Comparative Genomics Reveals a Significant Sequence Variability of Myticin Genes in Mytilus galloprovincialis



 and
and 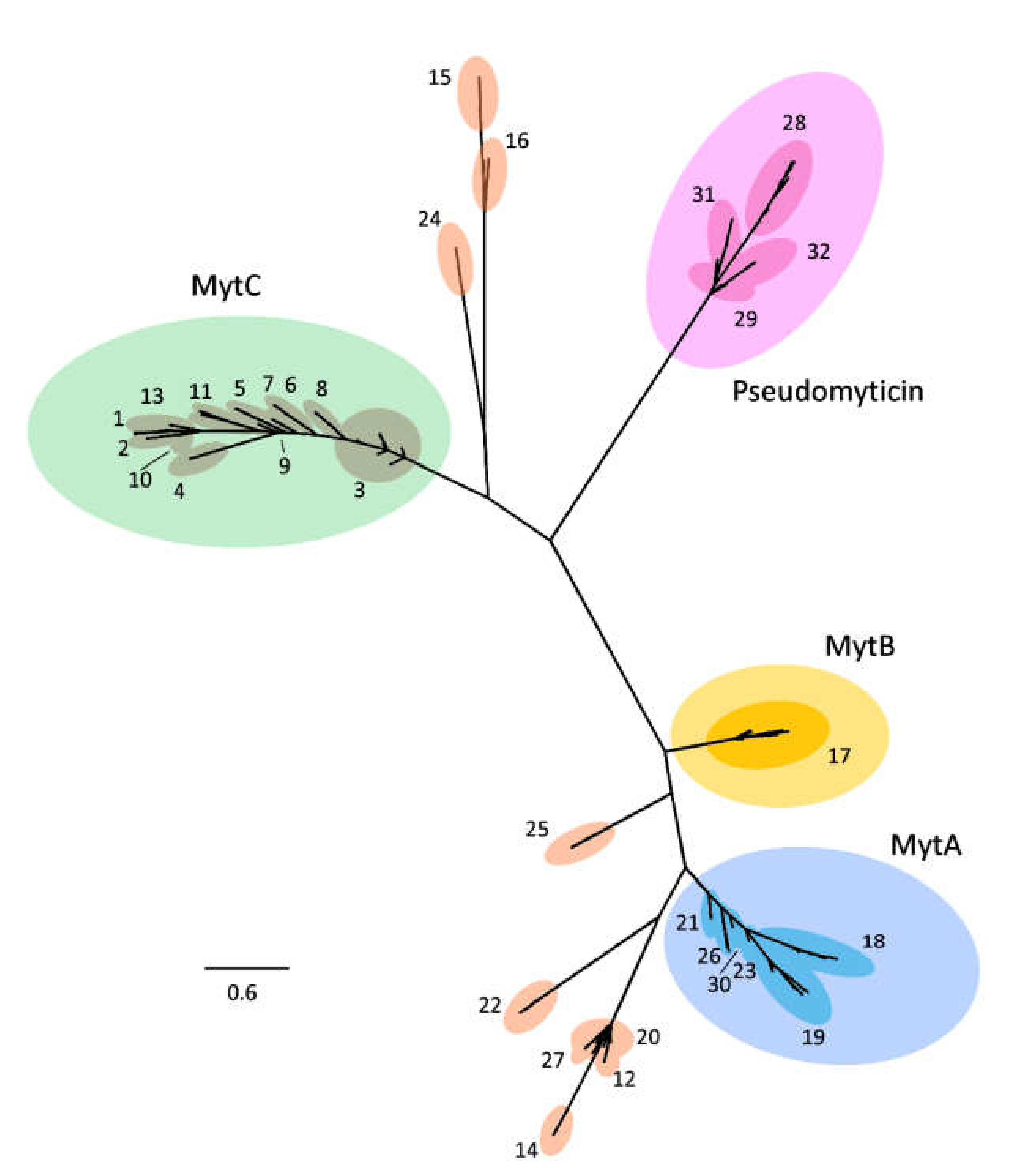{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Searching, Screening, and Identifying M. galloprovincialis Myticins
2.2. Phylogenetic Analysis
2.3. Isoelectrical Point
2.4. Positive and Negative Selection Analysis
2.5. Promoter Analysis
2.6. Genomic vs. Transcriptomic Data
2.7. Expression Analysis
3. Results
3.1. Searching, Screening, and Identifying M. galloprovincialis Myticins
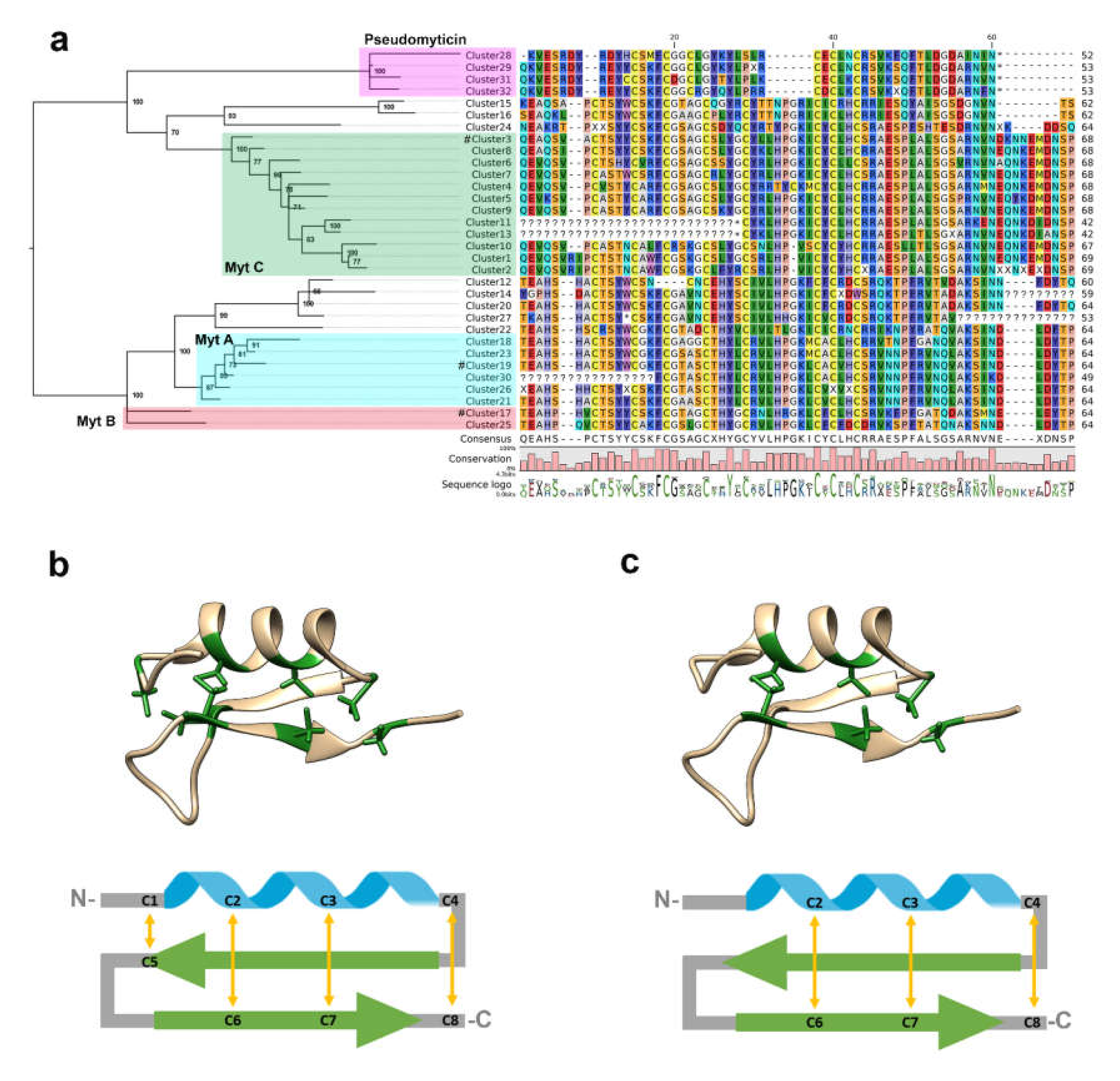
3.2. Phylogenetic Analysis
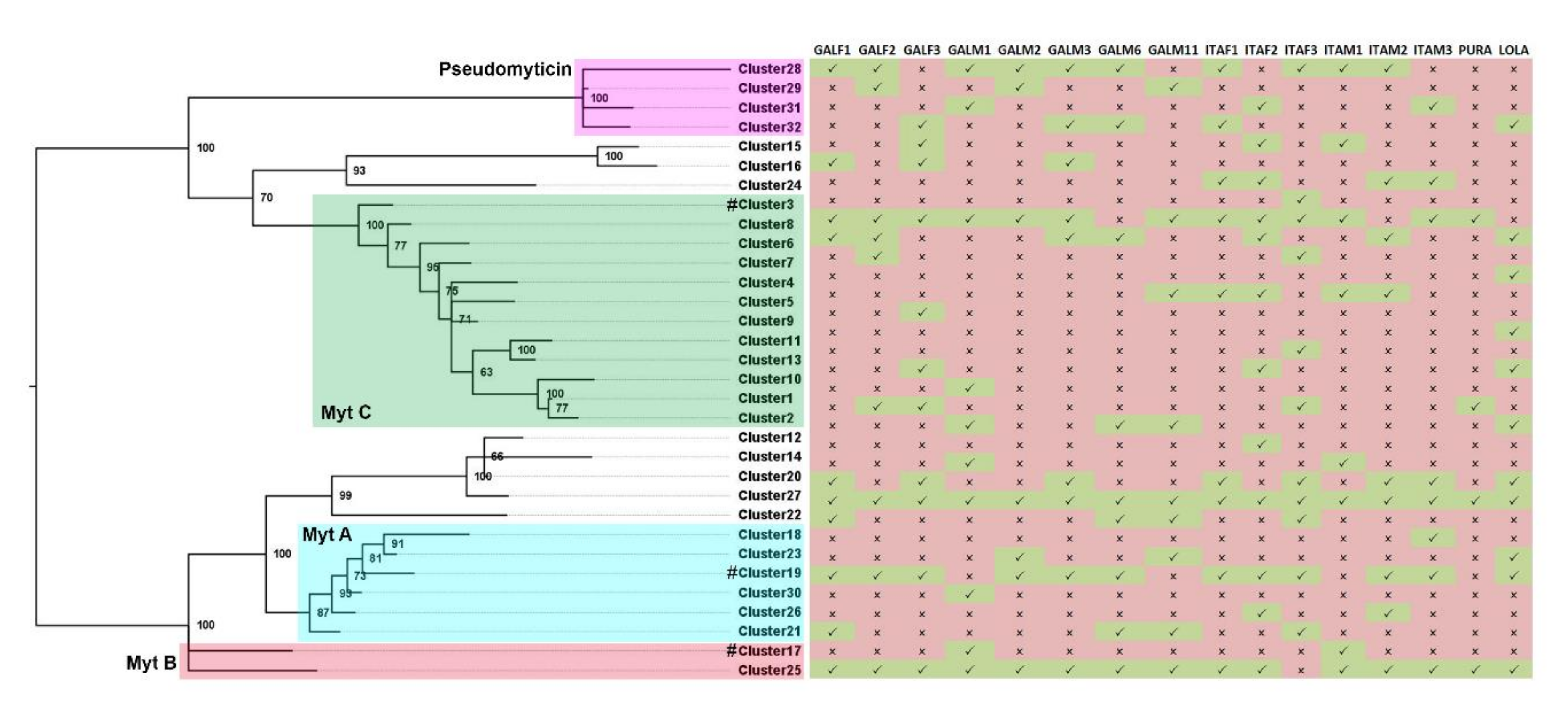
3.3. Presence/Absence Variation
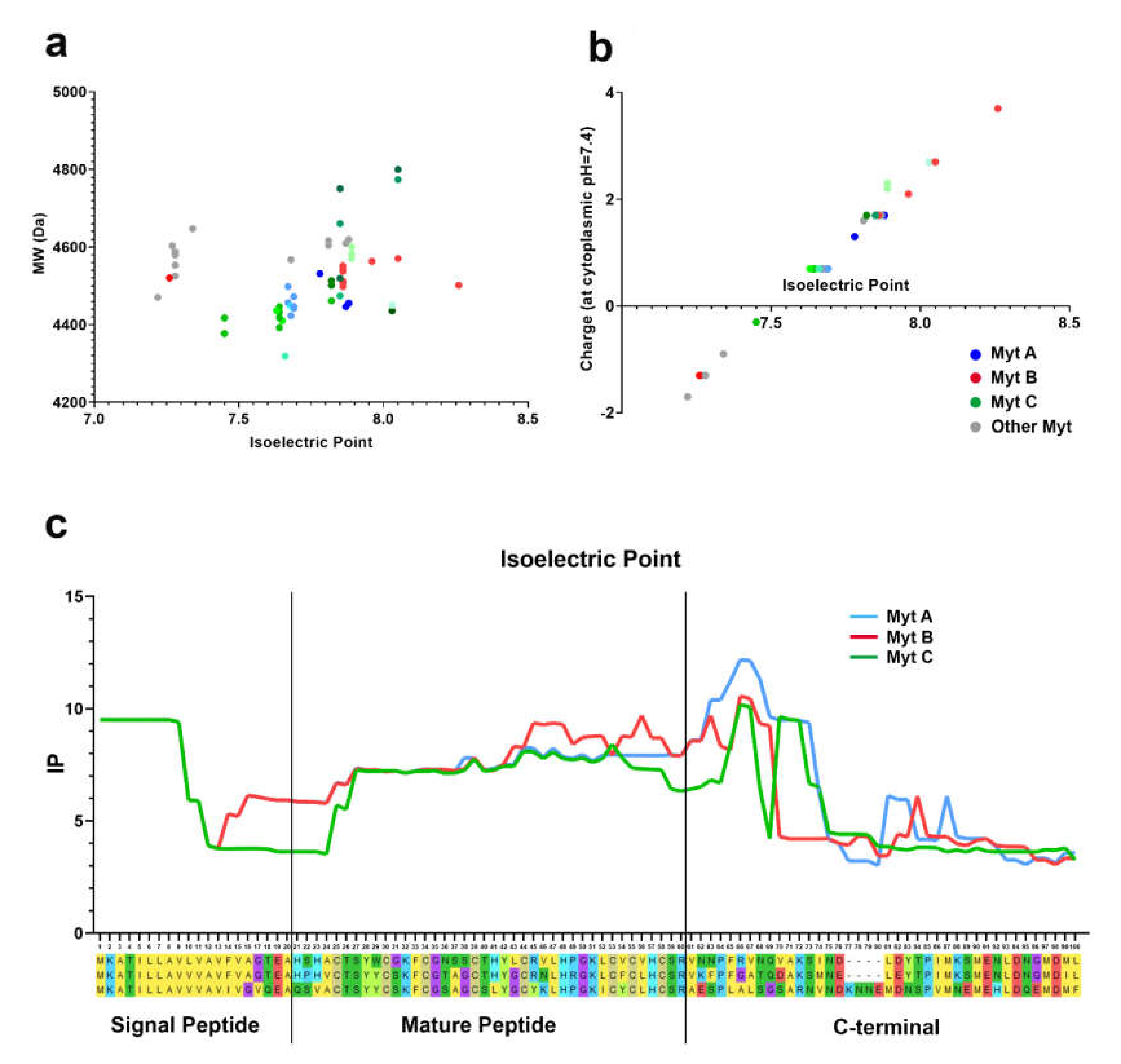
3.4. Isoelectric Point
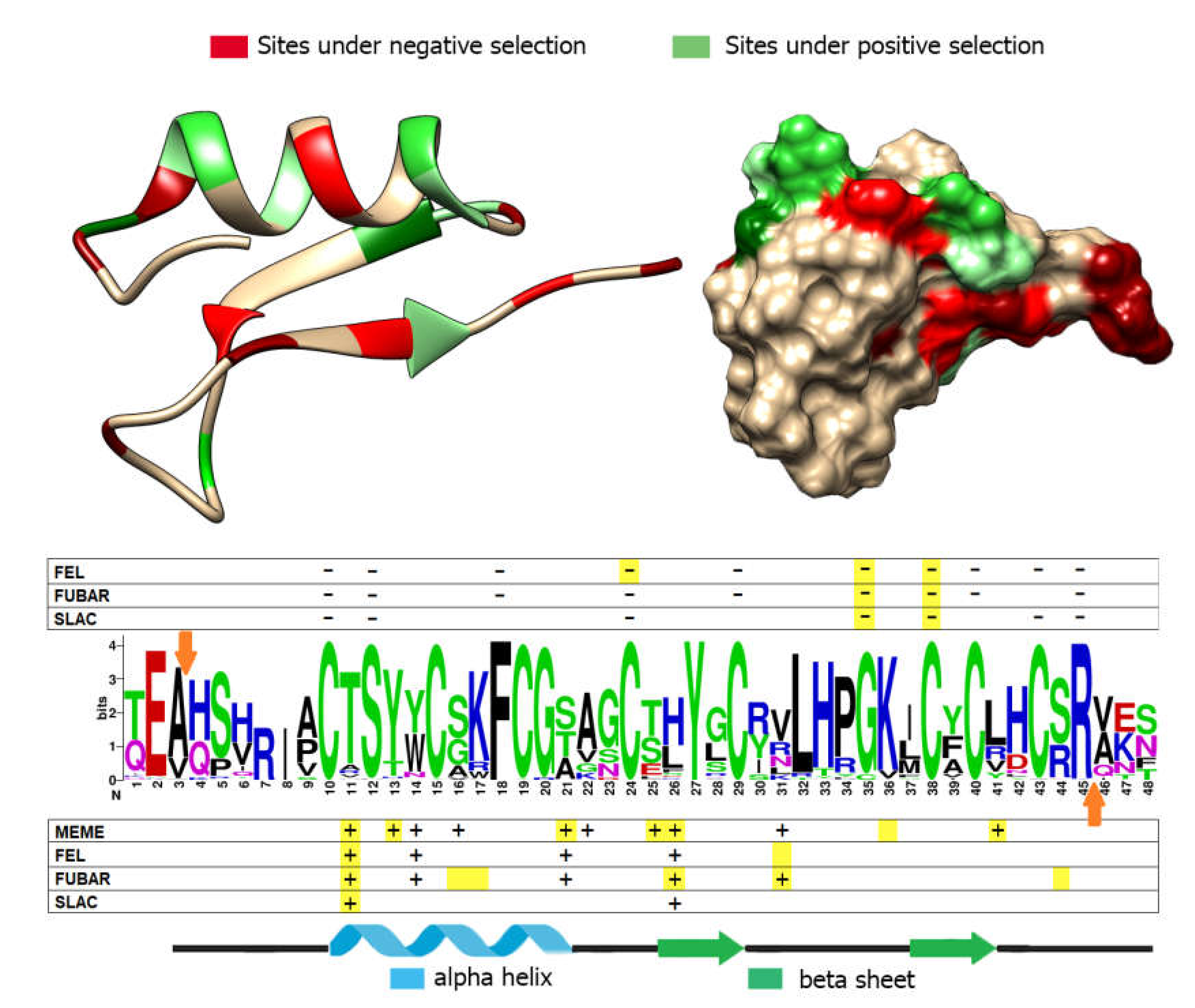
3.5. Positive and Negative Selection Analysis
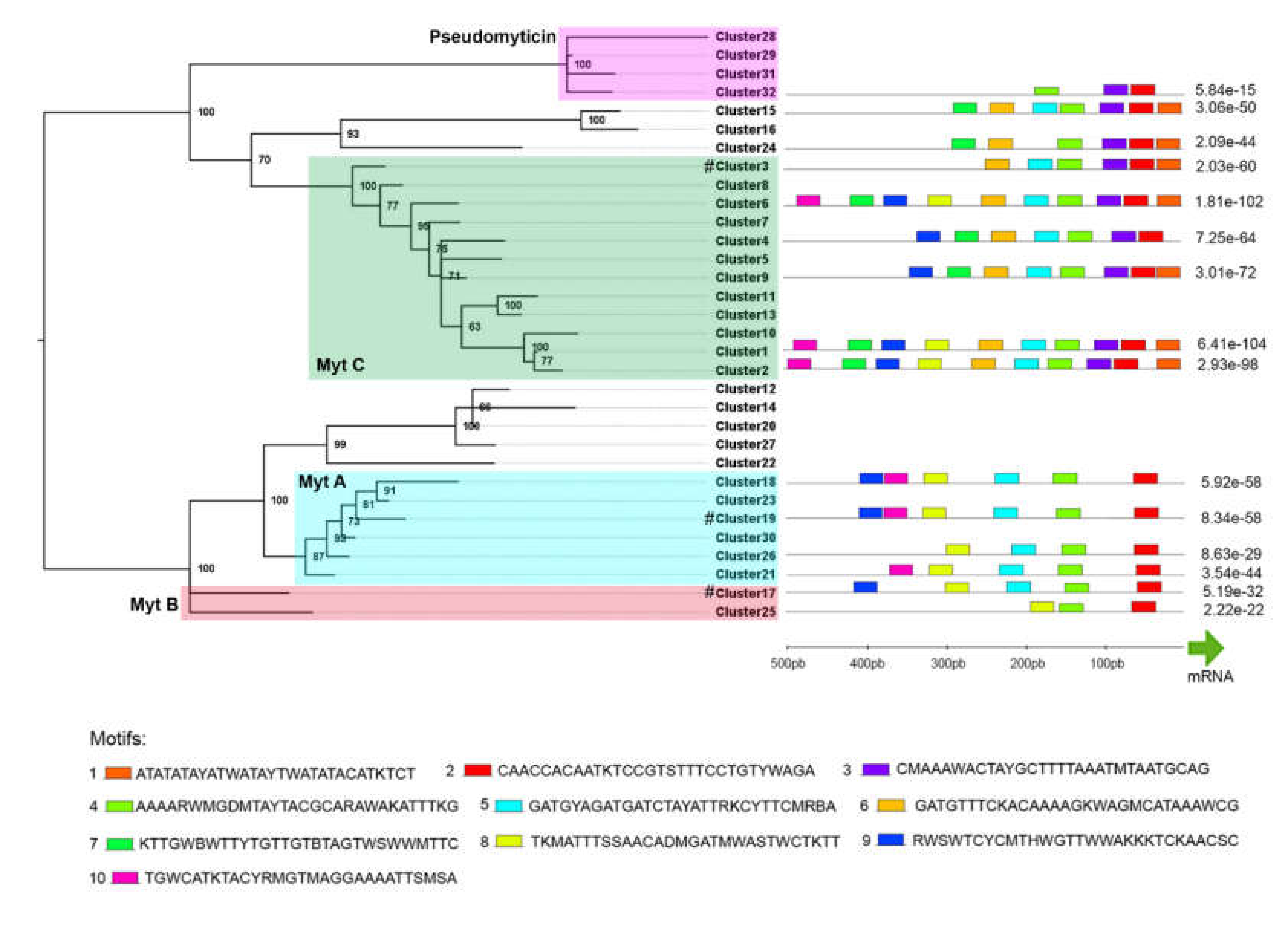
3.6. Promoter Analysis
3.7. Genomic vs. Transcriptomic data
3.8. Expression Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mitta, G.; Hubert, F.; Noël, T.; Roch, P. Myticin, a novel cysteine-rich antimicrobial peptide isolated from haemocytes and plasma of the mussel Mytilus galloprovincialis. Eur. J. Biochem. 1999, 265, 71–78. [Google Scholar] [CrossRef]
- Pallavicini, A.; del Mar Costa, M.; Gestal, C.; Dreos, R.; Figueras, A.; Venier, P.; Novoa, B. High sequence variability of myticin transcripts in hemocytes of immune-stimulated mussels suggests ancient host-pathogen interactions. Dev. Comp. Immunol. 2008, 32, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.M.; Dios, S.; Alonso-Gutierrez, J.; Romero, A.; Novoa, B.; Figueras, A. Evidence of high individual diversity on myticin C in mussel (Mytilus galloprovincialis). Dev. Comp. Immunol. 