Machine Learning to Identify Flexibility Signatures of Class A GPCR Inhibition


Abstract
1. Introduction
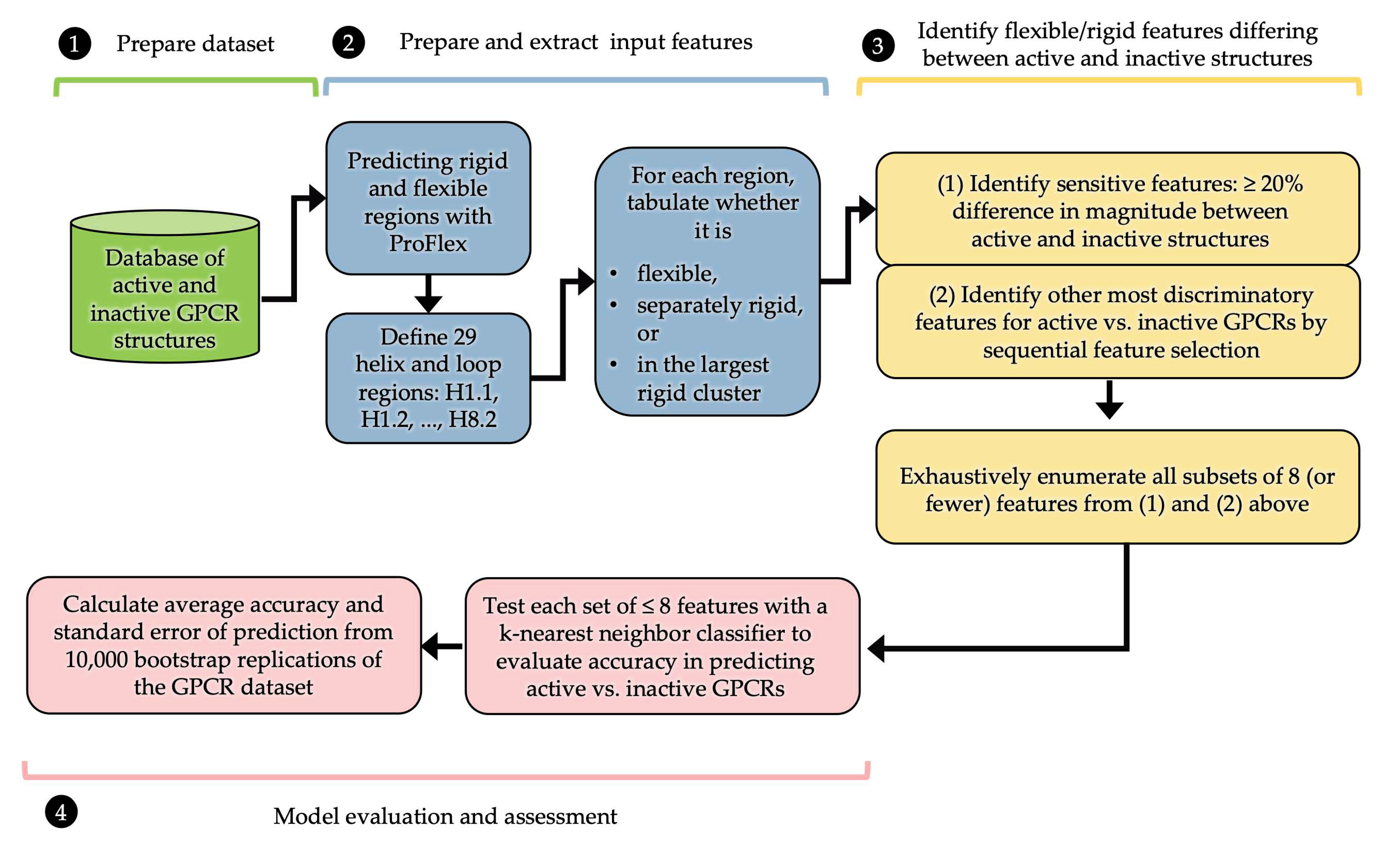
2. Materials and Methods
2.1. Selecting GPCR Structures
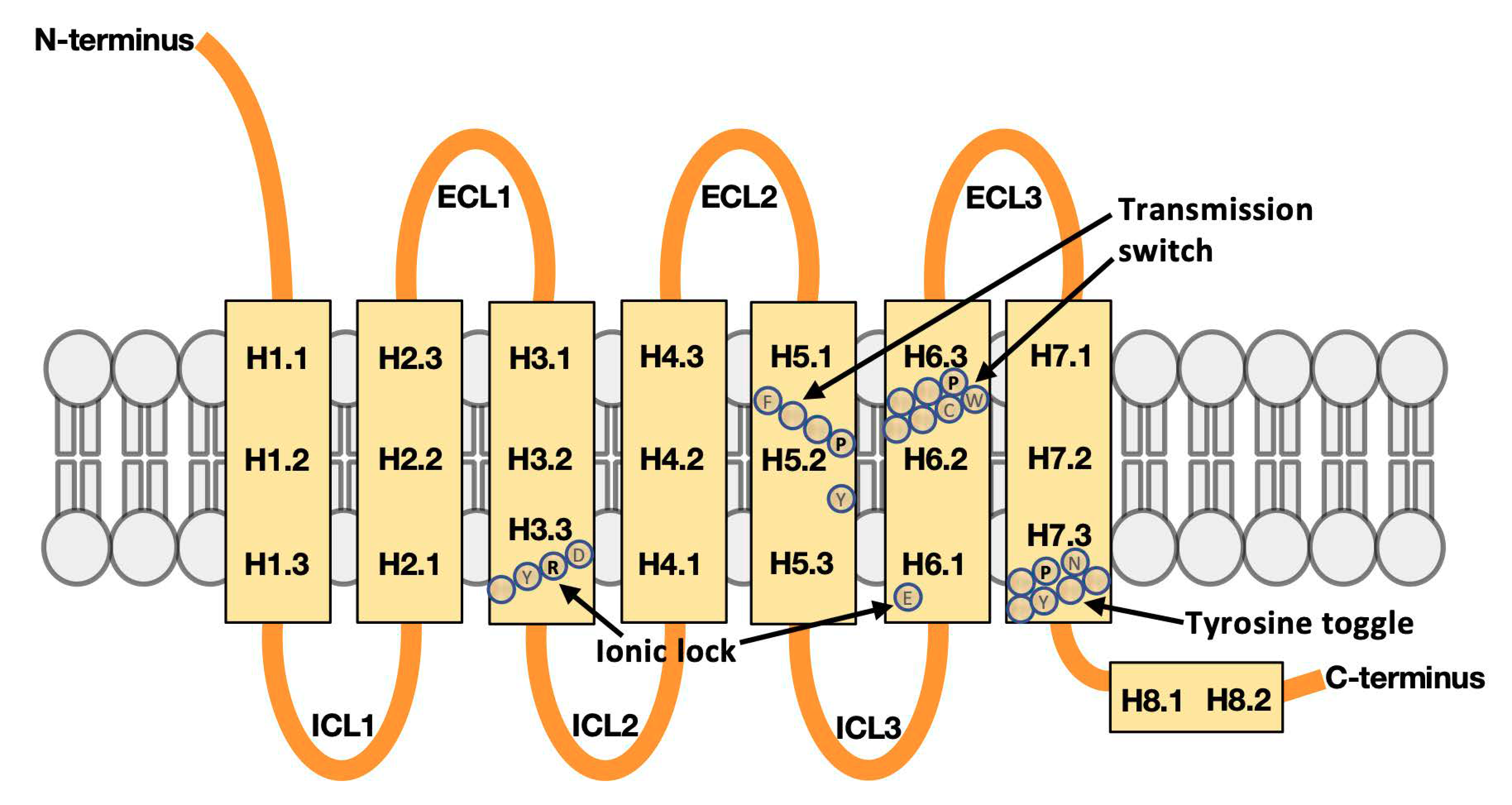
2.2. Defining Regions in GPCR Structures for Machine Learning
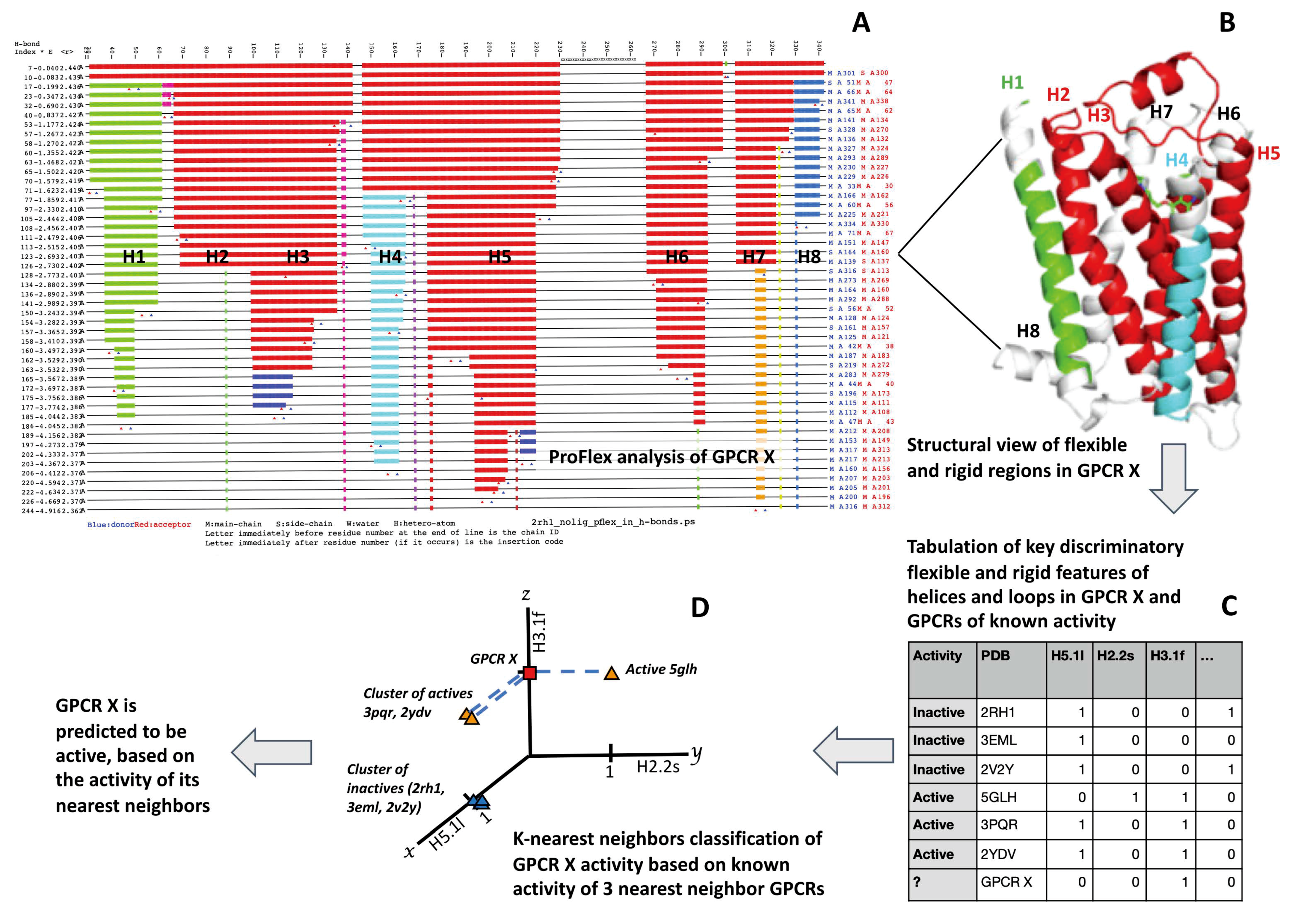
2.3. Performing and Interpreting ProFlex Analysis
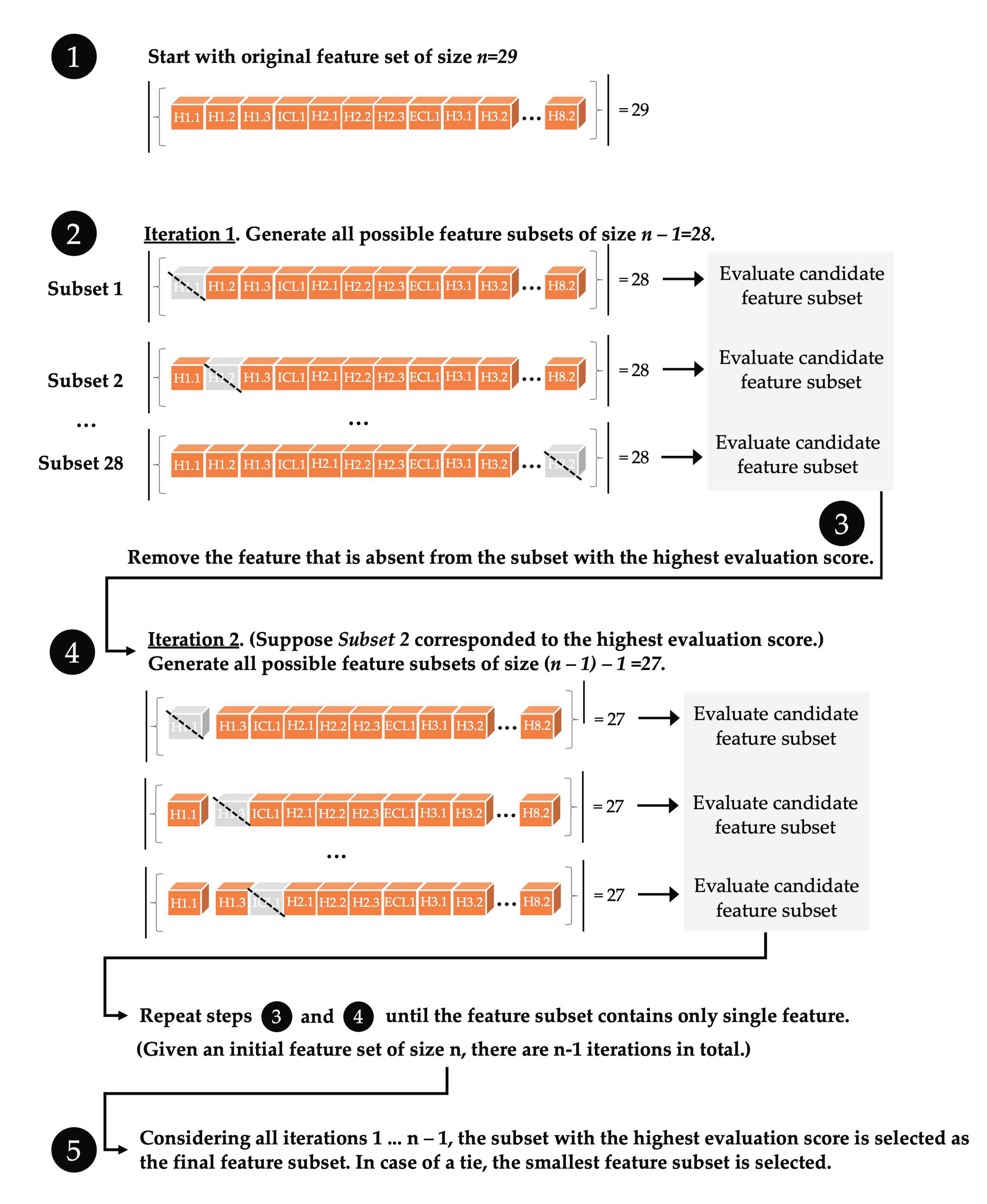
2.4. Machine Learning with ProFlex Features
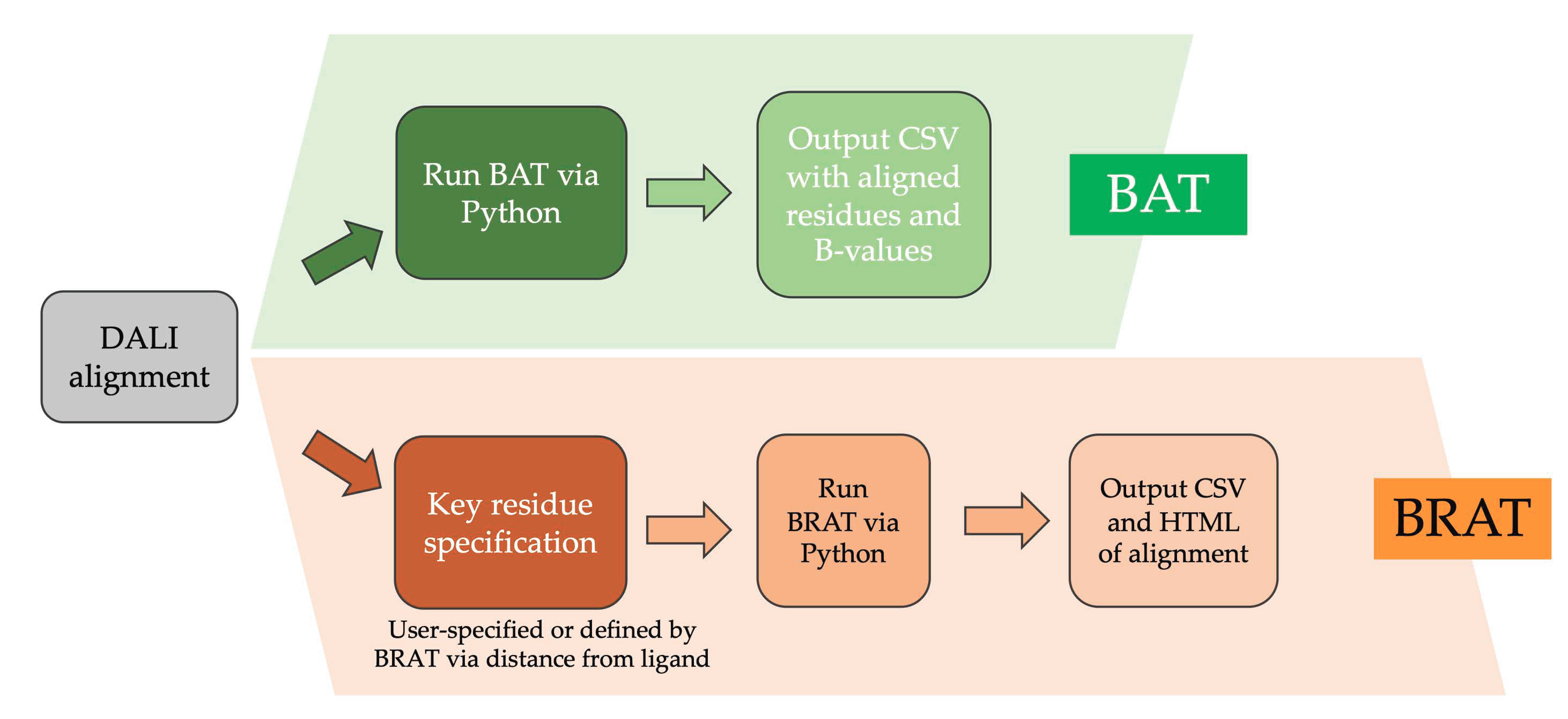
2.5. Comparing GPCR Regions and Numeric Properties with Alignment Visualization Tools
3. Results and Discussion
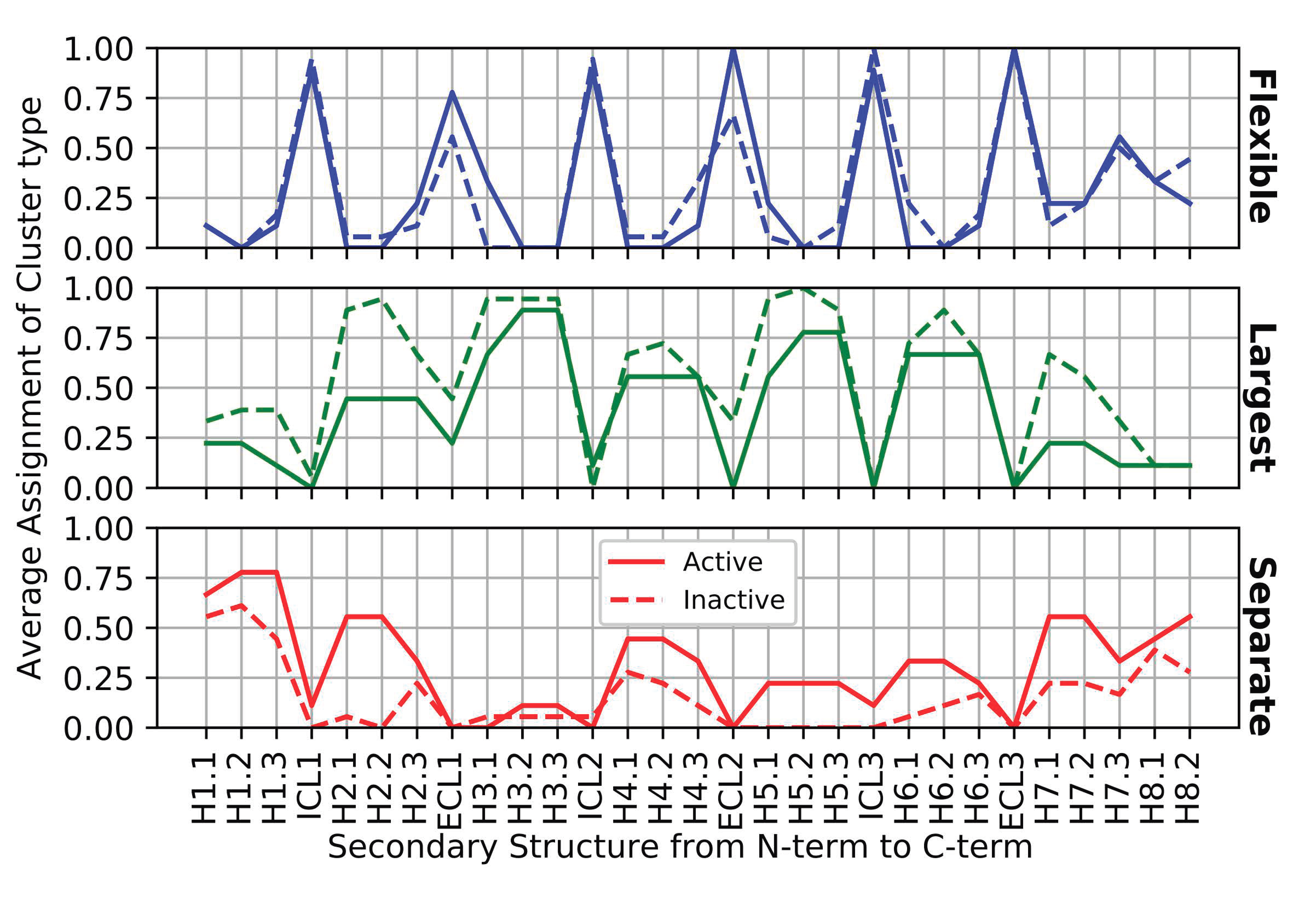
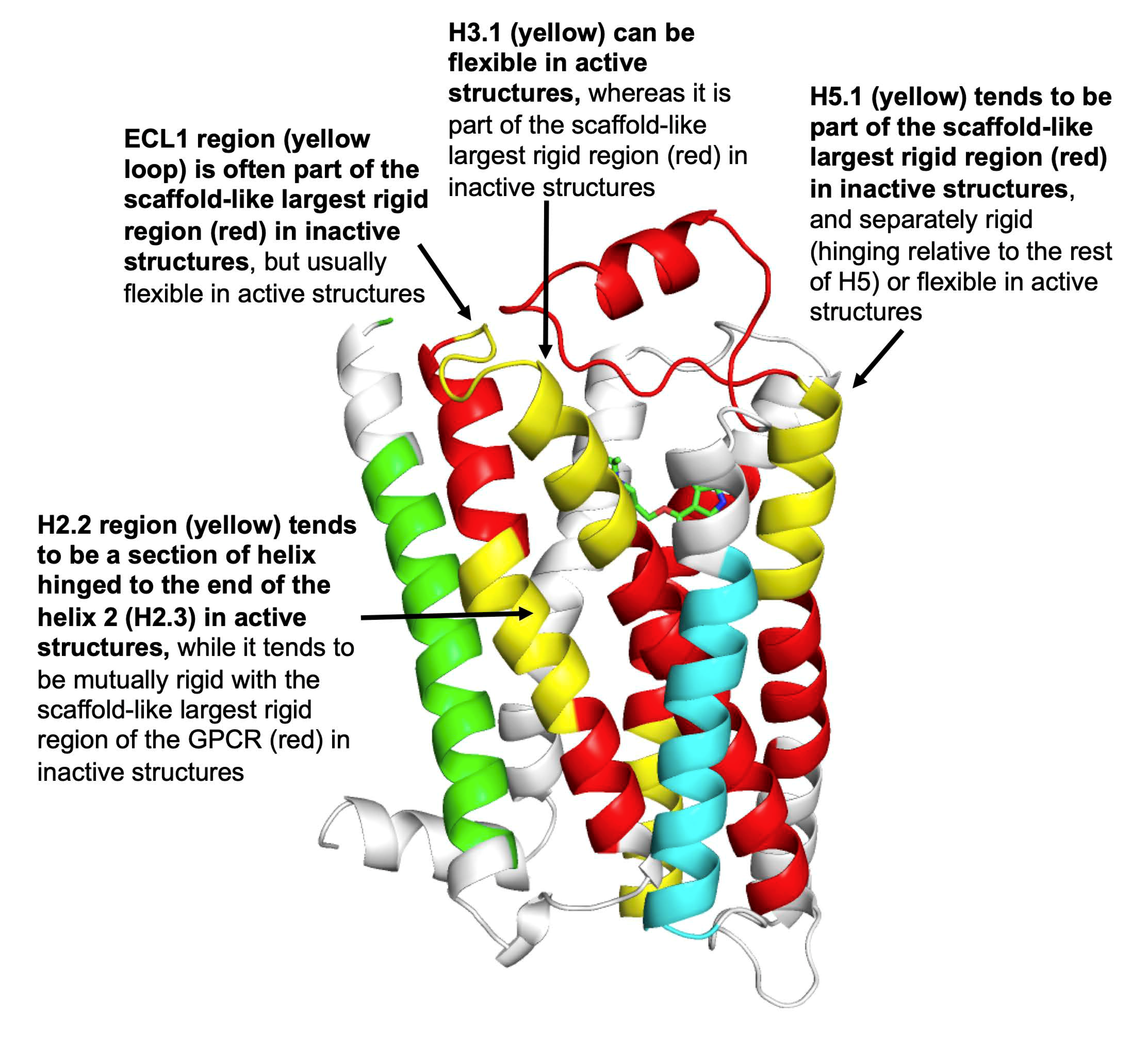
3.1. Identifying Key Flexibility Features for Predicting Activity
3.2. Accurate Classification of GPCR Activity Based on the Flexibility of Key Regions
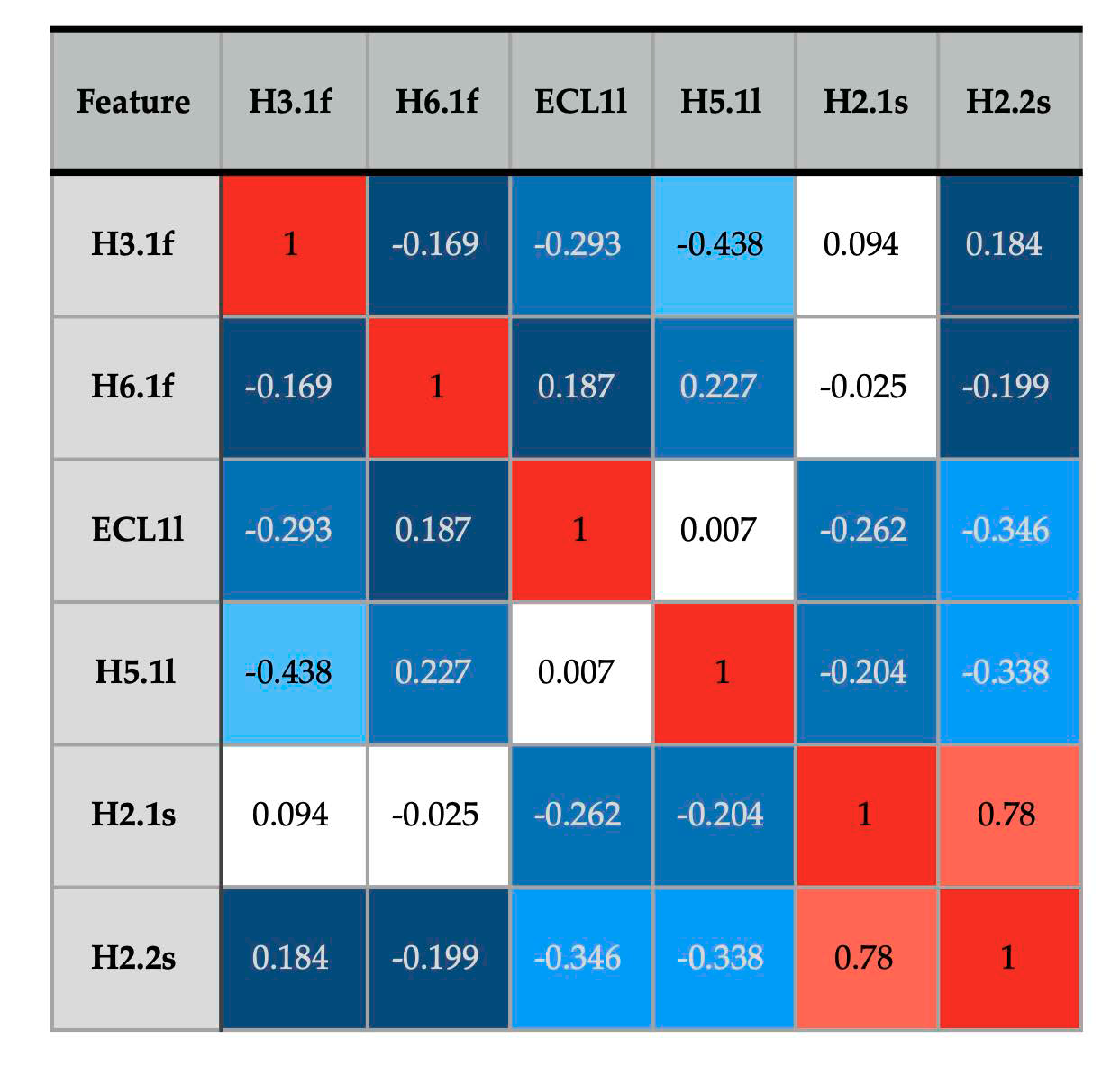
3.3. Patterns of Flexibility and Correlation between Activity-Predicting Features
