Carbamoyl-Phosphate Synthase 1 as a Novel Target of Phomoxanthone A, a Bioactive Fungal Metabolite

 ,
,  ,
, 
 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Purification of Mitochondria from HeLa Cells
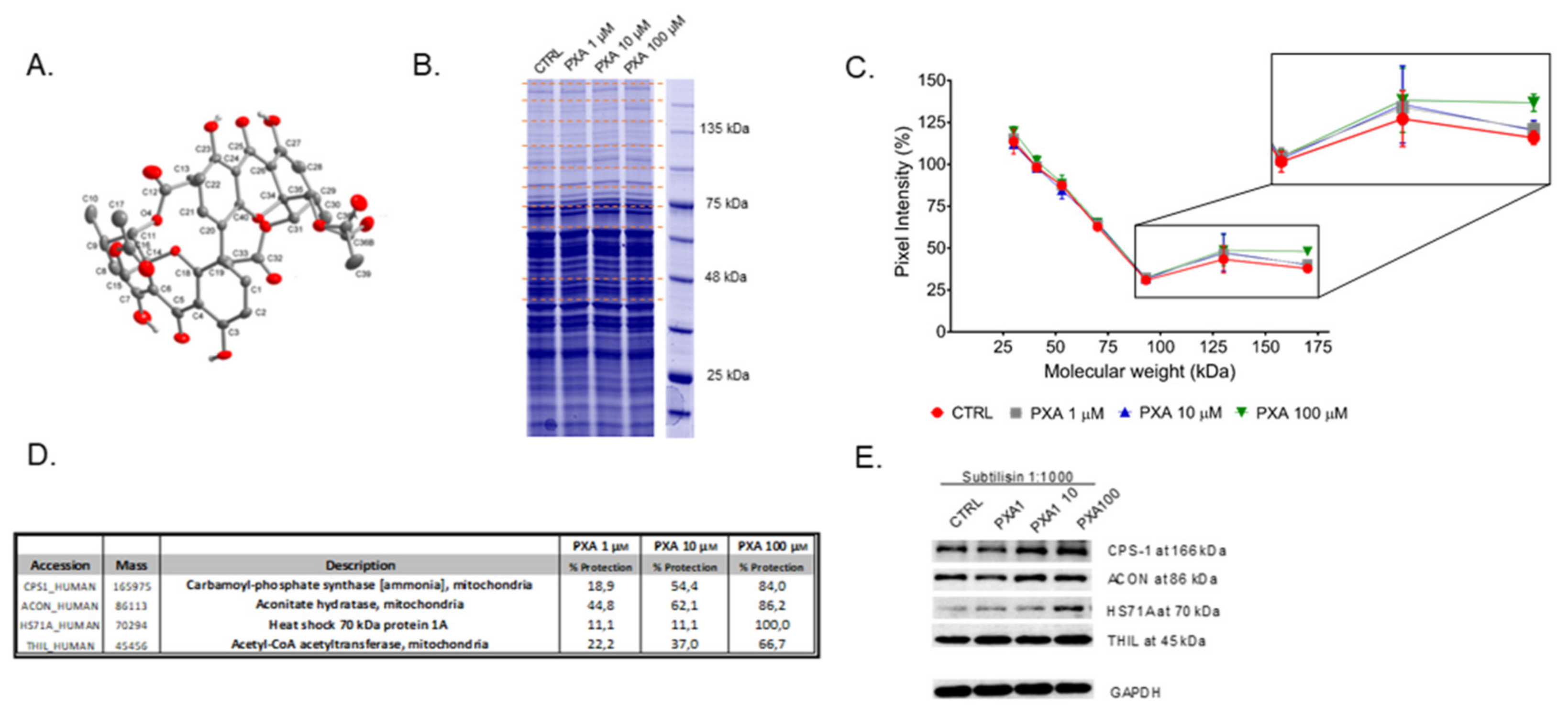
2.2. Identification of PXA Cellular Target(s) through DARTS
2.3. Western Blotting
2.4. T-LiP-MRM Analysis
2.5. In Silico Prediction of the PXA/CPS1 Complex
2.6. CPS1 In Vitro Activity Assay
3. Results and Discussion
3.1. Identification of PXA Cellular Target(s) through DARTS
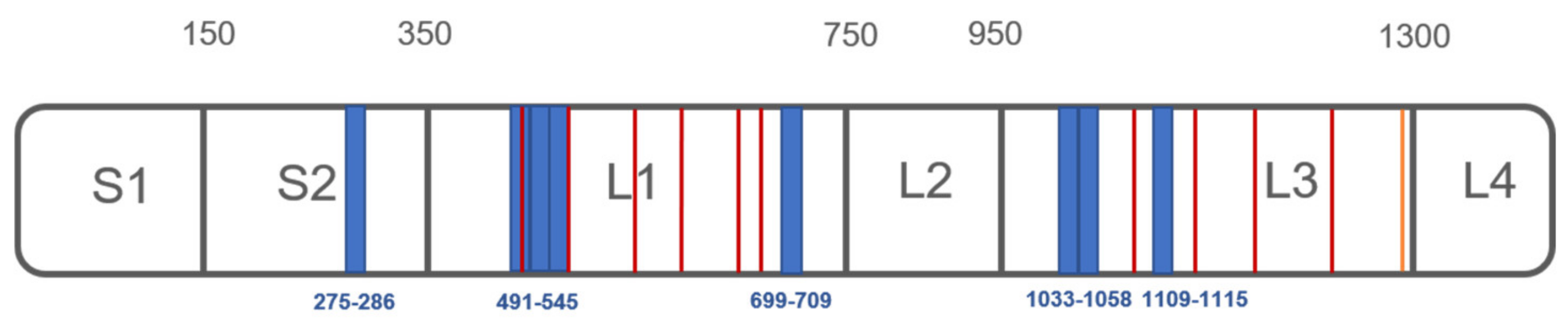
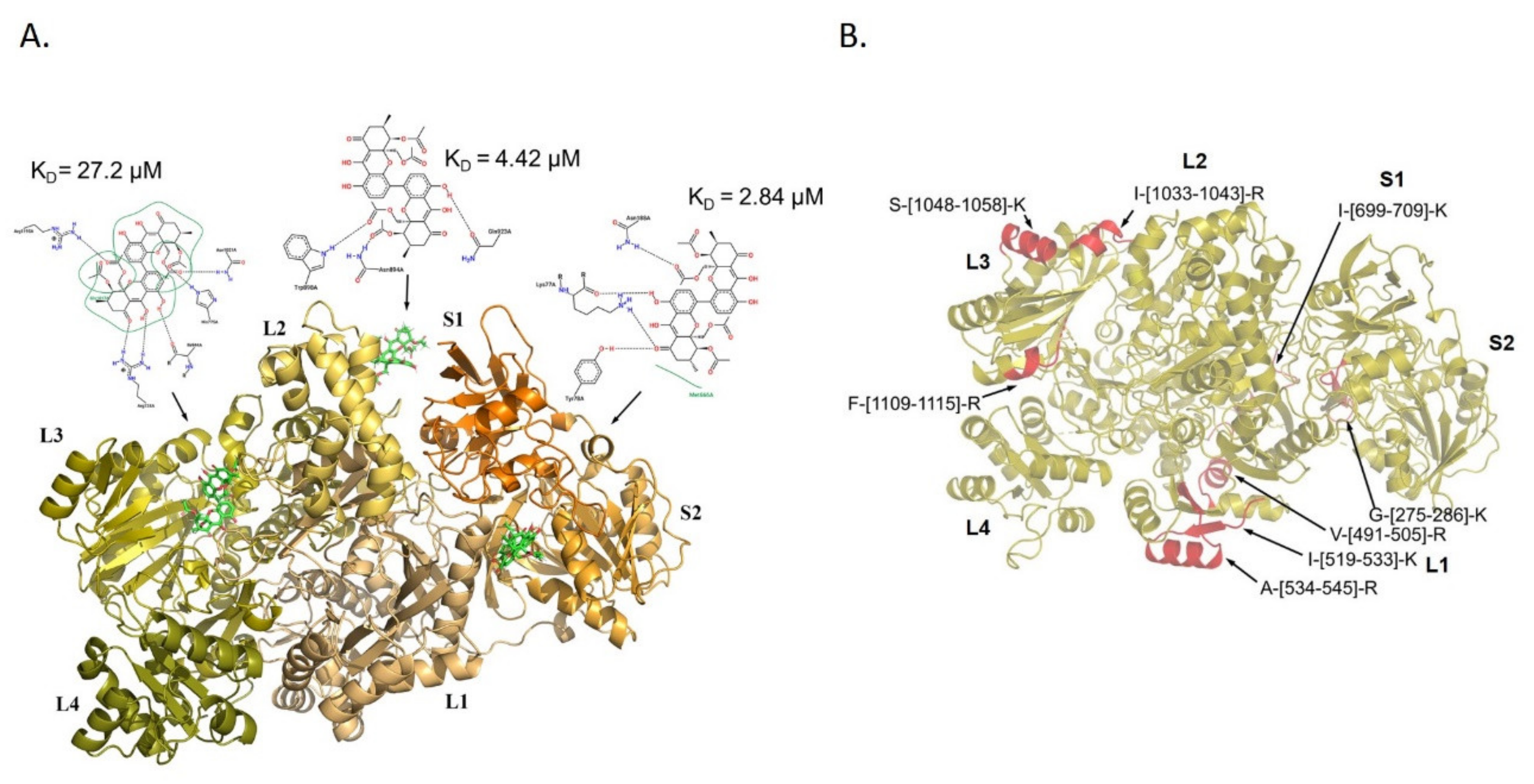
3.2. Analysis of PXA/Targets Interaction Area by t-LiP-MRM
3.3. Molecular Docking Analysis of PXA/CPS1 Complex
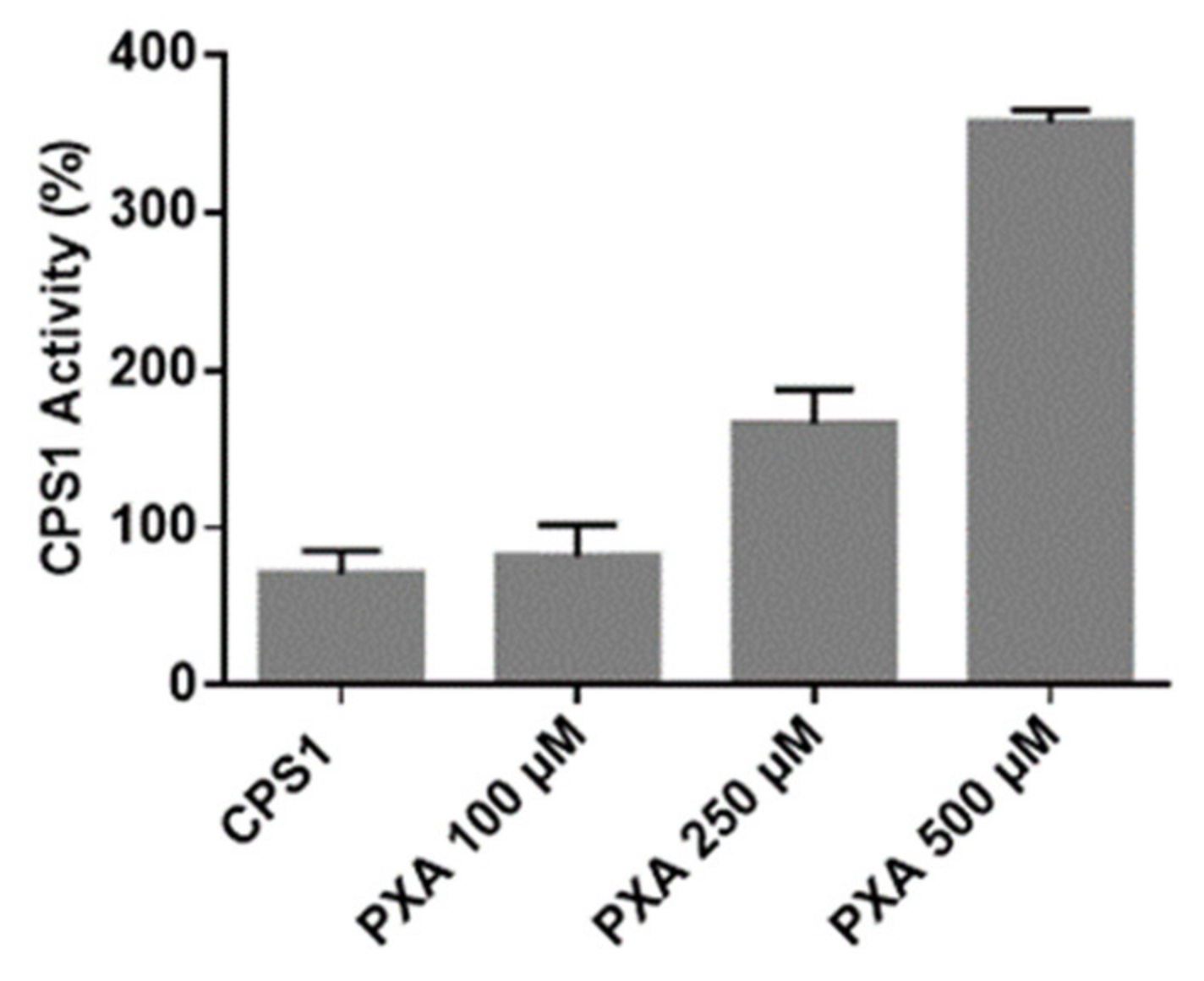
3.4. CPS-1 In Vitro Activity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Isaka, M.; Jaturapat, A.; Rukseree, K.; Danwisetkanjana, K.; Tanticharoen, M.; Thebtaranonth, Y. Phomoxanthones A and B, novel xanthone dimers from the endophytic fungus Phomopsis species. J. Nat. Prod. 2001, 64, 1015–1018. [Google Scholar] [CrossRef] [PubMed]
- Rönsberg, D.; Debbab, A.; Mándi, A.; Vasylyeva, V.; Böhler, P.; Stork, B.; Engelke, L.; Hamacher, A.; Sawadogo, R.; Diederich, M.; et al. Pro-Apoptotic and Immunostimulatory Tetrahydroxanthone Dimers from the Endophytic Fungus Phomopsis longicolla. J. Org. Chem. 2013, 78, 12409–12425. [Google Scholar] [CrossRef] [PubMed]
- Pavão, G.B.; Venâncio, V.P.; De Oliveira, A.L.L.; Hernandes, L.C.; Almeida, M.; Antunes, L.M.G.; Debonsi, H.M. Differential genotoxicity and cytotoxicity of phomoxanthone A isolated from the fungus Phomopsis longicolla in HL60 cells and peripheral blood lymphocytes. Toxicol. Vitr. 2016, 37, 211–217. [Google Scholar] [CrossRef]
