Peroxisome Proliferator Activator Receptor Gamma Coactivator-1α Overexpression in Amyotrophic Lateral Sclerosis: A Tale of Two Transgenics




Abstract
1. Introduction
2. Materials and Methods
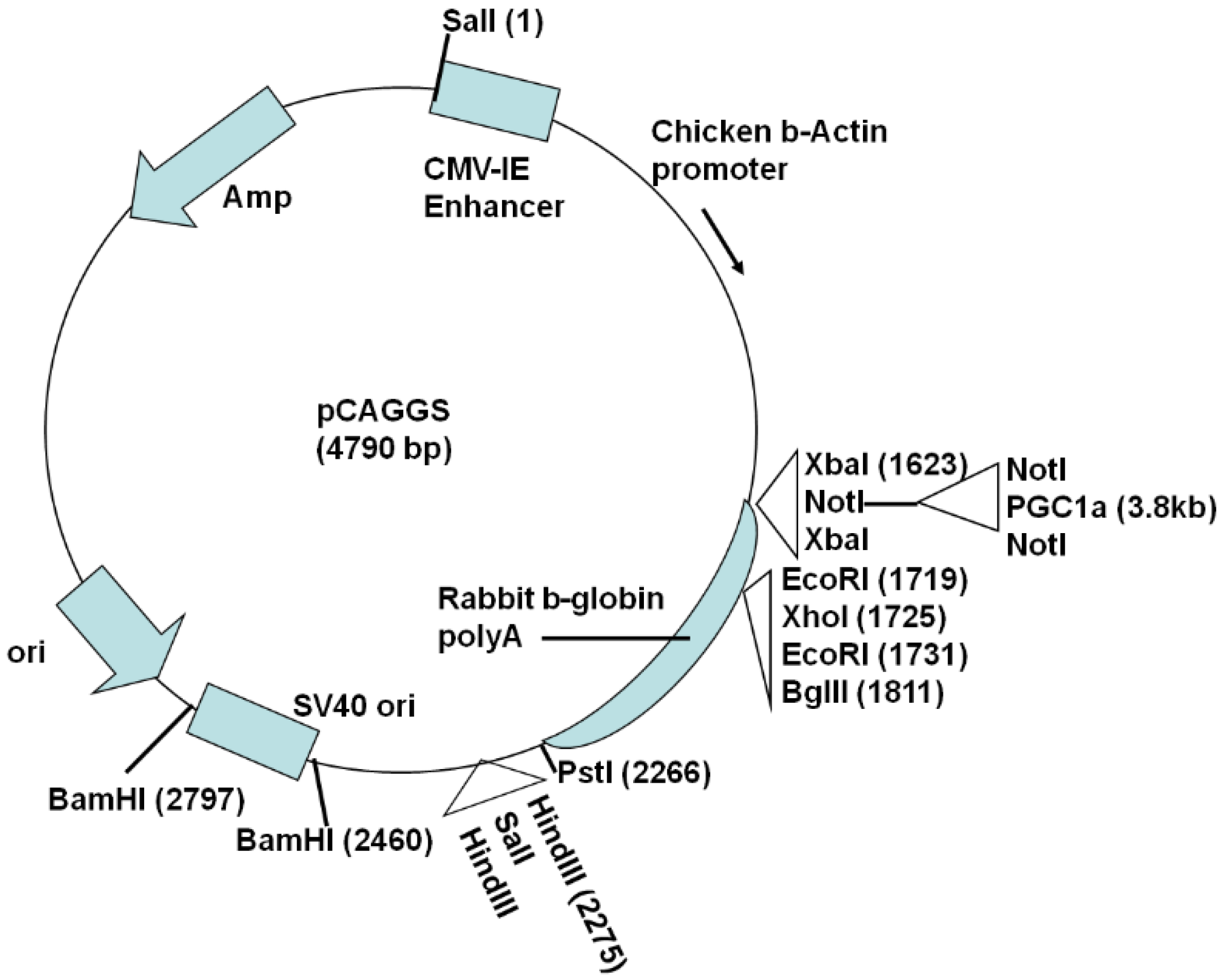
2.1. Generation of Actin-PGC-1α Mice and Crossing with SOD1G93A Mice
2.2. Analysis of Tissue Expression of Exogenous and Endogenous PGC-1α and Sirtuins
2.3. Assessment of Motor Performance, Body Weight, and Survival
2.4. Statistics
3. Results
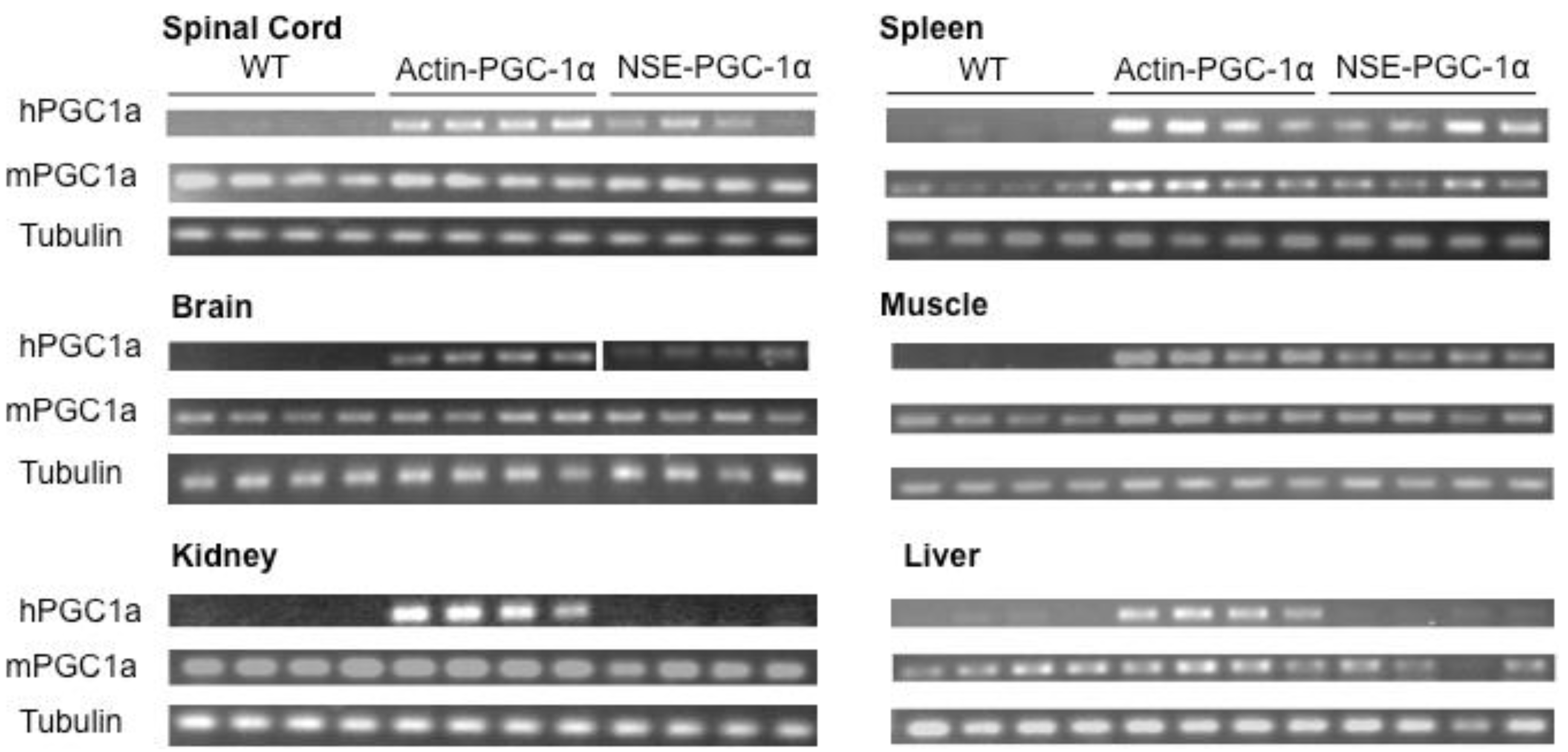
3.1. Assessment of Tissue Transgene Expression in NSE-PGC-1α and Actin-PGC-1α Mice
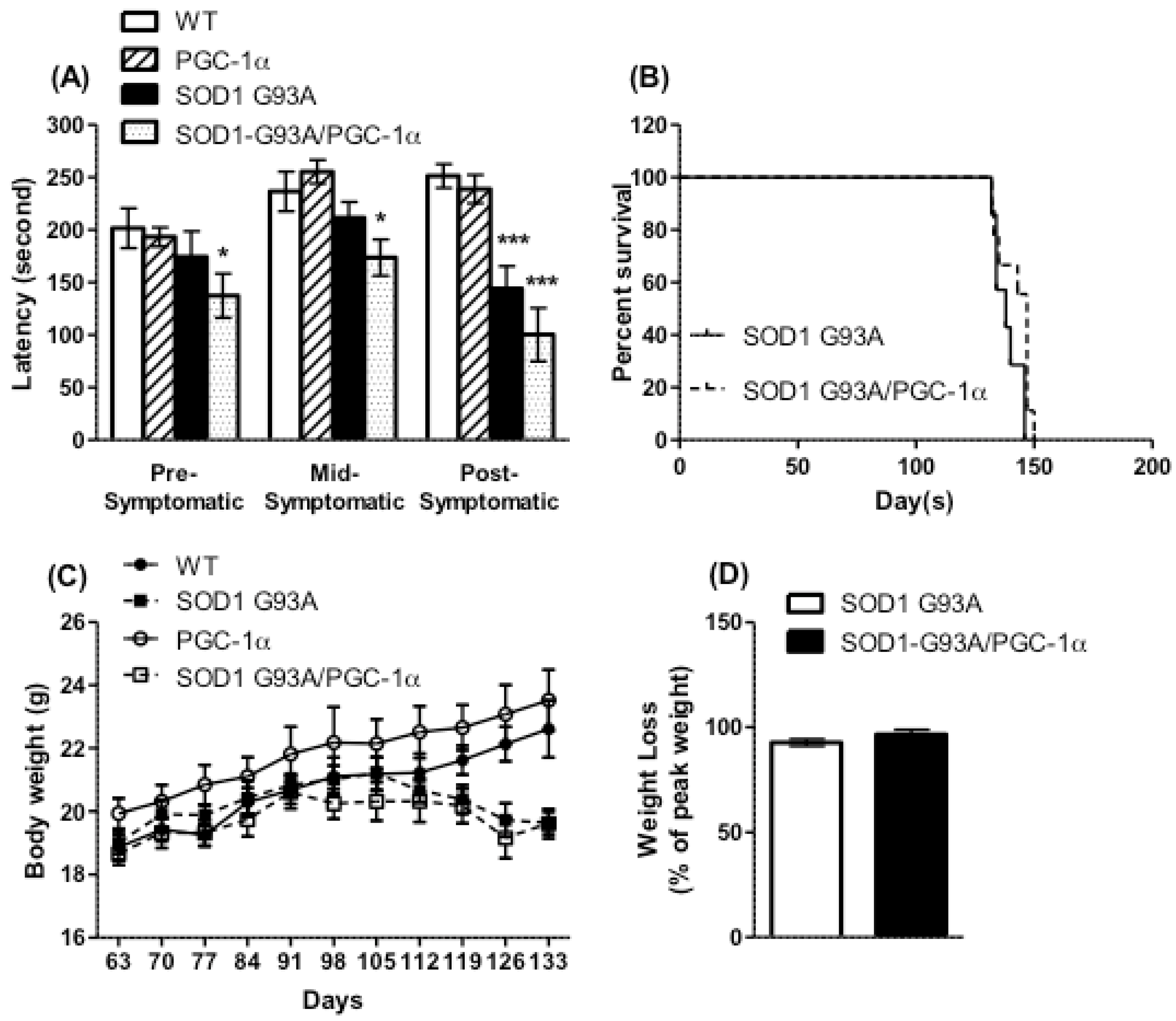
3.2. Motor Performance, Survival, and Body Weight Were Not Affected by Actin-PGC-1α Overexpression
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | amyotrophic lateral sclerosis, |
| fALS | familial ALS, |
| sALS | sporadic ALS, |
| SOD1 | superoxide dismutase 1, |
| PGC-1α | peroxisome proliferator-activated receptor γ coactivator-1 α, |
| PPAR-γ | peroxisome proliferator-activated receptor γ, |
| NSE | neuron-specific enolase, |
References
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Shi, P.; Gal, J.; Kwinter, D.M.; Liu, X.; Zhu, H. Mitochondrial dysfunction in amyotrophic lateral sclerosis. Biochimica et Biophysica Acta—Mol. Basis Dis. 2010, 1802, 45–51. [Google Scholar] [CrossRef]
- Bowling, A.C.; Schulz, J.B.; Brown, R.H.; Beal, M.F. Superoxide Dismutase Activity, Oxidative Damage, and Mitochondrial Energy Metabolism in Familial and Sporadic Amyotrophic Lateral Sclerosis. J. Neurochem. 1993, 61, 2322–2325. [Google Scholar] [CrossRef]
- Browne, S.E.; Bowling, A.C.; Baik, M.J.; Gurney, M.; Brown, R.H., Jr.; Beal, M.F. Metabolic Dysfunction in Familial, but Not Sporadic, Amyotrophic Lateral Sclerosis. J. Neurochem. 