein-coupled receptor |
| HIV | human immunodeficiency virus |
| INSOLU | insolubility |
| INSATU | instauration |
| LDLRA | low-density lipoprotein receptor domain |
| LIPO | lipophilicity |
| NMR | nuclear magnetic resonance |
| PDB | Protein Data Bank |
| PLpro | SARS-CoV2 papain-like protease |
| POLAR | polarity |
| SARS-CoV2 | Severe Acute Respiratory Syndrome Coronavirus type 2 |
| SARS-CoV | Severe Acute Respiratory Syndrome Coronavirus type 1 |
| SPD | serine protease domain |
| Tmprss2 | transmembrane serine protease type II |
References
- Graham, R.L.; Baric, R.S. SARS-CoV-2: Combating Coronavirus Emergence. Immunity 2020. [Google Scholar] [CrossRef] [PubMed]
- Singhal, T. A Review of Coronavirus Disease-2019 (COVID-19). Indian J. Pediatr. 2020, 87, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.F.; Chien, C.S.; Yarmishyn, A.A.; Lin, Y.Y.; Luo, Y.H.; Lin, Y.T.; Lai, W.Y.; Yang, D.M.; Chou, S.J.; Yang, Y.P.; et al. A Review of SARS-CoV-2 and the Ongoing Clinical Trials. Int. J. Mol. Sci. 2020, 21, 2657. [Google Scholar] [CrossRef] [PubMed]
- Jan, H.; Faisal, S.; Khan, A.; Khan, S.; Usman, H.; Liaqat, R.; Shah, S.A. COVID-19: Review of Epidemiology and Potential Treatments Against 2019 Novel Coronavirus. Discoveries (Craiova) 2020, 8, e108. [Google Scholar] [CrossRef] [PubMed]
- Gildenhuys, S. Expanding our understanding of the role polyprotein conformation plays in the coronavirus life cycle. Biochem. J. 2020, 477, 1479–1482. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Brindisi, M.; Shahabi, D.; Chapman, M.E.; Mesecar, A.D. Drug Development and Medicinal Chemistry Efforts toward SARS-Coronavirus and Covid-19 Therapeutics. ChemMedChem 2020. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020. [Google Scholar] [CrossRef]
- Li, C.; Xu, B.H. The viral, epidemiologic, clinical characteristics and potential therapy options for COVID-19: A review. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 4576–4584. [Google Scholar] [CrossRef]
- Zhao, N.; Zhou, Z.L.; Wu, L.; Zhang, X.D.; Han, S.B.; Bao, H.J.; Shu, Y.; Shu, X.G. An update on the status of COVID-19: A comprehensive review. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 4597–4606. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Yiu, C.B.; Wong, K.Y. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CL (pro)) structure: Virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Res 2020, 9, 129. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Tao, H.; Yan, Y.; Huang, S.Y.; Xiao, Y. Molecular Mechanism of Evolution and Human Infection with SARS-CoV-2. Viruses 2020, 12, 428. [Google Scholar] [CrossRef] [PubMed]
- Mengist, H.M.; Fan, X.; Jin, T. Designing of improved drugs for COVID-19: Crystal structure of SARS-CoV-2 main protease M(pro). Signal Transduct. Target. Ther. 2020, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Ortega, J.T.; Serrano, M.L.; Pujol, F.H.; Rangel, H.R. Unrevealing sequence and structural features of novel coronavirus using in silico approaches: The main protease as molecular target. EXCLI J. 2020, 19, 400–409. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Savarino, A. Expanding the frontiers of existing antiviral drugs: Possible effects of HIV-1 protease inhibitors against SARS and avian influenza. J. Clin. Virol. 2005, 34, 170–178. [Google Scholar] [CrossRef]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- McCreary, E.K.; Pogue, J.M. Coronavirus Disease 2019 Treatment: A Review of Early and Emerging Options. Open Forum Infect. Dis. 2020, 7, ofaa105. [Google Scholar] [CrossRef]
- Meo, S.A.; Klonoff, D.C.; Akram, J. Efficacy of chloroquine and hydroxychloroquine in the treatment of COVID-19. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 4539–4547. [Google Scholar] [CrossRef]