2009, 33, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Vera, M.; Martínez, P.; Poisa-Beiro, L.; Figueras, A.; Novoa, B. Genomic organization, molecular diversification, and evolution of antimicrobial peptide myticin-C genes in the mussel (Mytilus galloprovincialis). PLoS ONE 2011, 6, e24041. [Google Scholar] [CrossRef] [PubMed]
- Balseiro, P.; Falcó, A.; Romero, A.; Dios, S.; Martínez-López, A.; Figueras, A.; Estepa, A.; Novoa, B. Mytilus galloprovincialis myticin C: A chemotactic molecule with antiviral activity and immunoregulatory properties. PLoS ONE 2011, 6, e23140. [Google Scholar] [CrossRef] [PubMed]
- Novoa, B.; Romero, A.; Álvarez, Á.L.; Moreira, R.; Pereiro, P.; Costa, M.M.; Dios, S.; Estepa, A.; Parra, F.; Figueras, A. Antiviral Activity of Myticin C Peptide from Mussel: An Ancient Defense against Herpesviruses. J. Virol. 2016, 90, 7692–7702. [Google Scholar] [CrossRef] [PubMed]
- Fiorito, F.; Amoroso, M.G.; Lambiase, S.; Serpe, F.P.; Bruno, T.; Scaramuzzo, A.; Maglio, P.; Fusco, G.; Esposito, M. A relationship between environmental pollutants and enteric viruses in mussels (Mytilus galloprovincialis). Environ. Res. 2019, 169, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.; del Mar Costa, M.; Forn-Cuni, G.; Balseiro, P.; Chamorro, R.; Dios, S.; Figueras, A.; Novoa, B. Occurrence, seasonality and infectivity of Vibrio strains in natural populations of mussels Mytilus galloprovincialis. Dis. Aquat. Organ. 2014, 108, 149–163. [Google Scholar] [CrossRef]
- Benabdelmouna, A.; Garcia, C.; Ledu, C.; Lamy, P.; Maurouard, E.; Dégremont, L. Mortality investigation of Mytilus edulis and Mytilus galloprovincialis in France: An experimental survey under laboratory conditions. Aquaculture 2018, 495, 831–841. [Google Scholar] [CrossRef]