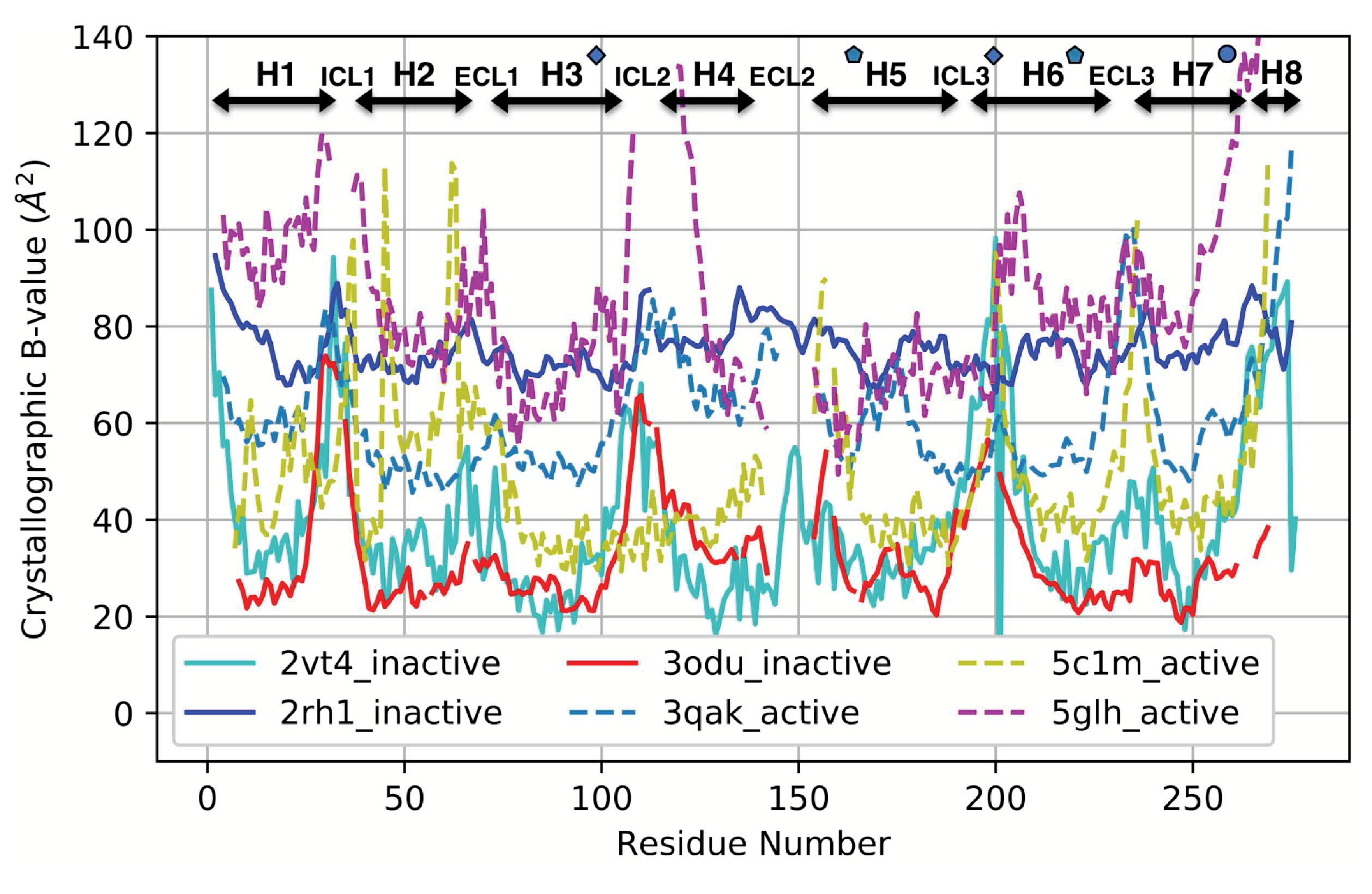
3.4. Comparison with a Crystallographic Measure of Flexibility for Active and Inactive GPCRs
3.5. Using the BRAT and BAT Alignment Visualization Tools to Identify Corresponding Sites and Quantitative Features from Structural Alignments of Sequence-Diverse Homologs
4. Conclusions
- By providing a software approach not previously used to assess protein activity, ProFlex, that predicts rigid and flexible regions and their coupling within a single protein structure. This makes it unnecessary to compare protein structures, which may have a different underlying mechanism of activation. In addition, it is unnecessary to provide user-defined hypotheses regarding regions important for (in)activation. Such hypotheses can bias towards prior knowledge, and limit the understanding of regions involved in activity.
- Additional utilities developed here in Python, BAT and BRAT, facilitate visualizing structurally-equivalent residues in key protein regions of interest, such as binding sites