- Maradey-Romero, C.; Fass, R. New and future drug development for gastroesophageal reflux disease. J. Neurogastroenterol. Motil. 2014, 20, 6–16. [Google Scholar] [CrossRef]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Ortega, J.T.; Serrano, M.L.; Suarez, A.I.; Baptista, J.; Pujol, F.H.; Cavallaro, L.V.; Campos, H.R.; Rangel, H.R. Antiviral activity of flavonoids present in aerial parts of Marcetia taxifolia against Hepatitis B virus, Poliovirus, and Herpes Simplex Virus in vitro. EXCLI J. 2019, 18, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Macarthur, M.W.; Moss, D.S.; Thornton, J.M. Procheck—A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Pedretti, A.; Villa, L.; Vistoli, G. VEGA--an open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J. Comput. Aided Mol. Des. 2004, 18, 167–173. [Google Scholar] [CrossRef]
- Ortega, J.T.; Jastrzebska, B. The Retinoid and Non-Retinoid Ligands of the Rod Visual G Protein-Coupled Receptor. Int. J. Mol. Sci. 2019, 20, 6218. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Linares, I.; Perez-Sanchez, H.; Cecilia, J.M.; Garcia, J.M. High-Throughput parallel blind Virtual Screening using BINDSURF. BMC Bioinform. 2012, 13 (Suppl. 14), S13. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Baez-Santos, Y.M.; St John, S.E.; Mesecar, A.D. The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antiviral. Res. 2015, 115, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Freedberg, D.E.; Conigliaro, J.; Sobieszczyk, M.E.; Markowitz, D.D.; Gupta, A.; O’Donnell, M.R.; Li, J.; Tuveson, D.A.; Jin, Z.; Turner, W.C.; et al. Famotidine Use is Associated with Improved Clinical Outcomes in Hospitalized COVID-19 Patients: A Retrospective Cohort Study. medRxiv 2020. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Zhang, B.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Liu, F.; Li, C.; Li, Y.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020. [Google Scholar] [CrossRef]
- Lai, S.T. Treatment of severe acute respiratory syndrome. Eur. J. Clin. Microbiol. Infect. Dis. 2005, 24, 583–591. [Google Scholar] [CrossRef]
- Lim, J.; Jeon, S.; Shin, H.Y.; Kim, M.J.; Seong, Y.M.; Lee, W.J.; Choe, K.W.; Kang, Y.M.; Lee, B.; Park, S.J. Case of the Index Patient Who Caused Tertiary Transmission of COVID-19 Infection in Korea: The Application of Lopinavir/Ritonavir for the Treatment of COVID-19 Infected Pneumonia Monitored by Quantitative RT-PCR. J. Korean Med. Sci. 2020, 35, e79. [Google Scholar] [CrossRef]
- Khan, S.A.; Zia, K.; Ashraf, S.; Uddin, R.; Ul-Haq, Z. Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. J. Biomol. Struct. Dyn. 2020, 1–10. [Google Scholar] [CrossRef]
- Rut, W.Z.; Zmudzinski, M.; Lyu, Z.; Nayak, D.; Snipas, S.J.; Bekes, M.; Huang, T.T.; Olsen, S.K.; Drag, M. Activity profiling of SARS-CoV-2-PLpro protease provides structural framework for anti-COVID-19 drug design. bioRxiv 2020. [Google Scholar] [CrossRef]
- Rimanshee, A.; Amit, D.; Vishal, P.; Mukesk, K. Potential inhibitors against papain-like protease of novel coronavirus (SARS-CoV-2) from FDA approved drugs. ChemRxiv Preprint 2020. [Google Scholar] [CrossRef]
- Ortega, J.T.; Serrano, M.L.; Pujol, F.H.; Rangel, H.R. Role of changes in SARS-CoV-2 spike protein in the interaction with the human ACE2 receptor: An in silico analysis. EXCLI J. 2020, 19, 410–417. [Google Scholar] [CrossRef]
- Astuti, I.; Ysrafil. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes Metab. Syndr. 2020, 14, 407–412. [Google Scholar] [CrossRef] [PubMed]
- McKee, D.L.; Sternberg, A.; Stange, U.; Laufer, S.; Naujokat, C. Candidate drugs against SARS-CoV-2 and COVID-19. Pharmacol. Res. 2020, 157, 104859. [Google Scholar] [CrossRef] [PubMed]
- Bourinbaiar, A.S.; Fruhstorfer, E.C. The effect of histamine type 2 receptor antagonists on human immunodeficiency virus (HIV) replication: Identification of a new class of antiviral agents. Life Sci. 1996, 59, 365–370. [Google Scholar] [CrossRef]
- Bourinbaiar, A.S.; Fruhstorfer, E.C. Effect of the oral anti-ulcer agent, cimetidine, on HIV type 1 replication. AIDS Res. Hum. Retrovir. 1997, 13, 281–282. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.; Cherepanov, P. The structural biology of HIV-1: Mechanistic and therapeutic insights. Nat. Rev. Microbiol 2012, 10, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Arts, E.J.; Hazuda, D.J. HIV-1 antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161. [Google Scholar] [CrossRef] [PubMed]