- Garcia, C.; Haond, C.; Chollet, B.; Nerac, M.; Omnes, E.; Joly, J.P.; Dubreuil, C.; Serpin, D.; Langlade, A.; Le Gal, D.; et al. Descriptions of Mikrocytos veneroïdes n. sp. and Mikrocytos donaxi n. sp. (Ascetosporea: Mikrocytida: Mikrocytiidae), detected during important mortality events of the wedge clam Donax trunculus Linnaeus (Veneroida: Donacidae), in France between 2008 and 2011. Parasit. Vectors 2018, 11, 119. [Google Scholar] [CrossRef]
- Segarra, A.; Pepin, J.F.; Arzul, I.; Morga, B.; Faury, N.; Renault, T. Detection and description of a particular Ostreid herpesvirus 1 genotype associated with massive mortality outbreaks of Pacific oysters, Crassostrea gigas, in France in 2008. Virus Res. 2010, 153, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Moreira, R.; Pereiro, P.; Canchaya, C.; Posada, D.; Figueras, A.; Novoa, B. RNA-Seq in Mytilus galloprovincialis: Comparative transcriptomics and expression profiles among different tissues. BMC Genom. 2015, 16, 728. [Google Scholar] [CrossRef] [PubMed]
- Hubert, F.; Noel, T.; Roch, P. A member of the arthropod defensin family from edible Mediterranean mussels (Mytilus galloprovincialis). Eur. J. Biochem. 1996, 240, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Mitta, G.; Vandenbulcke, F.; Hubert, F.; Salzet, M.; Roch, P. Involvement of mytilins in mussel antimicrobial defense. J. Biol. Chem. 2000, 275, 12954–12962. [Google Scholar] [CrossRef]
- Sonthi, M.; Toubiana, M.; Pallavicini, A.; Venier, P.; Roch, P. Diversity of coding sequences and gene structures of the antifungal peptide mytimycin (MytM) from the Mediterranean mussel, Mytilus galloprovincialis. Mar. Biotechnol. 2011, 13, 857–867. [Google Scholar] [CrossRef]