2002, 71, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lillo, C.; Jonsson, P.A.; Velde, C.V.; Ward, C.M.; Miller, T.M.; Subramaniam, J.R.; Rothstein, J.D.; Marklund, S.; Andersen, P.M.; et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 2004, 43, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Mattiazzi, M.; D’Aurelio, M.; Gajewski, C.D.; Martushova, K.; Kiaei, M.; Beal, M.F.; Manfredi, G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem. 2002, 277, 29626–29633. [Google Scholar] [CrossRef]
- Kirkinezos, I.G.; Bacman, S.R.; Hernandez, D.; Oca-Cossio, J.; Arias, L.J.; Perez-Pinzon, M.A.; Bradley, W.G.; Moraes, C.T. Cytochrome c association with the inner mitochondrial membrane is impaired in the CNS of G93A-SOD1 mice. J. Neurosci. 2005, 25, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Song, Y.; Kincaid, B.; Bossy, B.; Bossy-Wetzel, E. Mutant SOD1G93A triggers mitochondrial fragmentation in spinal cord motor neurons: Neuroprotection by SIRT3 and PGC-1α. Neurobiol. Dis. 2013, 51, 72–81. [Google Scholar] [CrossRef]
- Dal Canto, M.C.; Gurney, M.E. Development of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosis. Am. J. Pathol. 1994, 145, 1271–1279. [Google Scholar]
- Onesto, E.; Colombrita, C.; Gumina, V.; Borghi, M.O.; Dusi, S.; Doretti, A.; Fagiolari, G.; Invernizzi, F.; Moggio, M.; Tiranti, V.; et al. Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol. Commun. 2016, 4, 47. [Google Scholar] [CrossRef]
- Wiedemann, F.R.; Winkley, K.; Kuznetsov, A.V.; Bartels, C.; Vielhaber, S.; Feistner, H.; Kunz, W.S. Impairment of mitochondrial function in skeletal muscle of patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 1998, 156, 65–72. [Google Scholar] [CrossRef]
- Allen, S.P.; Duffy, L.M.; Shaw, P.J.; Grierson, A.J. Altered age-related changes in bioenergetic properties and mitochondrial morphology in fibroblasts from sporadic amyotrophic lateral sclerosis patients. Neurobiol. Aging 2015, 36, 2893–2903. [Google Scholar] [CrossRef]