- Wang, C.; Engelke, L.; Bickel, D.; Hamacher, A.; Frank, M.; Proksch, P.; Gohlke, H.; Kassack, M.U. The tetrahydroxanthone-dimer phomoxanthone A is a strong inducer of apoptosis in cisplatin-resistant solid cancer cells. Bioorganic Med. Chem. 2019, 27, 115044. [Google Scholar] [CrossRef] [PubMed]
- Böhler, P.; Stuhldreier, F.; Anand1 ‡, R.; Kondadi1 ‡, A.K.; Schlütermann, D.; Berleth, N.; Deitersen, J.; Wallot-Hieke, N.; Wu, W.; Frank, M.; et al. The mycotoxin phomoxanthone A disturbs the form and function of the inner mitochondrial membrane. Cell Death Dis. 2018, 9, 286. [Google Scholar] [CrossRef] [PubMed]
- Rix, U.; Superti-Furga, G. Target profiling of small molecules by chemical proteomics. Nat. Methods 2008, 5, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Margarucci, L.; Monti, M.C.; Alessandra, T.; Riccio, R.; Casapullo, A. Chemical Proteomics Discloses Petrosapongiolide M, an Antiinflammatory Marine Sesterterpene, as a Proteasome Inhibitor. Angew. Chem. Int. Ed. 2010, 49, 3960–3963. [Google Scholar] [CrossRef]
- Margarucci, L.; Monti, M.C.; Cassiano, C.; Mozzicafreddo, M.; Angeletti, M.; Riccio, R.; Alessandra, T.; Casapullo, A. Chemical proteomics-driven discovery of oleocanthal as an Hsp90 inhibitor. Chem. Commun. 2013, 49, 5844. [Google Scholar] [CrossRef]
- Del Gaudio, F.; Pollastro, F.; Mozzicafreddo, M.; Riccio, R.; Minassi, A.; Monti, M.C. Chemoproteomic fishing identifies arzanol as a positive modulator of brain glycogen phosphorylase. Chem. Commun. 2018, 54, 12863–12866. [Google Scholar] [CrossRef]