- Rosani, U.; Varotto, L.; Rossi, A.; Roch, P.; Novoa, B.; Figueras, A.; Pallavicini, A.; Venier, P. Massively parallel amplicon sequencing reveals isotype-specific variability of antimicrobial peptide transcripts in Mytilus galloprovincialis. PLoS ONE 2011, 6, e26680. [Google Scholar] [CrossRef]
- Mitta, G.; Vandenbulcke, F.; Noel, T.; Romestand, B.; Beauvillain, J.C.; Salzet, M.; Roch, P. Differential distribution and defence involvement of antimicrobial peptides in mussel. J. Cell Sci. 2000, 113, 2759–2769. [Google Scholar]
- Costa, M.M.; Prado-Alvarez, M.; Gestal, C.; Li, H.; Roch, P.; Novoa, B.; Figueras, A. Functional and molecular immune response of Mediterranean mussel (Mytilus galloprovincialis) haemocytes against pathogen-associated molecular patterns and bacteria. Fish Shellfish Immunol. 2009, 26, 515–523. [Google Scholar] [CrossRef]
- Rey-Campos, M.; Moreira, R.; Valenzuela-Muñoz, V.; Gallardo-Escárate, C.; Novoa, B.; Figueras, A. High individual variability in the transcriptomic response of Mediterranean mussels to Vibrio reveals the involvement of myticins in tissue injury. Sci. Rep. 2019, 9, 3569. [Google Scholar] [CrossRef]
- Rey-Campos, M.; Moreira, R.; Romero, A.; Medina-Gali, R.M.; Novoa, B.; Gasset, M.; Figueras, A. Transcriptomic Analysis Reveals the Wound Healing Activity of Mussel Myticin C. Biomolecules 2020, 10, 133. [Google Scholar] [CrossRef]
- Gerdol, M.; Moreira, R.; Cruz, F.; Gómez-Garrido, J.; Vlasova, A.; Rosani, U.; Venier, P.; Naranjo-Ortiz, M.A.; Murgarella, M.; Balseiro, P.; et al. Massive gene presence/absence variation in the mussel genome as an adaptive strategy: First evidence of a pan-genome in Metazoa. BioRxiv 2019, 781377. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef]