- Konrad, C.; Kawamata, H.; Bredvik, K.; Arreguin, A.; Cajamarca, S.A.; Hupf, J.C.; Ravits, J.; Miller, T.M.; Maragakis, N.J.; Hales, C.M.; et al. Fibroblast bioenergetics to classify amyotrophic lateral sclerosis patients. Mol. Neurodegener. 2017, 12, 76. [Google Scholar] [CrossRef]
- Codron, P.; Cassereau, J.; Vourc’H, P.; Veyrat-Durebex, C.; Blasco, H.; Kane, S.; Procaccio, V.; Letournel, F.; Verny, C.; Lenaers, G.; et al. Primary fibroblasts derived from sporadic amyotrophic lateral sclerosis patients do not show ALS cytological lesions. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 446–456. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Villena, J.A. New insights into PGC-1 coactivators: Redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015, 282, 647–672. [Google Scholar] [CrossRef]
- Thau, N.; Knippenberg, S.; Korner, S.; Rath, K.J.; Dengler, R.; Petri, S. Decreased mRNA expression of PGC-1α and PGC-1αregulated factors in the SOD1G93A ALS mouse model and in human sporadic ALS. J. Neuropathol. Exp. Neurol. 2012, 71, 1064–1074. [Google Scholar] [CrossRef]
- Russell, A.; Wada, S.; Vergani, L.; Hock, M.B.; Lamon, S.; Léger, B.; Ushida, T.; Cartoni, R.; Wadley, G.D.; Hespel, P.; et al. Disruption of skeletal muscle mitochondrial network genes and miRNAs in amyotrophic lateral sclerosis. Neurobiol. Dis. 2013, 49, 107–117. [Google Scholar] [CrossRef]
- Liang, H.; Ward, W.F.; Jang, Y.C.; Bhattacharya, A.; Bokov, A.F.; Jernigan, A.; Richardson, A.; Van Remmen, H. PGC-1α protects neurons and alters disease progression in an amyotrophic lateral sclerosis mouse model. Muscle Nerve 2011, 44, 947–956. [Google Scholar] [CrossRef]
- Zhao, W.; Varghese, M.; Yemul, S.; Pan, Y.; Cheng, A.; Marano, P.; Hassan, S.; Vempati, P.; Chen, F.; Qian, X.; et al. Peroxisome proliferator activator receptor gamma coactivator-1alpha (PGC-1α) improves motor performance and survival in a mouse model of amyotrophic lateral sclerosis. Mol. Neurodegener. 2011, 6, 51. [Google Scholar] [CrossRef]