- Sayer, J.M.; Louis, J.M. Interactions of different inhibitors with active-site aspartyl residues of HIV-1 protease and possible relevance to pepsin. Proteins 2009, 75, 556–568. [Google Scholar] [CrossRef]
- Delaney, J.S. ESOL: Estimating aqueous solubility directly from molecular structure. J. Chem. Inf. Comput. Sci. 2004, 44, 1000–1005. [Google Scholar] [CrossRef]
- Ali, J.; Camilleri, P.; Brown, M.B.; Hutt, A.J.; Kirton, S.B. Revisiting the general solubility equation: In silico prediction of aqueous solubility incorporating the effect of topographical polar surface area. J. Chem. Inf. Model. 2012, 52, 420–428. [Google Scholar] [CrossRef]
- Bohnert, T.; Gan, L.S. Plasma protein binding: From discovery to development. J. Pharm. Sci. 2013, 102, 2953–2994. [Google Scholar] [CrossRef]
- Howard, M.L.; Hill, J.J.; Galluppi, G.R.; McLean, M.A. Plasma protein binding in drug discovery and development. Comb. Chem. High Throughput Screen. 2010, 13, 170–187. [Google Scholar] [CrossRef] [PubMed]
- Hsu, A.; Isaacson, J.; Brun, S.; Bernstein, B.; Lam, W.; Bertz, R.; Foit, C.; Rynkiewicz, K.; Richards, B.; King, M.; et al. Pharmacokinetic-pharmacodynamic analysis of lopinavir-ritonavir in combination with efavirenz and two nucleoside reverse transcriptase inhibitors in extensively pretreated human immunodeficiency virus-infected patients. Antimicrob. Agents Chemother. 2003, 47, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Dhama, K.; Sharun, K.; Tiwari, R.; Dadar, M.; Malik, Y.S.; Singh, K.P.; Chaicumpa, W. COVID-19, an emerging coronavirus infection: Advances and prospects in designing and developing vaccines, immunotherapeutics, and therapeutics. Hum. Vaccin. Immunother. 2020, 1–7. [Google Scholar] [CrossRef]
- Yan, Y.; Shin, W.I.; Pang, Y.X.; Meng, Y.; Lai, J.; You, C.; Zhao, H.; Lester, E.; Wu, T.; Pang, C.H. The First 75 Days of Novel Coronavirus (SARS-CoV-2) Outbreak: Recent Advances, Prevention, and Treatment. Int. J. Environ. Res. Public Health 2020, 17, 2323. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, S.M.; Yu, X.H.; Tang, S.L.; Tang, C.K. Coronavirus disease 2019 (COVID-19): Current status and future perspectives. Int. J. Antimicrob. Agents 2020, 105951. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 14. [Google Scholar] [CrossRef]
- Mirza, M.U.; Froeyen, M. Structural elucidation of SARS-CoV-2 vital proteins: Computational methods reveal potential drug candidates against main protease, Nsp12 polymerase and Nsp13 helicase. J. Pharm. Anal. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ton, A.T.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol. Inform. 2020. [Google Scholar] [CrossRef]
- Prajapat, M.; Sarma, P.; Shekhar, N.; Avti, P.; Sinha, S.; Kaur, H.; Kumar, S.; Bhattacharyya, A.; Kumar, H.; Bansal, S.; et al. Drug targets for corona virus: A systematic review. Indian J. Pharmacol. 2020, 52, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, Y. Potential interventions for novel coronavirus in China: A systematic review. J. Med. Virol. 2020, 92, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.P.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S.; et al. Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases. ACS Cent. Sci. 2020, 6, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Te, H.S.; Randall, G.; Jensen, D.M. Mechanism of action of ribavirin in the treatment of chronic hepatitis C. Gastroenterol. Hepatol. (NY) 2007, 3, 218–225. [Google Scholar]
- Jaspe, R.C.; Ortega, J.; Zambrano, J.L.; Pujol, F.H. [Present and future of therapy against hepatitis C]. Investig. Clin. 2016, 57, 93–107. [Google Scholar]
- Manns, M.P.; Wedemeyer, H.; Cornberg, M. Treating viral hepatitis C: Efficacy, side effects, and complications. Gut 2006, 55, 1350–1359. [Google Scholar] [CrossRef]