- Guindon, S.; Gascuel, O. A simple, fast and accurate method to estimate large phylogenies by maximum-likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.X. Linear invariants under Jukes’ and Cantor’s one-parameter model. J. Theor. Biol. 1995, 173, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree, A Graphical Viewer of Phylogenetic Trees; University of Edinburgh: Edinburgh, UK, 2009. [Google Scholar]
- Bendtsen, J.D.; Nielsen, H.; Heijne, G.; Brunak, S. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 2004, 340, 783–795. [Google Scholar] [CrossRef]
- Kozlowski, L.P. IPC—Isoelectric Point Calculator. Biol. Direct. 2016, 11, 55. [Google Scholar] [CrossRef]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting Individual Sites Subject to Episodic Diversifying Selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Frost, S.D. Not So Different After All: A Comparison of Methods for Detecting Amino Acid Sites Under Selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Domeneghetti, S.; Franzoi, M.; Damiano, N.; Norante, R.; El Halfawy, N.M.; Mammi, S.; Marin, O.; Bellanda, M.; Venier, P. Structural and Antimicrobial Features of Peptides Related to Myticin C, a Special Defense Molecule from the Mediterranean Mussel Mytilus galloprovincialis. J. Agric. Food Chem. 2015, 63, 9251–9259. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved Splice Site Detection in Genie. J. Comp. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef]
- Zia, A.; Moses, A.M. Towards a theoretical understanding of false positives in DNA motif finding. BMC Bioinform. 2012, 13, 151. [Google Scholar] [CrossRef]
- Bailey, T.L.; Bodén, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Greco, S.; Gerdol, M.; Edomi, P.; Pallavicini, A. Molecular Diversity of Mytilin-Like Defense Peptides in Mytilidae (Mollusca, Bivalvia). Antibiotics 2020, 9, 37. [Google Scholar] [CrossRef]