or switch regions, for proteins that are sufficiently divergent that the corresponding residues cannot be defined with high confidence from sequence alignment.
- Although ProFlex can analyze a ligand-bound protein structure as input, in our machine learning approach, no data about the ligand or its contacts are used. Instead, ProFlex pinpoints rigid regions created by constraints within the protein’s covalent, hydrogen bond, and hydrophobic contact network, as well as separate internally rigid regions that can move relative to the protein scaffold region, followed by flexible regions.
- The flexibility and rigidity pattern within a protein structure defined by ProFlex can be used to create a set of features—segments of the protein labeled by their flexible, independently rigid, or mutually rigid state within the structure—that machine learning techniques such as feature selection and a classifier can use to focus down to the most discerning subset of features for predicting activity.
- The resulting KNN classifier of active or inactive state can drive experimental protein and ligand design, by pinpointing specific flexibility features that are more prevalent in active versus inactive structures of the protein. The KNN classifier is also intuitive, since it uses the focused feature set to identify proteins of known activity or inactivity with the most similar features to the user’s protein. This approach can also help group proteins according to similarity in the flexibility of motifs underlying (in)activation.
- The GPCR activity classifier using the identified six flexibility features has high accuracy: 96% correct prediction in leave-one-out cross-validation across the set of 18 inactive and 9 active GPCR structures, and 82% correct prediction when measured on held-out test sets across 10,000 iterations of bootstrap sampling. The most-predictive features colocalize around the ligand binding site proximal to the extracellular surface of the membrane protein, and thus add information to