- Al-Bari, M.A.A. Targeting endosomal acidification by chloroquine analogs as a promising strategy for the treatment of emerging viral diseases. Pharmacol. Res. Perspect. 2017, 5, e00293. [Google Scholar] [CrossRef]
- Vincent, M.J.; Bergeron, E.; Benjannet, S.; Erickson, B.R.; Rollin, P.E.; Ksiazek, T.G.; Seidah, N.G.; Nichol, S.T. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2005, 2, 69. [Google Scholar] [CrossRef]
- Liu, J.; Cao, R.; Xu, M.; Wang, X.; Zhang, H.; Hu, H.; Li, Y.; Hu, Z.; Zhong, W.; Wang, M. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020, 6, 16. [Google Scholar] [CrossRef]
- Gautret, P.; Lagier, J.C.; Parola, P.; Hoang, V.T.; Meddeb, L.; Mailhe, M.; Doudier, B.; Courjon, J.; Giordanengo, V.; Vieira, V.E.; et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: Results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents 2020, 105949. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Chemical Structure |
|---|---|
| Name: Famotidine Chemical formula: C8H15N7O2S3 Molecular Weight: 337.44 g/mol |  |
| Name: Lopinavir Chemical formula: C37H48N4O5 Molecular Weight: 628.80 g/mol |  |
| Name: Nafamostat Chemical formula: C19H17N5O2 Molecular Weight: 347.37 g/mol |  |
| Name: Ribavirin Chemical formula: C8H12N4O5 Molecular Weight: 244.20 g/mol |  |
| Name: SARS-CoV(1) Chemical formula: C25H27FN2O Molecular Weight: 390.49 g/mol |  |
| Name: SARS-CoV(2) Chemical formula: C20H20N2O Molecular Weight: 304.38 g/mol |  |
| Name: Darunavir Chemical formula: C27H37N3O7S Molecular Weight: 547.7 g/mol |  |
| Name: Camostat Chemical formula: C20H22N4O5 Molecular Weight: 398.4 g/mol |  |
| Compound | Pubmed ID | Binding Free Energy (kcal/mol) |
|---|---|---|
| Famotidine | 24883443 | −6.4 |
| Lopinavir | 92727 | −9.0 |
| Darunavir | 213039 | −8.3 |
| Compound | Pubmed ID | Binding Free Energy (kcal/mol) |
|---|---|---|
| Famotidine | 24883443 | −5.0 |
| Ribavirin | 37542 | −6.1 |
| SARS-CoV(1) | 44828571 | −6.5 |
| SARS-CoV(2) | 73659185 | −6.6 |
| Compound | Pubmed ID | Binding Free Energy (kcal/mol) |
|---|---|---|
| Famotidine | 24883443 | −5.8 |
| Nafamostat | 4413 | −7.6 |
| Camostat | 2536 | −6.3 |
| Compound | Pubmed ID | Binding Free Energy (kcal/mol) |
|---|---|---|
| Famotidine | 24883443 | −6.4 |
| Lopinavir | 92727 | −10.4 |
| Darunavir | 213039 | −10.5 |
| Solubility Predictor | Famotidine | Ribavirin | Lopinvavir | Nafamostat |
|---|---|---|---|---|
| Log S (ESOL) | −1.25 | −0.21 | −6.64 | −3.40 |
| Solubility | 5.60 × 10−2 M | 6.19 × 10−1 M | 2.31 × 10−7 M | 4.00 × 10−4 M |
| Class | Very soluble | Very soluble | Poorly soluble | Soluble |
| Log S (Ali) | −3.88 | −0.65 | −8.21 | −4.61 |
| Solubility | 1.32 × 10−4 M | 2.24 × 10−1 M | 6.10 × 10−9 M | 2.46 × 10−5 M |
| Class | Soluble | Very soluble | Poorly soluble | Moderately soluble |
| Log S (SILICOS-IT) | −1.26 | 1.76 | −10.05 | −5.35 |
| Solubility | 5.43 × 10−2 M | 5.73 × 101 M | 8.85 × 10−11 M | 4.46 × 10−6 M |
| Class | Soluble | Soluble | Insoluble | Moderately soluble |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortega, J.T.; Serrano, M.L.; Jastrzebska, B. Class A G Protein-Coupled Receptor Antagonist Famotidine as a Therapeutic Alternative against SARS-CoV2: An In Silico Analysis. Biomolecules 2020, 10, 954. https://doi.org/10.3390/biom10060954
Ortega JT, Serrano ML, Jastrzebska B. Class A G Protein-Coupled Receptor Antagonist Famotidine as a Therapeutic Alternative against SARS-CoV2: An In Silico Analysis. Biomolecules. 2020; 10(6):954. https://doi.org/10.3390/biom10060954
Chicago/Turabian StyleOrtega, Joseph T., Maria Luisa Serrano, and Beata Jastrzebska. 2020. "Class A G Protein-Coupled Receptor Antagonist Famotidine as a Therapeutic Alternative against SARS-CoV2: An In Silico Analysis" Biomolecules 10, no. 6: 954. https://doi.org/10.3390/biom10060954
APA StyleOrtega, J. T., Serrano, M. L., & Jastrzebska, B. (2020). Class A G Protein-Coupled Receptor Antagonist Famotidine as a Therapeutic Alternative against SARS-CoV2: An In Silico Analysis. Biomolecules, 10(6), 954. https://doi.org/10.3390/biom10060954