- Nijland, P.G.; Witte, M.E.; van het Hof, B.; van der Pol, S.; Bauer, J.; Lassmann, H.; van der Valk, P.; de Vries, H.E.; van Horssen, J. Astroglial PGC-1alpha increases mitochondrial antioxidant capacity and suppresses inflammation: Implications for multiple sclerosis. Acta Neuropathol. Commun. 2014, 2, 170. [Google Scholar] [CrossRef]
- Da Cruz, S.; Parone, P.A.; Lopes, V.S.; Lillo, C.; McAlonis-Downes, M.; Lee, S.K.; Vetto, A.P.; Petrosyan, S.; Marsala, M.; Murphy, A.N.; et al. Elevated PGC-1α Activity Sustains Mitochondrial Biogenesis and Muscle Function without Extending Survival in a Mouse Model of Inherited ALS. Cell Metab. 2012, 15, 778–786. [Google Scholar] [CrossRef]
- Gurney, M.; Pu, H.; Chiu, A.; Canto, M.D.; Polchow, C.; Alexander, D.; Caliendo, J.; Hentati, A.; Kwon, Y.; Deng, H.-X.; et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Muller, F.L.; Liu, J.; Jernigan, A.; Borchelt, D.; Richardson, A.; Van Remmen, H. MnSOD deficiency has a differential effect on disease progression in two different ALS mutant mouse models. Muscle Nerve 2008, 38, 1173–1183. [Google Scholar] [CrossRef]
- Boillée, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Cleveland, D.W. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef]
- Liu, Y.; Okada, T.; Nomoto, T.; Ke, X.; Kume, A.; Ozawa, K.; Xiao, S. Promoter effects of adeno-associated viral vector for transgene expression in the cochlea in vivo. Exp. Mol. Med. 2007, 39, 170–175. [Google Scholar] [CrossRef]
- Klein, R.L.; Hamby, M.E.; Gong, Y.; Hirko, A.C.; Wang, S.; Hughes, J.A.; King, M.A.; Meyer, E.M. Dose and promoter effects of adeno-associated viral vector for green fluorescent protein expression in the rat brain. Exp. Neurol. 2002, 176, 66–74. [Google Scholar] [CrossRef]
- Eschbach, J.; Schwalenstöcker, B.; Soyal, S.M.; Bayer, H.; Wiesner, D.; Akimoto, C.; Nilsson, A.-C.; Birve, A.; Meyer, T.; Dupuis, L.; et al. PGC-1 is a male-specific disease modifier of human and experimental amyotrophic lateral sclerosis. Hum. Mol. Genet. 2013, 22, 3477–3484. [Google Scholar] [CrossRef]