- Chang, J.; Kim, Y.; Kwon, H.J. Advances in identification and validation of protein targets of natural products without chemical modification. Nat. Prod. Rep. 2016, 33, 719–730. [Google Scholar] [CrossRef]
- Hwang, H.-Y.; Kim, T.Y.; Szász, M.A.; Dome, B.; Malm, J.; Marko-Varga, G.; Kwon, H.J. Profiling the Protein Targets of Unmodified Bio-Active Molecules with Drug Affinity Responsive Target Stability and Liquid Chromatography/Tandem Mass Spectrometry. Proteomics 2020, 20, e1900325. [Google Scholar] [CrossRef] [PubMed]
- Morretta, E.; Esposito, R.; Festa, C.; Riccio, R.; Casapullo, A.; Monti, M.C. Discovering the Biological Target of 5-epi-Sinuleptolide Using a Combination of Proteomic Approaches. Mar. Drugs 2017, 15, 312. [Google Scholar] [CrossRef] [PubMed]
- Morretta, E.; Tosco, A.; Festa, C.; Mozzicafreddo, M.; Monti, M.C.; Casapullo, A. Crellastatin A, a PARP-1 Inhibitor Discovered by Complementary Proteomic Approaches. ChemMedChem 2020, 15, 317–323. [Google Scholar] [CrossRef]
- Feng, Y.; De Franceschi, G.; Kahraman, A.; Soste, M.; Melnik, A.; Boersema, P.J.; De Laureto, P.P.; Nikolaev, Y.; Oliveira, A.P.; Picotti, P. Global analysis of protein structural changes in complex proteomes. Nat. Biotechnol. 2014, 32, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Schopper, S.; Kahraman, A.; Leuenberger, P.; Feng, Y.; Piazza, I.; Müller, O.; Boersema, P.J.; Picotti, P. Measuring protein structural changes on a proteome-wide scale using limited proteolysis-coupled mass spectrometry. Nat. Protoc. 2017, 12, 2391–2410. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Tomas, H.; Havli, J.; Olsen, J.V.; Mann, M.; Havli\[Sbreve], J. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006, 1, 2856–2860. [Google Scholar] [CrossRef]