- McInerney, J.O.; McNally, A.; O’Connell, M.J. Why prokaryotes have pangenomes. Nat. Microbiol. 2017, 2, 17040. [Google Scholar] [CrossRef]
- Hirsch, C.N.; Foerster, J.M.; Johnson, J.M.; Sekhon, R.S.; Muttoni, G.; Vaillancourt, B.; Peñagaricano, F.; Lindquist, E.; Pedraza, M.A.; Barry, K.; et al. Insights into the maize pan-genome and pan-transcriptome. Plant Cell 2014, 26, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Conrad, D.F.; Pinto, D.; Redon, R.; Feuk, L.; Gokcumen, O.; Zhang, Y.; Aerts, J.; Andrews, T.D.; Barnes, C.; Campbell, P.; et al. Origins and functional impact of copy number variation in the human genome. Nature 2010, 464, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Onsins, S.; Aguadé, M. Molecular evolution of the Cecropin multigene family in Drosophila. functional genes vs. pseudogenes. Genetics 1998, 150, 157–171. [Google Scholar] [PubMed]
- Hanson, M.A.; Lemaitre, B.; Unckless, R.L. Dynamic Evolution of Antimicrobial Peptides Underscores Trade-Offs Between Immunity and Ecological Fitness. Front. Immunol. 2019, 10, 2620. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.S.; Mitta, G.; Chavanieu, A.; Calas, B.; Sanchez, J.F.; Roch, P.; Aumelas, A. Solution structure and activity of the synthetic four-disulfide bond Mediterranean mussel defensin (MGD-1). Biochemistry 2000, 39, 14436–14447. [Google Scholar] [CrossRef] [PubMed]
- Roch, P.; Yang, Y.; Toubiana, M.; Aumelas, A. NMR structure of mussel mytilin, and antiviral-antibacterial activities of derived synthetic peptides. Dev. Comp. Immunol. 2008, 32, 227–238. [Google Scholar] [CrossRef]
- Tincu, J.A.; Taylor, S.W. Antimicrobial Peptides from Marine Invertebrates. Antimicrob. Agents Chemother. 2004, 48, 3645–3654. [Google Scholar] [CrossRef]