- Herrera-Marcos, L.V.; Sancho-Knapik, S.; Gabás-Rivera, C.; Barranquero, C.; Gascón, S.; Romanos, E.; Martínez-Beamonte, R.; Navarro, M.A.; Surra, J.C.; Arnal, C.; et al. Pgc1a is responsible for the sex differences in hepatic Cidec/Fsp27β mRNA expression in hepatic steatosis of mice fed a Western diet. Am. J. Physiol. Metab. 2020, 318, E249–E261. [Google Scholar] [CrossRef]
- Capllonch-Amer, G.; Sbert-Roig, M.; Galmes-Pascual, B.M.; Proenza, A.M.; Llado, I.; Gianotti, M.; Garcia-Palmer, F.J. Estradiol stimulates mitochondrial biogenesis and adiponectin expression in skeletal muscle. J. Endocrinol. 2014, 221, 391–403. [Google Scholar] [CrossRef]
- Xiang, Z.; Valenza, M.; Cui, L.; Leoni, V.; Jeong, H.-K.; Brilli, E.; Zhang, J.; Peng, Q.; Duan, W.; Reeves, S.A.; et al. Peroxisome-proliferator-activated receptor gamma coactivator 1 α contributes to dysmyelination in experimental models of Huntington’s disease. J. Neurosci. 2011, 31, 9544–9553. [Google Scholar] [CrossRef]
- Kiaei, M.; Kipiani, K.; Chen, J.; Calingasan, N.Y.; Beal, M.F. Peroxisome proliferator-activated receptor-gamma agonist extends survival in transgenic mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2005, 191, 331–336. [Google Scholar] [CrossRef]
- Schütz, B.; Reimann, J.; Dumitrescu-Ozimek, L.; Kappes-Horn, K.; Landreth, G.E.; Schurmann, B.; Zimmer, A.; Heneka, M.T. The oral antidiabetic pioglitazone protects from neurodegeneration and amyotrophic lateral sclerosis-like symptoms in superoxide dismutase-G93A transgenic mice. J. Neurosci. 2005, 25, 7805–7812. [Google Scholar] [CrossRef]
- Shibata, N.; Kawaguchi-Niida, M.; Yamamoto, T.; Toi, S.; Hirano, A.; Kobayashi, M. Effects of the PPARγ activator pioglitazone on p38 MAP kinase and IκBα in the spinal cord of a transgenic mouse model of amyotrophic lateral sclerosis. Neuropathology 2008, 28, 387–398. [Google Scholar] [CrossRef]