- Hanwell, M.; Curtis, D.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Chemin- 2012, 4, 17. [Google Scholar] [CrossRef]
- Mozzicafreddo, M.; Cuccioloni, M.; Bonfili, L.; Cecarini, V.; Palermo, F.A.; Cocci, P.; Mosconi, G.; Capone, A.; Ricci, I.; Eleuteri, A.M.; et al. Environmental pollutants directly affect the liver X receptor alpha activity: Kinetic and thermodynamic characterization of binding. J. Steroid Biochem. Mol. Boil. 2015, 152, 1–7. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- De Cima, S.; Polo, L.M.; Diez-Fernandez, C.; Martínez, A.I.; Cervera, J.; Fita, I.; Rubio, V. Structure of human carbamoyl phosphate synthetase: deciphering the on/off switch of human ureagenesis. Sci. Rep. 2015, 5, 16950. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.; Taylor, R. Development and validation of a genetic algorithm for flexible docking 1 1Edited by F. E. Cohen. J. Mol. Boil. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins: Struct. Funct. Bioinform. 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Stierand, K.; Maaß, P.C.; Rarey, M. Molecular complexes at a glance: automated generation of two-dimensional complex diagrams. Bioinform. 2006, 22, 1710–1716. [Google Scholar] [CrossRef] [PubMed]
- Lomenick, B.; Hao, R.; Jonai, N.; Chin, R.M.; Aghajan, M.; Warburton, S.; Wang, J.; Wu, R.P.; Gomez, F.; Loo, J.A.; et al. Target identification using drug affinity responsive target stability (DARTS). Proc. Natl. Acad. Sci. USA 2009, 106, 21984–21989. [Google Scholar] [CrossRef] [PubMed]