the switch regions characterized by others (such as ionic lock and tyrosine toggle), which are close to the intracellular interface with signaling partners. One of the six flexibility features, the third of helix 5 proximal to the extracellular interface, is adjacent to but non-overlapping with the transmission switch previously defined. Thus, the ProFlex-defined activation motif provides a direct connection between flexibility changes in the protein induced by ligand binding to those previously characterized in the transmission switch involving movements of helices 5 and 6 during activation.
- This approach can help clarify how ligand binding generates an active state in the protein. For instance, one could first use the KNN classifier step in this protocol to identify which GPCRs of known active/inactive state have the most similar flexibility state across the six key regions, relative to the user’s GPCR in complex with a designed or other test ligand. Then, the protein–protein and protein–ligand contacts in the six key regions can be compared between the user’s complex and the most similar GPCR complexes. This analysis can suggest ligand functional group changes (making or breaking specific protein contacts) to enhance the ability to inactivate (or activate) the GPCR.
- This intuitive feature-based classification of activity through machine learning is equally applicable to other protein families and other kinds of data. For instance, instead of ProFlex flexibility, one could test whether a subset of features defined as the presence/absence of specific residue–residue contacts (such as intraprotein hydrogen bonds, salt bridges, aromatic interactions, and/or ligand contacts) predict an active or inactive state. Because the feature selection and classifier can test many more combinations than a person could readily perform by synthesis/mutagenesis, and without bias, new information may result that usefully narrows the spectrum of experiments by homing in on key features of activation.