- Bechinger, B.; Gorr, S.U. Antimicrobial Peptides: Mechanisms of Action and Resistance. J. Dent. Res. 2017, 96, 254–260. [Google Scholar] [CrossRef]
- Padhi, A.; Verghese, B. Molecular diversity and evolution of myticin-C antimicrobial peptide variants in the Mediterranean mussel, Mytilus galloprovincialis. Peptides 2008, 29, 1094–1101. [Google Scholar] [CrossRef]
- Vallecillo-Viejo, I.C.; Liscovitch-Brauer, N.; Diaz Quiroz, J.F.; Montiel-Gonzalez, M.F.; Nemes, S.E.; Rangan, K.J.; Levinson, S.R.; Eisenberg, E.; Rosenthal, J.J.C. Spatially regulated editing of genetic information within a neuron. Nucleic Acids Res. 2020, 48, 3999–4012. [Google Scholar] [CrossRef]
- Moreira, R.; Pereiro, P.; Balseiro, P.; Milan, M.; Pauletto, M.; Bargelloni, L.; Novoa, B.; Figueras, A. Revealing Mytilus galloprovincialis transcriptomic profiles during ontogeny. Dev. Comp. Immunol. 2018, 84, 292–306. [Google Scholar] [CrossRef] [PubMed]
- De Boer, C.G.; Vaishnav, E.D.; Sadeh, R.; Abeyta, E.L.; Friedman, N.; Regev, A. Deciphering eukaryotic gene-regulatory logic with 100 million random promoters. Nat. Biotechnol. 2020, 38, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Andersson, R.; Sandelin, A. Determinants of enhancer and promoter activities of regulatory elements. Nat. Rev. Genet. 2020, 21, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Fuda, N.J.; Ardehali, M.B.; Lis, J.T. Defining mechanisms that regulate RNA polymerase II transcription in vivo. Nature 2009, 461, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Savinkova, L.K.; Ponomarenko, M.P.; Ponomarenko, P.M.; Drachkova, I.A.; Lysova, M.V.; Arshinova, T.V.; Kolchanov, N.A. TATA box polymorphisms in human gene promoters and associated hereditary pathologies. Biochemistry 2009, 74, 117–129. [Google Scholar] [CrossRef]