- Lomenick, B.; Olsen, R.W.; Huang, J. Identification of Direct Protein Targets of Small Molecules. ACS Chem. Boil. 2010, 6, 34–46. [Google Scholar] [CrossRef]
- Diez-Fernandez, C.; Häberle, J. Targeting CPS1 in the treatment of Carbamoyl phosphate synthetase 1 (CPS1) deficiency, a urea cycle disorder. Expert Opin. Ther. Targets 2017, 21, 391–399. [Google Scholar] [CrossRef]
- Moonen, R.M.; Paulussen, A.D.C.; Souren, N.Y.; Kessels, A.G.H.; Rubio-Gozalbo, M.E.; Villamor, E. Carbamoyl Phosphate Synthetase Polymorphisms as a Risk Factor for Necrotizing Enterocolitis. Pediatr. Res. 2007, 62, 188–190. [Google Scholar] [CrossRef]
- Pekkala, S.; Martínez, A.I.; Barcelona, B.; Yefimenko, I.; Finckh, U.; Rubio, V.; Cervera, J. Understanding carbamoyl-phosphate synthetase I (CPS1) deficiency by using expression studies and structure-based analysis. Hum. Mutat. 2010, 31, 801–808. [Google Scholar] [CrossRef][Green Version]
- Laemmle, A.; Hahn, D.; Hu, L.; Rüfenacht, V.; Gautschi, M.; Leibundgut, K.; Nuoffer, J.-M.; Häberle, J. Fatal hyperammonemia and carbamoyl phosphate synthetase 1 (CPS1) deficiency following high-dose chemotherapy and autologous hematopoietic stem cell transplantation. Mol. Genet. Metab. 2015, 114, 438–444. [Google Scholar] [CrossRef]
- Pierson, D.L. A rapid colorimetric assay for carbamyl phosphate synthetase I. J. Biochem. Biophys. Methods 1980, 3, 31–37. [Google Scholar] [CrossRef]
- Qin, T.; Iwata, T.; Ransom, T.T.; Beutler, J.A.; Porco, J.A. Syntheses of Dimeric Tetrahydroxanthones with Varied Linkages: Investigation of “Shapeshifting” Properties. J. Am. Chem. Soc. 2015, 137, 15225–15233. [Google Scholar] [CrossRef] [PubMed]