Author Contributions
Funding
Conflicts of Interest
References
- Hawkins, P.C.D.; Skillman, A.G.; Nicholls, A. Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 2007, 50, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W. Efficient drug lead discovery and optimization. Acc Chem Res. 2009, 42, 724–733. [Google Scholar] [CrossRef]
- Zavodszky, M.I.; Rohatgi, A.; Van Voorst, J.R.; Yan, H.; Kuhn, L.A. Scoring ligand similarity in structure-based virtual screening. J. Mol. Recognit. 2009, 22, 280–292. [Google Scholar] [CrossRef]
- Katritch, V.; Cherezov, V.; Stevens, R.C. Structure-function of the G protein–coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 531–556. [Google Scholar] [CrossRef] [PubMed]
- Kobilka, B.K.; Deupi, X. Conformational complexity of G-protein-coupled receptors. Trends Pharmacol. Sci. 2007, 28, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Venkatakrishnan, A.J.; Deupi, X.; Lebon, G.; Tate, C.G.; Schertler, G.F.; Madan Babu, M. Molecular signatures of G-protein-coupled receptors. Nature 2013, 494, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Latorraca, N.R.; Venkatakrishnan, A.J.; Dror, R.O. GPCR dynamics: Structures in motion. Chem. Rev. 2017, 117, 139–155. [Google Scholar] [CrossRef]
- Jacobs, D.J.; Rader, A.J.; Kuhn, L.A.; Thorpe, M.F. Protein flexibility predictions using graph theory. Proteins Struct. Funct. Genet. 2001, 44, 150–165. [Google Scholar] [CrossRef]
- Maxwell, J.C.L. On the calculation of the equilibrium and stiffness of frames. Philos. Mag. 1864, 27, 294–299. [Google Scholar] [CrossRef]
- Jacobs, D.J.; Hendrickson, B. An algorithm for two-dimensional rigidity percolation: The pebble game. J. Comput. Phys. 1997, 137, 364–365. [Google Scholar] [CrossRef]
- Hespenheide, B.M.; Rader, A.J.; Thorpe, M.F.; Kuhn, L.A. Identifying protein folding cores from the evolution of flexible regions during unfolding. J. Mol. Graph. Model. 2002, 21, 195–207. [Google Scholar] [CrossRef]
- Raschka, S. MLxtend: Providing machine learning and data science utilities and extensions to Python’s scientific computing stack. J. Open Source Softw. 2019, 3, 638. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Trzaskowski, B.; Latek, D.; Yuan, S.; Ghoshdastider, U.; Debinski, A.; Filipek, S. Action of molecular switches in GPCRs—Theoretical and experimental studies. Curr. Med. Chem. 2012, 19, 1090–1109. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Raschka, S. Automated discovery of GPCR ligands. Curr. Opin. Struct. Biol. 2019, 55, 17–24. [Google Scholar] [CrossRef]
- Holm, L.; Laakso, L.M. Dali server update. Nucleic Acids Res. 2016, 44, W351–W355. [Google Scholar] [CrossRef]
- Rose, P.W.; Prlić, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Di Costanzo, L.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB Protein Data Bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar]
- Kuhn, L.A. The Prediction and Characterization of Transmembrane Protein Sequences. Ph.D. Thesis, University of Pennsylvania, Philadelphia, PA, USA, 1989. [Google Scholar]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. A model recognition approach to the prediction of all-helical membrane protein structure and topology. Biochemistry 1994, 33, 3038–3049. [Google Scholar] [CrossRef]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins Struct. Funct. Bioinform. 2009, 77, 114–122. [Google Scholar] [CrossRef]
- Raschka, S.; Bemister-Buffington, J.; Kuhn, L.A. Detecting the native ligand orientation by interfacial rigidity: SiteInterlock. Proteins Struct. Funct. Bioinform. 2016, 84, 1888–1901. [Google Scholar] [CrossRef] [PubMed]
- Tanford, C. The Hydrophobic Effect, 2nd ed.; Wiley/Interscience: New York, NY, USA, 1980. [Google Scholar]
- Ferri, F.J.; Pudil, P.; Hatef, M.; Kittler, J. Comparative study of techniques for large-scale feature selection. Mach. Intell. Pattern Recognit. 1994, 16, 403–413. [Google Scholar]
- Pudil, P.; Novovičová, J.; Kittler, J. Floating search methods in feature selection. Pattern Recognit. Lett. 1994, 15, 1119–1125. [Google Scholar] [CrossRef]
- Park, J.; Scheerer, P.; Hofmann, K.; Choe, H.W.; Ernst, O. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature 2008, 454, 183–187. [Google Scholar] [CrossRef]
- Mathews, B.W. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta-Protein Struct. 1975, 405, 442–451. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB ID | Activity * | Chain | Structure Description | Ligand Name | Organism | Resolution (Å) | R(free) | R(work) |
|---|---|---|---|---|---|---|---|---|
| 2VT4 | 0 | A | Beta1 adrenergic receptor | 4-{[(2s)-3-(Tert-butylamino)-2- hydroxypropyl]oxy}- 3h-indole-2-carbonitrile | Meleagris gallopavo | 2.7 | 0.27 | 0.21 |
| 3ODU | 0 | A | CXCR4 chemokine receptor | (6,6-Dimethyl-5,6-dihydro- imidazo[2,1-b][1,3]thiazol-3-yl)methyl n,n’-dicyclohexylimidothiocarbamate | Homo sapiens | 2.5 | 0.28 | 0.24 |
| 3V2Y | 0 | A | Lyso-phospholipid sphingosine 1-phosphate receptor | {(3r)-3-Amino-4-[(3-hexylphenyl)amino]- 4-oxobutyl}phosphonic acid | Homo sapiens | 2.8 | 0.27 | 0.23 |
| 3VW7 | 0 | A | Human protease-activated receptor 1 (PAR1) | Ethyl [(1r,3ar,4ar,6r,8ar,9s,9as)-9-{(e)- 2-[5-(3-fluorophenyl)pyridin-2-yl]- ethenyl}-1-methyl-3-oxododecahydro-naphtho[2,3-c]furan-6-yl]carbamate | Homo sapiens | 2.2 | 0.24 | 0.22 |
| 3EML | 0 | A | A2A adenosine receptor | 4-{2-[(7-Amino-2-furan-2-yl[1,2,4]tria- zolo[1,5-a][1,3,5]triazin-5-yl)amino]ethyl}phenol | Homo sapiens | 2.6 | 0.23 | 0.20 |