- Dupuis, L.; Dengler, R.; Heneka, M.T.; Meyer, T.; Zierz, S.; Kassubek, J.; Fischer, W.; Steiner, F.; Lindauer, E.; Otto, M.; et al. A randomized, double blind, placebo-controlled trial of pioglitazone in combination with riluzole in amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e37885. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Orientation | Sequence (5′–3′) |
|---|---|---|
| Genotyping Primers | ||
| pCAGGS Human PGC-1α | Forward Reverse | GGTTCGGCTTCTGGCGTG CCACAGGGAGACTGTCTAGTGTC |
| Mouse IL-2 | Forward Reverse | CTAGGCCACAGAATTGAAAGATCT GTAGGTGGAAATTCTAGCATCATCC |
| qRT-PCR Primers | ||
| Human PGC-1α | Forward Reverse | CAGGCAGTAGATCCTCTTCAAG TCCTCGTAGCTGTCATACCTG |
| Mouse PGC-1α | Forward Reverse | AAGGTCCCCAGGCAGTAGAT CATAGCTGTCGTACCTGGGC |
| Mouse tubulin | Forward Reverse | TAGCAGAGATCACCAATGCC GGCAGCAAGCCATGTATTTA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varghese, M.; Zhao, W.; Trageser, K.J.; Pasinetti, G.M. Peroxisome Proliferator Activator Receptor Gamma Coactivator-1α Overexpression in Amyotrophic Lateral Sclerosis: A Tale of Two Transgenics. Biomolecules 2020, 10, 760. https://doi.org/10.3390/biom10050760
Varghese M, Zhao W, Trageser KJ, Pasinetti GM. Peroxisome Proliferator Activator Receptor Gamma Coactivator-1α Overexpression in Amyotrophic Lateral Sclerosis: A Tale of Two Transgenics. Biomolecules. 2020; 10(5):760. https://doi.org/10.3390/biom10050760
Chicago/Turabian StyleVarghese, Merina, Wei Zhao, Kyle J. Trageser, and Giulio M. Pasinetti. 2020. "Peroxisome Proliferator Activator Receptor Gamma Coactivator-1α Overexpression in Amyotrophic Lateral Sclerosis: A Tale of Two Transgenics" Biomolecules 10, no. 5: 760. https://doi.org/10.3390/biom10050760
APA StyleVarghese, M., Zhao, W., Trageser, K. J., & Pasinetti, G. M. (2020). Peroxisome Proliferator Activator Receptor Gamma Coactivator-1α Overexpression in Amyotrophic Lateral Sclerosis: A Tale of Two Transgenics. Biomolecules, 10(5), 760. https://doi.org/10.3390/biom10050760