- Elsässer, B.; Krohn, K.; Flörke, U.; Root, N.; Aust, H.-J.; Draeger, S.; Schulz, B.; Antus, S.; Kurtán, T. X-ray Structure Determination, Absolute Configuration and Biological Activity of Phomoxanthone A. Eur. J. Org. Chem. 2005, 2005, 4563–4570. [Google Scholar] [CrossRef]
- Kim, J.; Raushel, F.M. Access to the carbamate tunnel of carbamoyl phosphate synthetase. Arch. Biochem. Biophys. 2004, 425, 33–41. [Google Scholar] [CrossRef] [PubMed]
- De Chiara, F.; Heebøll, S.; Marrone, G.; Montoliu, C.; Hamilton-Dutoit, S.; Ferrandez, A.; Andreola, F.; Rombouts, K.; Grønbæk, H.; Felipo, V.; et al. Urea cycle dysregulation in non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 905–915. [Google Scholar] [CrossRef]
- Eriksen, P.L.; Vilstrup, H.; Rigbolt, K.; Suppli, M.P.; Sørensen, M.; Heebøll, S.; Veidal, S.S.; Knop, F.K.; Thomsen, K.L. Non-alcoholic fatty liver disease alters expression of genes governing hepatic nitrogen conversion. Liver Int. 2019, 39, 2094–2101. [Google Scholar] [CrossRef]
- Subramanian, P.; Jayakumar, M.; Jayapalan, J.; Hashim, O.H. Chronotherapeutic effect of fisetin on expression of urea cycle enzymes and inflammatory markers in hyperammonaemic rats. Pharmacol. Rep. 2014, 66, 1037–1042. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Q1_m/z | Peptide | rt | Fold Change at PXA 1 µM | p | Fold Change at PXA 10 µM | p |
|---|---|---|---|---|---|---|
| 663.36 | G-[275-286]-K | 6,7 min | 1.73 | 0.0001 | 1.74 | 0.0003 |
| 804.4 | V-[491-505]-R | 9,58 min | 1.77 | 0.0013 | 1.84 | 0.0008 |