| 2RH1 | 0 | A | Beta2-adrenergic receptor | (2s)-1-(9h-Carbazol-4-yloxy)- 3-(isopropylamino)propan-2-ol | Homo sapiens | 2.4 | 0.23 | 0.20 |
| 1GZM | 0 | A | Bovine rhodopsin | retinal | Bos taurus | 2.6 | 0.24 | 0.20 |
| 4DKL | 0 | A | Mu-opioid receptor | Methyl 4-{[(5beta,6alpha)-17-(cyclopropylmethyl)- 3,14-dihydroxy-4,5-epoxymorphinan- 6-yl]amino}-4-oxobutanoate | Mus musculus | 2.8 | 0.28 | 0.23 |
| 3PBL | 0 | A | Dopamine D3 receptor | Eticlopride | Homo sapiens | 2.9 | 0.27 | 0.24 |
| 4DJH | 0 | A | Kappa opioid receptor | JDTic | Homo sapiens | 2.9 | 0.27 | 0.23 |
| 4MBS | 0 | A | CCR5 chemokine receptor | Maraviroc | Homo sapiens | 2.7 | 0.26 | 0.22 |
| 4S0V | 0 | A | OX2 orexin receptor | Suvorexant | Homo sapiens | 2.5 | 0.24 | 0.20 |
| 4U15 | 0 | A | M3 muscarinic receptor | Tiotropium | Rattus norvegicus | 2.8 | 0.26 | 0.23 |
| 4XNW | 0 | A | Purinergic receptor P2Y1 | MRS2500 | Homo sapiens | 2.7 | 0.27 | 0.22 |
| 4YAY | 0 | A | Angiotensin receptor | ZD7155 | Homo sapiens | 2.9 | 0.27 | 0.23 |
| 4Z35 | 0 | A | Lysophosphatidic acid receptor 1 | ONO-9910539 | Homo sapiens | 2.9 | 0.27 | 0.28 |
| 5CXV | 0 | A | M1 muscarinic acetylcholine receptor | Tiotropium | Homo sapiens | 2.7 | 0.28 | 0.23 |
| 5T1A | 0 | A | CC chemokine receptor 2 (CCR2) | BMS-681 | Homo sapiens | 2.8 | 0.27 | 0.23 |
| 3QAK | 1 | A | A2A adenosine receptor | 6-(2,2-Diphenylethylamino)- 9-[(2r,3r,4s,5s)-5- (ethylcarbamoyl)-3,4-dihydroxy-oxolan-2-yl]- n-[2-[(1-pyridin-2-ylpiperidin- 4-yl)carbamoylamino]ethyl]purine- 2-carboxamide | Homo sapiens | 2.7 | 0.27 | 0.22 |
| 4IAR | 1 | A | 5-HT1b | Ergotamine | Homo sapiens | 2.7 | 0.26 | 0.22 |
| 4PXZ | 1 | A | Purinergic receptor P2Y12 receptor | 2-(Methylsulfanyl)adenosine 5’- (trihydrogen diphosphate) | Homo sapiens | 2.5 | 0.23 | 0.20 |
| 2YDV | 1 | A | A2A receptor | n-Ethyl-5’-carboxamido adenosine | Homo sapiens | 2.6 | 0.26 | 0.23 |
| 3PQR | 1 | A | Metarhodopsin II | retinal | Bos taurus | 2.8 | 0.25 | 0.22 |
| 5C1M | 1 | A | Mu-opioid receptor | (2s,3s,3ar,5ar,6r,11br,11cs)-3a-Methoxy-3,14 -dimethyl-2-phenyl-2,3,3a,6,7,11c-hexahydro- 1h-6,11b-(epiminoethano)- 3,5a-methanonaphtho[2,1-g]indol-10-ol | Mus musculus | 2.1 | 0.22 | 0.19 |
| 4XES | 1 | A | Neurotensin receptor | Neurotensin chain B | Rattus norvegicus | 2.6 | 0.28 | 0.23 |
| 5GLH | 1 | A | Endothelin receptor type B | Endothelin-1 peptide chain B | Homo sapiens | 2.8 | 0.28 | 0.23 |
| 5TVN | 1 | A | 5-HT2b receptor | LSD | Homo sapiens | 2.9 | 0.26 | 0.21 |
| Feature Set | Leave One Out Accuracy | Bootstrap Mean Accuracy | Standard Error |
|---|---|---|---|
| ECL1l, H2.2s, H3.1f, H5.1l | 96.3% | 79.6% | 14.0% |
| ECL1l, H2.1s, H2.2s, H3.1f, H5.1l, H6.1f | 96.3% | 81.7% | 12.6% |
| ECL1l, H2.2l, H2.2s, H3.1f, H5.1l, H6.1f | 96.3% | 81.1% | 12.5% |
| ECL1, H2.1l, H2.2s, H3.1f, H5.1l, H6.1f | 96.3% | 80.8% | 12.5% |
| Dummy classifier: always predicts inactive | 66.6% | 60.4% | 15.5% |
| PDB Entry | Structurally Aligned Residues | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Chain ID, Residue Number | C39 | C40 | D41 | D42 | D43 | D44 | D45 | D46 | D47 | … | |
| 2vt4 | Residue Name | GLN | TRP | GLU | ALA | GLY | MET | SER | LEU | LEU | … |
| Flexibility Index | 87.52 | 65.84 | 70.48 | 55.27 | 56.21 | 45.47 | 40.01 | 35.27 | 41.46 | … | |
| Chain ID, Residue Number | - | A32 | A33 | A34 | A35 | A36 | A37 | A38 | A39 | … | |
| 2rh1 | Residue Name | - | TRP | VAL | VAL | GLY | MET | GLY | ILE | VAL | … |
| Flexibility Index | - | 94.6 | 91.1 | 87.62 | 86.08 | 84.86 | 82.58 | 80.88 | 79.67 | … | |
| Chain ID, Residue Number | - | - | - | - | - | - | - | B43 | B44 | … | |
| 3odu | Residue Name | - | - | - | - | - | - | - | THR | ILE | … |
| Flexibility Index | - | - | - | - | - | - | - | 27.28 | 25.37 | … | |
| Chain ID, Residue Number | - | - | - | A7 | A8 | A9 | A10 | A11 | A12 | … | |
| 3qak | Residue Name | - | - | - | SER | VAL | TYR | ILE | THR | VAL | … |
| Flexibility Index | - | - | - | 69.69 | 67.31 | 61.19 | 59.49 | 60.78 | 58.29 | … | |
| Chain ID, Residue Number | - | - | - | - | - | - | B72 | B73 | B74 | … | |
| 5c1m | Residue Name | - | - | - | - | - | - | ARG | ASP | VAL | … |
| Flexibility Index | - | - | - | - | - | - | 34.11 | 42.58 | 40.51 | … | |
| Chain ID, Residue Number | - | - | - | A102 | A103 | A104 | A105 | A106 | A107 | … | |
| 5glh | Residue Name | - | - | - | TYR | ILE | ASN | THR | VAL | VAL | … |
| Flexibility Index | - | - | - | 3.03 | 92.12 | 99.68 | 98.95 | 1.22 | 94.85 | … | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bemister-Buffington, J.; Wolf, A.J.; Raschka, S.; Kuhn, L.A. Machine Learning to Identify Flexibility Signatures of Class A GPCR Inhibition. Biomolecules 2020, 10, 454. https://doi.org/10.3390/biom10030454
Bemister-Buffington J, Wolf AJ, Raschka S, Kuhn LA. Machine Learning to Identify Flexibility Signatures of Class A GPCR Inhibition. Biomolecules. 2020; 10(3):454. https://doi.org/10.3390/biom10030454
Chicago/Turabian StyleBemister-Buffington, Joseph, Alex J. Wolf, Sebastian Raschka, and Leslie A. Kuhn. 2020. "Machine Learning to Identify Flexibility Signatures of Class A GPCR Inhibition" Biomolecules 10, no. 3: 454. https://doi.org/10.3390/biom10030454
APA StyleBemister-Buffington, J., Wolf, A. J., Raschka, S., & Kuhn, L. A. (2020). Machine Learning to Identify Flexibility Signatures of Class A GPCR Inhibition. Biomolecules, 10(3), 454. https://doi.org/10.3390/biom10030454