- Hasegawa, Y.; Struhl, K. Promoter-specific dynamics of TATA-binding protein association with the human genome. Genome Res. 2019, 29, 1939–1950. [Google Scholar] [CrossRef]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417. [Google Scholar] [CrossRef]
- Juven-Gershon, T.; Kadonaga, J.T. Regulation of gene expression via the core promoter and the basal transcriptional machinery. Dev. Biol. 2010, 339, 225–229. [Google Scholar] [CrossRef]
- Cramer, P. Organization and regulation of gene transcription. Nature 2019, 573, 45–54. [Google Scholar] [CrossRef]
- Müller, F.; Tora, L. Chromatin and DNA sequences in defining promoters for transcription initiation. Biochim. Biophys. Acta 2014, 1839, 118–128. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rey-Campos, M.; Novoa, B.; Pallavicini, A.; Gerdol, M.; Figueras, A. Comparative Genomics Reveals a Significant Sequence Variability of Myticin Genes in Mytilus galloprovincialis. Biomolecules 2020, 10, 943. https://doi.org/10.3390/biom10060943
Rey-Campos M, Novoa B, Pallavicini A, Gerdol M, Figueras A. Comparative Genomics Reveals a Significant Sequence Variability of Myticin Genes in Mytilus galloprovincialis. Biomolecules. 2020; 10(6):943. https://doi.org/10.3390/biom10060943
Chicago/Turabian StyleRey-Campos, Magalí, Beatriz Novoa, Alberto Pallavicini, Marco Gerdol, and Antonio Figueras. 2020. "Comparative Genomics Reveals a Significant Sequence Variability of Myticin Genes in Mytilus galloprovincialis" Biomolecules 10, no. 6: 943. https://doi.org/10.3390/biom10060943
APA StyleRey-Campos, M., Novoa, B., Pallavicini, A., Gerdol, M., & Figueras, A. (2020). Comparative Genomics Reveals a Significant Sequence Variability of Myticin Genes in Mytilus galloprovincialis. Biomolecules, 10(6), 943. https://doi.org/10.3390/biom10060943