| 795.43 | I-[519-533]-K | 12,45 min | 2.57 | 0.0338 | 2.40 | 0.0473 |
| 653.85 | A-[534-545]-R | 9,13 min | 1.30 | 0.0016 | 1.25 | 0.0033 |
| 582.37 | I-[699-709]-K | 11,24 min | 1.23 | 0.0179 | 1.20 | 0.0422 |
| 615.83 | I-[1033-1043]-R | 8,04 min | 1.44 | 0.0011 | 1.41 | 0.0013 |
| 611.34 | S-[1048-1058]-K | 13,44 min | 1.88 | 0.0001 | 1.85 | 0.0001 |
| 433.22 | F-[1109-1115]-R | 5,05 min | 1.34 | 0.0235 | 1.28 | 0.0315 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ceccacci, S.; Deitersen, J.; Mozzicafreddo, M.; Morretta, E.; Proksch, P.; Wesselborg, S.; Stork, B.; Monti, M.C. Carbamoyl-Phosphate Synthase 1 as a Novel Target of Phomoxanthone A, a Bioactive Fungal Metabolite. Biomolecules 2020, 10, 846. https://doi.org/10.3390/biom10060846
Ceccacci S, Deitersen J, Mozzicafreddo M, Morretta E, Proksch P, Wesselborg S, Stork B, Monti MC. Carbamoyl-Phosphate Synthase 1 as a Novel Target of Phomoxanthone A, a Bioactive Fungal Metabolite. Biomolecules. 2020; 10(6):846. https://doi.org/10.3390/biom10060846
Chicago/Turabian StyleCeccacci, Sara, Jana Deitersen, Matteo Mozzicafreddo, Elva Morretta, Peter Proksch, Sebastian Wesselborg, Björn Stork, and Maria Chiara Monti. 2020. "Carbamoyl-Phosphate Synthase 1 as a Novel Target of Phomoxanthone A, a Bioactive Fungal Metabolite" Biomolecules 10, no. 6: 846. https://doi.org/10.3390/biom10060846
APA StyleCeccacci, S., Deitersen, J., Mozzicafreddo, M., Morretta, E., Proksch, P., Wesselborg, S., Stork, B., & Monti, M. C. (2020). Carbamoyl-Phosphate Synthase 1 as a Novel Target of Phomoxanthone A, a Bioactive Fungal Metabolite. Biomolecules, 10(6), 846. https://doi.org/10.3390/biom10060846